

Abstract
Epigenetic association studies have demonstrated differential promoter methylation in the core circadian genes in breast cancer cases relative to cancer-free controls. The current pilot study aims to investigate whether epigenetic changes affecting breast cancer risk could be caused by circadian disruption through exposure to light at night. Archived DNA samples extracted from whole blood of 117 female subjects from a prospective cohort conducted in Denmark were included in this study. A polymerase chain reaction (PCR)-based method was used for detection of gene-promoter methylation, whereas genome-wide methylation analysis was performed using the Illumina Infinium Methylation Chip. Long-term shiftwork resulted in the same promoter hypomethylation of CLOCK and hypermethylation of CRY2, as was previously observed in breast cancer case-control studies. Genome-wide methylation analysis further discovered widespread methylation alterations in shiftworkers, including changes in many methylation- and cancer-relevant genes. Pathway analysis of the genes with altered methylation patterns revealed several cancer-related pathways. One of the top three networks generated was designated as “DNA replication, recombination, and repair, gene expression, behavior” with ESR1 (estrogen receptor α) featured most prominently in the network, underscoring the potential breast cancer relevance of the genes differentially methylated in long-term shiftworkers. These results, although exploratory, demonstrate the first evidence of the cancer-relevant epigenetic effects of night shiftwork, which warrant further investigation. Considering there are millions of shiftworkers worldwide, understanding the effects of this exposure may lead to novel strategies for cancer prevention and new policies regulating shiftwork.
Keywords: Cancer, Circadian genes, Genome-wide methylation, Shiftwork
INTRODUCTION
The hypothesis that circadian disruption caused by “light-at-night” in modern societies might explain, in part, the elevated risk of breast cancer in industrialized societies was first proposed 20 yrs ago by Stevens (1987, 2009), with the related prediction that shiftworkers would be at increased risk of developing breast cancer (Stevens et al., 1992). In 2007, the International Agency for Research on Cancer (IARC) assembled an expert panel to assess the role of shiftwork in human cancer development and concluded that “shift work involving circadian disruption is probably carcinogenic to humans,” based on sufficient evidence from animal studies and limited evidence in human breast cancer (Straif et al., 2007). This finding has not gone unnoticed by policymakers. In Denmark, for example, women who develop breast cancer and have a long history of shiftwork can have the disease acknowledged as a work-related illness and be compensated accordingly (Hansen, 2010). Although several theories have been advanced to explain the observed association between shiftwork and breast cancer, including alterations in melatonin levels and changes in circadian gene expression (Straif et al., 2007), the underlying mechanisms have yet to be fully elucidated. Besides genetic variants, epigenetic alterations have been demonstrated to play an important role in tumorigenesis. However, no previous study has been conducted to assess the genome-wide epigenetic impact of shiftwork and the influence of these epigenetic changes on cancer-related genes and biological pathways. In the current pilot study, we test the hypothesis that exposure to shiftwork may have epigenetic consequences using samples from the Danish “Diet, Cancer and Health” prospective cohort.
METHODS
Study Population
All research methods and procedures were performed in accordance with the Helsinki Declaration of Human Studies and the ethical standards of the Journal (Portaluppi et al., 2010). Approval for the proposed study was obtained from the Danish Data Protection Agency (no. 2001-41-2603, 2008-41-2773), and from the regional ethical committee (no. H-KF-01-345/93, add. 55). All participants signed an informed consent document. Study subjects were previously participants in the Danish “Diet, Cancer and Health” prospective cohort, established between December 1993 and May 1997, with the primary purpose of studying the role of diet and nutrition in cancer. Participants were born in Denmark, free of any cancer, and aged between 50 and 64 yrs at the time of invitation. These women were examined to obtain anthropometrical measures and biological samples, including blood, urine, fat tissue, and toenail clippings. The baseline questionnaire included questions on food consumption, micronutrients (e.g., folate intake), lifestyle factors (e.g., smoking and drinking habits, sun exposure, physical activity, and medical anamnesis), reproductive history, education level, and exposures to selected occupational agents (e.g., asbestos) or job tasks (e.g., welding). A total of 117 female participants were available for the current pilot analysis based on the availability of blood DNA samples for methylation analysis, and information on past history of shiftwork as described below. More information regarding the parent cohort is available elsewhere (Tjonneland et al., 2007).
Exposure Assessment
Information on night shiftwork was obtained by telephone interviews conducted from 2003 to 2005, in which work history was reviewed, including periods of night shiftwork, defined as work starting at 19:00 h or later and ending before 09:00 h. Among the 117 participants included in this study, 19 were classified as having a history of long-term night shiftwork (defined as having ≥10 yrs of night shiftwork exposure), and 98 subjects had never been exposed to night shiftwork (day workers). Dietary data were collected at baseline using a 192-item food-frequency questionnaire mailed to each participant. A description of the food questionnaire has been published previously (Overvad et al., 1991). For each participant, average daily micronutrient intake, including folate intake, was calculated using the Food Calc software tool (www.ibt.ku.dk/jesper/foodcalc). Day workers and shiftworkers did not differ according to age (mean years ± SEM: day workers = 55.17 ± 0.33, shiftworkers = 55.26 ± 0.91; p = .9168) or by total folate intake (mean ± SEM μg/day: day workers = 373.2 ±14.57, shiftworkers = 367.4 ± 34.75; p = .8720).
Methylation Analysis of CLOCK and CRY2 Promoters
A standard DNA isolation protocol was used to extract DNA from archived blood samples collected at the time of recruitment of the cohort. Real-time polymerase chain reaction (PCR)-based methods were used for methylation analyses of the CLOCK and CRY2 promoters, as previously described (Hoffman et al., 2010a, 2010b). Specifically, genomic DNA was bisulfite treated using the EZ DNA Methylation Kit (Zymo Research, Orange, CA) according to the manufacturer’s protocol, which converts unmethylated cytosines into uracil, and leaves methylated cytosines unchanged. Following the treatment, quantitative PCR was performed using the primers described in our previous publications and the Power SYBR Green Kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol, in order to distinguish methylated from unmethylated DNA sequences. In order to assign a quantitative measure to the level of methylation, a methylation index (MI) was calculated for each sample using the formula: MI = [1/(1 + 2−(CTu–CTme)] × 100%, as previously described (Lu et al., 2007), where CTu is the average cycle threshold (CT) obtained from duplicate qPCRs using the unmethylated primer pair, and CTme is the average CT obtained using the methylated primer pair. Any sample that failed to amplify or had unreliable replicates (SEM >1 cycle) for any primer pair was removed from further analysis.
Genome-Wide Methylation Assay
Ten long-term shiftworkers were randomly selected from among the 19 total shiftworkers for genome-wide methylation analysis. Ten day workers were then selected via direct matching on age (± 2 yrs) and total folate intake (± 55 μg/day). A total of 50 ng of genomic DNA from each individual was used in the Illumina Infinium Methylation Assay, which covers 27 578 CpG sites spanning 14 495 genes. Tested CpG sites are located within the proximal promoter region of each gene, ranging from 0 to 1499 bp from the transcription start site (average ± SEM distance = 389 ± 34 bp). A methylation index was calculated for each site, which is a continuous variable between 0 and 1, representing the ratio of the intensity of the methylated signal to the total intensity. Zero corresponds to a completely unmethylated site, whereas 1 corresponds to a completely methylated site. All array data have been uploaded to the Gene Expression Omnibus (GEO) database, and can be accessed via their Web site (www.ncbi.nlm.nih.gov/geo) (accession number pending).
Pathway-Based Network Analysis
To further explore biological pathways affected by long-term exposure to shiftwork, differentially methylated genes identified by the methylation assay were analyzed for functional interrelatedness using the Ingenuity Pathway Analysis (IPA) software tool. IPA utilizes an extensive database of manually curated functional interactions drawn from peer-reviewed publications (Calvano et al., 2005). The p value for each network is defined as the likelihood of obtaining a network of the same number of genes randomly given the genes in the uploaded file, i.e., the set of genes displaying differential methylation between day and long-term shiftworkers, using Fisher’s exact test based on the hypergeometric distribution.
Statistical Analysis
SAS version 9.1 (SAS Institute, Cary, NC) was used to assess the association between shiftworker status and promoter methylation of CLOCK and CRY2. Since folate is important in mediating one-carbon metabolism and the synthesis of methyl donor groups (Kim, 2004), folate intake may influence DNA methylation levels and thereby act as a potential confounder. As such, both age and folate intake were included as covariates in all logistic regression models. A two-sided α of .05 was used to designate statistical significance for all analyses. Subjects were divided into tertiles of “high,” “mid,” or “low” CLOCK or CRY2 methylation based on the distribution of methylation values among the day workers. These classifications were then used in an unconditional logistic regression model to derive the odds ratios (ORs) and 95% confidence intervals (CIs) for associations between CLOCK and CRY2 promoter methylation and shiftworker status. The Infinium methylation data were analyzed using Illumina’s GenomeStudio software, and the mean methylation level in shiftworkers was compared to that of day workers. To control for multiple comparisons, adjustments were made using the Benjamini-Hochberg method in order to obtain an adjusted p value (designed as the false discovery rate, or Q value) for each observation. Genes were defined as differentially methylated if the Q values obtained were <.05, corresponding to an absolute DiffScore of >13.
RESULTS
Long-Term Shiftwork Affects Promoter Methylation in the Circadian Genes CLOCK and CRY2
We first investigated whether exposure to night shiftwork can modify CLOCK and CRY2 promoter methylation patterns. Our previous case-control studies have demonstrated significant epigenetic associations between core human circadian genes CLOCK (Hoffman et al., 2010a) and CRY2 (Hoffman et al., 2010b) and breast cancer risk. As these earlier findings indicated opposite oncogenic functions for CLOCK and CRY2 (Hoffman et al., 2010b), we predicted that shiftwork exposure would have opposite epigenetic consequences for these genes. As expected, shiftworkers had relatively lower levels of CLOCK methylation, and relatively higher levels of CRY2 methylation, compared to day workers (Supplementary Figure 1). We conducted logistic regression analyses, adjusting for potential confounding by age and total folate intake. In order to maximize statistical power, the largest tertile was used as the referent category, i.e. “low methylation” for CLOCK and “high methylation” for CRY2, and nonreferent categories were combined. Again, as expected, long-term shiftworkers were significantly more likely to have lower methylation indices (MIs) for CLOCK (OR: 0.36, 95% CI: 0.13–1.00) and higher MIs for CRY2 (OR: 0.32, 95% CI: 0.11–0.93) compared to day workers (Figure 1). Detailed results of logistic regression analyses of CLOCK and CRY2 methylation levels in day workers and long-term shiftworkers are provided in the Supplementary Table 1.
FIGURE 1.
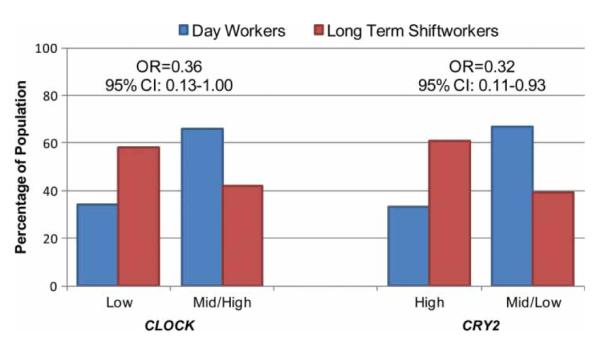
Distribution of day workers and long-term shiftworkers with different methylation status at the CLOCK and CRY2 gene promoters. Subjects were classified into tertiles based on the level of methylation among the day workers, and the tertile with the most shiftworkers was used as the reference category (“low methylation” for CLOCK, “high methylation” for CRY2). Long-term shiftworkers were significantly more likely to have low methylation indices (MIs) at the promoter site of the CLOCK gene (OR = 0.36, 95% CI: 0.13–1.00, p = .0499; mid/high vs. low tertile), and high methylation at the CRY2 promoter (OR = 0.32, 95% CI: 0.11–0.93, p = .0359; low/mid vs. high tertile) relative to day workers. Both age and folate intake were included as covariates in the analysis.
Long-Term Shiftwork Affects Methylation Patterns Genome-Wide
Whole-genome methylation analysis using 10 pairs of subjects identified differential methylation between day workers and long-term shiftworkers in 5409 CpG sites, corresponding to 4752 genes after adjusting for multiple comparisons. Interestingly, 3593 CpG sites, representing 66.4% of all differentially methylated loci, were hypermethylated, whereas only 1816 CpG sites (33.6%) were hypomethylated, which may suggest that the number of genes epigenetically inactivated greatly outnumbered the number of epigenetically activated genes in long-term shiftworkers. This analysis also helped confirm the existence of differential methylation of both CRY2 and CLOCK, as described above. The chip contained two CpG probes in the promoter region of CLOCK, and each probe showed decreased methylation in long-term shiftworkers (p = .0032 and p = .0009, after correcting for multiple comparisons). One of the two CpG sites of the CRY2 promoter exhibited increased methylation in long-term shiftworkers (adjusted p = .0068). The fact that these results are consistent with the findings from the qPCR analysis, which was conducted using the whole study population, provides support that the sample used for the whole-genome assay was representative.
Pathway Analysis of Differentially Methylated Genes
All 4752 genes fitting our significance criteria for differential methylation were tested for functional interrelatedness using the Ingenuity Pathway Analysis software tool. Supplementary Table 2 lists the top five networks identified as significantly associated with differentially methylated genes by IPA software. Interestingly, among the top three networks was a pathway designated as “DNA replication, recombination, and repair, gene expression, behavior” (p = 10−17), with estrogen receptor α (ESR1) featuring most prominently in the network (Figure 2). Details for each differentially methylated gene within this network, including fold change of methylation and a short description of relevant functions, are listed in Table 1. Briefly, 35 genes were present in the network, 24 of which were significantly hypermethylated whereas 10 were significantly hypomethylated in long-term shift relative to day workers. Several cancer-relevant genes were featured, including CDKN2A (p16), BCAS3 (breast carcinoma amplified sequence 3), and ESR1 (estrogen receptor α). Notable methylation-related genes in the network include MBD1 (methyl-CpG binding domain protein 1), MBD2, MBD3, and MECP2 (methyl CpG binding protein 2 [Rett syndrome]). In addition, HDAC2 (histone deacetylase 2), which encodes a protein responsible for the deacetylation of lysine residues at the N-terminal regions of core histones, and two DNA repair genes, ERCC3 (excision repair cross-complementing rodent repair deficiency, complementation group 3) and MGMT (O-6-methylguanine-DNA methyltransferase), are also included in the network.
FIGURE 2.

Network of interactions significantly enriched for genes displaying altered methylation patterns in long-term shiftworkers relative to day workers (p = 10−17). This network was designated as “DNA replication, recombination, and repair, gene expression, behavior,” determined by the IPA software. Of the 35 genes present in the network, 24 were significantly hypermethylated (in red), whereas only 10 were significantly hypomethylated (in green). (A color version of this figure is available in the online edition of this article.)
TABLE 1.
Genes in one of the top networks (p = 10−17) exhibiting different methylation patterns between long-term shiftworkers and day workers
| Symbol | Accession no. | Gene name and brief function | Fold change | Adjusted p value |
|---|---|---|---|---|
| BCAS3 | NM_017679.2 | Breast carcinoma-amplified sequence 3, metastasis associated antigen of breast cancer |
+1.81 | 2.63E-04 |
| CCNH | NM_001239.2 | Cyclin H, the catalytic subunit of the CDK-activating kinase enzymatic complex |
+2.22 | 1.85E-05 |
| CDK7 * | NM_001799.2 | Cyclin-dependent knase 7, regulator of cell cycle progression | +2.16 | 1.54E-03 |
| CDKN2A * | NM_058195.2 | Cyclin-dependent kinase inhibitor 2A, tumor suppressor | +1.51 −1.49 | 9.91E-03 3.39E-02 |
| CRYBB2 | NM_000496.1 | Crystalline, beta B2 | −1.10 | 8.22E-04 |
| DDX17 | NM_006386.3 | Dead box polypeptide 17, putative RNA helicases | +2.39 | 1.27E-03 |
| DDX21 | NM_004728.2 | Dead box polypeptide 21 | +2.53 | 8.57E-04 |
| DUSP9 | NM_001395.1 | Dual specificity phosphatase 9 | −1.21 | 6.38E-03 |
| EIF5A | NM_001970.3 | Eukaryotic translation initiation factor 5A | +2.15 | 9.73E-06 |
| ERCC3 | NM_000122.1 | Excision repair cross-complementing rodent repair deficiency, complementation group 3 |
−1.42 | 2.98E-02 |
| ESR1 | NM_000125.2 | Estrogen receptor 1, ligand-dependent nuclear receptor | +1.52 | 1.64E-04 |
| GPAM | NM_020918.2 | Glycerol-3-phosphate acyltransferase | +2.47 | 1.95E-08 |
| GTF2H2 | NM_001515.2 | General transcription factor IIH, polypeptide 2, transcription regulator | +3.03 | 1.60E-09 |
| HDAC2 | NM_001527.1 | Histone deacetylase 2, forms transcriptional repressor complexes | +2.31 | 1.35E-02 |
| HSPA13 | NM_006948.4 | Heat shock protein 70-kDa family, member 13 | +2.10 | 2.42E-04 |
| IER3 | NM_003897.2 | Immediate early response 3 | +1.77 | 3.40E-02 |
| KLK3 | NM_001648.2 | Kallikrein-related peptidase 3 | −1.18 | 7.62E-05 |
| MBD1 | NM_015845.2 | Methyl-CpG binding domain protein 1, transcriptional repressor | +2.20 | 1.90E-06 |
| MBD2 | NM_003927.3 | Methyl-CpG binding domain protein 2, transcription repressor | +1.64 | 4.21E-02 |
| MBD3 | NM_003926.5 | Methyl-CpG binding domain protein 3 | −1.05 | 1.95E-02 |
| MECP2 | NM_004992.2 | Methyl-CpG binding protein2, transcription regulator | +1.21 | 3.53E-02 |
| MGMT * | NM_002412.2 | O-6-methylguanine-DNA methyltransferase | −1.07 | 2.14E-03 |
| MNAT1 | NM_002431.2 | Ménage a trois homolog1, cyclin H assembly factor | +1.52 | 2.37E-04 |
| NCOA2 | NM_006540.2 | Nuclear receptor coactivator 2 | −1.31 | 2.62E-02 |
| OXT | NM_000915.2 | Oxytocin, prepropeptide | +1.91 | 1.05E-02 |
| PDZK1 | NM_002614.3 | PDZ domain containing 1, transporter | −1.07 | 1.34E-04 |
| PES1 | NM_014303.2 | Pescadillo homolog1, containing BRCT domain | +1.91 | 1.01E-02 |
| PPP6R2 | NM_014678.3 | Protein phosphatase 6, regulatory subunit 2 | +1.76 | 1.61E-02 |
| RPL5 | NM_000969.3 | Ribosomal protein L5 | +1.73 | 9.18E-03 |
| SLC39A8 | NM_022154.5 | Solute carrier family 39, member 8 | +2.11 | 5.85E-07 |
| STRN | NM_003162.2 | Striatin, calmodulin, binding protein | +2.71 | 1.77E-06 |
| TADA3 | NM_006354.2 | Transcriptional adaptor 3, transcription regulator | +1.44 | 2.02E-03 |
| TM4SF1 | NM_014220.2 | Transmembrane 4 L six family member 1 | −1.10 | 4.10E-02 |
| TMOD1 | NM_003275.1 | Tropomodulin 1 | −1.05 | 6.98E-04 |
Genes containing more than one CpG site with differential methylation. Fold change was averaged if multiple CpG sites had the same direction of methylation change.
As an additional quality control measure, we selected two genes from the top network, one hypomethylated (ERCC3) and one hypermethylated (GTF2H2) in long-term shiftworkers, and reevaluated their methylation status using methylation-specific qPCR. These data showed good agreement in the magnitude of changes observed by both qPCR and the array, and the same direction of methylation changes was observed in the comparison between day workers and long-term shiftworkers. Individual data for these genes, in addition to the data from CLOCK and CRY2 from both the qPCR analysis and the array, are shown in Supplemental Figure 2.
DISCUSSION
Our findings, although exploratory, support the hypothesis that long-term exposure to shiftwork can alter epigenetic patterns. We tested this hypothesis on two cancer-relevant circadian genes, CLOCK and CRY2, as findings from our earlier investigations had demonstrated that breast cancer patients tended to have low levels of CLOCK methylation (Hoffman et al., 2010a) and high levels of CRY2 methylation (Hoffman et al., 2010b). Significant methylation changes in these genes were noted in the current study, and for both genes the direction of the changes in shiftworkers was identical to that observed in breast cancer patients. As transcriptional regulators, circadian genes mediate the expression of many cancer-related genes and play various roles in cancer-relevant pathways, such as DNA repair (Kang et al., 2010). As such, the epigenetic impact of shiftwork on the activity and function of core circadian regulators may provide a missing link in the relationship between breast cancer and night shiftwork. The observed epigenetic impact of shiftwork is consistent with findings from a recent study showing an association between gene-specific methylation changes in the glucocorticoid receptor and length of shiftwork among employees of an Italian chemical plant (Bollati et al., 2010).
Moreover, our genome-wide methylation analysis revealed that the methylation patterns of more than 5000 CpG sites were altered in long-term shiftworkers. Interestingly, nearly twice as many CpG sites were hypermethylated in shiftworkers as were hypomethylated. These data are consistent with the general consensus that most tumors undergo widespread loss of methylation at the global level, but exhibit hypermethylation at CpG-rich, gene-associated regions, such as those included on the array chip (Ruike et al., 2010). Notably, many of the differentially methylated CpG sites were located near the promoter sequences of methylation-related and cancer-relevant genes.
Pathway analyses further showed that the genes with altered methylation formed a cancer-related network involved in DNA replication and repair, which included several important breast cancer-relevant genes, as well as genes important for regulating methylation patterns. For example, CDKN2A (p16), a well-established tumor suppressor, has been shown to have increased promoter methylation in breast tumor tissue (Celebiler Cavusoglu et al., 2010). The ESR1 gene encodes for estrogen receptor α, which plays an important role in breast tumorigenesis by mediating estrogen signaling and defines the hormone-responsive phenotype of breast cancer (Bai & Gust, 2009). The ESR1 gene is frequently hypermethylated in breast tumor tissue (Parrella et al., 2004), which is consistent with our finding that shiftworkers had approximately 50% more methylation in the ESR1 promoter than day workers. MBD2, encoding methyl-CpG-binding domain 2, is a major methylation-related gene and functions as a transcriptional repressor. Genetic variants in MBD2 have been associated with breast cancer risk (Zhu et al., 2005), a finding that is also consistent with our results showing a 64% increase in MBD2 promoter methylation in shift relative to day workers. Finally, excessive HDAC activity has been shown to induce hypoacetylation of histone and nonhistone protein substrates, thereby altering gene-expression patterns and cellular behavior associated with malignant transformations. HDAC2 has been previously shown to play a role in breast cancer progression (Suzuki et al., 2009), and we note a more than 2-fold increase in HDAC2 methylation in shift versus day workers.
Although our data suggest that long-term night shiftwork may result in epigenetic alterations in cancer-related genes and biological pathways, the molecular mechanisms underlying this epigenetic phenomenon remain obscure. An important limitation of our study is that our findings are based on a relatively small number of subjects, and, thus, further confirmation and investigation through additional studies are warranted. It is possible that occupational exposures could have confounded our analysis, as the long-term shiftworkers included in the current study worked in several different occupations. However, our limited sample size made it infeasible to interrogate this issue, which will be examined in future studies using larger samples. An additional limitation is the lack of information on morning or evening preference of the subjects, which previous studies have suggested could have a genetic component. Although no previous studies have linked epigenetic alterations to morningness/eveningness, if such an association exists, it could operate as a potential confounder, as people with a preference toward eveningness could be more likely to engage in shiftwork. It was also not feasible to examine the correlation between changes in methylation and changes in gene expression in the current study, because RNA samples were not isolated in the parent cohort. Moreover, a potential concern is whether the observed epigenetic changes in surrogate tissue (peripheral blood lymphocytes) accurately reflect changes in the target tissue. However, a recent large-scale case-control study of breast cancer detected a significant association between the methylation of several estrogen receptor α target (ERT) genes measured in peripheral blood lymphocytes (PBLs) and human breast cancer risk (Widschwendter et al., 2008), suggesting that methylation in PBLs may serve as a reasonable surrogate indicator of breast tissue-specific methylation in association analyses.
In summary, the findings from this pilot study have the potential to break new ground in clock-cancer research by elucidating a novel mechanism through which exposure to long-term shiftwork may influence epigenetic phenotypes. A broader understanding of the relationship between shiftwork and methylation changes could have widespread implications, perhaps most directly in the areas of environmental oncology, cancer epigenetics, and occupational health. Considering that millions of women worldwide are being exposed to ill-timed light through exposure to shiftwork, improving our understanding of the effects of this exposure, including developing a more complete understanding of the relationship between shiftwork and altered methylation, is needed and may lead to new policies for shiftworkers as well as novel strategies for cancer prevention.
Supplementary Material
Supplementary Table 1. Logistic regression analysis of CLOCK and CRY2 methylation levels in day workers and long-term shiftworkers.
Supplementary Table 2. Top five networks identified as significantly associated with differentially methylated genes by the IPA software.
Supplementary Figure 1. Scatter plot of methylation indices for CLOCK and CRY2 in day and long-term shiftworkers.
Supplementary Figue 2. Methylation measurement of CLOCK, CRY2, ERCC3, and GTF2H2 by methylation-specific qPCR. These data showed good agreement in the magnitude of changes observed by both qPCR and the Illumina Infinium Methylation Assay. Primers of methylation-specific PCR for CLOCK and CRY2 can be found in our previous publications (see Methods for references) and for GTF2H2 and ERCC3 are: GTF2H2: M.Left: 5′-TTTTGTGAATTTTAGTTGGAATATC-3′, M.Right: 5′-CGCCTAACTAACCAAACGAC-3′, U.Left: 5′ TTTTGTGAATTTTAGTTGGAATATT-3′U.Right: 5′-CCACCTAACTAACCAAACAAC-3′, ERCC3: M.Left: 5′- AGTAGGAGTTTTTAGGGTTTTTTTC-3′, M.Right: 5′- GCTCTACCTCGAAACTACTCCG -3′, U.Left: 5′- GGAGTTTTTAGGGTTTTTTTT-3′, U.Right: 5′- CCACTCTACCTCAAAACTACTCCA-3′.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Environmental Health Sciences (NIEHS) grant ES018915 and funds from Yale University. The Diet, Cancer and Health cohort was funded by the Danish Cancer Society.
Footnotes
Declaration of Interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Bai Z, Gust R. Breast cancer, estrogen receptor and ligands. Arch. Pharm. (Weinheim) 2009;342:133–149. doi: 10.1002/ardp.200800174. [DOI] [PubMed] [Google Scholar]
- Bollati V, Baccarelli A, Sartori S, Tarantini L, Motta V, Rota F, Costa G. Epigenetic effects of shiftwork on blood DNA methylation. Chronobiol. Int. 2010;27:1093–1094. doi: 10.3109/07420528.2010.490065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- Celebiler Cavusoglu A, Sevinc AI, Saydam S, Canda T, Baskan Z, Kilic Y, Sakizli M. Promoter methylation and expression changes of CDH1 and P16 genes in invasive breast cancer and adjacent normal breast tissue. Neoplasma. 2010;57:465–472. doi: 10.4149/neo_2010_05_465. [DOI] [PubMed] [Google Scholar]
- Hansen J. Women with night shift work and breast cancer: the situation in Denmark. J. Epidemiol. Community Health. 2010;64(12):1025–1026. doi: 10.1136/jech.2009.101691. [DOI] [PubMed] [Google Scholar]
- Hoffman AE, Yi CH, Zheng T, Stevens RG, Leaderer D, Zhang Y, Holford TR, Hansen J, Paulson J, Zhu Y. CLOCK in breast tumorigenesis: genetic, epigenetic, and transcriptional profiling analyses. Cancer Res. 2010a;70:1459–1468. doi: 10.1158/0008-5472.CAN-09-3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AE, Zheng T, Yi CH, Stevens RG, Ba Y, Zhang Y, Leaderer D, Holford T, Hansen J, Zhu Y. The core circadian gene Cryptochrome 2 influences breast cancer risk, possibly by mediating hormone signaling. Cancer Prev. Res. (Phila. Pa.) 2010b;3:539–548. doi: 10.1158/1940-6207.CAPR-09-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TH, Lindsey-Boltz LA, Reardon JT, Sancar A. Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc. Natl. Acad. Sci. U. S. A. 2010;107:4890–4895. doi: 10.1073/pnas.0915085107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YI. Folate and DNA methylation: a mechanistic link between folate deficiency and colorectal cancer? Cancer Epidemiol. Biomarkers Prev. 2004;13:511–519. [PubMed] [Google Scholar]
- Lu L, Katsaros D, de la Longrais IA, Sochirca O, Yu H. Hyper-methylation of let-7a-3 in epithelial ovarian cancer is associated with low insulin-like growth factor-II expression and favorable prognosis. Cancer Res. 2007;67:10117–10122. doi: 10.1158/0008-5472.CAN-07-2544. [DOI] [PubMed] [Google Scholar]
- Overvad K, Tjonneland A, Haraldsdottir J, Ewertz M, Jensen OM. Development of a semiquantitative food frequency questionnaire to assess food, energy and nutrient intake in Denmark. Int. J. Epidemiol. 1991;20:900–905. doi: 10.1093/ije/20.4.900. [DOI] [PubMed] [Google Scholar]
- Parrella P, Poeta ML, Gallo AP, Prencipe M, Scintu M, Apicella A, Rossiello R, Liguoro G, Seripa D, Gravina C, Rabitti C, Rinaldi M, Nicol T, Tommasi S, Paradiso A, Schittulli F, Altomare V, Fazio VM. Nonrandom distribution of aberrant promoter methylation of cancer-related genes in sporadic breast tumors. Clin. Cancer Res. 2004;10:5349–5354. doi: 10.1158/1078-0432.CCR-04-0555. [DOI] [PubMed] [Google Scholar]
- Portaluppi F, Smolensky MH, Touitou Y. Ethics and methods for biological rhythm research on animals and human beings. Chronobiol. Int. 2010;27:1911–1929. doi: 10.3109/07420528.2010.516381. [DOI] [PubMed] [Google Scholar]
- Ruike Y, Imanaka Y, Sato F, Shimizu K, Tsujimoto G. Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing. BMC Genomics. 2010;11:137. doi: 10.1186/1471-2164-11-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens RG. Electric power use and breast cancer: a hypothesis. Am. J. Epidemiol. 1987;125:556–561. doi: 10.1093/oxfordjournals.aje.a114569. [DOI] [PubMed] [Google Scholar]
- Stevens RG. Electric light causes cancer? Surely you’re joking, Mr. Stevens. Mutat. Res. 2009;682:1–6. doi: 10.1016/j.mrrev.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Stevens RG, Davis S, Thomas DB, Anderson LE, Wilson BW. Electric power, pineal function, and the risk of breast cancer. FASEB J. 1992;6:853–860. doi: 10.1096/fasebj.6.3.1740235. [DOI] [PubMed] [Google Scholar]
- Straif K, Baan R, Grosse Y, Secreton B, El Ghissassi F, Bouvard V, Altieri A, Benbrahim-Tallaa L, Cogliano V. Carcinogenicity of shiftwork, painting, and fire-fighting. Lancet Oncol. 2007;8:1065–1066. doi: 10.1016/S1470-2045(07)70373-X. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Chen YY, Scott GK, Devries S, Chin K, Benz CC, Waldman FM, Hwang ES. Protein acetylation and histone deacetylase expression associated with malignant breast cancer progression. Clin. Cancer Res. 2009;15:3163–3171. doi: 10.1158/1078-0432.CCR-08-2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjonneland A, Olsen A, Boll K, Stripp C, Christensen J, Engholm G, Overvad K. Study design, exposure variables, and socioeconomic determinants of participation in Diet, Cancer and Health: a population-based prospective cohort study of 57,053 men and women in Denmark. Scand. J. Public Health. 2007;35:432–441. doi: 10.1080/14034940601047986. [DOI] [PubMed] [Google Scholar]
- Widschwendter M, Apostolidou S, Raum E, Rothenbacher D, Fiegl H, Menon U, Stegmaier C, Jacobs IJ, Brenner H. Epigenotyping in peripheral blood cell DNA and breast cancer risk: a proof of principle study. PLoS ONE. 2008;3:e2656. doi: 10.1371/journal.pone.0002656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Brown HN, Zhang Y, Holford TR, Zheng T. Genotypes and haplotypes of the methyl-CpG-binding domain 2 modify breast cancer risk dependent upon menopausal status. Breast Cancer Res. 2005;7:R745–R752. doi: 10.1186/bcr1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Logistic regression analysis of CLOCK and CRY2 methylation levels in day workers and long-term shiftworkers.
Supplementary Table 2. Top five networks identified as significantly associated with differentially methylated genes by the IPA software.
Supplementary Figure 1. Scatter plot of methylation indices for CLOCK and CRY2 in day and long-term shiftworkers.
Supplementary Figue 2. Methylation measurement of CLOCK, CRY2, ERCC3, and GTF2H2 by methylation-specific qPCR. These data showed good agreement in the magnitude of changes observed by both qPCR and the Illumina Infinium Methylation Assay. Primers of methylation-specific PCR for CLOCK and CRY2 can be found in our previous publications (see Methods for references) and for GTF2H2 and ERCC3 are: GTF2H2: M.Left: 5′-TTTTGTGAATTTTAGTTGGAATATC-3′, M.Right: 5′-CGCCTAACTAACCAAACGAC-3′, U.Left: 5′ TTTTGTGAATTTTAGTTGGAATATT-3′U.Right: 5′-CCACCTAACTAACCAAACAAC-3′, ERCC3: M.Left: 5′- AGTAGGAGTTTTTAGGGTTTTTTTC-3′, M.Right: 5′- GCTCTACCTCGAAACTACTCCG -3′, U.Left: 5′- GGAGTTTTTAGGGTTTTTTTT-3′, U.Right: 5′- CCACTCTACCTCAAAACTACTCCA-3′.