

SUMMARY
Astrocytes are proposed to participate in brain energy metabolism by supplying substrates to neurons from their glycogen stores and from glycolysis. However, the molecules involved in metabolic sensing and the molecular pathways responsible for metabolic coupling between different cell types in the brain are not fully understood. Here we show that a recently cloned bicarbonate (HCO3−) sensor, soluble adenylyl cyclase (sAC), is highly expressed in astrocytes and becomes activated in response to HCO3− entry via the electrogenic NaHCO3 cotransporter (NBC). Activated sAC increases intracellular cAMP levels, causing glycogen breakdown, enhanced glycolysis, and the release of lactate into the extracellular space, which is subsequently taken up by neurons for use as an energy substrate. This process is recruited over a broad physiological range of [K+]ext and also during aglycemic episodes, helping to maintain synaptic function. These data reveal a molecular pathway in astrocytes that is responsible for brain metabolic coupling to neurons.
INTRODUCTION
Astrocytes alone make and store glycogen in the mammalian adult brain (Cataldo and Broadwell, 1986). By recruiting this energy store, astrocytes can deliver lactate (and possibly pyruvate) to neurons for fuel, helping maintain axonal and synaptic function (Izumi et al., 1997; Magistretti and Pellerin, 1999; Wender et al., 2000), particularly during brief periods of aglycemia (Wender et al., 2000) or during intense neuronal activation (Brown et al., 2003; Magistretti et al., 1993; Wyss et al., 2011). The importance of astrocyte-to-neuron lactate transport has been demonstrated by the recent report demonstrating that it is required for long-term memory formation in vivo (Suzuki et al., 2011). Although astrocytes can release lactate in response to glutamate uptake (Magistretti, 2006; Magistretti et al., 1999; Wender et al., 2000), here we describe another molecular pathway that leads to glycogen metabolism and lactate efflux as a result of metabolic or neuronal activity.
Soluble adenylyl cyclase (sAC) is sensitive to bicarbonate (HCO3−) and is posited to be a metabolic sensor (Zippin et al., 2001); however, its cellular distribution and function in the brain have not been identified. Due to their relationship to pH, HCO3− and HCO3−-sensitive enzymes represent a potentially effective way by which cells can initiate cellular cascades to meet metabolic demands that are often accompanied by changes in acid/base homeostasis. HCO3−-mediated sAC activation increases the production of the second messenger cAMP (Chen et al., 2000). In astrocytes, high levels of cAMP lead to the breakdown of glycogen (Sorg and Magistretti, 1992) and the production of lactate that can serve as an alternative energy source to neurons. Thus, new enzymes that lead to cAMP generation in astrocytes may be critical for mobilizing metabolic support for neurons during periods of intense neural activity or glucose deprivation.
A well-studied mechanism that increases astrocyte intracellular HCO3− is the electrogenic transport of HCO3− in response to small elevations in extracellular K+ ([K+]ext) caused by local neural activity (Chesler, 1990; Ransom, 1992). This transport occurs via the NaHCO3 cotransporter (NBC, SLC4a4) (Bevensee et al., 2000; Boyarsky et al., 1993; Pappas and Ransom, 1994; Schmitt et al., 2000), a protein that is highly expressed in astrocytes (Cahoy et al., 2008). In addition, astrocytes also express other HCO3−-relevant enzymes such as carbonic anhydrase (Cahoy et al., 2008). We reasoned that HCO3−-sensitive sAC, if present in astrocytes, could provide an important link for coupling neuronal activity to the metabolic protection provided by the breakdown of glycogen and subsequent release of lactate from astrocytes.
Here we show that in the brain, HCO3−-sensitive sAC is highly expressed in astrocytes. HCO3− activation of this enzyme, by either high [K+]ext or aglycemia, increases intracellular cAMP, which leads to glycogen breakdown and the delivery of lactate to neurons for use as an energy substrate.
RESULTS
Astrocytes Express Bicarbonate-Sensitive Soluble Adenylyl Cyclase
We used several approaches to determine whether HCO3−-sensitive sAC is expressed in the brain and, if so, in which cell types it resides. Immunohistochemical staining showed that GFAP-labeled astrocyte somata and major processes, including endfeet, expressed sAC (using R21, anti-sAC monoclonal antibody) (Figure 1A, top), whereas MAP-2-labeled neuronal somata and dendrites revealed no specific sAC staining (Figure 1B). As a control for the specificity of labeling, immunohistochemical staining using R21 in the presence of a sAC blocking peptide that corresponds to the epitope identified by R21 (Hallows et al., 2009) showed no sAC labeling in rat brain slices (Figure 1A, bottom). Western blotting (with R21 antibody) results confirmed that sAC protein was expressed in both rat brain slices and cultured astrocytes (Figure 1C) and, in the presence of sAC-blocking peptide, antigen-antibody interaction was disrupted (Figure 1C).
Figure 1. Expression of Bicarbonate-Responsive Soluble Adenylyl Cyclase in Astrocytes In Situ and In Vitro.

(A and B) Immunohistochemical staining shows that GFAP-labeled astrocyte somata and major processes, including endfeet, expressed sAC (using R21, anti-sAC monoclonal antibody) (A), whereas MAP-2-labeled neuronal somata and dendrites revealed no specific sAC staining (B).
(C) Western blots using sAC antibody (R21) show that sAC protein is found in both rat brain slices and cultured astrocytes. Blocking peptide disrupts antigen-antibody interaction between R21 and sAC.
(D) RT-PCR results showing sAC mRNA is expressed in rat brain slices and cultured astrocytes.
(E) sAC-immunoreactivity is found in glial processes in the stratum radiatum of the hippocampal CA1 region. Representative electron micrographs from a male wild-type mouse (left) demonstrate sAC-immunoperoxidase labeling in astrocytes (yellow outline) identified by glial filaments (gf) and other glial processes (GP), but not in a male Sacytm1Lex/Sacytm1Lex mouse (right; yellow highlight, unlabeled GP). S, synapse.
(F) Quantification of the number of glial profiles (n = 3 mice per condition, ***p < 0.001). Error bars indicate SEM.
RT-PCR results confirmed that sAC mRNA was expressed in both rat brain slices and cultured astrocytes (Figure 1D). Several splice variants of sAC have been reported in different tissues (Farrell et al., 2008). Using further RT-PCR experiments with cultured astrocytes, we demonstrated that astrocytes expressed all the different reported splice variants of sAC. These include sAC, which is encoded by exons 1–5 (see Figure S1A available online), sACsomatic, which has a unique start site up-stream of exons 5–13 (Farrell et al., 2008) (Figure S1B), sACfl, which is encoded by all 32 of the known exons (Buck et al., 1999; Jaiswal and Conti, 2001) (indicated by the top band in Figure S1C), and sACt, which is encoded by exons 9–13 but skips exon 12, resulting in an early stop codon (indicated by the bottom band in Figure S1C).
Finally, we used immunoelectron microscopy to examine the distribution of sAC in the hippocampus region of wild-type and Sacytm1Lex/Sacytm1Lex (genetic deletion of the exon 2 through exon 4 catalytic domain, sAC-C1 knockout [KO]) mice (Esposito et al., 2004; Hess et al., 2005) to further verify subcellular expression of sAC protein in the brain. In the stratum radiatum of the hippocampus, sAC immuneperoxidase labeling was observed in glial processes from wild-type (WT) mice, but not in male Sacytm1Lex/Sacytm1Lex mice (Figure 1E). The number of glial processes stained with R21 antibody was significantly reduced in sAC-C1 KO animals compared to wild-type animals (WT: 191.0 ± 18.0/411 μm2 versus sAC-C1 KO: 6.9 ± 4.6/411 μm2). The quantification is shown in Figure 1F. These data show that astrocytes, as opposed to neurons, are the predominant site for sAC expression in the hippocampus.
sAC Increases cAMP in Cultured Astrocytes
Because of their selective permeability to K+, astrocytes are exquisitely sensitive to the changes in [K+]ext, which occur as a result of changes in neuronal depolarization generated by synaptic activity and neuronal spiking. Physiological increases in [K+]ext of only a few millimolar cause astrocyte depolarization and permit HCO3− entry through the electrogenic NBC, resulting in intracellular alkalinization (Pappas and Ransom, 1994). If increases in [K+]ext activate sAC via HCO3− influx, we predict that there should be a corresponding increase in cAMP that would be inhibited by DIDS, a blocker of NBC. Therefore, we examined the effect of elevated [K+]ext on the production of cAMP in cultured astrocytes expressing a cAMP sensor (GFPnd-EPAC(dDEP)-mCherry) (van der Krogt et al., 2008) using Försters resonance energy transfer (FRET) confocal imaging (green fluorescent protein [GFP] donor/mCherry acceptor) (Figure S2). Elevating [K+]ext from 2.5 mM to 5 or 10 mM progressively increased the cAMP sensor FRET ratio, indicating a rise in intracellular cAMP (control: 0.32% ± 0.27%, n = 13; 5 mM K+: 9.60% ± 1.06%, n = 11, p < 0.001; 10 mM K+: 18.70% ± 1.12%, n = 9, p < 0.001; Figures 2A–2C). Several lines of experiments confirmed that this rise in cAMP was due to sAC activation by HCO3− entry. The increase in the cAMP sensor FRET ratio normally observed in high [K+]ext was significantly inhibited by the sAC-selective inhibitor 2-hydroxyestrone (2-OH, 20 μM) (Hess et al., 2005; Schmid et al., 2007; Steegborn et al., 2005) (3.82% ± 1.09%, n = 13, p < 0.001; Figures 2A–2C) and was prevented by inhibiting the electrogenic NBC with DIDS (450 μM) (0.71% ± 0.60%, n = 9, p < 0.001; Figures 2B and 2C). Furthermore, the cAMP sensor FRET ratio increased when the external solution was changed from HCO3−-free (replaced with HEPES buffered) to one containing HCO3−, which should increase sAC activity (6.51% ± 1.79%, n = 13, p < 0.001; Figure 2C). As a control for our FRET-cAMP measurement and to provide a comparison with other stimulators of cAMP synthesis, we measured the cAMP sensor FRET ratio when we increased cAMP via sAC-independent pathways by directly stimulating transmembrane adenylyl cyclases (tmACs) with forskolin (25 μM) (31.3% ± 1.8% increase in the cAMP sensor FRET ratio, n = 5; Figure 2C) or the beta-adrenergic agonist isoproterenol (100 μM) (Figure S3). The isoproterenol-induced increase in the cAMP sensor FRET ratio was blocked by the selective tmAC antagonist 2′, 5′-dideoxyadenosine (DDA, 50 μM) (Figure S3), confirming that these cAMP changes are due to tmAC activity. Together, these data demonstrate that K+-induced HCO3− entry through NBC activates sAC and leads to the generation of physiologically significant levels of cAMP in cultured astrocytes.
Figure 2. Activation of sAC Increases cAMP Concentration in Astrocytes In Vitro Detected by a FRET Sensor.
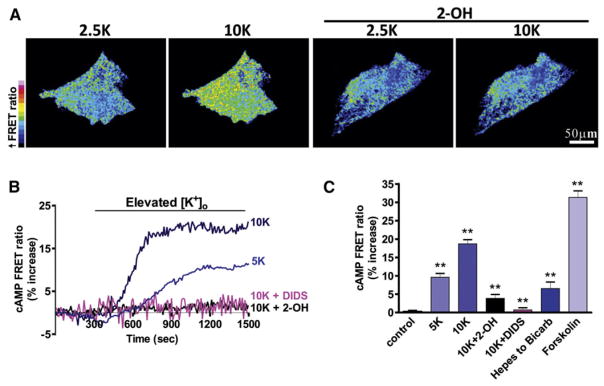
(A) Pseudocolored cultured astrocytes expressing a cAMP FRET sensor. High [K+]ext increased the cAMP concentration (increased FRET ratio), which was blocked by the sAC-selective inhibitor 2-OH (20 μM).
(B) Elevated [K+]ext (to 5 or 10 mM) increased FRET ratios over time, indicating increased intracellular cAMP levels, which were blocked by 2-OH or the NBC inhibitor DIDS (450 μM).
(C) Summary data showing the increase of the FRET ratio either by changing the external solution from HCO3−-free (with HEPES buffered) to one containing HCO3− (with regular aCSF solution) or by adding forskolin (25 μM) to stimulate tmACs. Error bars indicate SEM.
sAC Increases cAMP in Astrocytes in Brain Slices
We examined whether HCO3−-sensitive sAC was functionally active in astrocytes in brain slices by directly measuring the sAC-dependent production of cAMP using ELISA. We first used two-photon microscopy to image the pH-sensitive dye 2′,7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein (BCECF)/AM to confirm previous reports that high [K+]ext causes widespread astrocyte alkalinization by HCO3− entry (Bevensee et al., 2000; Boyarsky et al., 1993; Pappas and Ransom, 1994; Schmitt et al., 2000) (Figure S4). To provide definitive evidence that the high K+-induced increase in cAMP in the brain was due to activation of sAC, we compared cAMP responses between wild-type and sAC-C1 KO mice. The cAMP levels were significantly increased by raising [K+]ext to 10 mM only in brain slices from wild-type mice (2.5 K+: 6.03 ± 0.26 pmol/ml, n = 7; 10 mM K+: 8.94 ± 0.29 pmol/ml, n = 7, p < 0.001; Figure 3A); in brain slices from KO mice, there was no change in cAMP when [K+]ext was raised to 10 mM (2.5 K+: 6.21 ± 0.44 pmol/ml, n = 7; 10 mM K+: 6.03 ± 0.59 pmol/ml, n = 7, p > 0.05; Figure 3A). Next, we examined whether the increase in cAMP in high [K+]ext required HCO3− by comparing the increase when NaHCO3 was removed and brain slices were maintained in a HEPES buffer. In control rat brain slices, raising [K+]ext to 10 mM for 20 min significantly increased the cAMP level (2.5 mM K+: 4.3 ± 0.5 pmol/ml, n = 4; 10 mM K+: 7.5 ± 0.2 pmol/ml, n = 4, p < 0.001; Figure 3B). Similar to our observations in cultured astrocytes, this increase in cAMP was dependent upon extracellular HCO3− and was not observed in matched brain slices in HEPES (2.5 K+: 4.4 ± 0.4 pmol/ml, n = 4; 10 K+: 4.5 ± 0.2 pmol/ml, n = 4, p > 0.05; Figure 3B). The high K+-induced increase in cAMP was significantly reduced by the sAC-specific inhibitors 2-OH (4.6 ± 0.4 pmol/ml, n = 5, p < 0.001; Figure 3C) and KH7 (10 μM) (Hess et al., 2005) (4.5 ± 0.6 pmol/ml, n = 5, p < 0.001; Figure 3C) but not by the tmAC inhibitor DDA (9.2 ± 0.6 pmol/ml, n = 5, p > 0.05; Figure 3C). As a negative control for 2-OH, we also determined that 17β-estradiol, an estrogen parent compound that is ineffective on sAC (Hallows et al., 2009), did not reduce the high K+-induced increase in cAMP (17β-estradiol, 20 μM, 9.1 ± 1.3 pmol/ml, n = 5, p > 0.05; Figure 3C). Furthermore, 2-OH had no effect on cAMP production mediated by the activation of beta-adrenoceptors using isoproterenol (100 μM) or norepinephrine (NE, 10 μM) (Figure 3D), receptors that signal via tmACs, confirming that under these conditions, 2-OH is specific for sAC. Very high levels of intracellular [Ca2+] could activate sAC, but the EC50 of calcium for sAC activation is 750 μM (Litvin et al., 2003), well above normal elevations in [Ca2+]i used for cell signaling. We observed that there were no calcium signals detected in astrocytes when 10 mM K+ was bath applied (Figure S5), ruling out [Ca2+]i as the trigger in these experiments. These data indicate that functional sAC protein, which is expressed in astrocytes in this region of the brain, produces cAMP when HCO3− entry is triggered by high [K+]ext.
Figure 3. Activation of sAC Increases cAMP Concentration In Situ.
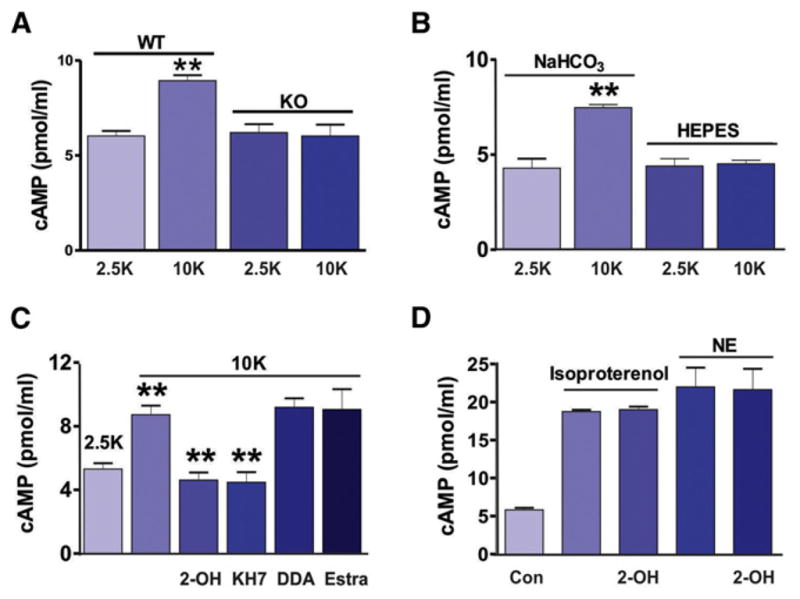
(A) Raising [K+]ext to 10 mM significantly increased the cAMP level in brain slices from the wild-type mice but had no effect in brain slices from sAC KO mice.
(B) ELISA showed high [K+]ext increased [cAMP] in rat brain slices only in the presence of HCO3−.
(C) ELISA demonstrating the increase of cAMP in high [K+]ext was reduced by sAC inhibitors, 2-OH (20 μM), or KH7 (10 μM) but not by the tmAC inhibitor DDA (50 μM). An inert estrogen parent compound, 17β-estradiol (a negative control for 2-OH), had no effect on the high K+-induced increase in cAMP.
(D) ELISA showed 2-OH has no effect on cAMP production by the activation of beta-adrenoceptors using isoproterenol (100 μM) or norepinephrine (NE, 10 μM).
sAC Induces Glycogenolysis and Lactate Production
Glycogen in the brain is only stored in astrocytes (Brown, 2004; Brown et al., 2005; Magistretti, 2006) and some neurotransmitters such as vasoactive intestinal peptide, noradrenaline, and adenosine promote astrocytic glycogenolysis in the brain (Sorg and Magistretti, 1991). In addition, glycogenolysis in brain tissue was previously reported to be promoted by high [K+]ext (Hof et al., 1988) through an unknown mechanism. Because astrocytes do not express the enzyme glucose-6-phosphatase (Brown and Ransom, 2007; Dringen and Hamprecht, 1993; Magistretti et al., 1993), they cannot generate free glucose from glycogen; therefore, in astrocytes, glycogen breakdown induced by increased cAMP (Pellerin et al., 2007) results in pyruvate, followed by lactate. We tested the hypothesis that sAC was responsible for coupling K+ increases to glycogen breakdown in astrocytes and for the production and release of lactate. Raising [K+]ext to 10 mM for 30 min significantly reduced cellular glycogen levels (26.7% ± 6.5%, n = 6, p < 0.001; Figure 4A) compared to control condition (2.5 mM K+: 100%, n = 6). This effect was significantly inhibited by the sAC inhibitor 2-OH (90.5% ± 10.6%, n = 6, p < 0.001; Figure 4A) but not by the tmAC antagonist DDA (32.7% ± 7.5%, n = 5, p > 0.05; Figure 4A). Superfusate measurements of lactate release revealed that brain slices exposed to high [K+]ext showed elevated lactate levels (2.5 mM K+: 30.7 ± 3.1 μM, n = 7; 10 mM K+: 69.0 ± 5.2 μM, n = 6, p < 0.001; Figure 4B), which were blocked by 2-OH (32.1 ± 3.6 μM, n = 6, p < 0.001) and KH7 (26.7 ± 7.2 μM, n = 4, p < 0.001; Figure 4B) but not by DDA (61.3 ± 9.6 μM, n = 6, p > 0.05; Figure 4B). Furthermore, the increase in lactate by high [K+]ext was dose dependent with applications of 2.5, 5, 7.5, and 10 mM K+ (Figure 4C).
Figure 4. High [K+]ext Induces Glycogen Breakdown and Increased Lactate Production via sAC Activation.

(A) High [K+]ext stimulated glycogen breakdown in brain slices, which was inhibited by the sAC inhibitor 2-OH (20 μM) but not by the tmAC inhibitor DDA (50 μM).
(B) High [K+]ext increased lactate release from brain slices, which was blocked by KH7 (10 μM) and 2-OH but not DDA.
(C) Lactate release from slices in response to different [K+]ext showed dose dependency.
(D) Direct measurements using a lactate enzyme-based electrode showed the rapid time course of lactate release from brain slices. A transient increase of lactate was induced by 5 mM [K+]ext and the addition of 10 mM [K+]ext led to a further augmentation. High [K+]ext increased the cytosolic astrocyte NADH signal.
(E) Hippocampal arteriole and astrocytes showing NADH (top) and colocalization with the astrocyte marker SR-101 (bottom).
(F) NADH fluorescence changes in response to high [K+]ext in four astrocytes (from E).
(G) Summary of NADH fluorescence changes in response to high [K+]ext (top) and block with 2-OH (bottom). Application of 10 mM [K+]ext increased the NADH signal (top), which was reduced by sAC inhibition with 2-OH (bottom).
We verified and extended these findings by taking direct measurements of the time course of lactate release from brain slices using a lactate enzyme-based electrode. An immediate and transient increase of lactate was induced by 5 mM [K+]ext and subsequent addition of 10 mM [K+]ext led to a further augmentation, demonstrating dose dependency and rapid efflux of lactate when [K+]ext changes (n = 3; Figure 4D). Finally, we confirmed the role of glycolysis in the production of lactate from glycogen using the glycolytic inhibitor iodoacetate (IA, 200 μM) and the lactate dehydrogenase (LDH) inhibitor oxamate (2.5 mM) (Gordon et al., 2008; Pellerin and Magistretti, 2004; Takano et al., 2007). In astrocytes, LDH converts pyruvate and NADH to lactate and NAD+ (Pellerin and Magistretti, 2004; Takano et al., 2007). In high [K+]ext, we observed significantly less lactate in the presence of IA (29.8 ± 4.3 μM, n = 5, p < 0.001) or oxamate (39.0 ± 5.1 μM, n = 5, p < 0.001; Figure S6) compared to 10 mM K+ alone. These data show that sAC is a critical enzyme linking elevations in [K+]ext to glycogenolysis and lactate production in astrocytes.
sAC Increases Astrocytic Glycolysis
The glycolytic metabolism that follows glycogenolysis generates NADH in the step in which pyruvate is formed. NADH is an endogenous electron carrier with fluorescent properties that allow relative changes in metabolic processes to be visualized. Two-photon excitation of NADH provides a sensitive, subcellular measure of both oxidative metabolism (punctate NADH fluorescence from mitochondria) and glycolytic metabolism (diffuse NADH fluorescence from the cytosol) in situ (Gordon et al., 2008; Kasischke et al., 2004). We examined whether an elevation in [K+]ext that stimulated glycogen breakdown and glycolysis within astrocytes would transiently increase NADH and be apparent as an increase in cytosolic NADH fluorescence. The increase in NADH would probably be transient as NADH is in turn converted to nonfluorescent NAD+ when pyruvate is converted to lactate. We observed, as previously reported (Gordon et al., 2008; Kasischke et al., 2004), that astrocytes showed bright, intracellularly diffuse NADH fluorescence in the soma and endfeet (Figure 4E, top). Figure 4E (bottom) shows colocalization of NADH fluorescence with the astrocyte marker SR-101. NADH fluorescence changes were observed in response to high [K+]ext in four astrocytes (same astrocytes as in bottom panel of Figure 4E) (Figure 4F). Application of 10 mM [K+]ext transiently increased the cytosolic astrocyte NADH signal (118.1% ± 3.4%, 28 cells; Figure 4G, top), which was reduced by sAC inhibition with 2-OH (102.3% ± 2.9%, 18 cells; Figure 4G, bottom; p < 0.0001). These data show that high [K+]ext initiates a sAC-mediated metabolic process within the cytosol of astrocytes that is indicative of glycogen breakdown and subsequent glycolysis.
sAC Provides an Energy Substrate during Glucose Deprivation
Astrocyte-derived lactate can be delivered to neurons for use as an alternative energy substrate (Pellerin and Magistretti, 1994). Lactate leaves astrocytes via monocarboxylate transporter subtypes 1 and 4 (MCT1 and MCT4) and enters neurons via MCT2 (Debernardi et al., 2003; Pierre et al., 2002). To test the hypothesis that neurons take up the extracellular lactate released as a consequence of high [K+]ext and thus sAC activation, we utilized α-cyano-4-hydroxycinnamate (4-CIN), an MCT inhibitor that is effective at concentrations under 250 μM in selectively blocking neuronal uptake of exogenous or endogenous lactate in rat hippocampal slices (Erlichman et al., 2008; Izumi and Zorumski, 2009; Schurr et al., 1999). At higher concentrations such as 2 mM, 4-CIN has additional effects by inhibiting pyruvate uptake into mitochondria (Halestrap and Armston, 1984; Izumi et al., 1997; Izumi and Zorumski, 2009). In brain slices treated with 4-CIN (100 μM), application of 10 mM [K+]ext significantly increased extracellular lactate (81.0 ± 6.4 μM, n = 5; + 4-CIN: 121.7 ± 7.0 μM, n = 7 p < 0.001; Figure 5A), suggesting that when neuronal lactate uptake is inhibited by 4-CIN, more lactate is free to diffuse out of the brain slice into the superfusate.
Figure 5. sAC-Dependent Glycogen Breakdown and Lactate Delivery from Astrocytes to Neurons during Hypoglycemia.
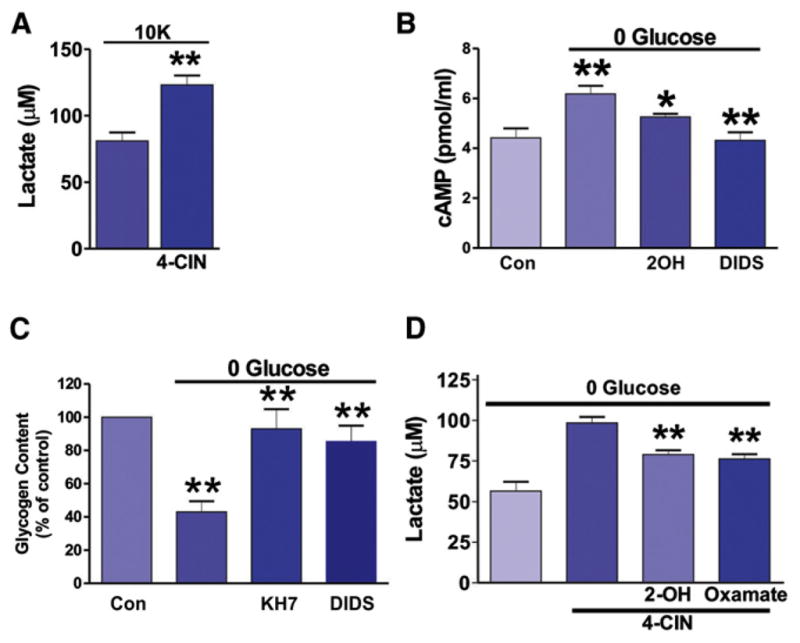
(A) Inhibition of neuronal uptake of lactate with 4-CIN increased extracellular lactate from brain slices in high [K+]ext.
(B) Removing glucose from the aCSF for 15 min increased cAMP levels, which were inhibited by 2-OH (20 μM) and DIDS (450 μM).
(C) Depletion of extracellular glucose significantly decreased glycogen content in rat brain slices and this was significantly inhibited by KH7 and DIDS.
(D) 4-CIN in the absence of glucose increased extracellular lactate compared to glucose deprivation alone, which was inhibited by 2-OH (20 μM) and oxamate (2.5 mM).
Previous studies have demonstrated that when extracellular glucose levels are reduced, lactate is produced by astrocytes and provided to neurons to promote neuronal viability (Aubert et al., 2005; Izumi et al., 1997; Wender et al., 2000). Furthermore, aglycemia is associated with alkalinization (Bengtsson et al., 1990; Brown et al., 2001), which could subsequently activate sAC. Therefore, we tested the hypothesis that aglycemia recruits sAC to initiate the astrocyte-neuron lactate shuttle. We first examined whether aglycemic condition induced astrocyte alkalinization. We used two-photon laser scanning microscopy to image the pH-sensitive dye BCECF/AM to monitor astrocytic intracellular pH change in aglycemic condition. We found that applying aglycemic solution induced a gradual alkalinization of intracellular pH in astrocytes (Figure S7). Next, we examined the effect of aglycemia in brain slices on the production of cAMP. We detected increased cAMP in slices when exposed to aglycemic aCSF (control; 10 mM glucose: 4.4 ± 0.4 pmol/ml, n = 6; 0 glucose: 6.2 ± 0.3 pmol/ml, n = 7, p < 0.01; Figure 5B) and this increase was significantly inhibited by 2-OH (5.3 ± 0.1 pmol/ml, n = 7, p < 0.05; Figure 5B) and DIDS (4.3% ± 0.3%, n = 7, p < 0.01; Figure 5B), indicating bicarbonate-sensitive sAC is activated by glucose-free condition.
We further tested the hypothesis that sAC was responsible for coupling aglycemia to glycogen breakdown in astrocytes and for the production and release of lactate. Depleting extracellular glucose for 30 min significantly reduced glycogen levels in brain slices (control: 100%, n = 11; 0 glucose: 43.0% ± 6.6%, n = 12, p < 0.01; Figure 5C). This effect was prevented by sAC inhibition with KH7 (85.5% ± 9.4%, n = 7, p < 0.01; Figure 5C) and NBC antagonist DIDS (93.1% ± 11.8%, n = 7, p < 0.01; Figure 5C). Treating with 4-CIN in the absence of glucose significantly increased extracellular lactate (98.5 ± 3.6 μM, n = 4) compared to glucose deprivation alone (56.5 ± 5.7 μM, n = 4, p < 0.001; Figure 5D), an effect that was partially inhibited by 2-OH (79.0 ± 2.6 μM, n = 6, p < 0.001; Figure 5D) or oxamate (76.3 ± 2.9 μM, n = 3, p < 0.001; Figure 5D), suggesting sAC and LDH involvement, respectively.
To further explore whether this sAC-dependent lactate shuttle has functional consequences to the maintenance of neuronal activity when the supply of glucose is compromised, we recorded field excitatory postsynaptic potentials (fEPSPs) in the stratum radiatum of the CA1 region during aglycemia in the presence or absence of 2-OH. Basal fEPSPs were not effected by 2-OH alone (Figure 6A). Glucose deprivation slowly depressed fEPSP amplitude (T1/2 [time to half amplitude] = 31.1 ± 3.3 min; Figure 6A, open circles) (Fowler, 1993; Schurr et al., 1988). In 2-OH, glucose deprivation caused a significantly faster decline of fEPSPs (T1/2 = 15.0 ± 1.4 min, p < 0.001; Figure 6A, filled circles), with poorer recovery compared to glucose deprivation alone. fEPSP traces from corresponding time points are shown in Figure 6B. The depression of fEPSP by inhibiting sAC with 2-OH was rescued by the addition of exogenous lactate (5 mM) (T1/2 = 23.4 ± 1.9 min; + 2-OH: T1/2 = 22.1 ± 2.3 min, p > 0.05; Figure 6C). fEPSP traces from corresponding time points are shown in Figure 6D. These data suggest that aglycemia stimulates the sAC-dependent breakdown of glycogen in astrocytes, leading to the generation and release of lactate to provide an energy substrate for neurons to maintain synaptic function.
Figure 6. sAC Activation during Hypogly-cemia Protects Synaptic Function by Lactate Release.

(A) Glucose deprivation (open circle) decreased fEPSP amplitude, which recovered when glucose was reintroduced. fEPSP amplitude declined more rapidly to a greater extent and did not recover as well when sAC was inhibited with 2-OH (filled circle).
(B) fEPSP traces from corresponding time points in (A).
(C) Glucose deprivation (open circle) in the presence of exogenous lactate (5 mM). The adverse effect of 2-OH (filled circle) on fEPSP decline and recovery in hypoglycemia was no longer observed in the presence of lactate (5 mM).
(D) fEPSP traces from corresponding time points in (C). Error bars indicate SEM.
DISCUSSION
Here we report a mechanism in brain metabolic coupling in which astrocytes respond to an increase in [K+]ext or aglycemia by the activation of HCO3−-sensitive sAC, an enzyme that is abundantly expressed in these cells within the brain. sAC activation leads to an increase in intracellular cAMP, which, in turn, triggers glycogen breakdown within astrocytes and the subsequent generation and release of lactate so that neurons are provided with additional energy substrates (see Figure 7 diagram). Our data show that this mechanism is recruited to help meet energy demand and maintain synaptic operation during moderate K+ challenges and during drastic reduction in levels of glucose, the brain’s most important fuel.
Figure 7. A Summary Diagram Showing High [K+]ext-Mediated Activation of sAC via HCO3− Influx through NBCs and Its Physiological Role.

A diagram showing high [K+]ext-mediated activation of sAC via HCO3− influx through NBCs and its role in increasing intracellular cAMP concentration, initiating glycogenolysis and producing lactate, which can be used as a neuronal fuel source.
With respect to K+ handling, our data show that the ability of astrocytes to respond to small changes in [K+]ext goes beyond the simple maintenance of ionic homeostasis to which astrocytes are prescribed and instead reflects a broader functional significance in the coordination of energy utilization in the brain. K+-mediated HCO3− entry and sAC activation represent an elegant solution of detecting the needs of neurons by virtue of the fact that action potentials and synaptic potentials require new fuel substrates to supply energy production required for Na+/K+ ATPase activity (Alle et al., 2009; Attwell and Laughlin, 2001). Elevation in intracellular free [Ca2+] in astrocytes is unlikely to have a role in K+-triggered metabolic coupling because the EC50 of calcium for sAC activation is 750 μM (Litvin et al., 2003), well above normal elevations in [Ca2+]i used for cell signaling. We did not detect calcium signals in astrocytes when 10 mM K+ was bath applied (Figure S5). In addition, the threshold for [K+]ext to evoke an elevation in [Ca2+]i in astrocytes is approximately 25 mM (Duffy and MacVicar, 1994), much higher than the 5–10 mM used here. In addition to the roles of elevations in [K+]ext in promoting HCO3− entry into astrocytes through NBCs, our work also suggests that NBCs play a role in sAC activation in response to glucose deprivation. Hypoglycemia in the brain is accompanied by an extracellular alkaline pH change (Bengtsson et al., 1990; Brown et al., 2001). Our results suggest that the alkaline shift during aglycemia leads to sAC activation followed by the increased lactate production that we have observed. The sensitivity of the cAMP increase and the glycogen breakdown to DIDS in aglycemia suggest that HCO3− entry via NBCs plays a predominant role in activating sAC during aglycemia as compared to intracellular HCO3− production.
Our data expand upon a body of evidence showing the existence of an astrocyte-neuron lactate shuttle that is initiated by glutamate transport into astrocytes. Glutamate uptake is coupled to Na+, resulting in an intracellular Na+ load and enhanced Na+/K+-ATPase activity. The need for more ATP to drive Na+/K+ pumps increases glycolysis, leading to the production and release of lactate, which is subsequently taken up by neurons for fuel (Magistretti et al., 1999; Pellerin and Magistretti, 1994). The HCO3−-sensitive sAC mechanism described here may work in concert with this original shuttle model, whereby neural activity produces an elevation in extracellular glutamate and K+, both of which then act independently through their respective mechanisms to augment lactate release for neurons. Finally, our results shed light on the importance of the astrocyte store of glycogen as an energy reserve. Previous data have shown that glycogen provides an important alternative energy source during ischemic-like conditions to prolong survival of neurons and integrity of axons (Brown and Ransom, 2007; Wender et al., 2000). Our data add to this concept, suggesting that glycogen stores can be recruited by moderate elevations in [K+]ext as well as more severe aglycemic challenges. Therefore, the unique presence of bicarbonate-responsive sAC in astrocytes and its critical role in controlling lactate levels through glycogenolysis demonstrate that this molecular pathway may be an essential process in the maintenance or optimization of total brain energy metabolism during both physiological and pathophysiological conditions. Targeting this pathway may provide a site of intervention for the treatment of perturbed energy metabolism in the brain.
EXPERIMENTAL PROCEDURES
Hippocampal Slice Preparation
Sprague-Dawley rats (postnatal days 18–28) were anaesthetized with halothane and decapitated according to protocols approved by the University of British Columbia committee on animal care. Brains were rapidly extracted and placed into ice-cold dissection medium containing the following: 87 mM NaCl, 2.5 mM KCl, 2 mM NaH2PO4, 7 mM MgCl2, 25 mM NaHCO3, 0.5 mM CaCl2, 25 mM d-glucose, and 75 mM sucrose saturated with 95% O2/5% CO2. Hippocampal slices (transverse, 400 μm thick) were cut using a vibrating tissue slicer (VT1000S, Leica) and recovered for 1 hr at 24°C in aCSF containing the following: 119 mM NaCl, 2.5 mM KCl, 1.3 mM MgSO4, 26 mM NaHCO3, 2.5 mM CaCl2, and 10 mM d-glucose, and aerated with 95% O2/5% CO2.
Two-Photon Imaging
Images were acquired at depths between 50 and 100 μm into the brain slice in order to avoid unhealthy tissue at more superficial depths. SR-101 and NADH epifluorescence were separated by a dichroic mirror reflecting wavelengths below 510 nm. The NADH signal was collected with an external photomultiplier tube (PMT) detector after passing through a 450 nm (30 nm band pass) emission filter, while SR-101 was collected by a separate PMT after passing through 630 nm (60 nm band pass). The laser power necessary for NADH excitation was ~30 mW (after the objective). To reduce photo damage, we acquired a single NADH image every 30 s, which provided a stable NADH baseline and adequate time resolution for measuring NADH changes in the long-duration high [K+]ext experiments. Continuous scanning was possible during the afferent stimulation experiments as they occurred over a shorter time frame. SR-101 and BCECF epifluorescence were separated by a dichroic mirror reflecting wavelengths below 575 nm. The BCECF signal was collected with an external PMT detector after passing through a 535 nm (30 nm band pass) emission filter, while SR-101 was collected by a separate PMT after passing through a 630 nm (60 nm band pass) emission filter. For aglycemia experiments, which were done at 30°C, a gradual and steady decrease in baseline fluorescence occurred in control solutions due to the efflux of BCECF. We compensated for the steady dye efflux by normalizing signals to the rate of decrease during baseline as previously described (Beierlein et al., 2004; Zhang et al., 2006).
Immunohistochemistry
Free-floating sections (16 μm horizontal sections) were processed for immunostaining as described previously (Ryu and McLarnon, 2009). The primary antibodies used for immunostaining were as follows: anti-microtubule-associated protein-2 (MAP-2, Chemicon, 1:2,000), anti-glial fibrillary acidic protein (GFAP, Sigma, 1:2,000), and anti-soluble adenylyl cyclase (sAC, R21, 1:1,000). Alexa Fluor 543 anti-mouse or Alexa Fluor 488 anti-rabbit IgG (1:1,000) secondary antibodies (Invitrogen) were used for immunofluorescent staining. For immunostaining using R21 antibody, rat hippocampal brain sections were pretreated with 0.1% SDS for 5 min at room temperature to denature the protein. As a negative control experiment, primary antibody was omitted during the immunostaining. For preabsorption of R21 antibody, 2× volume of blocking peptide was added to the aliquot of R21 antibody (200× ratio peptide:ab in a molar basis), then incubated overnight at 4°C with a gentle orbital shaking. Then, the subsequent preabsorbed antibody was used for immunohistochemistry.
Immunoelectron Microscopy
Adult wild-type or Sacytm1Lex/Sacytm1Lex male mice were anesthetized with sodium pentobarbital (150 mg/kg), perfused with 3.75% acrolein and 2% paraformaldehyde in 0.1 M phosphate buffer, and processed for electron microscopy as previously described (Mitterling et al., 2010). For primary antibody incubation, 40-μm-thick free-floating sections containing the dorsal hippocampus were incubated with a monoclonal anti-sAC antibody, R21 (0.6 μg/ml), in 0.1% bovine serum albumin in 0.1 M Tris saline (pH 7.6) for 1 day at room temperature and an additional 3 days at 4°C. The primary antibodies were visualized by the immunoperoxidase method. Sections were analyzed on a Tecnai Biotwin transmission electron microscope (FEI) equipped with an AMT digital camera. Profiles were identified by the morphological criteria as previously described (Peters et al., 1991). For the quantitative analysis, ten random nonoverlapping micrographs (36 μm2 per micrograph) were taken from the tissue-plastic interface of stratum radiatum of the dorsal hippocampus of each animal (n = 3 per condition).
Western Blotting
Cultured rat astrocytes and rat hippocampal brain slices were used for western blotting. Cells and brain slices were homogenized using lysis buffer containing the following: 100 mM Tris (pH 7.0), 2 mM EGTA, 5 mM EDTA, 30 mM NaF, 20 mM sodium pyrophosphate, and 0.5% NP40 with phosphatase and protease inhibitor cocktail (Roche). The homogenates were then centrifuged at 13,000 ×g (20 min, 4°C) to remove cellular debris, and then protein concentrations of the crude lysates were determined by performing a Bradford assay with the DC Protein Assay dye (Bio-Rad). The protein samples were diluted with 1×Laemmli sample buffer and boiled for 5 min. After SDS/PAGE, proteins were transferred to PVDF membranes, blocked in 5% milk for 1 hr at room temperature, rinsed with Tris-buffered saline with 0.1% Tween 20 (TBST) and incubated with mouse anti-sAC monoclonal antibody (R21, 1:2,500) overnight at 4°C. After four washes with TBST, the membranes were incubated with the anti-mouse secondary antibody conjugated to horseradish peroxidase (1:10,000) for 1 hr at room temperature. The membranes were then washed three to four times (15 min) with TBST, and bands were visualized using enhanced chemiluminescence (ECL, Amersham Bioscience).
RT-PCR
Total RNAs were extracted from hippocampal brain slices and cultured astrocytes using TRIzol reagent (GIBCO-BRL) and were subjected to DNase I treatment and complementary DNA synthesis was carried out using M-MLV reverse transcriptase (GIBCO-BRL). Reverse transcriptase was omitted as a negative control. PCR primers (Pastor-Soler et al., 2003) are all intron spanning and sequences and expected product sizes are as follows: sAC sense 5′-CATGAGTAAGGAATGGTGGTACTC-3′; antisense 5′-AGGGTTACGTTGC CTGATACAATT-3′ (110 bp); β-actin sense 5′-GTGGGGCGCCCCAGGCA CCA-3′ and antisense 5′-GTCCTTAATGTCACGCACGATTTC-3′ (526 bp). Primers used to amplify sAC splice variants were as follows: sAC; i.e., from exons 1 to 5: sense 5′-ATGAGTGCCCGAAGGCAGGAATTACAG-3′ antisense 5′-TGCTCTCTGATCCG GAATCCT-3′; sACt from sACfl splice variants; i.e., from exons 9 to 13: sense 5′-TGCAAACCCACTGCTTGCTTGC-3′ antisense 5′-ACTCGGCTGCAGTTCGTCA T-3′; sACsomatic, which starts at the alternate promoter upstream from exon 5; i.e., upstream 5 to exon 13: sense 5′-CTCA CGCTTTGGAAAGTGCCG-3′ antisense 5′-ACTCGGCTGCAGTTCGTCAT-3′. PCR conditions were as follows: initial denaturation at 95°C for 6 min followed by 35 cycles of denaturation at 95°C for 45 s, annealing at 58°C for 1 min and extension at 72°C for 1 min. A final extension was carried out at 72°C for 10 min. β-actin was used as the reaction standard. The amplified DNAs were identified using 1.5% agarose gels containing ethidium bromide and visualized under UV light.
Astrocyte Cultures and FRET Imaging
Rat astrocyte cultures (prepared from postnatal day 1 Sprague-Dawley rats) were cultured on a coverslip coated with poly-D-lysine using MEM with 15% horse serum and pen/strep (100 U). Cells were transfected with a GFPnd-EPAC(dDEP)-mCherry (van der Krogt et al., 2008) construct using a calcium phosphate transfection kit (Amersham). Imaging was carried out 24–48 hr after transfection in MEM with no phenol red using a confocal laser-scanning microscope (Zeiss LSM510-Axioskop-2 fitted with a 40×-W/1.0 numerical aperture objective lens) directly coupled to an argon laser (488 nm). Emissions of GFP and mCherry were collected through 502–537 nm and 588–625 nm band-pass filters, respectively. Quantification of all fluorescence signals were performed with Zeiss LSM (version 2.8) software and ImageJ.
Glycogen Measurement
Intracellular glycogen levels were measured as described previously (Brown et al., 2003). In brief, hippocampal brain sections were immediately immersed in ice-cold ethanol (85%)/30 mM HCl (15%) and then stored at −20°C until assays were performed. Tissues were transferred to 600 μl ice-cold 0.1 M NaOH/0.01% SDS plus 81 μl 1M HCl and homogenized. We used 30 μl of homogenate to determine protein concentrations of the lysates by performing a Bradford assay with the DC Protein Assay dye (Bio-Rad). Tissue homogenates were divided into two 300 μl fractions and incubated for 1 hr at 37°C in the presence and absence of amyloglucosidase (EC3.2.1.3). Known concentrations of glucose were used to make a standard curve. Glucose levels were measured using a Glucose Assay Kit (Sigma). This kit is based on the formation of NADH from NAD. NADH fluorescence was measured (excitation at 360 nm, emission at 415 nm) using Gemini Fluorescence Microplate Reader Systems (Molecular Devices).
Lactate and cAMP Measurements
Extracellular lactate levels were measured using Lactate Assay Kit (Biomedical Research Service Centre, SUNY Buffalo) (Gordon et al., 2008). This kit is based on the reduction of the tetrazolium salt INT in an NADH-coupled enzymatic reaction to formazan, which exhibits an absorption maximum at 492 nm. For cAMP measurement, hippocampal brain slices were lysed in 0.1 M HCl and centrifuged for 10 min at 600 × g. The supernatants were collected and intra-cellular cAMP levels were quantified using Correlate-EIA Direct cAMP Assay Kit (Assay Designs). In both lactate and cAMP measurements, hippocampal brain slices were treated in the presence of tetrodotoxin (TTX) (1 μM) and a nonspecific phosphodiesterase inhibitor, 1-Methyl-3-isobutylxanthine (IBMX) (100 μM), to minimize neuronal network activation and preserve cAMP levels, respectively. Real-time lactate measurements within slices were acquired with a calibrated enzyme-based lactate electrode using the FAST16 mKII amperometry system (Quanteon).
Electrophysiology
Slices were transferred to a recording chamber located on an upright microscope (Axioskop, Zeiss) and perfused with aCSF (2 ml/min) aerated with 95% O2/5% CO2. fEPSPs were evoked by stimulation of the Schaffer collateral pathway using a bipolar tungsten-stimulating electrode. fEPSPs were recorded with glass micropipettes filled with aCSF (4–6 MΩ), positioned in stratum radiatum of the CA1 region, and signals were acquired via an Axopatch 200B amplifier (Molecular Devices). Baseline synaptic responses were established by evoking fEPSPs every 30 s (0.03 Hz) for at least 20 min. Data were analyzed using Clampfit 9.0 (Molecular Devices). Trains of electrical stimulation (20 Hz, 30 s at 5V) of the Schaffer collaterals were performed with a concentric bipolar electrode (Frederick Haer) positioned ~100 μm upstream of the NADH imaging area using a Grass S88× stimulator.
Data Collections and Statistical Analysis
Images were collected using 512 × 512 pixels and the scanning frame rate was 393.2 ms or 983.4 ms, depending on the area scanned, and 8 line averaging was utilized. For intracellular pH imaging, both BCECF and SR-101 fluorescence signals were defined as ΔF/F = [(F1 − B1) − (F0 − B0)]/(F0 − B0), where F1 and F0 are fluorescence inside of the cell plasma at any given time point and at the beginning of the experiment, respectively, and B1 and B0 are the back-ground fluorescence at the same time point and at the beginning of the experiment, respectively. Background values were taken from an adjacent area of the imaged cell. Normalized BCECF values were calculated as [ΔF/FBCECF]/[ΔF/FSR101] at the same time point. Quantification of BCECF and SR-101 fluorescence was performed with Zeiss LSM (version 2.8) software and ImageJ. Experimental values are presented as the mean ± SEM, expressed in percent from 100% baseline. The “n” value represents the number of experiments conducted for analysis. Statistical analyses were performed using a two-tailed Student’s t test or ANOVA. p < 0.05 was accepted as statistically significant (*p < 0.05, **p < 0.01).
Drugs
Sodium-Oxamate (2.5 mM), Sodium-Iodoacetate (200 μM), and 4-CIN were applied continuously with different concentrations of [K+]ext. TTX (1 μM) (Alamone Laboratories) and IBMX (100 μM) (Sigma) were always present during the release assays. KH7, 2-OH (Steraloids), and DDA (Sigma) were simultaneously applied to brain slices with different concentrations of [K+]ext.
Supplementary Material
Acknowledgments
This work was supported by an operating grant from the Canadian Institutes of Health Research and Transatlantic Networks of Excellence Program from the Foundation Leducq. B.A.M. is a Canada Research Chair. H.B.C. is supported by postdoctoral fellowships from the Arthur and June Wilms fellowship and the Heart and Stroke Foundation of Canada. G.R.J.G. is supported by fellowships from the Alberta Heritage Foundation for Medical Research and MSFHR and Natural Sciences and Engineering Research Council of Canada (NSERC). N.Z. was supported by a doctoral award from MSFHR. L.R.L. and J.B. are supported by grants from the National Institutes of Health. We are grateful to Dr. Kees Jalink for providing a cAMP FRET construct (GFPnd-EPAC(dDEP)-mCherry) for cAMP FRET imaging. We thank Xiling Zhou for providing cultured astrocytes for FRET imaging.
Footnotes
Supplemental Information includes seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.neuron.2012.08.032.
References
- Alle H, Roth A, Geiger JR. Energy-efficient action potentials in hippocampal mossy fibers. Science. 2009;325:1405–1408. doi: 10.1126/science.1174331. [DOI] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- Aubert A, Costalat R, Magistretti PJ, Pellerin L. Brain lactate kinetics: Modeling evidence for neuronal lactate uptake upon activation. Proc Natl Acad Sci USA. 2005;102:16448–16453. doi: 10.1073/pnas.0505427102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierlein M, Gee KR, Martin VV, Regehr WG. Presynaptic calcium measurements at physiological temperatures using a new class of dextran-conjugated indicators. J Neurophysiol. 2004;92:591–599. doi: 10.1152/jn.00057.2004. [DOI] [PubMed] [Google Scholar]
- Bengtsson F, Boris-Möller F, Hansen AJ, Siesjö BK. Extracellular pH in the rat brain during hypoglycemic coma and recovery. J Cereb Blood Flow Metab. 1990;10:262–269. doi: 10.1038/jcbfm.1990.43. [DOI] [PubMed] [Google Scholar]
- Bevensee MO, Schmitt BM, Choi I, Romero MF, Boron WF. An electrogenic Na(+)-HCO(−)(3) cotransporter (NBC) with a novel COOH-terminus, cloned from rat brain. Am J Physiol Cell Physiol. 2000;278:C1200–C1211. doi: 10.1152/ajpcell.2000.278.6.C1200. [DOI] [PubMed] [Google Scholar]
- Boyarsky G, Ransom B, Schlue WR, Davis MB, Boron WF. Intracellular pH regulation in single cultured astrocytes from rat forebrain. Glia. 1993;8:241–248. doi: 10.1002/glia.440080404. [DOI] [PubMed] [Google Scholar]
- Brown AM. Brain glycogen re-awakened. J Neurochem. 2004;89:537–552. doi: 10.1111/j.1471-4159.2004.02421.x. [DOI] [PubMed] [Google Scholar]
- Brown AM, Ransom BR. Astrocyte glycogen and brain energy metabolism. Glia. 2007;55:1263–1271. doi: 10.1002/glia.20557. [DOI] [PubMed] [Google Scholar]
- Brown AM, Sickmann HM, Fosgerau K, Lund TM, Schousboe A, Waagepetersen HS, Ransom BR. Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J Neurosci Res. 2005;79:74–80. doi: 10.1002/jnr.20335. [DOI] [PubMed] [Google Scholar]
- Brown AM, Tekkök SB, Ransom BR. Glycogen regulation and functional role in mouse white matter. J Physiol. 2003;549:501–512. doi: 10.1113/jphysiol.2003.042416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Wender R, Ransom BR. Ionic mechanisms of aglycemic axon injury in mammalian central white matter. J Cereb Blood Flow Metab. 2001;21:385–395. doi: 10.1097/00004647-200104000-00007. [DOI] [PubMed] [Google Scholar]
- Buck J, Sinclair ML, Schapal L, Cann MJ, Levin LR. Cytosolic adenylyl cyclase defines a unique signaling molecule in mammals. Proc Natl Acad Sci USA. 1999;96:79–84. doi: 10.1073/pnas.96.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Broadwell RD. Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions. II Choroid plexus and ependymal epithelia, endothelia and pericytes. J Neurocytol. 1986;15:511–524. doi: 10.1007/BF01611733. [DOI] [PubMed] [Google Scholar]
- Chen Y, Cann MJ, Litvin TN, Iourgenko V, Sinclair ML, Levin LR, Buck J. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science. 2000;289:625–628. doi: 10.1126/science.289.5479.625. [DOI] [PubMed] [Google Scholar]
- Chesler M. The regulation and modulation of pH in the nervous system. Prog Neurobiol. 1990;34:401–427. doi: 10.1016/0301-0082(90)90034-e. [DOI] [PubMed] [Google Scholar]
- Debernardi R, Pierre K, Lengacher S, Magistretti PJ, Pellerin L. Cell-specific expression pattern of monocarboxylate transporters in astrocytes and neurons observed in different mouse brain cortical cell cultures. J Neurosci Res. 2003;73:141–155. doi: 10.1002/jnr.10660. [DOI] [PubMed] [Google Scholar]
- Dringen R, Hamprecht B. Differences in glycogen metabolism in astroglia-rich primary cultures and sorbitol-selected astroglial cultures derived from mouse brain. Glia. 1993;8:143–149. doi: 10.1002/glia.440080302. [DOI] [PubMed] [Google Scholar]
- Duffy S, MacVicar BA. Potassium-dependent calcium influx in acutely isolated hippocampal astrocytes. Neuroscience. 1994;61:51–61. doi: 10.1016/0306-4522(94)90059-0. [DOI] [PubMed] [Google Scholar]
- Erlichman JS, Hewitt A, Damon TL, Hart M, Kurascz J, Li A, Leiter JC. Inhibition of monocarboxylate transporter 2 in the retrotrapezoid nucleus in rats: a test of the astrocyte-neuron lactate-shuttle hypothesis. J Neurosci. 2008;28:4888–4896. doi: 10.1523/JNEUROSCI.5430-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Jaiswal BS, Xie F, Krajnc-Franken MA, Robben TJ, Strik AM, Kuil C, Philipsen RL, van Duin M, Conti M, Gossen JA. Mice deficient for soluble adenylyl cyclase are infertile because of a severe sperm-motility defect. Proc Natl Acad Sci USA. 2004;101:2993–2998. doi: 10.1073/pnas.0400050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell J, Ramos L, Tresguerres M, Kamenetsky M, Levin LR, Buck J. Somatic ‘soluble’ adenylyl cyclase isoforms are unaffected in Sacy tm1Lex/Sacy tm1Lex ‘knockout’ mice. PLoS ONE. 2008;3:e3251. doi: 10.1371/journal.pone.0003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler JC. Glucose deprivation results in a lactate preventable increase in adenosine and depression of synaptic transmission in rat hippocampal slices. J Neurochem. 1993;60:572–576. doi: 10.1111/j.1471-4159.1993.tb03187.x. [DOI] [PubMed] [Google Scholar]
- Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456:745–749. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Armston AE. A re-evaluation of the role of mitochondrial pyruvate transport in the hormonal control of rat liver mitochondrial pyruvate metabolism. Biochem J. 1984;223:677–685. doi: 10.1042/bj2230677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallows KR, Wang H, Edinger RS, Butterworth MB, Oyster NM, Li H, Buck J, Levin LR, Johnson JP, Pastor-Soler NM. Regulation of epithelial Na+ transport by soluble adenylyl cyclase in kidney collecting duct cells. J Biol Chem. 2009;284:5774–5783. doi: 10.1074/jbc.M805501200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess KC, Jones BH, Marquez B, Chen Y, Ord TS, Kamenetsky M, Miyamoto C, Zippin JH, Kopf GS, Suarez SS, et al. The “soluble” adenylyl cyclase in sperm mediates multiple signaling events required for fertilization. Dev Cell. 2005;9:249–259. doi: 10.1016/j.devcel.2005.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof PR, Pascale E, Magistretti PJ. K+ at concentrations reached in the extracellular space during neuronal activity promotes a Ca2+-dependent glycogen hydrolysis in mouse cerebral cortex. J Neurosci. 1988;8:1922–1928. doi: 10.1523/JNEUROSCI.08-06-01922.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Benz AM, Katsuki H, Zorumski CF. Endogenous monocarboxylates sustain hippocampal synaptic function and morphological integrity during energy deprivation. J Neurosci. 1997;17:9448–9457. doi: 10.1523/JNEUROSCI.17-24-09448.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Zorumski CF. Glial-neuronal interactions underlying fructose utilization in rat hippocampal slices. Neuroscience. 2009;161:847–854. doi: 10.1016/j.neuroscience.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal BS, Conti M. Identification and functional analysis of splice variants of the germ cell soluble adenylyl cyclase. J Biol Chem. 2001;276:31698–31708. doi: 10.1074/jbc.M011698200. [DOI] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- Litvin TN, Kamenetsky M, Zarifyan A, Buck J, Levin LR. Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate. J Biol Chem. 2003;278:15922–15926. doi: 10.1074/jbc.M212475200. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ. Neuron-glia metabolic coupling and plasticity. J Exp Biol. 2006;209:2304–2311. doi: 10.1242/jeb.02208. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L. Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos Trans R Soc Lond B Biol Sci. 1999;354:1155–1163. doi: 10.1098/rstb.1999.0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L, Rothman DL, Shulman RG. Energy on demand. Science. 1999;283:496–497. doi: 10.1126/science.283.5401.496. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Sorg O, Yu N, Martin JL, Pellerin L. Neurotransmitters regulate energy metabolism in astrocytes: implications for the metabolic trafficking between neural cells. Dev Neurosci. 1993;15:306–312. doi: 10.1159/000111349. [DOI] [PubMed] [Google Scholar]
- Mitterling KL, Spencer JL, Dziedzic N, Shenoy S, McCarthy K, Waters EM, McEwen BS, Milner TA. Cellular and subcellular localization of estrogen and progestin receptor immunoreactivities in the mouse hippocampus. J Comp Neurol. 2010;518:2729–2743. doi: 10.1002/cne.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas CA, Ransom BR. Depolarization-induced alkalinization (DIA) in rat hippocampal astrocytes. J Neurophysiol. 1994;72:2816–2826. doi: 10.1152/jn.1994.72.6.2816. [DOI] [PubMed] [Google Scholar]
- Pastor-Soler N, Beaulieu V, Litvin TN, Da Silva N, Chen Y, Brown D, Buck J, Levin LR, Breton S. Bicarbonate-regulated adenylyl cyclase (sAC) is a sensor that regulates pH-dependent V-ATPase recycling. J Biol Chem. 2003;278:49523–49529. doi: 10.1074/jbc.M309543200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55:1251–1262. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Neuroscience. Let there be (NADH) light Science. 2004;305:50–52. doi: 10.1126/science.1100428. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster Hd. The Fine Structure of the Nervous System: Neurons and Their Supporting Cells. 3. Oxford: Oxford University Press; 1991. [Google Scholar]
- Pierre K, Magistretti PJ, Pellerin L. MCT2 is a major neuronal monocarboxylate transporter in the adult mouse brain. J Cereb Blood Flow Metab. 2002;22:586–595. doi: 10.1097/00004647-200205000-00010. [DOI] [PubMed] [Google Scholar]
- Ransom BR. Glial modulation of neural excitability mediated by extracellular pH: a hypothesis. Prog Brain Res. 1992;94:37–46. doi: 10.1016/s0079-6123(08)61737-9. [DOI] [PubMed] [Google Scholar]
- Ryu JK, McLarnon JG. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J Cell Mol Med. 2009;13(9A):2911–2925. doi: 10.1111/j.1582-4934.2008.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid A, Sutto Z, Nlend MC, Horvath G, Schmid N, Buck J, Levin LR, Conner GE, Fregien N, Salathe M. Soluble adenylyl cyclase is localized to cilia and contributes to ciliary beat frequency regulation via production of cAMP. J Gen Physiol. 2007;130:99–109. doi: 10.1085/jgp.200709784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt BM, Berger UV, Douglas RM, Bevensee MO, Hediger MA, Haddad GG, Boron WF. Na/HCO3 cotransporters in rat brain: expression in glia, neurons, and choroid plexus. J Neurosci. 2000;20:6839–6848. doi: 10.1523/JNEUROSCI.20-18-06839.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr A, Miller JJ, Payne RS, Rigor BM. An increase in lactate output by brain tissue serves to meet the energy needs of glutamate-activated neurons. J Neurosci. 1999;19:34–39. doi: 10.1523/JNEUROSCI.19-01-00034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr A, West CA, Rigor BM. Lactate-supported synaptic function in the rat hippocampal slice preparation. Science. 1988;240:1326–1328. doi: 10.1126/science.3375817. [DOI] [PubMed] [Google Scholar]
- Sorg O, Magistretti PJ. Characterization of the glycogenolysis elicited by vasoactive intestinal peptide, noradrenaline and adenosine in primary cultures of mouse cerebral cortical astrocytes. Brain Res. 1991;563:227–233. doi: 10.1016/0006-8993(91)91538-c. [DOI] [PubMed] [Google Scholar]
- Sorg O, Magistretti PJ. Vasoactive intestinal peptide and noradrenaline exert long-term control on glycogen levels in astrocytes: blockade by protein synthesis inhibition. J Neurosci. 1992;12:4923–4931. doi: 10.1523/JNEUROSCI.12-12-04923.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegborn C, Litvin TN, Hess KC, Capper AB, Taussig R, Buck J, Levin LR, Wu H. A novel mechanism for adenylyl cyclase inhibition from the crystal structure of its complex with catechol estrogen. J Biol Chem. 2005;280:31754–31759. doi: 10.1074/jbc.M507144200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144:810–823. doi: 10.1016/j.cell.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, Nedergaard M. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci. 2007;10:754–762. doi: 10.1038/nn1902. [DOI] [PubMed] [Google Scholar]
- van der Krogt GN, Ogink J, Ponsioen B, Jalink K. A comparison of donor-acceptor pairs for genetically encoded FRET sensors: application to the Epac cAMP sensor as an example. PLoS ONE. 2008;3:e1916. doi: 10.1371/journal.pone.0001916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender R, Brown AM, Fern R, Swanson RA, Farrell K, Ransom BR. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J Neurosci. 2000;20:6804–6810. doi: 10.1523/JNEUROSCI.20-18-06804.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B. In vivo evidence for lactate as a neuronal energy source. J Neurosci. 2011;31:7477–7485. doi: 10.1523/JNEUROSCI.0415-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LL, Pathak HR, Coulter DA, Freed MA, Vardi N. Shift of intracellular chloride concentration in ganglion and amacrine cells of developing mouse retina. J Neurophysiol. 2006;95:2404–2416. doi: 10.1152/jn.00578.2005. [DOI] [PubMed] [Google Scholar]
- Zippin JH, Levin LR, Buck J. CO(2)/HCO(3)(−)-responsive soluble adenylyl cyclase as a putative metabolic sensor. Trends Endocrinol Metab. 2001;12:366–370. doi: 10.1016/s1043-2760(01)00454-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.