

Abstract
The BIO14.6 hamster carries a mutation in the delta sarcoglycan gene causing muscular dystrophy and cardiomyopathy. The disease can be prevented by systemic delivery of delta sarcoglycan cDNA using adeno-associated viruses (AAVs). However, all AAVs also target the liver, raising concerns about their therapeutic efficacy in human applications. We compared the AAV2/8 with the chimeric AAV2/2i8, in which the 585-QQNTAP-590 motif of the AAV8 serotype was added to the heparan sulfate receptor footprint of the AAV2 strain. Both vectors carrying the human delta sarcoglycan cDNA were delivered into 24 14-day-old BIO14.6 hamsters. We followed transgene expression in muscle and liver for 7 months. We detected a sustained ectopic expression of delta sarcoglycan in the liver when using AAV2/8 but not AAV2/2i8. Genomic copies of AAV2/2i8 were not detectable in the liver, while at least 100-fold more copies of AAV2/8 were counted. In contrast, the hamster skeletal muscle expressed more delta sarcoglycan using AAV2/2i8 and were still healthy after 7 months at the lower dosage. We conclude that this chimeric vector is a robust option for safer and longer-term diseased muscle targeting.
Rotundo and colleagues generate a chimeric adeno-associated virus (AAV) 2/8 vector called AAV2/2i8, in which the 585-QQNTAP-590 motif of the AAV8 serotype is added to the heparan sulfate receptor footprint of the AAV2 strain. They show that muscle delivery of AAV2/2i8 works in the BIO14.6 hamster at lower dosages than AAV2/8 and that this occurs without any liver targeting, even within the context of intraperitoneal injections.
Introduction
The BIO14.6 hamster displays an absence of delta sarcoglycan (DSG) from the muscle membrane (Nigro et al., 1997), followed by a deficiency of alpha, beta, and gamma sarcoglycan, reproducing the human limb-girdle muscular dystrophy type 2F (LGMD2F) phenotype (Nigro et al., 1996a). This is the most severe form of LGMD, with onset in early childhood (Piluso et al., 2010; Blain and Straub, 2011; Nigro et al., 2011). In the last few years, viral gene delivery has become a promising experimental therapeutic strategy in humans. Recombinant adeno-associated viruses (AAVs) have been identified as one of the best options for gene delivery as they provide significantly longer transgene expression and do not exhibit pathogenicity in humans (Berns and Linden, 1995; Hasbrouck and High, 2008; Mingozzi and High, 2011). In previous studies, we demonstrated that long-life hamster disease rescue was achieved using systemic administration of AAV2/1, 2/8, or 2/9 (Vitiello et al., 2009) and that steroid treatment was unfavorable (Rotundo et al., 2011).
Before proposing systemic use in humans, it will be necessary to adapt the AAV vector tropism to particular cells in order to enhance infection while reducing transduction of nontarget tissues and unwanted vector dissemination (Shi et al., 2001). Even if transgene expression can be restricted by using target-specific promoters (Lu, 2004; Wang et al., 2008), the presence of virus particles outside the targeted tissue raises a number of issues. Almost all AAVs tested so far targeted the liver (Zincarelli et al., 2008; Paneda et al., 2009). This suggests that long-term effects of capsid sequences on liver tissue cannot be ruled out.
Clinical trials of AAV Factor IX treatment in hemophilia B patients have shown that therapeutic levels of transgenes decreased steadily with a parallel increment of transaminases in the liver during the weeks following treatment (Manno et al., 2006). This result is very likely due to a cellular immune response to specific epitopes in the AAV capsid. Consequently, only low systemic AAV dosages have been authorized to date. However, such dosages are not effective to correct most genetic defects (Manno et al., 2006).
Muscle tissue tropism displayed by various AAVs, such as AAV2/8 (Wang et al., 2005) and AAV2/9, is beneficial for gene transfer by systemic delivery. However, these AAVs all have a propensity to accumulate within the liver (Zincarelli et al., 2008). It is thus necessary to develop a new generation of engineered AAV strains using molecular genetic tools (Yang et al., 2011; Asokan et al., 2012).
AAV2 serotype is able to infect both dividing and quiescent cells with a tropism for different tissues and organs such as the liver. Liver targeting is mediated by the region around residue 520 or residues 560–590, corresponding to the heparan sulfate (HS) binding site (Hileman et al., 1998; Summerford and Samulski, 1998; Wu et al., 2000; Shi et al., 2001; Levy et al., 2009) that acts as a primary receptor.
To develop AAV vectors with improved tropism for clinical applications, Asokan and colleagues (Asokan et al., 2010) reengineered the heparan sulfate receptor footprint on the AAV2 capsid surface. The heparan sulfate footprint on the AAV2 capsid is composed of the basic amino acid residues R484, R487, K527, K532, R585, and R588, which form a continuous basic patch (Levy et al., 2009). The R585 and R588 residues within the so-called GH loop form the inner walls of the spikes located on the icosahedral threefold axis, while the other residues occupy the floor surrounding these regions (Levy et al., 2009). Mutations of either R585 or R588 disrupt the basic cluster and prevent heparan sulfate binding (Opie et al., 2003).
AAV2/8 ubiquitously transduces muscle and liver tissue with high efficacy, while AAV2/2 preferentially transduces liver. Substituting 585-RGNRQA-590 residues in the AAV2 capsid with 585-QQNTAP-590 motif of the AAV8 results in disruption of the continuous basic patch formed by the cluster of arginine and lysine residues. This chimeric vector was called AAV2i8, but for consistency, we will designate it as AAV2/2i8 henceforth. In mice, AAV2/2i8 vector transduces cardiac and skeletal muscle tissue with high efficacy similar to that of AAV2/8 and is liver-detargeted (Asokan, 2010).
The peculiar tissue tropism of AAV2/2i8 compared to any naturally occurring AAV is relevant for gene therapy in muscular dystrophies, which requires the transduction of a wide range of muscle types after systemic administration (Gregorevic et al., 2004).
Materials and Methods
Mutagenesis and vector production
The chimeric 2i8 capsid was generated from an AAV2 capsid protein using the QuickChange II site-directed mutagenesis kit (Stratagene). For 585-QQNTAP-590 mutation the mutagenic sense primer was 5′-ctaccaacctccagcaacagaacacagcaccagctaccgcagatg-3′ (Asokan et al., 2010).
AAV vector construction and production
The human δ-SG gene was cloned into the plasmid pAAV-2/1CMV-EGFP. A recombinant AAV vector containing human DSG cDNA, driven by the cytomegalovirus (CMV) promoter, was constructed by standard cloning protocols and was packaged into the AAV2/8 and AAV2/2i8 serotypes. The resulting pAAV2/1-CMV-delta-SG was transfected into subconfluent 293 cells along with the pAd-Helper and AAV2/8 and AAV2/2i8 packaging plasmids, as described (Gao et al., 2002). The recombinant AAV2/8 and AAV2/2i8 vectors were purified by two rounds of caesium chloride (CsCl). Vector titers were assessed by real-time polymerase chain reaction (PCR) (ABI 7900 Real Time PCR System) as described (Gao et al., 2000), obtaining a concentration of 3.2×1012 and 2×1012 GC/ml for two AAV2/8 preparations, as well as 1.6×1012 and 2.7×1012 GC/ml for two AAV2/2i8 preparations. To minimize possible variations caused by AAV preparation, all AAV batches were pooled together and used in animal treatment.
Experimental animals
All animals were male Syrian hamsters, either BIO14.6 strain hamsters (cardiomyopathic) or BIO golden Syrian control hamsters. They were purchased from Bio Breeders, Inc., (www.biobreeders.com), which guarantees a homogeneous pure genetic background. All procedures on wild-type (WT) BIO golden Syrian and dystrophic BIO14.6 hamsters were approved by the “Ministero della Salute” committee in Rome, Italy. The study was carried out in conformity with the European Commission Directive 86/609/EEC.
Protocol design
We tested different AAV serotypes and AAV dosages as follows. All hamsters were intraperitoneally injected using a standard volume of 600 μL. Group AAV2/8max (n=8): We injected into 2-week-old BIO14.6 hamsters at a dosage of 5×1013 genomic copies (GC)/kg, corresponding to about 1×1012 GC/hamster. Group AAV2/8mid (n=8): We injected AAV2/8-CMV-hSCGD into 2-week-old BIO14.6 hamsters at a dosage of 1.5×1013 GC/kg, corresponding to about 3.3×1011 GC/hamster. Group AAV2/8min (n=8): We injected AAV2/8-CMV-hSCGD into 2-week-old BIO14.6 hamsters at a dosage of 5×1012 GC/kg, corresponding to about 1×1011 GC/hamster. Group AAV2/2i8max (n=8): We injected AAV2/2i8-CMV-hSCGD into 2-week-old BIO14.6 hamsters at a dosage of 5×1013 GC/kg, corresponding to about 1×1012 GC/hamster. Group AAV2/2i8mid (n=8): We injected AAV2/2i8-CMV-hSCGD into 2-week-old BIO14.6 hamsters at a dosage of 1.5×1013 GC/kg, corresponding to about 3.3×1011 GC/hamster. Group AAV2/2i8min (n=8): We injected AAV2/2i8-CMV-hSCGD into 2-week-old BIO14.6 hamsters at a dosage of 5×1012/kg, corresponding to about 1×1011 GC/hamster.
AAV vector genome distribution
Genomic DNA was extracted from 100 mg of snap-frozen tissue (liver and muscle) after lysis in a buffer containing 10 mmol/l Tris, 10 mmol/l ethylenediaminetetraacetic acid (EDTA), 0.6% sodium dodecyl sulfate (SDS), 200 ng/μl proteinase K (Qiagen). RNAseA (Qiagen) was added at a final concentration of 10 μg/ul, and samples were incubated at 37°C for 1 hr followed by double phenol/chlorophorm extraction. DNA was then precipitated by adding 2.5 volumes of ethyl alcohol (EtOH) and incubating at −80°C for 2 hr. Samples were centrifuged at 14,000 rpm for 1 hr. DNA pellets were washed in 70% EtOH and resuspended in water.
Real-time PCR analysis was performed on 100 ng genomic DNA using a set of primers and Taqman probes specific for the viral genome with Taqman universal PCR master mix (Applied Biosystems) and standard cycles (Tessitore et al., 2008). All the reactions were performed in triplicate.
Western blot
The hamsters were euthanized by inhalation in a CO2 chamber, a procedure performed by expert technicians. Tissues were rapidly explanted, frozen in liquid nitrogen, and stored at −70°C. For muscle (gastrocnemius, soleus, and quadriceps) and liver, about 100 mg of tissue was homogenized in a lysis assay buffer (Urea 8 M, SDS 4%, 125 mM Tris HCl pH 6.8). Samples from similarly treated animals and from the same tissue were pooled together. About 50 μg of protein extract was separated on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane. After blocking in 10% nonfat dry milk in Tween Tris-buffered saline (TTBS-1X) (10 mM Tris-HCl, 150 mM NaCl, 0.05% Tween-20) for 1 hr, the membranes were incubated with primary antibodies in TTBS 1X at room temperature for 1.5 hr. The monoclonal antibody recognizing a human epitope of DSG (NCL-d-SARC; Novocastra Laboratories) was used at 1:25 dilution, while the monoclonal antibody anti-tubulin (Sigma) was used at 1:5,000 dilution.
Following primary antibody incubation and rinses, membranes were incubated with the secondary antibody, goat anti-mouse immunoglobulin conjugated with horseradish (Sigma) at 1:10,000 dilution in 0.5% dry milk and TTBS 1X. After 45 min of antibody incubation and five washes with TTBS 1X buffer, the DSG protein band was visualized with a chemiluminescence reagent (SuperSignal® West FEMTO Max. Sensitivity Substrate, Euroclone) and processed by ChemiDoc-It Imaging System. Band intensity was determined using Total Lab 1D software (TotalLab Limited). After background subtraction, values of band intensity were normalized with respect to the internal control (tubulin) and expressed as a difference compared to the sample showing the highest expression level.
Histology
The tissue samples were collected at 2 and 7 months of age. The samples were processed by cryosections at 7- to 10-mm thickness. Cryosections of the muscular tissues (gastrocnemius and quadriceps) were fixed in 4% PFA, then washed in phosphate buffered saline (PBS) 1X buffer (10 mM Tris-HCl, 200 mM NaCl, 0.05% NP 40, 0.05% Tween 20) and stained in hematoxylin for 4 min and in eosin for 6 min. Cryosections were dried in ethanol, fixed in xylene, and mounted with the EUKITT mounting kit (O.Kindler GmbH & CO).
Immunofluorescence
For immunofluorescence (IF) staining, unfixed muscle cryosections were immediately blocked in 10% goat serum and PBS at room temperature for 1 hr. Monoclonal antibodies against alpha- and beta-SG (Novocastra Laboratories) were diluted at 1:100 in 10% goat serum, whilst a polyclonal antibody to delta-SG was diluted at 1:1,000 in 10% horse serum–PBS and incubated with the cryosections for 1 hr and 30 min at room temperature. After three washes, the sections were incubated with Cy-3-labeled anti-mouse or anti-rabbit secondary antibodies (Jackson Immuno Research Laboratories) at 1:300 and 1:400 dilutions respectively in 10% horse serum PBS. After three washes, the samples were mounted in Vectashield mounting with 4',6-diamidino-2-phenylindole (DAPI). All sections were photographed with a Zeiss Axioplan fluorescence microscope, using the Axio Vision 4.5 Program at a magnification of 10× and 20×.
Immuno response
The presence of neutralizing antibodies directed at the human DSG transgene in the sera of treated BIO14.6 hamsters was determined using an enzyme-linked immunosorbent assay (ELISA), with conditions similar to those proposed by Tessitore and colleagues for detecting anti-ARSB immunoglobulin G antibodies (Tessitore et al., 2008). Briefly, the recombinant human DSG was produced and purified as previously reported (Nigro et al., 1996b). Serial dilutions from 1:50 to 1:5,000 of hamster serum were assayed. Biotinylated anti-hamster immunoglobulin G antibodies (Vector Laboratories) were used as secondary antibodies. Binding was revealed by the addition of extravidin–horseradish peroxidase (Sigma-Aldrich) followed by incubation with o-phenylenediamine dihydrochloride substrate (Sigma-Aldrich). The level of enzyme-linked immunosorbent assay optical density (OD) observed in an assay represents a quantitative measure of antibody affinity. For OD units up to 0.2, the antibody reactivity was classified as either low or nil and for values >1.2 OD units, it was classified as high-affinity antibody reactivity. Serum from nontreated hamsters was used as a negative control. No reaction against DSG was observed.
Results
Experimental design
To compare AAV2/8 and AAV2/2i8, we intraperitoneally injected AAV2/8-CMV-dSG and AAV2/2i8-CMV-dSG into newborn (postnatal day 14) male hamsters at three different doses (max=5×1013 GC/Kg; mid=1.5×1013 GC/Kg; and min=5×1012 GC/Kg). Figure 1 shows the experimental timelines of six different hamster groups. We measured the following parameters at different times after treatment: i) liver targeting; ii) muscle targeting; iii) expression of other components of the sarcoglycan complex; iv) persistence of AAVs; and v) muscle rescue. As in previous studies (Vitiello et al., 2009; Rotundo et al., 2011), we did not observe any immune response against DSG.
FIG. 1.

Timeline of experimental procedures for different hamster groups. Two-week old BIO14.6 hamsters (n=8 for each group) were intraperitoneally injected with delta sarcoglycan (DSG) in AAV2/8 or AAV2/2i8 vectors at the following dosages: AAV2/8max at 5×1013 GC/Kg; AAV2/8mid at 1.5×1013 GC/Kg; AAV2/8min at 5×1012 GC/Kg; AAV2/2i8max at 5×1013 GC/Kg; AAV2/2i8mid at 1.5×1013 GC/Kg; AAV2/2i8min at 5×1012 GC/Kg. Untreated BIO14.6 (n=4) and golden Syrian (n=4) hamsters were used as negative and healthy controls respectively. Treated (n=4) and untreated (n=2) animals were sacrificed at 2 and 7 months (STOP marks), respectively. AAV, adeno-associated virus.
Liver detargeting
To test the effectiveness of liver detargeting, we analyzed liver proteins by Western blotting at 2 months (n=4) and 7 months (n=4) after injection, using a monoclonal antibody (NCL-d-SARC) against human DSG (Fig. 1, STOP marks). NCL-d-SARC cannot detect hamster protein in control animals, as it recognizes a human-specific N-terminal epitope. We observed that DSG was expressed in the AAV2/8max and AAV2/8mid groups, but not in the AAV2/2i8max and AAV2/2i8mid groups, both at 2 and 7 months (Fig. 2a and b), confirming liver detargeting for the AAV2/2i8 serotype. No expression was observed in liver of the AAV2/8min and AAV2/2i8min groups.
FIG. 2.
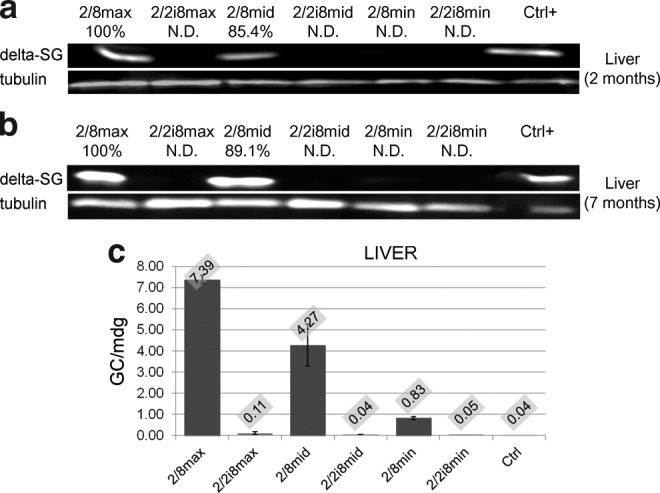
AAV transduction to liver: (a) Expression of DSG assessed by Western blot analysis in liver in 2/8max (5x1013 GC/Kg); 2/2i8max (5×1013 GC/Kg); 2/8mid (1.5×1013 GC/Kg); 2/2i8mid (1.5×1013 GC/Kg); 2/8min (5x1012 GC/Kg); and 2/2i8min (5×1012 GC/Kg) at 2 months after treatment (Ctrl+; internal laboratory control of Ab specificity). (b) Expression of DSG assessed by Western blot analysis in the liver in 2/8max (5×1013 GC/Kg); 2/2i8max (5×1013 GC/Kg); 2/8mid (1.5×1013 GC/Kg); 2/2i8mid (1.5×1013 GC/Kg); 2/8min (5×1012 GC/Kg); and 2/2i8min (5×1012 GC/Kg) groups at 7 months after treatment (Ctrl+; internal laboratory control of Ab specificity). After normalization (a, b), a percentage is reported that quantify protein expression compared to the sample showing the highest expression level. (c) AAV genome copies for molecules of diploid genome (mdg) extracted from liver of the same animal groups at 7 months after treatment. The mean absolute value is labeled at the top of each bar.
In addition, persistence of liver transduction in animals injected with AAV2/8, but not in animals treated with AAV2/2i8, was confirmed by the presence of detectable AAV genomic copies at 7 months (Fig. 2c). To exclude the possibility that the absence of expression in the liver could be due to inactivation of CMV promoter, we also measured AAV genomic copies. The hamsters receiving higher and middle vector doses of AAV2/8 and displaying a higher liver expression of the protein also exhibited higher AAV genome copy numbers. The hamsters receiving higher and middle vector doses of AAV2/2i8 and displaying no liver expression of the protein also exhibited lower AAV genome copy numbers (Fig. 2c). No expression in other tissues, including stomach and intestine, was observed for both vectors, as well as no significant difference was observed between animals of the same group at the same AAV dose (data not shown).
Muscle targeting
To test the expression of DSG, we analyzed muscle tissue extracts by Western blotting (Fig. 3), using a monoclonal antibody (NCL-d-SARC) against human DSG (Fig. 1, STOP marks). In skeletal muscle, the DSG expression was weaker in the AAV2/8 group than in the AAV2/2i8 group after 2 months (Fig. 3a). To confirm long-term transgene expression, we performed Western blot analyses from skeletal muscle tissue at 7 months after the treatment (Fig. 3b). To measure the number of AAV genomic copies, we performed real-time PCR assays on muscle samples (Fig. 3c). Again, we observed a higher level of AAV2/2i8 that paralleled the Western blot results. No difference was observed between animals of the same group injected with the same AAV dose (data not shown).
FIG. 3.
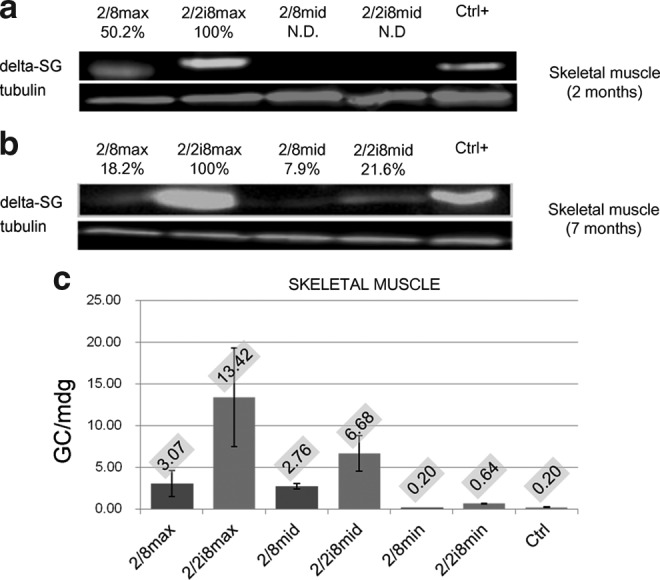
DSG expression in skeletal muscle. (a) Expression of DSG assessed by Western blotting analysis of pooled muscles in 2/8max (5×1013 GC/Kg); 2/2i8max (5×1013 GC/Kg), 2/8mid (1.5×1013 GC/Kg); and 2/2i8mid (1.5×1013 GC/Kg) groups at 2 months after treatment (Ctrl+; internal laboratory control of Ab specificity). (b) Expression of DSG assessed by Western blot analysis of pooled muscles in 2/8max (5×1013 GC/Kg); 2/2i8max (5×1013 GC/Kg); 2/8mid (1.5×1013 GC/Kg); and 2/2i8mid (1.5×1013 GC/Kg) groups at 7 months after treatment (Ctrl+; internal laboratory control of Ab specificity). After normalization (a, b), a percentage is reported that quantify protein expression compared to the sample showing the highest expression level. (c) AAV genome copies for molecules of diploid genome extracted from muscles of the same animal groups at 7 months after treatment. The mean absolute value is labeled at the top of each bar.
Immunofluorescent (IF) staining also confirmed Western blotting analyses. At 2 months after injection, skeletal muscle from AAV2/8max showed few positive fibers, while muscle from AAV2/2i8max presented approximately 100% positive fibers, a similar result to that of the WT group (using a polyclonal antibody) (Fig. 4a). Muscle of the AAV2/2i8mid group showed few positive fibers, whereas that of the AAV2/8mid group presented none (Fig. 4a). Expression levels using AAV2/2i8 also improved in samples from 7-month-old animals, obtaining the same results as AAV2/8 at two- to four-fold lower dosages (Fig. 4b).
FIG. 4.

Recovery of sarcoglycan complex expression in skeletal muscle. Immunofluorescence (IF) analysis of DSG expression in skeletal muscle from 2/8max, 2/2i8max, 2/8mid, 2/2i8mid, and control (+/−) groups is shown at 2 (a) and 7 (b) months after the treatment. (c) Rescue of sarcoglycan complex was assessed by IF analysis of beta sarcoglycan expression in the same muscle samples. Magnification 20×.
In addition to DSG, we tested the recovery of beta sarcoglycan as a measure of functional reconstitution of the whole sarcoglycan complex. By IF we showed that beta sarcoglycan was also recovered in skeletal muscle after 7 months (Fig. 4c).
Pathology rescue
We analyzed the skeletal muscle structure by evaluating necrotic areas using hematoxylin-eosin staining (Fig. 5a) and the presence of fibrosis using the Masson's Trichrome procedure (Fig. 5b). The time point was set at 7 months because fibrosis is a late-occurring event. The best results were observed in the skeletal muscle of animal groups treated with AAV2/2i8, which displayed an absence of necrotic areas and a homogeneous fiber dimension comparable with that of controls.
FIG. 5.

Recovery of skeletal muscle histology. (a) Hematoxilin-eosin staining on cryosections of skeletal muscle from the same groups, tested 7 months after treatment. (b) Masson's trichrome staining as carried out on the same animal groups. Magnification 20×.
Discussion
We previously reported the use of different combinations of AAV vectors for gene therapy in BIO14.6 hamsters (Vitiello et al., 2009). Muscle and heart rescue as well as increased lifespan was demonstrated with the use of systemic treatment (Zhu et al., 2005; Vitiello et al., 2009). However, we observed strong liver targeting that captured the majority of virus particles.
To search for novel AAV vectors with improved clinical value, we tested in vivo an AAV with the capsid proposed by Asokan and colleagues, which created the AAV2/2i8 chimera (Asokan et al., 2010). In mice, this chimeric vector was shown to maintain muscle tropism, but was no longer liver-targeted. The selective muscle tropism of chimeric 2i8 capsid, and its ability to evade sequestration by the liver, should allow for the precise control over vector biodistribution as well as cardiac or skeletal muscle-specific transgene expression.
Here we show that muscle delivery by AAV2/2i8 also works in the BIO14.6 hamster at lower dosages than AAV2/8. In addition, this occurs without any liver targeting, even using intraperitoneal AAV injections. The success of intraperitoneal delivery confirms previous observations in mice and could lead to the development of in utero gene transfer protocols (David et al., 2011).
At 2 and 7 months after treatment, AAV2/2i8 vector efficiently delivered DSG into skeletal muscle but not in liver of BIO14.6 hamsters, as confirmed by the absence of AAV genomic copies in liver at the end of treatment. In addition, a higher protein level was detected by Western blotting and IF analysis in several muscles (including gastrocnemius, soleus, and quadriceps) by comparing AAV2/2i8 results with AAV2/8. The rescue of sarcoglycan complex and its long-term expression suggest normal fiber organization as highlighted by the absence of necrotic areas and a homogeneous dimension of fibers with fewer centralized nuclei (not shown).
Given that the cardiomyopathy may be a major issue in BIO14.6 hamsters, we also measured an improvement in ejection fraction at 6.5 months, as previously shown (Vitiello et al., 2009; Rotundo et al., 2011). However, in contrast to skeletal muscle, no significant differences were observed using AAV2/8max or AAV2/2i8max (data not shown). To compare and detect any differences in lifespan, we would need long-life testing.
We are using the intraperitoneal delivery in hamsters, because this is similarly efficacious as the intravenous delivery (Dane et al., 2012), but with our settings i) it was safer and the animals were not stressed; ii) it can be easily used in newborn animals or before birth; iii) the reproducibility was higher. Our experimental conditions (a different animal model, use of non–muscle-specific CMV promoter and AAV injection intraperitoneally in close anatomical proximity to the liver) were favorable to liver targeting. Despite this, our transgene was not delivered to the liver, transduced muscle, and was highly expressed at 7 months with significant therapeutic efficacy. Recently, novel-engineered AAV2/9 variants were tested in mice and displayed 10- to 25-fold lower gene transfer efficacy in liver, while transducing cardiac and skeletal muscle as effectively as AAV2/9 (Pulicherla et al., 2011). As previously reported (Vitiello et al., 2009), the results obtained in our laboratory using AAV2/9 were unsatisfactory in BIO14.6 and were in contrast with reported observations on the ability of AAV2/9 in mice to transcend vasculature and transduce myocardium (Pacak et al., 2006). In 2-week-old hamsters, AAV2/9 serotype showed much weaker transduction than AAV2/8, and this result was confirmed using different AAV preparations (Vitiello et al., 2009).
We conclude that AAV2/2i8 is equally effective in mice and hamsters, which are evolutionarily distant, and may be useful in long-term clinical applications. This particular AAV leads to a significant overall improvement in young animals with muscular dystrophy and could be translated to the treatment of human muscular pathologies, including those with congenital onset.
Acknowledgments
AAV vectors were produced by the TIGEM AAV Vector Core Facility. We are grateful to Marco Corona, Emanuela Giordano, and Daniele Di Napoli for animal housing and veterinary support. We thank Vincenzo Russo for the echocardiography. We also thank Manuela Dionisi, Anna Cuomo, and Rosalba Erpice for sequence analyses.
This study was supported by grants from Telethon, Italy (TIGEM-TVNP01TELC), FP7/2007-2013, under grant agreement no. 223143 (project acronym: TECHGENE), and the “Ministero della Salute” (Ricerca Finalizzata RF-MUL-2007-666195). We thank Aon Benfield Italia S.p.A. - Milan for the generous support. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Disclosure Statement
No competing financial interests exist.
References
- Asokan A. Reengineered AAV vectors: old dog, new tricks. Discov Med. 2010;9:399–403. [PMC free article] [PubMed] [Google Scholar]
- Asokan A. Conway J.C. Phillips J.L., et al. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat. Biotechnol. 2010;28:79–82. doi: 10.1038/nbt.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asokan A. Schaffer D.V. Samulski R.J. The AAV vector toolkit: poised at the clinical crossroads. Mol. Ther. 2012;20:699–708. doi: 10.1038/mt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns K.I. Linden R.M. The cryptic life style of adeno-associated virus. Bioessays. 1995;17:237–245. doi: 10.1002/bies.950170310. [DOI] [PubMed] [Google Scholar]
- Blain A.M. Straub V.W. Delta-Sarcoglycan-deficient muscular dystrophy: from discovery to therapeutic approaches. Skelet. Muscle. 2011;1:13. doi: 10.1186/2044-5040-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dane A.P. Wowro S.J. Cunningham S.C. Alexander I.E. Comparison of gene transfer to the murine liver following intraperitoneal and intraportal delivery of hepatotropic AAV pseudo-serotypes. Gene Ther. 2012 doi: 10.1038/gt.2012.67. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- David A.L. Mcintosh J. Peebles D.M., et al. Recombinant adeno-associated virus-mediated in utero gene transfer gives therapeutic transgene expression in the sheep. Hum. Gene Ther. 2011;22:419–426. doi: 10.1089/hum.2010.007. [DOI] [PubMed] [Google Scholar]
- Gao G. Qu G. Burnham M.S., et al. Purification of recombinant adeno-associated virus vectors by column chromatography and its performance in vivo. Hum Gene Ther. 2000;11:2079–2091. doi: 10.1089/104303400750001390. [DOI] [PubMed] [Google Scholar]
- Gao G.P. Alvira M.R. Wang L., et al. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. U S A. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic P. Blankinship M.J. Allen J.M., et al. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasbrouck N.C. High K.A. AAV-mediated gene transfer for the treatment of hemophilia B: problems and prospects. Gene Ther. 2008;15:870–875. doi: 10.1038/gt.2008.71. [DOI] [PubMed] [Google Scholar]
- Hileman R.E. Fromm J.R. Weiler J.M. Linhardt R.J. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays. 1998;20:156–167. doi: 10.1002/(SICI)1521-1878(199802)20:2<156::AID-BIES8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Levy H.C. Bowman V.D. Govindasamy L., et al. Heparin binding induces conformational changes in Adeno-associated virus serotype 2. J Struct. Biol. 2009;165:146–156. doi: 10.1016/j.jsb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y. Recombinant adeno-associated virus as delivery vector for gene therapy–a review. Stem Cells Dev. 2004;13:133–145. doi: 10.1089/154732804773099335. [DOI] [PubMed] [Google Scholar]
- Manno C.S. Pierce G.F. Arruda V.R., et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- Mingozzi F. High K.A. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet. 2011;12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- Nigro V. De Sa Moreira E. Piluso G., et al. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the delta-sarcoglycan gene. Nat. Genet. 1996a;14:195–198. doi: 10.1038/ng1096-195. [DOI] [PubMed] [Google Scholar]
- Nigro V. Piluso G. Belsito A., et al. Identification of a novel sarcoglycan gene at 5q33 encoding a sarcolemmal 35 kDa glycoprotein. Human Mol. Gen. 1996b;5:1179–1186. doi: 10.1093/hmg/5.8.1179. [DOI] [PubMed] [Google Scholar]
- Nigro V. Okazaki Y. Belsito A., et al. Identification of the Syrian hamster cardiomyopathy gene. Human Mol. Gen. 1997;6:601–607. doi: 10.1093/hmg/6.4.601. [DOI] [PubMed] [Google Scholar]
- Nigro V. Aurino S. Piluso G. Limb girdle muscular dystrophies: update on genetic diagnosis and therapeutic approaches. Curr. Opin. Neurol. 2011;24:429–436. doi: 10.1097/WCO.0b013e32834aa38d. [DOI] [PubMed] [Google Scholar]
- Opie S.R. Warrington K.H., Jr. Agbandje-Mckenna M., et al. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J. Virol. 2003;77:6995–7006. doi: 10.1128/JVI.77.12.6995-7006.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacak C.A. Mah C.S. Thattaliyath B.D., et al. Recombinant Adeno-Associated Virus Serotype 9 Leads to Preferential Cardiac Transduction In Vivo. Circulation Research. 2006;99:e3–e9. doi: 10.1161/01.RES.0000237661.18885.f6. [DOI] [PubMed] [Google Scholar]
- Paneda A. Vanrell L. Mauleon I., et al. Effect of adeno-associated virus serotype and genomic structure on liver transduction and biodistribution in mice of both genders. Hum. Gene Ther. 2009;20:908–917. doi: 10.1089/hum.2009.031. [DOI] [PubMed] [Google Scholar]
- Piluso G. Aurino S. Cacciottolo M., et al. Mendelian bases of myopathies, cardiomyopathies, and neuromyopathies. Acta Myol. 2010;29:1–20. [PMC free article] [PubMed] [Google Scholar]
- Pulicherla N. Shen S. Yadav S., et al. Engineering liver-detargeted AAV9 vectors for cardiac and musculoskeletal gene transfer. Mol. Ther. 2011;19:1070–1078. doi: 10.1038/mt.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotundo I.L. Faraso S. De Leonibus E., et al. Worsening of cardiomyopathy using deflazacort in an animal model rescued by gene therapy. PLoS One. 2011;6:e24729. doi: 10.1371/journal.pone.0024729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W. Arnold G.S. Bartlett J.S. Insertional mutagenesis of the adeno-associated virus type 2 (AAV2) capsid gene and generation of AAV2 vectors targeted to alternative cell-surface receptors. Hum. Gene Ther. 2001;12:1697–1711. doi: 10.1089/104303401750476212. [DOI] [PubMed] [Google Scholar]
- Summerford C. Samulski R.J. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 1998;72:1438–1445. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessitore A. Faella A. O'malley T., et al. Biochemical, pathological, and skeletal improvement of mucopolysaccharidosis VI after gene transfer to liver but not to muscle. Mol. Ther. 2008;16:30–37. doi: 10.1038/sj.mt.6300325. [DOI] [PubMed] [Google Scholar]
- Vitiello C. Faraso S. Sorrentino N.C., et al. Disease rescue and increased lifespan in a model of cardiomyopathy and muscular dystrophy by combined AAV treatments. PLoS One. 2009;4:e5051. doi: 10.1371/journal.pone.0005051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B. Li J. Fu F.H., et al. Construction and analysis of compact muscle-specific promoters for AAV vectors. Gene therapy. 2008;15:1489–1499. doi: 10.1038/gt.2008.104. [DOI] [PubMed] [Google Scholar]
- Wang Z. Zhu T. Qiao C., et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- Wu P. Xiao W. Conlon T., et al. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J. Virol. 2000;74:8635–8647. doi: 10.1128/jvi.74.18.8635-8647.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L. Li J. Xiao X. Directed evolution of adeno-associated virus (AAV) as vector for muscle gene therapy. Methods Mol. Biol. 2011;709:127–139. doi: 10.1007/978-1-61737-982-6_8. [DOI] [PubMed] [Google Scholar]
- Zhu T. Zhou L. Mori S., et al. Sustained whole-body functional rescue in congestive heart failure and muscular dystrophy hamsters by systemic gene transfer. Circulation. 2005;112:2650–2659. doi: 10.1161/CIRCULATIONAHA.105.565598. [DOI] [PubMed] [Google Scholar]
- Zincarelli C. Soltys S. Rengo G. Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]