

Abstract
Malaria continues to be a difficult disease to eradicate, largely due to the widespread populations it affects and to the resistance that malaria parasites have developed against once very potent therapies. The natural product artemisinin has been a boon for antimalarial chemotherapy as artemisinin combination therapy (ACT) has become the first line of chemotherapy. Because the threat of resistance is always on the horizon, it is imperative to continually identify new treatments, comprised of both advanced analogs of all antimalarial drugs, especially artemisinin, as well as the exploration of novel combinations, ideally with distinct mechanisms of action. Here we report, for the first time, synthesis of a series of two-carbon linked artemisinin-derived dimers, their unique structural features, and demonstration of their antimalarial efficacy via single oral dose administration in two 60-day survival studies of Plasmodium berghei-infected mice. Several of the new endoperoxide chemical entities consistently demonstrated excellent antimalarial efficacy, and combinations with two non-peroxide antimalarial drugs have been studied.
Introduction
As the landscape of therapeutic treatment advances, orally available small molecule inhibitors remain a prominent weapon in the fight against disease. Perhaps nowhere is this more important than in the treatment of malaria, whose parasites infected over 200 million people and killed ~655,000 people in 2010,1 though a recent report suggests deaths of greater than 1 million people in 2010.2 Prevention of malaria caused by the Plasmodium parasites has centered on the use of bed nets (of which implementation suffers from poor compliance)3 and attempts to develop a vaccine (which has recently demonstrated protection, albeit marginal),4 but the large number of people who still become infected continues to keep small molecule therapeutic treatment of paramount importance. The history of malaria therapy is long and dozens of small molecules have been the treatment of choice at some point.5 Well known examples include quinine, chloroquine, and atovaquone (compounds 1-3, respectively, Figure 1). Each of these molecules (and others) has fallen from prominence, due either to significant side effects or to the development of parasite resistance, or both.5 These chemical entities have been largely replaced by treatments containing the natural product artemisinin.
Figure 1.
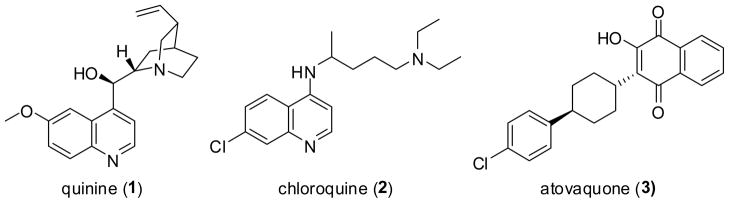
Antimalarial agents to which resistance has developed
Artemisinin (4a, Figure 2) could be considered one of the most significant natural product small molecule discoveries over the past 40 years. The molecule was first isolated from its natural source, Artemisia annua, in 1972 by a team under the direction of Dr. Youyou Tu.6 It contains a surprisingly stable endoperoxide linkage that is essential for the demonstrated activity in a variety of diseases, most notably malaria and, more recently, several types of cancers and cytomegalovirus.7 Unfortunately the molecule has poor bioavailability and is rapidly cleared from the body.8 First generation derivatives that attempt to circumvent this issue include lipid-soluble artemether (4b) and water-soluble artesunate (4c), and these molecules are now part of the first line of antimalarial treatment known as artemisinin combination therapy (ACT). Example combinations are artemether (4b) plus lumefantrine (6),9 artesunate (4c) plus mefloquine (7) and the recently FDA-approved artesunate (4c) plus pyronaridine (8, Figure 3).10 However, both artemether and artesunate still have significant metabolic liabilities and short half-lives, so most new candidate drug combinations pair an artemisinin-derived analog with a drug that has better physical properties and a longer duration of activity. Further structure-activity relationships (SAR) have been explored around this peroxide linkage and have ranged from functionalization of the natural product11–15 to completely synthetic peroxide structures.16–19 The pursuit of highly efficacious artemisinin derivatives and novel drug combinations continues.
Figure 2.
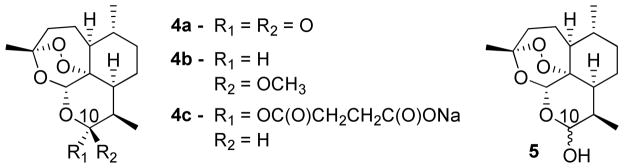
artemisinin, currently used first generation derivatives, and dihydroartmesinin
Figure 3.
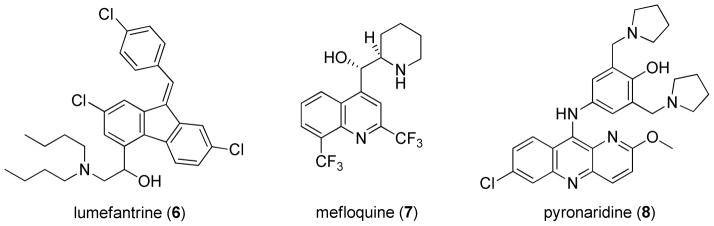
currently used therapeutic drugs in ACT
Synthesis of artemisinin-derived dimers
Efforts to improve the pharmacokinetic properties of artemisinin are ongoing. A wide variety of functional groups have been incorporated in order to improve absorption and increase the half-life of the molecule in vivo while also maintaining efficacy.19 An alternative approach has been to synthetically tether two artemisinin units together to form dimeric molecules, the rationale being that, at a minimum, for every molecule that survives the metabolism and excretion processes, two of the required endoperoxide pharmacophores will be delivered to the sight of action. However, dimer trioxanes are often more than simply twice as potent as the corresponding monomers, and therefore an as yet unknown explanation must exist. Many examples of artemisinin dimers exist with varying lengths of the linker between the two units, usually tethering at the C10 position (position numbered in Figure 2). The most successful examples include linker lengths of 5,20 4,21 or 322, 23 carbon atoms (example structures 9, 10, and 11, respectively, Figure 4), with three-carbon analogs generally showing the greatest efficacy in vivo. There is, to the best of our knowledge, only one example of a two-atom linked dimer (12, Figure 4)24 and only one example of a single-atom linked dimer (13, Figure 4).25 Here we report the synthesis of a series of novel two-carbon linked dimers, their absolute stereochemical configuration, and their antimalarial efficacies. Furthermore, several of the best dimers have been screened in combination with two adjuvants in order to establish a diverse set of potential drug combinations.
Figure 4.
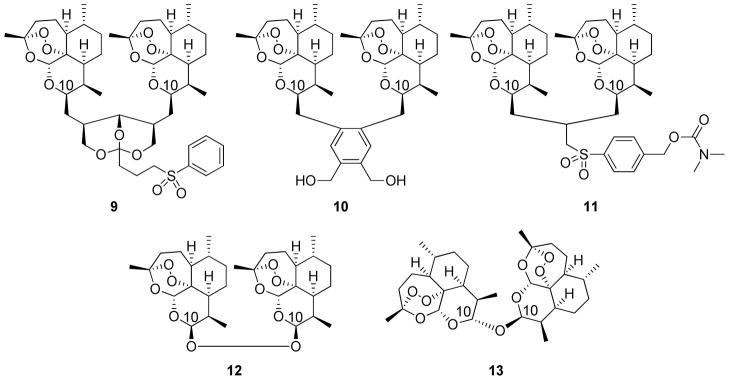
Representative artemisinin dimers of varying linker length
Synthesis of two-carbon linked dimers
The currently used drugs artemether (4b) and artesunate (4c) contain a C10 acetal linkage. It is generally accepted that this linkage is a significant metabolic liability. Both molecules 4b and 4c are rapidly catabolized to dihydroartemisinin (DHA, 5, Figure 2) and excreted following glucuronidation. Methodology has been previously developed to replace the C10 acetal linkage with a carbon-carbon bond,26, 27 and these molecules are called C10-carba analogs. These analogs have shown increased stability in simulated stomach conditions as compared to the first generation acetals artemether (4b) and artesunate (4c),28 but have yet to translate into a marketed drug. By adapting established methodology, we set out to synthesize a two-carbon linked artemisinin dimer that might have distinct benefits over dimers of greater linker lengths.
The main obstacle in synthesizing such a molecule is the large steric interaction that would likely occur between two artemisinin units joined through such a short distance. Established methodology follows a pathway using a Lewis acid activation of an appropriately functionalized artemisinin precursor (Scheme 1). In this pathway, artemisinin is functionalized with an appropriate leaving group (LG) at the C10 position (general structure 14, Scheme 1) which is thought to form oxocarbenium ion (15) in situ after Lewis-acid activation. Treatment with a nucleophile can then proceed through either one of two pathways – ideally nucleophilic addition occurs at C10, providing desired products of general structure 16. However, the C9 proton is also easily accessible and elimination is a major competing pathway, particularly with harder nucleophiles, leading to the undesired elimination product (17).
Scheme 1.
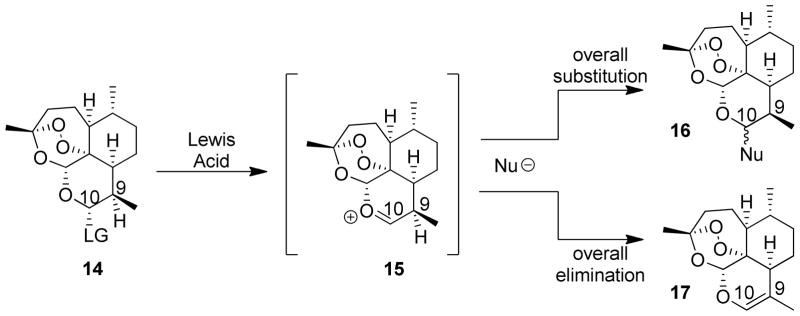
Nucleophilic substitution at C10 and competing elimination pathway
With this reactivity in mind, we envisioned a two-carbon linker unit that could be made nucleophilic in successive steps. The acetylene unit became the focus because substituted acetylides have previously achieved substitution at the C10 position by reacting an electrophilic C10 fluoride analog with nucleophilic trimethylaluminum-activated acetylenes.29 However, this methodology was low-yielding and the C10 fluoride is difficult to manipulate. As such, a more robust method was required. Organometallic reagents have achieved substitution at the C10 position,29 and the commercially available ethynyl Grignard would allow installation of the acetylene unit. Initial attempts to introduce the acetylene unit at the C10 position using the common electrophilic precursor dihydroartemisinin acetate (DHA-OAc, 19, Scheme 2) provided almost exclusively 17, likely due to the highly basic (and weakly nucleophilic) nature of the Grignard acetylide reagent. Efforts were still futile upon addition of zinc(II) chloride (ZnCl2), in an attempt to form a more nucleophilic zinc-magnesium hybrid species (likely [{Mg2Cl3(Et2O)6}+{Zn(acetylene)3}−]) in situ.30, 31 A new precursor containing a nucleophile assisting leaving group32 was synthesized in order to form a chelating intermediate that would direct the organometallic reagent to the C10 position. Using standard procedure, the artemisinin lactone was reduced using diisobutylaluminum hydride (DIBALH) at −78 °C. The resulting oxyanion can be trapped with the appropriate electrophile (in this case 2,4-dimethoxybenzoyl chloride, 18) directly in situ to provide exclusively the desired intermediate 20 (Scheme 2). The 2,4-dimethoxybenzoate group is an electron rich moiety to which ZnCl2 or other Lewis acidic species can strongly chelate. The previously described zinc-magnesium hybrid nucleophile would be directed to the C10 position upon coordination to the benzoate. Simultaneous addition of the Lewis acid ZnCl2 and ethynylmagnesium chloride to a stirring solution of 20 at −5 °C provided the previously unreported C10 β-alkyne (21). The best results were achieved with a 3.2:1.0 ratio of the Grignard reagent to ZnCl2, which is consistent with previous studies that utilized similar reagents.30, 31
Scheme 2.
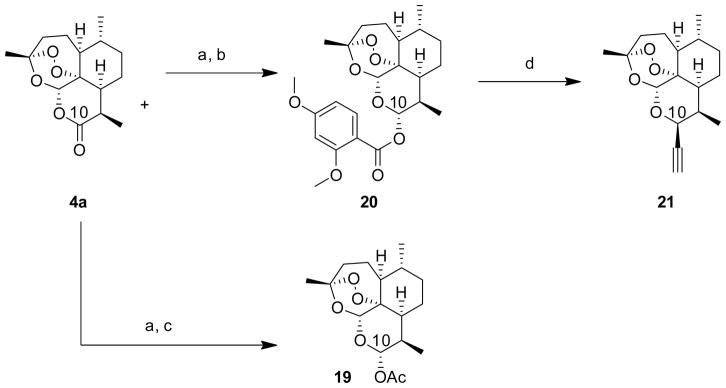
Preparation of DHA-OAc (19) and C10 β-alkyne (21)
a) DIBALH, CH2Cl2, −78 °C, b) DMAP, py, 2,4 dimethoxybenzoyl chloride (18), 98% over 2 steps; c) DMAP, py, Ac2O, 98% over 2 steps; d) 1.1 eq ZnCl2, 3.5 eq ethynylmagnesium chloride, Et2O, 74%.
With 21 prepared in only two steps and in 73% overall yield from artemisinin, the two-carbon linked dimer system was within reach. Attempts to convert 21 into a Grignard reagent (and into other organometallic reagents, for example, a dimethylaluminate29 or a tri-n-butylstannane, not shown) and then simply to repeat the coupling procedure with 20 led to decomposition of starting material. It was hypothesized that the large bulk of the artemisinin prevented attachment to a second artemisinin via a straight-on attack that would be required of a linear alkyne, so efforts shifted to conversion of the alkyne into alternate functionality.
Ziffer has established that trimethylsilyl (TMS) enol ethers could react with 19 to form C10 carba analogs in good yields,27 and we believed that if the alkyne could be converted into a TMS enol ether then similar reactivity would be achieved. Treatment of 21 with p-toluenesulfonic acid (pTsOH) and mercury(II) acetate [(HgOAc)2] in water and acetone afforded the C10 methyl ketone (22) in excellent yield (Scheme 3). Subsequent conversion into TMS enol ether (23) was accomplished by treatment of 22 with a solution of freshly prepared lithium diisopropylamide (LDA) in tetrahydrofuran (THF) in the presence of chlorotrimethylsilane (TMSCl) and triethylamine (Et3N) at −78 °C. Compound 23 is surprisingly stable to silica gel and can be isolated and characterized as a clean material, but it must be used immediately. The final step for the formation of the two-carbon linked dimer system was then achieved by following established protocol.22, 26 Slow addition of tin(IV) chloride (SnCl4) into a solution of 23 and 19 at −78 °C gave, for the first time, the two-carbon linked dimer ketone (24) in good yield. Compound 24 is prepared in five linear steps starting from artemisinin in 36% overall yield, and has been prepared on 500 mg scale.
Scheme 3.

synthesis of two-carbon linked artemisinin dimer ketone (24)
a) pTsOH, Hg(OAc)2, acetone: H2O, rt, 90%; b) TMSOTf, Et3N, THF, −78 °C to rt, 81%; c) DHA-OAc (19), SnCl4, CH2Cl2, −78 °C, 69%.
Analog synthesis
A series of oxime derivatives were prepared from 24 in order to add diversity to this chemical series and to probe SAR. The oxime functionality was desirable because it is already contained in known drugs,33 has shown activity against protozoa,34, 35 and evidence suggests that it is a suitable “water-soluble prodrug” that might unveil the ketone under biological conditions.36–39 The initial series of analogs, prepared by treating 24 with the appropriate commercially available hydroxylamine in pyridine, consisted of the parent oxime (25), the O-methyl oxime (26), and the O-allyl oxime (27, Scheme 4a). Analogs containing bulkier alkyl groups were also prepared and included cyclopropylmethylene oxime (28), isobutyloxime (29), and tert-butyloxime (30). While two geometric isomers of the oxime would be expected, the Z-isomer was favored, and in some cases was the only isomer produced (stereochemical implications will be discussed, vide infra). Compound 25 was further converted into the O-propargyloxime (31), O-dimethyl carbamoyloxime (32), O-acetate oxime (33), and a pair of O-phosphate oximes (dimethyl 34, diethyl 35) upon treatment with sodium hydride (NaH) and the appropriate halide in THF at 0 °C (Scheme 4b).
Scheme 4.

preparation of two-carbon linked dimer oxime analogs from 24
a) RONH2-HCl, py, rt; b) R-X, NaH, THF, 0 °C.
Stereochemistry
Synthesis of the two-carbon linked dimer and its derivatives generates several stereochemical questions. Reduction of artemisinin at low temperatures leads exclusively to products with α-sterochemistry,40 as described in the synthesis of 19 and 20. Furthermore, nucleophilic displacement on these analogs has been shown to produce exclusively products with β-configuration,26, 29, 40 as demonstrated in the synthesis of 21. The proton NMR coupling constants between the C9/C10 protons are indicative of the stereochemistry at this location, as α-products position these protons in an axial-axial orientation resulting in larger coupling constants.41 For example, the C9/C10 coupling constant for 20 (protons are trans-diaxial) is 9.8 Hz but for 21 (protons are axial-equatorial) it is 5.7 Hz (see table S1). The C5 proton is affected by C10 stereochemistry as well. Conversion of 20 to 21 increases the chemical shift from 5.47 to 5.56 ppm, and it is hypothesized that the C5 proton is positioned to interact with the deshielded anisotropic cone of the alkyne in the β-orientation. Synthesis of 22 should not change the C10 stereochemistry, and indeed the C9/C10 coupling constant and C5 chemical shift support this hypothesis (J9,10 = 6.8 Hz; C5 δ = 5.65 ppm). The C5 proton is further deshielded, likely due to hydrogen-bonding interactions with the carbonyl oxygen. While it could be possible to epimerize the C10 position upon reaction with LDA to produce 23, the steric hindrance is likely too great. Proton NMR again confirms this hypothesis (J9,10 = 6.4 Hz). Finally, reaction of 23 with 19 should produce 24 with β-configuration at both C10 positions [referred to as C10 (adjacent to the carbonyl) and C10′ (on the second artemisinin unit) in Figure 5]. The chemical shifts and coupling constants for C10 and C10′ were 4.84 ppm (J = 6.3 Hz) and 4.57 (J = 6.2 Hz), respectively, suggesting that both positions were in the β-configuration; this hypothesis was confirmed by X-ray crystallography (see Figure S1). Interestingly, the C5 and C5′ protons had very different chemical shifts – 5.57 ppm and 5.30 ppm, respectively. This provides further evidence that the C5 proton is interacting with the carbonyl oxygen, an important observation for the oxime analogs (vide infra). Interestingly, the endoperoxides from both artemisinin units are exposed and locked on the outside of the caged structure, a feature that could be beneficial for antimalarial activity of these two-carbon linked dimers.
Figure 5.
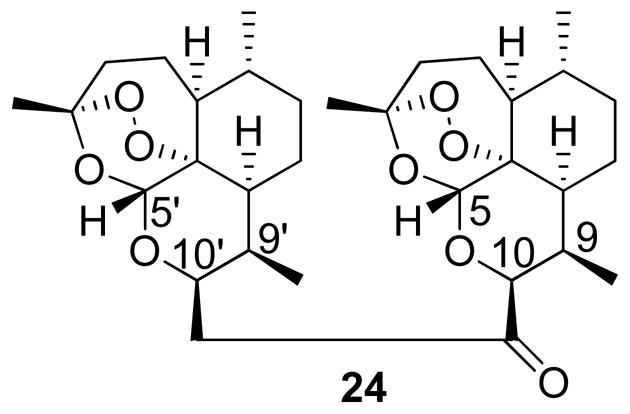
Carbon numbering for proton NMR analysis
Conversion of 24 into various oximes 25-30 raised an additional stereochemical question. The conditions employed could form either the E- or Z-isomer and could also epimerize the C10 position (because it is alpha to the carbonyl). The synthesis of each oxime from 24 generally formed one major product (yields shown in Scheme 4a, the less polar product by analytical TLC), and in some cases one minor product (yields shown in Scheme 4a, the more polar spot by analytical TLC). First, it should be noted that in all cases the C9-10 and C9′-10′ coupling constants are ~6 Hz, suggesting that the β-stereochemistry has been maintained at both positions. The C5 and C5′ and the C10 and C10′ protons from each molecule were again of interest. For example, the chemical shifts of the C5 and C5′ protons for the less polar oxime [designated (Z)-25] are 5.39 ppm and 5.37 ppm, respectively, and the C10 and C10′ protons are 5.68 ppm (J = 6.9 Hz) and 4.53 ppm (J = 5.9 Hz), respectively. The observation that the C5 proton is no longer deshielded suggests that the less polar compound is the Z-isomer – the hydroxyl group would be on the same side of the artemisinin unit, and the steric clash would force the C10-oxime bond to rotate out, thereby avoiding the interaction of the oxime nitrogen with the C5 proton (see Figure 6a). Furthermore, the C10 proton is drastically deshielded, shifting from 4.84 ppm in the starting ketone to 5.68 ppm. This suggests that the hydroxyl group is positioned to interact with the C10 proton. Conversely, the chemical shifts of the C5 and C5′ protons for the more polar oxime (minor product (E)-25) are 5.50 ppm and 5.43 ppm, respectively, and the C10 and C10′ were 5.17 ppm (J = 6.3 Hz) and 4.59 ppm (J = 6.3 Hz), respectively. In this polar isomer, the C10 and C10′ protons are both deshielded, which could be explained by interaction with the oxime nitrogen (for C10) and possibly the hydroxyl group (for C10′). This would be indicative of the E-isomer (see Figure 6b). The chemical shift trends for C5, C5′, C10, and C10′ are consistent for both the major and the minor isomers across the remainder of the oxime analogs (see table S1), suggesting that each major product has the same configuration (and conversely, each minor product as well). These hypotheses were confirmed by 2D NOESY NMR experiments on (Z)-26 and (E)-26, where the oxime methyl-C10 NOE was observed in (Z)-26 but not in (E)-26 (see Supporting Information). All analogs tested have Z-stereochemistry unless otherwise noted.
Figure 6.
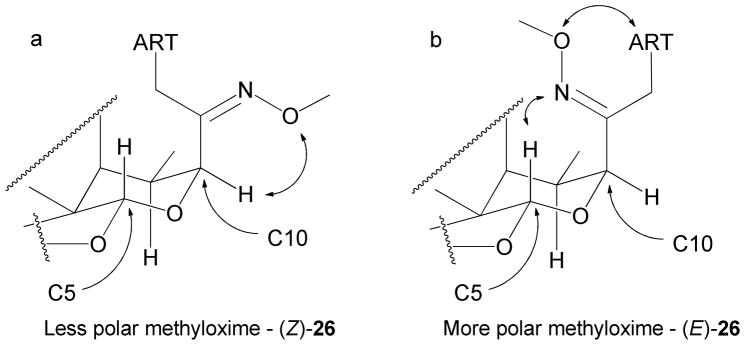
a) proposed interactions in the less polar dimer methyloxime (Z)-26; b) proposed interactions in the more polar dimer methyloxime (E)-26
Biological Evaluation
In vivo studies
Nearly all of the two-carbon linked dimer analogs were evaluated with Plasmodium berghei ANKA infected mice using a 60-day survival study. Each trioxane (0.64 mg) and mefloquine hydrochloride (1.92 mg) were dissolved in 100 μL of 7:3 Tween:EtOH mixture, and the mixture was further diluted with 965 μL of water (total volume of 1065 μL). Twenty four hours after infection with P. berghei ANKA strain (2 × 107 parasitized erythrocytes), each of four 5-week old C57BL/6J male mice (Jackson Laboratory, weighing approximately 20 g each) received 200 μL of the trioxane solution via oral gavage, corresponding to a dose of 6 mg/kg of trioxane and 18 mg/kg of mefloquine hydrochloride. Blood parasitemia levels at day three and duration of animal survival as compared to survival time of animals receiving no drug were monitored, both being widely accepted measures of a drug’s antimalarial efficacy. A select set were tested in the initial experiment, and these results are summarized in Table 1.
Table 1.
In Vivo Antimalarial Efficacy Using a Single Oral Dose of Trioxane Combined with Mefloquine Hydrochloride in P.berghei Infected Mice
| trioxane | single oral dose (mg/kg)
|
survival after infection (days) | avg survival | % parasitemia suppression1 | |
|---|---|---|---|---|---|
| trioxane | Mef-HCl | ||||
| 24 | 6 | 18 | 34, 39, 60, 60 | 48.3 | >99.9 |
| 25 | 6 | 18 | 30, 60, 60, 60 | 52.5 | >99.9 |
|
| |||||
| 27 | 6 | 18 | 44, 60, 60, 60 | 56.0 | >99.9 |
| 27 | 120 | 0 | 20, 21, 60, 60 | 40.3 | >99.9 |
|
| |||||
| 32 | 6 | 18 | 27, 60, 60, 60 | 51.8 | >99.9 |
| 33 | 6 | 18 | 13, 16, 20, 60 | 27.3 | >99.9 |
| 34 | 6 | 18 | 44, 60, 60, 60 | 56.0 | >99.9 |
| 35 | 6 | 18 | 16, 20, 30, 30 | 21.5 | >99.9 |
| controls: | |||||
| vehicle (no drug) | 0 | 0 | 6, 7, 8, 8 | 7.3 | 0 |
| artemether(4b) | 6 | 18 | 16, 24, 60, 60 | 40.0 | >99.9 |
| mefloquine | 0 | 18 | 17, 20, 20, 27 | 21.0 | >99.9 |
determined on day 3 post infection
The data in this study demonstrate the overall high efficacy of this class of compounds. Mice receiving the drug artemether (4b) in combination with mefloquine survived an average of 40.0 days. Nearly all of the compounds tested were definitively more efficacious, with two compounds (26 and 31) achieving complete cures of all four mice. The cured mice gained an average of 10.7 grams and 12.0 g for 26 and 31, respectively, as compared to an average of 7.7 gram for uninfected control mice receiving vehicle only, suggesting that the cured mice were exceptionally healthy. Another compound of note was 27, which was tested both in combination with mefloquine and as a single dose monotherapy (at 120 mg/kg, without mefloquine). The combination therapy was much more efficacious than artemether combined with mefloquine, extending the survival time to 56.0 days, while the single dose monotherapy was equally as efficacious as artemether plus mefloquine.
Encouraged by these initial results, a second mouse experiment was initiated to further evaluate several of the lead compounds. We also wished to evaluate an alternative adjuvant to establish a broader understanding of the efficacy of these compounds. As such, the lead compounds were screened in combination with lumefantrine and also as a single dose monotherapy at 150 mg/kg. As summarized in Table 2, all of the lead compounds outperformed the drug artemether (4b) which prolonged survival to an average of 38.5 days and 19.0 days in combination with mefloquine and lumefantrine, respectively. One mouse receiving dimer 26 and mefloquine died early in the experiment (day 16) while the other three lived to day 60, an average of 49.0 days. Compound 26, however, was marginally effective in combination with lumefantrine (26.3 day average) and not effective as a single dose monotherapy, with an average survival time (8.3 days) similar to that of infected mice receiving no drug (8.8 days). Compound 27 was efficacious in all three groups, extending survival time to 49.0 days, 41.3 days, and 46.8 days in combination with mefloquine, lumefantrine, or as a monotherapy, respectively. Compound 31 was once again the most efficacious compound. One mouse receiving mefloquine combination died at an extended time point (35 days) while the other three survived the entire 60 days, an average survival of 53.8 days. All four mice receiving lumefantrine combination survived the entire 60 days, with no parasitemia detected on day 60. These mice had gained as much weight as the uninfected control mice receiving vehicle only, again suggesting that the cured mice were healthy. The single dose monotherapy using 31 prolonged survival time to an average of 46.8 days, with three of the four mice having no detectable parasitemia on day 60, similar to the monotherapy results using 27. Interestingly, the dimer tert-butyl oxime (30) combined with mefloquine was not very efficacious, prolonging survival to only 23.8 days. As the tert-butyl moiety would be less susceptible to metabolic oxidation (that would lead to subsequent conversion to the free oxime and ultimately to the ketone via hydrolysis),37–39 these data suggest that the oxime analogs may be acting as prodrugs.
Table 2.
In Vivo Antimalarial Efficacy Using a Single Oral Dose of Trioxane Combined with Mefloquine Hydrochloride or Lumefantrine in P.berghei Infected Mice
| trioxane | single oral dose (mg/kg)
|
survival after infection (days) | avg survival | % parasitemia suppression1 | ||
|---|---|---|---|---|---|---|
| trioxane | adjuvant | dose | ||||
| E-26 | 6 | mefloquine | 18 | 16, 35, 37, 60 | 37.0 | >99.9 |
|
| ||||||
| 26 | 6 | mefloquine | 18 | 16, 60, 60, 60 | 49.0 | >99.9 |
| 26 | 6 | lumefantrine | 18 | 13, 16, 16, 60 | 26.3 | >99.9 |
| 26 | 150 | none | 0 | 7, 7, 9, 10 | 8.3 | 98 |
|
| ||||||
| 27 | 6 | mefloquine | 18 | 16, 60, 60, 60 | 49.0 | >99.9 |
| 27 | 6 | lumefantrine | 18 | 16, 29, 60, 60 | 41.3 | >99.9 |
| 27 | 150 | none | 0 | 7, 60, 60, 60 | 46.8 | >99 |
|
| ||||||
| 28 | 6 | mefloquine | 18 | 15, 29, 60, 60 | 41 | >99.9 |
| 30 | 6 | mefloquine | 18 | 18, 20, 20, 37 | 23.8 | >99.9 |
|
| ||||||
| 31 | 6 | mefloquine | 18 | 35, 60, 60, 60 | 53.8 | >99.9 |
|
| ||||||
| 31 | 150 | none | 0 | 7, 60, 60, 60 | 46.8 | >99.9 |
| controls: | ||||||
| vehicle (no drug) | 0 | 0 | 6, 7, 7, 15 | 8.8 | 0 | |
| artemether(4b) | 6 | mefloquine | 18 | 29, 29, 36, 60 | 38.5 | >99.9 |
| artemether(4b) | 6 | lumefantrine | 18 | 15, 16, 16, 29 | 19.0 | >99.9 |
| mefloquine | 0 | 18 | 15, 16, 30, 31 | 23.0 | >99.9 | |
| lumefantrine | 0 | 18 | 7, 7, 7, 35 | 14.0 | >99.9 | |
determined on day 3 post infection
Conclusion
Malaria remains an oppressive disease, especially in tropical regions. Antimalarial drugs that have long term chemical stability in vivo have been difficult to achieve due to the complexity of the parasitic life cycle and to the parasite’s ability to develop resistance (particularly when exposed to sub-lethal doses). Artemisinin combination therapy has been a staple of chemotherapy over the past 20 years, but the poor properties of most artemisinin-derived drugs make them susceptible to rapid clearance and therefore reduced efficacy. Our group has focused on the development of artemisinin-derived C10 carba analogs that might have improved physical properties to maintain an effective concentration in the blood stream, and this is demonstrated by their efficacy in in vivo experiments as compared to the currently used drug artemether (4b). We report here the synthesis and biological evaluation of a new series of artemisinin-derived two-carbon linked dimers, whose unique conformation places the endoperoxides of each artemisinin unit (necessary for biological activity) on the outside of a caged structure. This two-carbon linked dimer system has been further functionalized into a series of oxime derivatives in an effort to improve their behavior in vivo. Many of our new dimers are much more efficacious than artemether (4b) when administered as a single oral dose combined with either mefloquine or lumefantrine. In particular, the propargyl oxime (31) achieved a complete cure in two separate experiments, and in combination with both either adjuvant mefloquine or lumefantrine. The average survival time of all mice receiving this new trioxane dimer was significantly higher (56.9 days in combination with mefloquine over two experiments, 60.0 days in combination with lumefantrine over one experiment) than the survival time of all mice receiving artemether combination therapy (39.3 days in combination with mefloquine over two experiments, 19.0 days in combination with lumefantrine over one experiment). The efficient synthesis of these compounds and their high antimalarial efficacy make especially the curative propargyl oxime (31) a leading candidate for further drug development.
Experimental
All reactions were performed under argon in oven-dried or flame-dried glassware. Dichloromethane and tetrahydrofuran were dispensed from an LC Technology Solutions SPBT-1 bench top solvent purification system. All commercially available reagents were purchased from Sigma Aldrich and used as received. All experiments were monitored by analytical thin layer chromatography (TLC) performed on Silicycle silica gel 60 Å glass supported plates with 0.25 mm thickness. Flash chromatography was performed with EMD silica gel (40–63 μM). Automated purification was performed on a Teledyne Isco CombiFlash Rf purification system (referred to as “Isco”) using Teledyne RediSep Rf Gold High Performance pre-packed silica columns. Yields are not optimized. Infrared (IR) spectra were recorded on a Perkin Elmer 1600 FT-IR spectrometer. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance 400 MHz FT-NMR spectrometer (400 MHz for 1H, 100 MHz for 13C) or a Bruker Avance 300 MHz FT-NMR spectrometer (300 MHz for 1H, 75 MHz for 13C). The following abbreviations are used in the experimental section for the description of 1H NMR spectra: singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), broad singlet (bs), doublet of doublets (dd), doublet of triplets (dt), and doublet of quartets (dq). HPLC-MS (electrospray ionization) were acquired on an Agilent Technologies 6130 quadrupole spectrometer coupled to an Agilent Technologies 1200 series HPLC equipped with a Luna C18 3 micron 3×75 mm column. Elution gradient was 4 to 100% acetonitrile (modified with 0.05% TFA) in water over 5 min, unless otherwise noted. High resolution mass spectrum-electron ionization sprary (HRMS-ESI) were obtained on an Agilent Technologies 1200 series Dual Absorbance Detector HPLC system equipped with a Phenomenex Luna 75×3 mm, C18, 3Ym column at 45 °C (UV detection at 220nm, BW 8nm, and 254nm BW 8nm, flow rate: 0.8 mL/min (increasing), Injection volume: 1.0 YL, sample solvent: 100% Methanol, sample conc.: ~0.01 mg/mL, mobile phase A: Water with 0.1% acetic acid, mobile phase B: Acetonitrile with 0.1% acetic acid) coupled to a Agilent 6210 time-of-flight mass spectrometer (ion source: Duel ESI, min range: 115 m/z, max range: 1400 m/z, scan rate: 0.9 seconds, gas temp: 340°C, gas flow: 10 L/min, nebulizer: 50 PSI, ion polarity: positive, VCap: 3500 V, fragmentor: 175 V, skimmer1: 65 V, OctopoleRFPeak: 250 V, ref mass: enabled (Agilent P/N G1969-85001). Data were analyzed using Agilent Masshunter Workstation Data Acquisition (v B.02.00, Patch 1,2,3) and Agilent Masshunter Qualitative Analysis (v B.02.00, Build 2.0.197.7, Patch 3).
All dimers were >95% pure based on HPLC data unless otherwise noted.
Synthesis of C10-α-2,4-dimethoxybenzoate 20
To a 100 mL round bottom flask were added artemisinin (300 mg, 1.06 mmol, 1.0 eq) and CH2Cl2 (10 mL). The stirring solution was cooled to −78 °C for 20 min, at which time diisobutylaluminum hydride (DIBALH, 1.0 M in CH2Cl2, 1.275 mL, 1.27 mmol, 1.2 eq) was added dropwise over 15 min. The reaction mixture was stirred at −78 °C for 2 h (until complete consumption of starting material was seen by TLC). At that time, 4-dimethylaminopyridine (DMAP, 143 mg, 1.17 mmol, 1.1 eq) and pyridine (py, 0.5 mL, 6.38 mmol, 6 eq) were added, followed by 2,4-dimethoxybenzoyl chloride (1.07 mg, 5.31 mmol, 5 eq). The resulting reaction mixture was allowed to gradually warm to rt and stirred overnight. Upon completion, the reaction was quenched with sat. aq. NH4Cl and diluted with EtOAc. The organic layer was washed again with sat. aq. NH4Cl and brine, then separated, dried on MgSO4, filtered, and the solvent was removed under reduced pressure. The residue was purified directly on silica. Gradient elution (10–35% ethyl acetate in hexanes) afforded the desired product as a colorless solid: 421 mg, 98% yield; mp 127.8 °C; 1H NMR (400 MHz, CDCl3) δ 7.96-7.94 (d, 1 H, J = 8.6 Hz), 6.47-6.43 (m, 2 H), 5.97-5.95 (d, 1 H, J = 9.8 Hz), 5.47 (s, 1 H), 3.85 (s, 3 H), 3.83 (s, 3 H), 2.72-2.63 (m, 1 H), 2.40-2.31 (m, 1 H), 2.04-1.98 (m, 1 H), 1.91-1.83 (m, 1 H), 1.81-1.68 (m, 2 H), 1.67-1.60 (m, 1 H), 1.54-1.43 (m, 2 H), 1.40 (s, 3 H), 1.34-1.25 (m, 2 H), 1.15-0.88 (m, 7 H); 13C NMR (100 MHz, CDCl3) δ 164.5, 161.9, 134.5, 104.3, 98.8, 91.8, 91.5, 80.2, 55.8, 55.5, 52.9, 51.7, 45.4, 41.1, 37.2, 36.3, 34.1, 32.0, 25.9, 24.6, 22.0, 20.2, 12.2; HRMS (FAB) m/z for C24H33O8 (M + H)+ calc. = 449.2175, found = 449.2169; FT-IR (cm−1) 3010, 2939, 2876, 1732, 1652, 1608, 1575, 1558, 1540, 1506, 1457, 1418, 1375, 1330, 1267, 1248, 1211, 1164, 1131, 1107, 1086, 1030, 1016, 942, 926, 878, 861, 830, 752; [α]D 23=+26.27 (c=0.645, CHCl3); HPLC tr = 3.93 min.
Synthesis of β-C10-acetylene 21
To a 100 mL round bottom flask were added 20 (500 mg, 1.11 mmol, 1.0eq) and Et2O (30 mL). The slurry was stirred at rt for 10 min until complete dissolution was achieved, and the stirring solution was subsequently cooled to 0 °C. After 10 min, zinc(II) chloride (ZnCl2, 1.0 M in Et2O, 1.34 mL, 1.43 mmol, 1.2 eq) and ethynyl magnesium chloride (0.5 M in THF, 8.03 ml, 4.01 mmol, 3.6 eq) were added dropwise simultaneously from separate syringes. The reaction mixture was stirred at 0 °C for 3 h, and then gradually warmed to rt as the ice bath melted and stirred overnight. Upon completion, the reaction was quenched with H2O and diluted with EtOAc. The organic layer was washed with sat. aq. NH4Cl and brine, then extracted, dried on MgSO4, filtered, and the solvent was removed under reduced pressure. The residue was purified directly on silica. Gradient elution (0 to 20% EtOAc in hexanes) afforded the desired product as a white solid: 242 mg, 74% yield, mp 121.6 °C, 1H NMR (400 MHz, CDCl3) δ 5.56 (s, 1 H), 4.74-4.72 (dd, 1 H, J = 2.5, 5.8 Hz), 2.81-2.70 (m, 1 H), 2.53-2.52 (d, 1 H, J = 2.5 Hz), 2.41-2.30 (m, 1 H), 2.24-2.00 (m, 2 H), 1.92-1.83 (m, 1 H), 1.79-1.62 (m, 2 H), 1.59-1.48 (m, 2 H), 1.42 (s, 3 H), 1.39-1.20 (m, 3 H), 1.00-0.85 (m, 7 H); 13C NMR (100 MHz, CDCl3) δ 104.2, 89.4, 81.4, 80.8, 77.2, 67.1, 52.6, 45.3, 37.3, 36.2, 34.5, 29.8, 26.0, 24.6, 22.9, 20.3, 13.7; HRMS (FAB) m/z for C17H25O4 (M + H)+ calc. = 293.1747, found = 293.1744; FT-IR (cm−1) 2942, 2871, 1506, 1450, 1208, 1136, 1122, 1088, 1055; [α]D 23=+112.57 (c=0.390, CHCl3); HPLC tr = 3.84 min.
Synthesis of artemisinin monomer methyl ketone 22
To a round bottom flask were added monomer acetylene 21 (695 mg, 2.38 mmol, 1.0 eq), water (H2O, 200 μL, 5.0 eq) and acetone (15 mL). para-Toluenesulfonic acid (pTsOH, 597 mg, 1.0 eq) and mercuric acetate [Hg(OAc)2, 547 mg, 0.7 eq] were added to the stirring solution, and the resulting slurry was stirred at rt overnight. Upon completion the reaction mixture was filtered through a short pad of celite and concentrated in vacuo. The residue was purified directly on the Isco Combiflash automated purification system. Gradient elution (0 to 40% ethyl acetate in hexanes) afforded the desired product as a colorless oil: 664 mg, 90%, yield; 1H NMR (300 MHz, CDCl3) δ 5.65 (s, 1 H), 4.75 (d, J = 6.78 Hz, 1 H), 2.93 (ddd, J = 14.03, 7.54, 6.50 Hz, 1 H), 2.38 - 2.24 (m, J = 13.23, 10.69, 3.91, 3.91, 3.91 Hz, 1 H), 2.21 (s, 3 H), 2.08 – 1.98 (m, 1 H), 1.98 - 1.87 (m, 1 H), 1.76 - 1.70 (m, 1 H), 1.70 - 1.65 (m, 2 H), 1.65 - 1.60 (m, 1 H), 1.52 - 1.43 (m, 1 H), 1.40 (s, 3 H), 1.38 - 1.31 (m, 1 H), 1.31 - 1.24 (m, 2 H), 1.24 - 1.15 (m, 1 H), 1.02 (d, J = 7.68 Hz, 3 H), 0.95 (d, J = 5.98 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 210.2, 103.2, 90.1, 80.9, 79.9, 51.8, 44.2, 37.3, 36.4, 34.1, 30.0, 28.8, 25.9, 24.7, 23.7, 20.0, 13.5; ESI-HRMS m/z (M + H)+ for C17H27O5 calc. = 311.1853, found = 311.1854; FT-IR (cm−1) 2930, 2874, 1783, 1711, 1672, 1566, 1548, 1532, 1526, 1514, 1457, 1430, 1366, 1279, 1196, 1102, 1062, 1012, 957, 931, 883; [α]D 25 +70.37 (c = 0.18, CHCl3); HPLC tr = 3.66 min.
Synthesis of artemisinin monomer trimethylsilyl enol ether 23
To a round bottom flask were added artemisin monomer methyl ketone 22 (50 mg, 0.16 mmol, 1.0 eq) and THF (5 mL), and the stirring solution was cooled to −78 °C. Triethylamine (Et3N, 0.045 mL, 0.32 mmol, 2.0 eq) and trimethylsilyl trifluormethanesulfonate (TMSOTf, 0.058 mL, 0.32 mmol, 2.0 eq) were added, and the reaction mixture was stirred at −78 °C for 30 min before warming to rt. Upon completion, the reaction was quenched with sat. aq. NaHCO3 and the product was extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and conc in vacuo. The residue was purified directly on silica gel. Gradient elution (0 – 5% EtOAc in hexanes) afforded the desired compound as a colorless oil: 50 mg, 81% yield; 1H NMR (400 MHz, CDCl3) δ 5.57 (s, 1H), 4.54 (d, J=6.32 Hz, 1H), 4.37 (s, 1H), 4.14 (s, 1H), 2.83-2.65 (m, 1H), 2.29 (dt, J = 3.92, 13.96 Hz, 1H), 2.02-1.92 (m, 1H), 1.87 (tdd, J = 3.41, 6.32, 13.64 Hz, 1H), 1.70-1.47 (m, 4H), 1.42-1.34 (m, 5H), 1.30-1.15 (m, 2H), 0.91 (d, J = 6.57 Hz, 6H), 0.18 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 157.3, 103.1, 90.3, 89.6, 81.1, 74.7, 52.1, 44.4, 37.4, 36.5, 34.3, 26.0, 24.8, 23.3, 20.1, 13.9, 0.1.
Synthesis of dihydroartemisinin acetate 19 has been previously reported.20–22
Synthesis of artemisin two-carbon linked dimer ketone 24
To a round bottom flask were added artemisinin monomer trimethylsilyl enol ether 23 (36.5 mg, 0.095 mmol, 1.0 eq), dihydroartemisinin acetate 19 (31.0 mg, 0.095 mmol, 1.0 eq) and dicholoromethane (CH2Cl2, 1 mL). The stirring solution was cooled to −78 °C, and after 10 min tin IV chloride (SnCl4, 1.0 M in CH2Cl2, 0.095 mL, 0.095 mmol, 1.0 eq, further diluted to 1 mL) was added in one portion. The reaction mixture was stirred at −78 °C for 2 h. Analytical TLC showed 9,10-olefin (elimination product), artemisinin monomer methyl ketone (either residual from TMS enol ether starting material or unreacted enolate), and two new spots (one of which was desired product, the other has yet to be identified). The reaction was quenched with water and the products were extracted with CH2Cl2. The organic layer was washed with brine, dried, filtered, and concentrated under reduced pressure. The residue was purified directly on silica gel. Gradient elution (0 – 15% ethyl acetate in hexanes) afforded the desired product as a colorless solid: 38 mg, 69% yield; mp 135–137 °C; 1H NMR (300 MHz, CDCl3) δ 5.57 (s, 1 H), 5.30 (s, 1 H), 4.84 (ddd, J = 9.51, 6.17, 3.81 Hz, 1 H), 4.57 (d, J = 6.31 Hz, 1 H), 3.18 (dd, J = 15.82, 9.09 Hz, 1 H), 2.99 - 2.85 (m, J = 13.61, 7.58, 7.58, 6.03 Hz, 1 H), 2.76 (dt, J = 13.70, 7.35, 6.36 Hz, 1 H), 2.39 (dd, J = 15.71, 3.74 Hz, 1 H), 2.35 - 2.25 (m, 2 H), 2.07 - 2.02 (m, 1 H), 2.02 – 1.95 (m, 1 H), 1.94 - 1.83 (m, 2 H), 1.82 - 1.72 (m, 2 H), 1.72 - 1.62 (m, 3 H), 1.61 - 1.55 (m, 2 H), 1.53 - 1.43 (m, 1 H), 1.41 (s, 3 H), 1.37 (s, 3 H), 1.36 - 1.32 (m, 1 H), 1.31 - 1.26 (m, 2 H), 1.23 (d, J = 5.79 Hz, 2 H), 1.21 - 1.15 (m, 1 H), 1.10 (d, J = 7.68 Hz, 3 H), 0.95 (d, J = 5.98 Hz, 3 H), 0.92 (d, J = 6.26 Hz, 4 H), 0.84 (d, J = 7.49 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 211.9, 103.5, 103.2, 90.2, 88.9, 80.9, 80.8, 80.1, 71.6, 52.3, 52.1, 44.6, 44.3, 40.0, 37.5, 36.9, 36.5, 36.4, 34.4, 34.3, 30.6, 29.9, 25.9, 25.9, 24.7, 24.6, 23.9, 20.1, 13.7, 13.4; ESI-HRMS m/z (M + Na)+ for C32H48O9Na calc. = 599.3191, found = 99.3184; FT-IR (cm−1) 2937, 2872, 1712, 1651, 1549, 1532, 1525, 1514, 1457, 1194, 1098, 1053, 1018, 941, 876, 829, 753; [α]D 25 +93.98 (c = 0.26, CHCl3); HPLC (4–100% acetonitrile in water over 8 min) tr = 7.04 min.
Synthesis of dimer oximes Z-25 and E-25
To a round bottom flask were added dimer ketone 24 (15.0 mg, 0.03 mmol, 1.0 eq), hydroxyl amine hydrochloride (9.0 mg, 0.13 mmol, 5.0 eq), and pyridine (1 mL). The reaction mixture was stirred at rt overnight. Upon completion as detected by analytical TLC (both isomers were visualized), the reaction mixture was diluted with EtOAc and organic layer was washed twice with 10% citric acid. The aqueous layer was back extracted with EtOAc and the organic layers were pooled and washed with brine. The solvent was removed under reduced pressure and the residue was purified directly on silica gel. Gradient elution (0 to 40% ethyl acetate in hexanes) afforded the desired products as a colorless amorphous solid: Z-25 – 12.5 mg, 81% yield; 1H NMR (300 MHz, CDCl3) δ 5.68 (d, J = 6.90 Hz, 1 H), 5.39 (s, 1 H), 5.37 (s, 1 H), 4.53 (ddd, J = 9.13, 5.91, 3.30 Hz, 1 H), 3.48(s, 1 H), 2.92 (sxt, J = 7.68 Hz, 1 H), 2.82 (dd, J = 13.40, 7.48 Hz, 1 H), 2.54 (dd, J = 14.62, 9.15 Hz, 1 H), 2.45 (dd, J = 14.67, 3.33 Hz, 1 H), 2.37 - 2.24 (m, 2 H), 2.04 - 2.01 (m, 1 H), 2.01 – 1.94 (m, 2 H), 1.91 - 1.81 (m, 2 H), 1.81 - 1.77 (m, 1 H), 1.77 - 1.74 (m, 1 H), 1.74 - 1.69 (m, 1 H), 1.68 - 1.65 (m, 1 H), 1.65 - 1.61 (m, 1 H), 1.57 - 1.51 (m, 1 H), 1.51 - 1.44 (m, 1 H), 1.44 - 1.39 (m, 4 H), 1.39 - 1.36 (m, 3 H), 1.36 - 1.29 (m, 2 H), 1.29 - 1.24 (m, 3 H), 1.24 - 1.19 (m, 1 H), 0.98 - 0.93 (m, 7 H), 0.91 (d, J = 7.48 Hz, 4 H), 0.81 (d, J = 7.63 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 159.9, 103.4, 102.3, 90.4, 88.3, 81.1, 80.7, 74.1, 74.1, 69.6, 52.7, 51.2, 44.8, 43.2, 37.6, 37.3, 36.7, 36.6, 34.6, 34.2, 30.3, 29.5, 29.1, 26.1, 25.8, 25.1, 24.8, 24.6, 20.4, 19.9, 13.6, 12.7; ESI-HRMS m/z (M + H)+ for C32H50NO9 calc. = 592.3480, found = 592.3480; FT-IR (cm−1) 3412, 2968, 2942, 2874, 1660, 1453, 1376, 1204, 1123, 1091, 1054, 1011, 977, 946, 879, 754; [α]D 25 +45.69 (c = 0.625, CHCl3); HPLC (4–100% acetonitrile in water over 8 min) tr = 7.06 min.
E-25
1.4 mg, 9% yield; 1H NMR (300 MHz, CDCl3) δ 5.50 (s, 1H), 5.42 (s, 1H), 5.30 (s, 1H), 5.17 (d, J = 6.40 Hz, 1H), 4.67-4.52 (m, 1H), 2.87-2.74 (m, 2H), 2.62-2.55 (m, 2H), 2.37-2.25 (m, 2H), 2.05-1.79 (m, 4H), 1.76-1.57 (m, 4H), 1.55-1.18 (m, 14H), 1.09-0.78 (m, 16H); 13C NMR (101 MHz, CDCl3) δ 186.6, 160.4, 103.4, 102.9, 90.3, 88.5, 81.0, 81.0, 77.2, 73.8, 73.6, 53.4, 52.6, 51.9, 51.9, 44.7, 44.3, 37.4, 37.3, 36.6, 36.5, 34.6, 34.4, 31.0, 30.4, 26.8, 26.1, 25.8, 24.8, 24.8, 24.6, 24.4, 20.3, 20.1, 13.9, 13.3.
Synthesis of dimer O-methyl oximes Z-26 and E-26
The desired compound was prepared in a similar manner to dimer oxime 25 as described above, substituting O-methyl hydroxylamine hydrochloride for hydroxylamine hydrochloride. Gradient elution on silica gel (0 to 40% ethyl acetate in hexanes) afforded the desired product as a colorless amorphous solid: Z-26 – 11.9 mg, 75% yield; 1H NMR (300 MHz, CDCl3) δ 5.56 (d, J = 6.94 Hz, 1 H), 5.41 (s, 1 H), 5.35 (s, 1 H), 4.55 (ddd, J = 8.93, 5.89, 3.23 Hz, 1 H), 3.83 (s, 3 H), 3.49 (s, 1 H), 2.89 - 2.77 (m, 2 H), 2.55 (dd, J = 14.57, 9.00 Hz, 1 H), 2.41 (dd, J = 14.48, 3.23 Hz, 1 H), 2.31 (qd, J = 14.04, 4.38 Hz, 2 H), 2.04 - 2.01 (m, 1 H), 2.01 – 1.97 (m, 1 H), 1.97 - 1.93 (m, 1 H), 1.91 - 1.83 (m, 1 H), 1.83 - 1.77 (m, 1 H), 1.77 - 1.72 (m, 1 H), 1.72 - 1.68 (m, 1 H), 1.68 - 1.61 (m, 4 H), 1.58 - 1.53 (m, 1 H), 1.53 - 1.48 (m, 1 H), 1.48 - 1.41 (m, 1 H), 1.40 (s, 3 H), 1.38 (s, 3 H), 1.37 - 1.30 (m, 3 H), 1.30 - 1.21 (m, 4 H), 0.95 (dd, J = 5.97, 3.23 Hz, 7 H), 0.92 (d, J=7.48 Hz, 4 H), 0.77 (d, J=7.63 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 158.8, 103.3, 102.3, 90.3, 88.4, 81.1, 80.7, 74.2, 70.0, 61.6, 52.7, 51.2, 44.9, 43.2, 37.5, 37.3, 36.7, 36.6, 34.6, 34.3, 30.4, 30.0, 29.1, 26.1, 25.8, 25.0 24.7, 24.7, 24.6, 20.3, 19.9, 13.6, 12.7; ESI-HRMS m/z (M + H)+ for C33H52NO9 calc. = 606.3637, found = 606.3636; HPLC (4–100% acetonitrile in water over 8 min) tr = 7.54 min.
E-26
1.7 mg, 11% yield; 1H NMR (400 MHz, CDCl3) δ 5.62 (s, 1H), 5.35 (s, 1H), 5.09 (d, J = 6.26 Hz, 1H), 4.61-4.54 (m, 1H), 3.86 (s, 3H), 2.94-2.71 (m, 2H), 2.58 (dd, J = 7.83, 17.22 Hz, 1H), 2.45 (dd, J = 10.37, 13.11 Hz, 1H), 2.32 (dt, J = 3.72, 13.99 Hz, 2H), 2.06-1.96 (m, 2H), 1.94-1.77 (m, 4H), 1.71-1.61 (m, 4H), 1.59-1.48 (m, 6H), 1.46-1.28 (m, 8H), 1.27-1.22 (m, 2H), 1.16-1.05 (m, 1H), 1.00 (d, J = 7.43 Hz, 3H), 0.97-0.93 (m, J = 6.10, 6.10 Hz, 6H), 0.91 (d, J = 7.83 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 215.6, 159.8, 103.3, 103.0, 90.2, 88.4, 81.0, 80.9, 74.2, 73.9, 67.9, 52.6, 52.1, 44.7, 44.7, 37.4, 37.3, 36.6, 34.6, 34.5, 30.9, 30.4, 27.6, 26.0, 25.9, 24.7, 24.7, 24.3, 20.3, 20.1, 14.2, 13.3.
Synthesis of 2C-dimer O-allyl oxime 27
The desired compound was prepared in a similar manner to dimer oxime 25 as described above, substituting O-allyl hydroxylamine hydrochloride hydrate for hydroxylamine hydrochloride. The crude product consisted of a single oxime diastereomer. Gradient elution on silica gel (0 to 40% ethyl acetate in hexanes) afforded the desired diastereomerically pure product as a colorless amorphous solid: 8.2 mg, 75% yield; 1H NMR (300 MHz, CDCl3) δ 6.04 – 5.91 (m, J = 15.99, 10.61, 6.75, 5.43, 5.43, 5.18 Hz, 1 H), 5.63 (d, J = 6.85 Hz, 1 H), 5.40 (s, 1 H), 5.36 (s, 1 H), 5.26 (dd, J = 17.31, 1.71 Hz, 1 H), 5.13 (dd, J = 10.37, 1.52 Hz, 1 H), 4.62 - 4.52 (m, 2 H), 2.94 - 2.75 (m, 2 H), 2.55 (dd, J = 14.67, 8.51 Hz, 1 H), 2.46 (dd, J = 14.67, 3.91 Hz, 1 H), 2.38 - 2.24 (m, J=13.98, 13.98, 13.73, 4.38 Hz, 2 H), 2.05 – 1.94 (m, 3 H), 1.92 - 1.84 (m, 1 H), 1.83 - 1.76 (m, 1 H), 1.76 - 1.69 (m, 2 H), 1.69 - 1.60 (m, 3 H), 1.59 - 1.50 (m, 1 H), 1.50 - 1.41 (m, 1 H), 1.39 (d, J = 4.99 Hz, 6 H), 1.36 - 1.30 (m, 2 H), 1.30 - 1.19 (m, 3 H), 0.96 (d, J = 3.57 Hz, 3 H), 0.95 (d, J = 4.11 Hz, 3 H), 0.91 (d, J = 7.48 Hz, 3 H), 0.79 (d, J =7.63 Hz, 2 H); 13C NMR (100 MHz, CDCl3) δ 159.1, 134.8, 116.5, 103.3, 102.2, 90.4, 90.4, 88.6, 88.5, 81.1, 80.7, 74.6, 73.9, 70.0, 52.6, 51.2, 44.9, 43.2, 37.6, 37.3, 36.7, 36.6, 34.6, 34.3, 30.3, 30.1, 29.2, 26.1, 25.7, 25.1, 24.7, 20.3, 19.9, 13.5, 12.7; ESI-HRMS m/z (M + H)+ for C35H54NO9 calc. = 632.3793, found = 632.3797; FT-IR (cm−1) 2991, 2969, 2920, 1665, 1454, 1376, 1114, 1092, 1056, 1011, 983, 945, 880, 755; [α]D 25 +50.15 (c = 0.40, CHCl3); HPLC (4–100% acetonitrile in water over 8 min) tr = 7.65 min.
Synthesis of dimer O-cyclopropylmethyl oxime Z-28
The desired compound was prepared in a similar manner to dimer oxime 25 as described above, substituting O-methylcyclopropyl hydroxylamine hydrochloride for hydroxylamine hydrochloride. Gradient elution on silica gel (0 to 40% ethyl acetate in hexanes) afforded the desired diastereomerically pure product as a colorless amorphous solid: Z-28 – 9.9 mg, 74% yield; 1H NMR (400 MHz, CDCl3) δ 5.62 (d, J = 6.82 Hz, 1H), 5.40 (s, 1H), 5.36 (s, 1H), 4.61-4.56 (m, 1H), 3.91-3.81 (m, 2H), 2.94-2.77 (m, 2H), 2.55 (dd, J = 9.09, 14.91 Hz, 1H), 2.45 (dd, J = 4.04, 14.65 Hz, 1H), 2.37-2.25 (m, 2H), 2.08-1.95 (m, 4H), 1.91-1.62 (m, 9H), 1.57-1.51 (m, 1H), 1.50-1.37 (m, 6H), 1.37-1.21 (m, 6H), 1.17-1.07 (m, 1H), 0.94 (m, with three doublets, J = 4.55, 5.95, 7.58 Hz, 10H), 0.80 (d, J = 7.58 Hz, 3H), 0.49-0.44 (m, 2H), 0.28-0.23 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 158.6, 103.2, 102.2, 90.4, 88.6, 81.2, 80.7, 78.0, 73.9, 70.1, 52.6, 51.2, 44.8, 43.3, 37.6, 37.4, 36.8, 36.6, 34.6, 34.3, 30.3, 30.1, 29.2, 26.1, 25.7, 25.1, 24.8, 24.7, 24.7, 20.3, 19.9, 13.5, 12.6, 10.6, 2.9, 2.8; ESI-HRMS m/z (M + H)+ for C36H56NO9 calc. = 646.3950, found = 646.3962; FT-IR (cm−1) 3081, 2947, 2920, 2873, 1455, 1376, 1278, 1252, 1224, 1202, 1178, 1114, 1095, 1054, 1010, 943, 881, 823, 756; [α]D 25 +48.24 (c = 0.465, CHCl3).
Synthesis of dimer O-isobutyl oxime Z-29
The desired compound was prepared in a similar manner to dimer oxime Z-25 as described above, substituting O-isobutyl hydroxylamine hydrochloride for hydroxylamine hydrochloride. The crude product consisted of a single oxime diastereomer. Gradient elution on silica gel (0 to 40% ethyl acetate in hexanes) afforded the desired diastereomerically pure product as a colorless amorphous solid: 10.8 mg, 80% yield; 1H NMR (300 MHz, CDCl3) δ 5.63 (d, J = 6.78 Hz, 1H), 5.40 (s, 1H), 5.35 (s, 1H), 4.60-4.53 (m, 1H), 3.86 (dd, J = 6.31, 10.08 Hz, 1H), 3.77 (dd, J = 6.97, 9.98 Hz, 1H), 2.93-2.73 (m, 2H), 2.53 (dd, J = 9.00, 17.71 Hz, 1H), 2.46 (dd, J = 3.77, 15.82 Hz, 1H), 2.39-2.23 (m, 2H), 2.04-1.91 (m, 4H), 1.88-1.61 (m, 6H), 1.58-1.48 (m, 2H), 1.46-1.21 (m, 14H), 1.00-0.86 (m, 18H), 0.78 (d, J = 7.54 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 158.4, 103.2, 102.2, 90.5, 88.5, 81.2, 80.7, 80.4, 74.0, 72.2, 70.0, 52.6, 51.3, 44.9, 43.3, 37.6, 37.4, 36.8, 36.6, 35.6, 34.6, 34.3, 30.4, 30.0, 29.3, 28.0, 26.1, 25.7, 25.1, 24.8, 24.7, 20.3, 19.9, 19.3, 19.3, 13.5, 12.7; ESI-HRMS m/z (M + H)+ for C36H58NO9 calc. = 648.4106, found = 648.4100; FT-IR (cm−1) 2997, 2956, 2920, 2872, 2847, 1458, 1376, 1224, 1202, 1114, 1095, 1048, 1011, 945, 880, 756; [α]D 25 +40.65 (c = 0.515, CHCl3).
Synthesis of dimer O-tert-butyl oxime Z-30
The desired compound was prepared in a similar manner to dimer oxime Z-25 as described above, substituting O-tert-butyl hydroxylamine hydrochloride for hydroxylamine hydrochloride. The crude product consisted of a single oxime diastereomer. Gradient elution on silica gel (0 to 40% ethyl acetate in hexanes) afforded the desired diastereomerically pure product as a colorless amorphous solid: 10.5 mg, 72% yield; 1H NMR (400 MHz, CDCl3) δ 5.58 (d, J = 6.82 Hz, 1H), 5.38 (s, 1H), 5.36 (s, 1H), 4.67 (q, J = 6.32 Hz, 1H), 2.89-2.75 (m, 2H), 2.60 (dd, J = 7.07, 15.66 Hz, 1H), 2.52 (dd, J = 7.07, 15.92 Hz, 1H), 2.37-2.25 (m, 2H), 2.04-1.94 (m, 3H), 1.91-1.86 (m, 1H), 1.84-1.55 (m, 8H), 1.41-1.30 (m, 9H), 1.30-1.20 (m, 12H), 0.97 (d, J = 1.52 Hz, 3H), 0.95 (d, J = 1.77 Hz, 4H), 0.90 (d, J = 7.58 Hz, 4H), 0.76 (d, J=7.58 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 156.9, 103.0, 102.1, 90.5, 89.0, 81.3, 80.8, 77.9, 77.2, 73.4, 69.8, 52.5, 51.3, 44.8, 43.2, 37.1, 34.7, 34.3, 30.6, 30.2, 29.2, 27.6, 26.1, 25.7, 25.1, 24.8, 24.8, 20.3, 19.9, 13.2, 12.6;; ESI-HRMS m/z (M + Na)+ for C36H57NNaO9 calc. = 670.3926, found = 670.3932; FT-IR (cm−1) 2970, 2948, 2922, 2873, 2848, 1453, 1377, 1255, 1197, 1112, 1096, 1051, 1010, 964, 938, 882, 821, 756; [α]D 25 +31.91 (c = 0.525, CHCl3).
Synthesis of dimer propargyl oxime Z-31
To a 1 dram vial were added dimer oxime Z-25 (4.8 mg, 0.01 mmol, 1.0 eq) and tetrahydrofuran (THF, 1 mL). The stirring solution was cooled to 0 °C in an ice bath, and sodium hydride (NaH, 0.3 mg, 1.5 eq) was added as a dry powder, and the reaction mixture was stirred for 20 min. At that time, propargyl bromide (2 μL, 1.5 eq) was added and the reaction mixture was stirred overnight, gradually warming to room temperature. Upon completion the reaction mixture was quenched with water and diluted with EtOAc. The organic layer was washed with sat. aq. NH4Cl and brine, extracted, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The residue was purified directly on silica gel. Gradient elution (0 to 50% ethyl acetate in hexanes) afforded the desired product as a colorless amorphous solid: 4.1 mg, 60% yield; 1H NMR (300 MHz, CDCl3) δ 5.60 (d, J = 6.88 Hz, 1 H), 5.41 (s, 1 H), 5.36 (s, 1 H), 4.71 - 4.60 (m, J = 15.85, 3.47, 2.40, 2.40 Hz, 2 H), 4.58 (td, J = 5.73, 3.00 Hz, 1 H), 2.95 - 2.78 (m, 2 H), 2.58 (dd, J = 14.53, 8.59 Hz, 1 H), 2.51 - 2.44 (m, J = 14.53, 3.66 Hz, 1 H), 2.39 (t, J = 2.40 Hz, 1 H), 2.38 - 2.25 (m, J = 14.04, 14.04, 13.77, 4.39 Hz, 2 H), 2.07 – 1.94 (m, 2 H), 1.93 - 1.85 (m, 1 H), 1.85 - 1.78 (m, 1 H), 1.78 - 1.70 (m, 1 H), 1.70 - 1.65 (m, 1 H), 1.65 - 1.59 (m, 4 H), 1.59 - 1.49 (m, 2 H), 1.48 - 1.42 (m, 1 H), 1.40 (d, J = 6.25 Hz, 7 H), 1.38 - 1.30 (m, 4 H), 1.30 - 1.20 (m, 4 H), 0.97 (dd, J = 6.03, 3.06 Hz, 7 H), 0.93 (d, J = 7.52 Hz, 4 H), 0.82 (d, 3 H); 13C NMR (100 MHz, CDCl3) δ 160.4, 103.3, 102.3, 90.4, 88.5, 81.1, 80.7, 80.5, 74.0, 73.7, 70.0, 61.3, 52.6, 51.2, 44.9, 43.2, 37.6, 37.3, 36.7, 36.6, 34.6, 34.2, 30.3, 30.1, 29.2, 26.1, 25.7, 25.1, 24.7, 20.3, 19.9, 13.6, 12.7; ESI-HRMS m/z (M + H)+ for C35H52NO9 calc. = 630.3637, found = 630.3613; FT-IR (cm−1) 2955, 2872, 1631, 1455, 1375, 1051, 1008, 756; [α]D 25 +55.72 (c = 0.105, CHCl3).
Synthesis of dimer oxime dimethylcarbamate Z-32
The desired compound was prepared in a similar manner to propargyl oxime Z-31 as described above, substituting dimethylcarbamyl chloride for propargyl bromide. Gradient elution on silica gel (10 to 100% ethyl acetate in hexanes) afforded the desired diastereomerically pure product as a colorless amorphous solid: 4.1 mg, 76% yield; 1H NMR (300 MHz, CDCl3) δ 5.73 (d, J = 6.88 Hz, 1 H), 5.47 (s, 1 H), 5.38 (s, 1 H), 4.60 - 4.52 (m, 1 H), 2.98 (s, 6 H), 2.94 - 2.85 (m, 2 H), 2.85 - 2.78 (m, 1 H), 2.51 (dd, J = 13.96, 2.27 Hz, 1 H), 2.38 - 2.24 (m, 2 H), 2.08 – 1.95 (m, 3 H), 1.91 - 1.80 (m, 2 H), 1.80 - 1.72 (m, 2 H), 1.72 - 1.62 (m, 7 H), 1.56 - 1.46 (m, 2 H), 1.46 - 1.41 (m, 1 H), 1.41 - 1.37 (m, 8 H), 1.37 - 1.33 (m, 2 H), 1.33 - 1.18 (m, 5 H), 1.00 – 0.93 (m, 10 H), 0.86 (d, J = 7.64 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 156.5, 155.3, 103.4, 102.4, 90.7, 88.3, 81.1, 80.7, 73.9, 70.4, 52.7, 51.1, 44.8, 43.2, 37.5, 36.6, 36.6, 36.0, 35.9, 34.5, 34.1, 30.5, 30.3, 30.1, 25.7, 24.9, 24.6, 24.6, 20.3, 19.9, 13.7, 12.6; ESI-HRMS m/z (M + Na)+ for C35H54N2O10 calc. 685.3671, found = 685.3680.
Synthesis of dimer oxime acetate Z-33
The desired compound was prepared in a similar manner to propargyl oxime Z-31 as described above, substituting acetyl chloride for propargyl bromide. Gradient elution on silica gel (10 to 80% ethyl acetate in hexanes) afforded the desired product as a colorless amorphous solid: 8.5 mg, 99% yield; 1H NMR (300 MHz, CDCl3) δ 5.67 (d, J = 6.88 Hz, 1 H), 5.43 (s, 1 H), 5.37 (s, 1 H), 4.59 - 4.52 (m, 1 H), 2.92 - 2.81 (m, 2 H), 2.76 (dd, J = 14.02, 9.66 Hz, 1 H), 2.54 (dd, J = 14.02, 2.72 Hz, 1 H), 2.36 - 2.26 (m, 2 H), 2.16 (s, 3 H), 2.06 – 1.95 (m, 3 H), 1.87 (ddd, J = 10.22, 6.74, 3.38 Hz, 1 H), 1.84 - 1.81 (m, 1 H), 1.81 - 1.761 (m, 1 H), 1.76 - 1.70 (m, J = 13.46, 3.60, 3.60, 3.47 Hz, 1 H), 1.68 - 1.62 (m, 2 H), 1.53 (dt, J = 13.63, 4.46 Hz, 1 H), 1.49 - 1.43 (m, 1 H), 1.43 - 1.35 (m, 7 H), 1.35 - 1.29 (m, 3 H), 1.29 - 1.18 (m, 3 H), 1.00 – 0.95 (m, 6 H), 0.94 (d, J = 7.14 Hz, 6 H), 0.81 (d, J = 7.64 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 168.5, 168.04, 103.4, 102.4, 90.4, 88.3, 81.0, 80.7, 73.9, 70.4, 52.7, 51.1, 44.8, 43.2, 37.5, 36.6, 36.6, 34.5, 34.1, 30.4, 30.3, 30.0, 25.7, 25.0, 24.6, 24.6, 20.3, 19.9, 19.8, 13.7, 12.6; ESI-HRMS m/z (M + H)+ for C34H52NO10 calc. 634.3586, found = 634.3564.
Synthesis of dimer oxime dimethyl phosphate Z-34
To a 1 dram vial were added dimer oxime Z-25 (7.0 mg, 0.01 mmol, 1.0 eq) and dichloromethane (CH2Cl2, 1 mL), followed by triethylamine (Et3N, 3 μL, 0.02 mmol, 2.0 eq) and dimethyl chlorophosphate (3 μL, 0.02 mmol, 2.0 eq). The reaction mixture was stirred overnight at rt. Upon completion the reaction mixture is further diluted with DCM and washed with sat. aq. NH4Cl and brine. The organic layer was extracted, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The residue was purified directly on silica gel. Gradient elution (10 to 100% ethyl acetate in hexanes) afforded the desired product as a colorless amorphous solid: 2.8 mg, 37% yield; 1H NMR (300 MHz, CDCl3) δ 5.72 (d, J = 6.88 Hz, 1 H), 5.40 (s, 1 H), 5.38 (s, 1 H), 4.52 (ddd, J = 9.39, 6.02, 3.41 Hz, 1 H), 3.96 - 3.84 (m, 6 H), 2.97 - 2.89 (m, J = 15.28, 7.83, 7.58 Hz, 1 H), 2.89 - 2.83 (m, 1 H), 2.68 (dd, J = 14.08, 9.47 Hz, 1 H), 2.59 (dd, J = 14.15, 3.35 Hz, 1 H), 2.39 - 2.24 (m, 2 H), 2.06 – 1.97 (m, 3 H), 1.92 - 1.83 (m, 2 H), 1.83 - 1.79 (m, 1 H), 1.79 - 1.71 (m, 2 H), 1.71 - 1.62 (m, 10 H), 1.56 (ddd, J = 14.04, 4.82, 4.71 Hz, 1 H), 1.51 - 1.42 (m, 1 H), 1.40 (s, 4 H), 1.36 (s, 4 H), 1.35 - 1.30 (m, 2 H), 1.30 - 1.21 (m, 5 H), 0.97 (dd, J = 5.97, 3.88 Hz, 8 H), 0.93 (d, J = 7.52 Hz, 4 H), 0.85 (d, J = 7.58 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 168.5, 168.4, 103.2, 102.3, 90.4, 88.3, 81.0, 80.6, 73.44, 70.0, 64.6, 64.6, 54.8, 54.2, 52.7, 51.1, 44.7, 43.2, 37.5, 37.3, 36.7, 36.6, 34.6, 34.2, 30.2, 30.0, 29.7, 26.1, 25.7, 25.0, 24.8, 24.7, 20.3, 19.8, 13.5, 12.6; ESI-HRMS m/z (M + H)+ for C34H55NO12P calc. 700.3456, found = 700.3487.
Synthesis of dimer oxime diethyl phosphate Z-35
The desired compound was prepared in a similar manner to 2C-dimer-oxime-diethyl phosphate Z-34 as described above, substituting diethyl chlorophosphate for dimethyl chlorophosphate. Gradient elution on silica gel (10 to 100% ethyl acetate in hexanes) afforded the desired product as a colorless amorphous solid: 5.4 mg, 63% yield; 1H NMR (300 MHz, CDCl3) δ 5.70 (d, J = 6.92 Hz, 1 H), 5.39 (s, 1 H), 5.37 (s, 1 H), 4.52 (ddd, J = 9.15, 5.83, 3.44 Hz, 1 H), 4.35 - 4.17 (m, 4 H), 2.99 - 2.80 (m, 2 H), 2.75 - 2.64 (m, J = 14.13, 9.04 Hz, 1 H), 2.57 (dt, J = 14.18, 3.44 Hz, 1 H), 2.41 - 2.24 (m, 2 H), 2.06 – 1.95 (m, 3 H), 1.93 - 1.83 (m, 2 H), 1.83 - 1.76 (m, 2 H), 1.75 - 1.63 (m, 5 H), 1.61 - 1.50 (m, 1 H), 1.50 - 1.41 (m, 1 H), 1.41 - 1.33 (m, 14 H), 1.33 - 1.20 (m, 5 H), 1.01 - .95 (m, 7 H), 0.93 (d, J = 7.49 Hz, 4 H), 0.85 (d, J = 7.58 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 168.6, 168.4, 103.3, 102.3, 90.4, 88.3, 81.1, 80.6, 73.44, 70.0, 65.1, 65.0, 64.6, 64.6, 52.7, 51.1, 44.7, 43.2, 37.5, 37.3, 36.7, 36.6, 34.6, 34.2, 30.2, 30.0, 29.7, 26.1, 25.7, 25.0, 24.8, 24.7, 20.3, 19.8, 16.2, 13.5, 12.6; ESI-HRMS m/z (M + Na)+ for C36H58NNaO12P calc. 750.3589, found = 750.3618.
Supplementary Material
Acknowledgments
We thank the NIH (grant R37 AI 34885 to G. H.P.), the Johns Hopkins Malaria Research Institute, and the Bloomberg Family Foundation for financial support.
Abbreviations Used
- ACT
artemisinin combination therapy
- SAR
structure-activity relationships
- DHA
dihydroartemisinin
- LG
leaving group
- DHA-OAc
dihydroartemisinin acetate
- DIBALH
diisobutylaluminum hydride
- TMS
trimethylsilyl
- pTsOH
para-toluenesulfonic acid
- LDA
lithium diisopropylamide
Footnotes
Associated Content
Crystallographic information and tabular spectral data are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.W.H.O. [(August 13, 2012).];Malaria fact sheet No 94. http://www.who.int/mediacentre/factsheets/fs094/en/index.html.
- 2.Murray CJL, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet. 2012;379:413–431. doi: 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- 3.Frey C, Traore C, De Allegri M, Kouyate B, Muller O. Compliance of young children with ITN protection in rural Burkina Faso. Malaria J. 2006;5:1–8. doi: 10.1186/1475-2875-5-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The RTS, S Clincal; Trials; Partnership. First results of phase 3 trial of RTS S/AS01 malaria vaccine in African children. N Engl J Med. 2011;365:1863–1875. doi: 10.1056/NEJMoa1102287. [DOI] [PubMed] [Google Scholar]
- 5.Schlitzer M. Malaria chemotherapeutics. Part I: History of antimalarial drug development, currently used therapeutics, and drugs in clinical development. Chem Med Chem. 2007;2:944–986. doi: 10.1002/cmdc.200600240. [DOI] [PubMed] [Google Scholar]
- 6.Miller LH, Su X-z. Artemisinin: Discovery from the Chinese Herbal Garde. Cell. 2011;146:855–858. doi: 10.1016/j.cell.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He R, Mott BT, Rosenthal AS, Genna DT, Posner GH, Arav-Boger R. An artemisinin-derived dimer has highly potent anti-cytomegalovirus (CMV) and anti-cancer activities. PLoS One. 2011;6:e24334. doi: 10.1371/journal.pone.0024334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kyle DE, Teja-Isavadharm P, Li Q, Leo K. Pharmacokinetics and pharmacodynamics of qinghaosu derivatives: how do they impact on the choice of drug and the dosage regimens? Med Trop. 1998;58:38–44. [PubMed] [Google Scholar]
- 9.Coartem® GSK. [(September 6, 2012).]; http://www.coartem.com/coartem-riamet.htm.
- 10.M. M. V. Pyramax® (pyronaridine-artesunate) [September 6, 2012]; http://www.mmv.org/research-development/project-portfolio/pyramax%C2%AE-pyronaridine-artesunate.
- 11.Haynes RK, Fugmann B, Stetter J, Rieckmann K, Hans-Dietrich H, Chan HW, Cheung MK, Lam WL, Wong HN, Croft SL, Vivas L, Rattray L, Stewart L, Peters W, Robinson BL, Edstein MD, Kotecka B, Kyle DE, Beckermann B, Gerisch M, Radtke M, Schmuck G, Steinke W, Wollborn U, Schmeer K, Romer A. Artemisone - a highly active antimalarial drug of the artemisinin class. Angew Chem Int Ed. 2006;45:2082–2088. doi: 10.1002/anie.200503071. [DOI] [PubMed] [Google Scholar]
- 12.Araujo NCP, Barton V, Jones M, Stocks PA, Ward SA, Davies J, Bray PG, Shone AE, Cristiano MLS, O’Neill PM. Semi-synthetic and synthetic 1,2,4-trioxaquines and 1,2,4-trioxolaquines: synthesis, preliminary SAR and comparison with acridine endoperoxide conjugates. Bioorg Med Chem Lett. 2009;19:2038–2043. doi: 10.1016/j.bmcl.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Pacorel B, Leung SC, Stachulski AV, Davies J, Vivas L, Lander H, Ward SA, Kaiser M, Brun R, O’Neill PM. Modular Synthesis and in vitro and in vivo Antimalarial Assessment of C-10 Pyrrole Mannich Base Derivatives of Artemisinin. J Med Chem. 2010;53:633–640. doi: 10.1021/jm901216v. [DOI] [PubMed] [Google Scholar]
- 14.Chadwick J, Jones M, Mercer AE, Stocks PA, Ward SA, Park BK, O’Neill PM. Design, synthesis and antimalarial/anticancer evaluation of spermidine linked artemisinin conjugates designed to exploit polyamine transporters in Plasmodium falciparum and HL-60 cancer cell lines. Bioorg Med Chem. 2010;18:2586–2597. doi: 10.1016/j.bmc.2010.02.035. [DOI] [PubMed] [Google Scholar]
- 15.Begue JP, Bonnet-Delpon D. Fluoroartemisinins: metabolically more stable antimalarial artemisinin derivatives. Chem Med Chem. 2007;2:608–624. doi: 10.1002/cmdc.200600156. [DOI] [PubMed] [Google Scholar]
- 16.Singh C, Hassam M, Verma VP, Singh AS, Naikade NK, Puri SK, Maulik PR, Kant R. Bile acid-based 1,2,4-trioxanes: synthesis and antimalarial assessment. J Med Chem. 2012;55:10662–10673. doi: 10.1021/jm301323k. [DOI] [PubMed] [Google Scholar]
- 17.Dong Y, Chollet J, Matile H, Charman SA, Chiu FCK, Charman WN, Scorneaux B, Urwyler H, Tomas JS, Schuerer C, Snyder C, Dorn A, Wang X, Karle JM, Tang Y, Wittlin S, Brun R, Vennerstrom JL. Spiro and dispiro-1,2,4-trioxolanes as antimalarial peroxides: Charting a workable structure-activity relationship using simple prototypes. J Med Chem. 2005;48:4953–4961. doi: 10.1021/jm049040u. [DOI] [PubMed] [Google Scholar]
- 18.Charman SA, Arbe-Barnes S, Bathurst IC, Brun R, Campbell M, Charman WN, Chiu FCK, Chollet J, Craft JC, Creek DJ, Dong Y, Matile H, Maurer M, Morizzi J, Nguyen T, Papastogiannidis P, Scheurer C, Shackleford DM, KS, Stingelin L, Tang Y, Urwyler H, Wang X, White KL, Wittlin S, Zhou L, Vennerstrom JL. Synthetic ozonide drug candidate OZ439 offers new hope for a singledose cure of uncomplicated malaria. Proc Nat Acad Sci. 2011;108:4400–4405. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slack RD, Jacobine AM, Posner GH. Antimalarial peroxides: advances in drug discovery and design. Med Chem Commun. 2012:3. [Google Scholar]
- 20.Moon DK, Tripathi A, Sullivan D, Siegler MA, Parkin S, Posner GH. A single, low, oral dose of a 5-carbon-linked trioxane dimer orthoester plus mefloquine cures malaria-infected mice. Bioorg Med Chem Lett. 2011;21:2773–2775. doi: 10.1016/j.bmcl.2010.09.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paik IH, Xie S, Shapiro TA, Labonte T, Narducci-Sarjeant AA, Baege AC, Posner GH. Second generation, orally active, antimalarial, artemisinin-derived trioxane dimers with high stability, efficacy, and anticancer activity. J Med Chem. 2006;49:2731–2734. doi: 10.1021/jm058288w. [DOI] [PubMed] [Google Scholar]
- 22.Rosenthal AS, Chen X, Liu JO, West DC, Hergenrother PJ, Shapiro TA, Posner GH. Malaria-infected mice are cured by a single oral dose of new dimeric trioxane sulfones which are also selectively and powerfully cytotoxic to cancer cells. J Med Chem. 2009;52:1198–1203. doi: 10.1021/jm801484v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woodard LE, Chang W, Chen X, Liu JO, Shapiro TA, Posner GH. Malaria-infected mice live until at least day 30 after a new monomeric trioxane combined with mefloquine are administered together in a single low oral dose. J Med Chem. 2009;52:7458–7462. doi: 10.1021/jm9005934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Posner GH, Murray C, O’Dowd H, Xie S, Shapiro TA. Preparation of artemisinin analogs having antimalarial, antiproliferative, and antitumor activities. 2000042046 A1 20000720. PCT Int Appl WO. 2000
- 25.Galal AM, Ahmad MS, El-Feraly FS, McPhail AT. Preparation and characterization of a new artemisinin-derived dimer. J Nat Prod. 1996;59:917–920. [Google Scholar]
- 26.Pu YM, Ziffer H. Synthesis and antimalarial activities of 12β-Allyldeoxoartemisinin and its derivatives. J Med Chem. 1995;38:613–616. doi: 10.1021/jm00004a007. [DOI] [PubMed] [Google Scholar]
- 27.Ma J, Katz E, Kyle DE, Ziffer H. Syntheses and antimalarial activities of 10-substituted deoxoartemisinins. J Med Chem. 2000;43:4228–4232. doi: 10.1021/jm000195l. [DOI] [PubMed] [Google Scholar]
- 28.Jung M, Lee K, Kendrick H, Robinson BL, Croft SL. Synthesis, stability, and antimalarial activity of new hydrolytically stable and water-soluble (+)-deoxoartelinic acid. J Med Chem. 2002;45:4940–4944. doi: 10.1021/jm020244p. [DOI] [PubMed] [Google Scholar]
- 29.Posner GH, Ploypradith P, Parker MH, O’Dowd H, Woo SH, Northrop J, Krasavin M, Dolan P, Kensler TW, Xie S, Shapiro TA. Antimalarial, antiproliferative, and antitumor activities of artemisinin-derived, chemically robust, trioxane dimers. J Med Chem. 1999;42:4275–4280. doi: 10.1021/jm990363d. [DOI] [PubMed] [Google Scholar]
- 30.Hevia E, Chua JZ, Garcia-Alvarez P, Kennedy AR, McCall MD. Exposing the hidden complexity of stoichiometric and catalytic metathesis reactions by elucidation of Mg-Zn hybrids. Proc Nat Acad Sci. 2010;107:5294–5299. doi: 10.1073/pnas.0913307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hatano M, Suzuki S, Ishihara K. Highly chemoselective stoichiometric alkylation of ketones with Grignard reagent derived Zinc(II)ate complexes. Synlett. 2010;2:321–324. [Google Scholar]
- 32.Lepore SD, Mondal D. Recent advances in heterolytic nucleofugal leaving groups. Tetrahedron. 2007;63:5103–5122. doi: 10.1016/j.tet.2007.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Musilek K, Dolezal M, Gunn-Moore F, Kuca K. Design, evaluation and structure-activity relationship studies of the AChE reactivators against organophosphorus pesticides. Med Res Rev. 2009;31:548–575. doi: 10.1002/med.20192. [DOI] [PubMed] [Google Scholar]
- 34.Krivogorksy B, Grundt P, Yolken R, Jones-Brando L. Inhibition of Toxoplasma gondii by indirubin and tryptanthrin Analogs. Antimicrob Agents Chemother. 2008;52:4466–4469. doi: 10.1128/AAC.00903-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibson CL, Huggan JK, Kennedy A, Kiefer L, Lee JH, Suckling CJ, Clements C, Harvey AL, Hunter WN, Tulloch LB. Diversity oriented syntheses of fused pyrimidines designed as potential antifolates. Org Biomol Chem. 2009;7:1829–1842. doi: 10.1039/b818339b. [DOI] [PubMed] [Google Scholar]
- 36.Huttunen KM, Kumpulainen H, Leppanen J, Rautio J, Jarvinen T, Vepsalainen J. Efficient strategy to prepare water-soluble prodrugs of ketones. Synlett. 2006;5:701–704. [Google Scholar]
- 37.Venhuis BJ, Dijkstra D, Wustrow D, Meltzer LT, Wise LD, Johnson SJ, Wikstrom HV. Orally active oxime derivatives of the dopaminergic prodrug 6-(N,N-di-n-propylamino)-3,4,5,6,7,8-hexahydro-2H-naphthalen-1-one. Synthesis and pharmacological activity. J Med Chem. 2003;46:4136–4140. doi: 10.1021/jm0307786. [DOI] [PubMed] [Google Scholar]
- 38.Kumpulainen H, Mahonen N, Laitinen ML, Jaurakkajarvi M, Raunio H, Juvonen RO, Vepsalainen J, Jarvinen T, Rautio J. Evaluation of hydroxyimine as cytochrome P450-selective prodrug structure. J Med Chem. 2006;49:1207–1211. doi: 10.1021/jm0510124. [DOI] [PubMed] [Google Scholar]
- 39.Prokai L, Wu WM, Somogyi G, Bodor N. Ocular delivery of the β-Adrenergic antagonist alprenolol by sequential bioactivation of its methoxime analogue. J Med Chem. 1995;38:2018–2020. doi: 10.1021/jm00011a021. [DOI] [PubMed] [Google Scholar]
- 40.Haynes RK, Chan HW, Cheung MK, Lam WL, Soo MK, Tsang HW, Voerste A, Williams ID. C-10 Ester and ether derivatives of dihydroartemisinin - 10-α artesunate, preparation of authentic 10-β artesunate, and of other ester and ether derivatives bearing potential aromatic intercalating groups at C-10. Eur J Org Chem. 2002;1:113–132. [Google Scholar]
- 41.Karplus M. Contact Electron-Spin Coupling of Nuclear Magnetic Moments. J Chem Phys. 1959;30:11–15. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.