

Abstract
Adenosine is a signaling nucleoside that is generated in response to cellular injury and orchestrates the balance between tissue protection and the progression to pathological tissue remodeling. Adenosine deaminase (ADA)-deficient mice develop progressive airway inflammation and remodeling in association with adenosine elevations, suggesting that adenosine can promote features of chronic lung disease. Furthermore, pharmacological studies in ADA-deficient mice demonstrate that A2BR antagonism can attenuate features of chronic lung disease, implicating this receptor in the progression of chronic lung disease. This study examines the contribution of A2BR signaling in this model by generating ADA/A2BR double-knockout mice. Our hypothesis was that genetic removal of the A2BR from ADA-deficient mice would lead to diminished pulmonary inflammation and damage. Unexpectedly, ADA/A2BR double-knockout mice exhibited enhanced pulmonary inflammation and airway destruction. Marked loss of pulmonary barrier function and excessive airway neutrophilia are thought to contribute to the enhanced tissue damage observed. These findings support an important protective role for A2BR signaling during acute stages of lung disease.
Chronic lung diseases are characterized by persistent inflammation and tissue remodeling that contribute to progressive loss of lung function (1–3). Examples of chronic lung disease include asthma, chronic obstructive pulmonary disease and interstitial lung diseases where fibrosis is a detrimental component. The inflammatory responses seen in these disorders are diverse and involve the recruitment of multiple cell types that release soluble mediators that promote inflammation and subsequent tissue remodeling (1– 4). Although the signals associated with the genesis of inflammation and the control of tissue remodeling have been described, relatively little is known about the mechanisms that promote the chronic nature of these disorders.
Deregulated or overactive wound healing pathways are hypothesized to contribute to the excessive remodeling responses that are seen in chronic lung diseases (2, 5). Adenosine is a signaling molecule that is produced after injury and promotes processes important in wound healing such as angiogenesis, matrix production, and the regulation of inflammation (6). Consistent with the aforementioned hypothesis, there is evidence to suggest that adenosine may regulate features of chronic lung disease. Adenosine levels are elevated in the lungs of human patients (7, 8) and in animal models of chronic lung disease (9 –11). In addition, lowering elevated adenosine levels in models of chronic lung disease results in the resolution of established airway inflammation and remodeling (9, 10). Moreover, mice lacking adenosine deaminase (ADA),3 an enzyme that catalyzes the breakdown of adenosine, spontaneously develop features of chronic lung disease in association with adenosine elevations (12–14). These findings suggest that elevations of adenosine in the lung might activate pathways that promote the chronicity of certain lung disorders.
Adenosine orchestrates cellular responses by engaging cell surface adenosine receptors (AR), of which there are four (A1R, A2AR, A2BR, and A3R; Ref. 15). All of these receptors have been implicated in the regulation of pulmonary inflammation (16); however, the A2BR is emerging as a receptor that serves disparate roles in acute and chronic pulmonary disorders. Exposure of mice to hypoxia or ventilator-induced lung injury leads to elevations in adenosine in association with acute pulmonary inflammation and edema (17, 18). In these models, pharmacological blockade or genetic removal of the A2BR is associated with enhanced pulmonary disease, whereas A2BR activation promoted protection from hypoxia or ventilator-induced pulmonary injury. These findings suggest a protective role for the A2BR in processes associated with acute lung injury. In contrast, pharmacological blockade of the A2BR in the ADA-deficient model of chronic lung disease resulted in attenuation of pulmonary inflammation, fibrosis, and alveolar airway enlargement (19), suggesting that the A2BR serves a detrimental role in features of chronic lung disease. Understanding the specific roles of A2BR signaling in pulmonary disease processes will be essential for designing A2BR-based therapeutic approaches for pulmonary disorders.
The focus of the current study was to investigate further the involvement of the A2BR in features of chronic lung disease by genetically removing the A2BR from ADA-deficient mice. Our hypothesis was that as with the pharmacological blockade of the A2BR in this model, genetic removal of the A2BR would result in the attenuation of features of chronic lung disease. This hypothesis was found to be null in that all features of pulmonary inflammation and remodeling found in the ADA-deficient model were enhanced following the removal of the A2BR. This was associated with a precipitous loss of barrier function and neutrophilia in the lungs of ADA/A2BR double-knockout mice. This study provides important insight into the role of A2BR signaling in this model and supports the protective role for this receptor in acute lung injury processes.
Materials and Methods
Mice
ADA-deficient mice were generated and genotyped as previously described (20). Mice homozygous for the null Ada allele were designated ADA deficient (ADA−/−), whereas mice heterozygous for the null Ada allele were designated as ADA-competent mice (ADA+). ADA/A2BR double-knock-out mice were generated by mating ADA−/− mice with A2BR-deficient mice (A2BR−/−). All mice were congenic on a C57BL/6 background, and all phenotypic comparisons were performed among littermates. Animal care was in accordance with the Animal Care Committee at the University of Texas Health Science Center (Houston, TX). All mice were housed in ventilated cages equipped with microisolator lids and maintained under strict containment protocols.
ADA enzyme therapy
Polyethylene glycol-modified ADA (PEG-ADA) was generated by the covalent modification of purified bovine ADA with activated polyethylene glycol as described (21). ADA−/− mice received i.m. injections of PEG-ADA on postnatal days 1, 5, 9, 13, and 17 (0.625, 1.25, 2.5, 2.5, and 2.5 U, respectively) and i.p. injections on postnatal day 21 (5 U). Mice were sacrificed on day 14 after the last PEG-ADA injection (postnatal day 35).
Bronchial alveolar lavage (BAL) and histology
Mice were anesthetized with avertin, and lungs were lavaged four times with 0.3 ml of PBS; 0.95–1 ml of pooled lavage fluid was recovered. Total cell counts were determined using a hemocytometer, and aliquots were cytospun onto microscope slides and stained with Diff-Quick (Dade Behring) for cellular differentials. After lavage, the lungs were infused with 10% buffered formalin at 25 cm of pressure and fixed overnight at 4°C. Fixed lung samples were dehydrated and embedded in paraffin, and sections (5 μm) were collected on microscope slides and stained with H&E (Shandon-Lipshaw) according to the manufacturer’s instructions.
Immunohistochemistry
Rehydrated slides were quenched with 3% hydrogen peroxide, Ag retrieval performed (Dako), and endogenous avidin and biotin blocked with a Biotin Blocking System (Dako). Slides were incubated with a rat anti-mouse neutrophil Ab (AbD SeroTec, 1/500 dilution, overnight at 4°C). Sections were incubated with ABC Elite Streptavidin reagents and appropriate secondary Abs, then developed with 3,3′-diaminobenzidine (Sigma-Aldrich), and counterstained with methyl green.
RT-PCR and quantitative RT-PCR
Total RNA was isolated from whole-lung tissue using Trizol reagent (Invitrogen) and treated with RNase-free DNase (Invitrogen). Transcript levels were quantified using real-time quantitative RT-PCR. A2BR and β-actin transcripts were analyzed using TaqMan probes on the Smart Cycler (Cepheid). Cytokine, chemokine and α1 procollagen transcripts were analyzed using TaqMan probes or the SYBR Green method on the Smart Cycler (Cepheid). Primer sequences for the transcripts examined were the same as previously used (12, 13, 19, 22, 23). Specific transcript levels were determined either through comparison to a standard curve generated from the PCR amplification of template dilutions or normalized to 18S rRNA and presented as mean normalized transcript levels using the comparative Ct method (2ΔΔCt) (24).
Cytokine and chemokine analysis
Blood was collected using EDTA as an anticoagulant and centrifuged at 1200 rpm for 20 min to obtain plasma. BAL fluid was collected as described previously and was centrifuged to remove cells. Plasma and the BAL fluid supernatants were analyzed using a Milliplex mouse panel that simultaneously analyzed 32 of the most common cytokines and chemokines (Millipore).
Western blot analysis
Lungs were homogenized and lysed on ice with protein lysis buffer (1 M Tris (pH7.4), 1 M NaCl, 1% Triton X-100) freshly supplemented with 1× protease inhibitor mixture (Roche Diagnostics). A 50-μg portion of total protein was electrophoresed on 10% SDS-PAGE gels and transferred overnight at 4°C to Immobilon-P polyvinylidene difluoride (Millipore), and Western blotting was performed as described (25). The following Abs were used: rat monoclonal anti-E-selectin Ab (R&D Systems; 1/5000 dilution); rabbit polyclonal anti-ICAM-1 Ab (Santa Cruz Biotechnology; 1/200 dilution); rabbit monoclonal anti-IκB-αAb (abcam; 1/5000 dilution); and mouse monoclonal anti-β-actin Ab (Sigma-Aldrich; 1/5000 dilution). Secondary Abs included: anti-rabbit IgG-HRP (eBioscience; 1/10,000 dilution), anti-mouse IgG-HRP (eBioscience; 1/10,000 dilution), and anti-rat IgG-HRP (Sigma-Aldrich; 1/10,000 dilution). Signals were detected by chemiluminesce (Pierce Chemical).
Measurement of vascular permeability
BAL fluid supernatants were assayed for total protein content by Bradford assay (BioRad). To assess pulmonary edema, lungs were weighed and dried. Weight ratios before and after drying were used to indicate lung water content. Lung vascular permeability was quantified by i.p. administration of Evans blue dye (0.2 ml of 0.5% in PBS). Four hours later, mice were perfused with PBS, and lungs were harvested. Organ Evans blue concentrations were quantified after formamide extraction (55°C over-night) by measuring OD610 with subtraction of reference OD at 450 nm. Evans blue dye contents were determined through comparison to a standard curve generated from dye dilutions.
Mucus index
The extent of mucus production in bronchial airways was determined by quantifying the amount of periodic acid-Schiff (PAS)-stained material using Image-Pro Plus analysis software (Media Cybernetics). PAS-stained material was identified on digitized images, and the pixel intensities of each color channel (red, blue, and green) were averaged. This was repeated for each image, and the values were averaged and used to determine the area (M) and intensity (I) of PAS-stained material in bronchial airways. In addition, the area (A) of the total epithelium (including PAS-stained material) was determined. The mucus index was determined using the equation (M × I)/A. Final indices were results of an average of 10 images per lung encompassing large and small bronchial airways. All quantitative studies were performed blinded.
Airway size measurement and collagen measurements
Alveolar airway size was determined in pressure-infused lungs by measuring mean chord lengths on H&E-stained lung sections (12). Representative images were digitized, and a grid consisting of 53 black lines at 10.5-μm intervals was overlaid on the image. This line grid was subtracted from the lung images using Image-Pro Plus image analysis software (Media Cybernetics), and the resultant lines were measured and averaged to determine the mean chord length of the alveolar airways. The final mean chord lengths represent averages from 10 nonoverlapping images of each lung specimen. All quantitative studies were performed blinded. The Sircol assay (Biocolor) was used to measure collagen levels in clarified BAL fluid.
TUNEL analysis
End labeling of exposed 3′-OH ends of DNA fragments in paraffin-embedded tissue was undertaken with the ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (Chemicon), using the instructions provided by the manufacturer. After staining, a minimum of 300 cells were visually evaluated in each section. The labeled cells were expressed as a percentage of total nuclei.
Whole-mount immunohistochemistry for CD31 on tracheas
Procedures for tracheal whole mount immunohistochemistry were as previously described (26). Tracheas were fixed using zinc fixative (BD Pharmingen) for 24 h, permeabilized using PBS containing 1% Triton X-100, and reacted with Abs to CD31 followed by peroxidase detection.
Statistics
Values are expressed as mean ± SEM. As appropriate, groups were compared by ANOVA; follow-up comparisons between groups were conducted using a two-tailed Student t test. A p value of ≤0.05 was considered to be significant.
Results
Elevated A2BR expression in the lungs of ADA-deficient mice
Increased levels of the A2BR have been noted in inflamed lung tissue (9, 10). Quantitative RT-PCR was performed to determine whether levels of the A2BR were elevated in the lungs of ADA-deficient (ADA−/−) mice in conjunction with the inflammation and damage seen in this model. Results demonstrated a mean 2.5-fold increase of A2BR transcripts in whole-lung RNA extracts from ADA−/− mice (Fig. 1). These findings demonstrate that A2BR transcripts are elevated in the lungs of ADA−/− mice exhibiting pulmonary inflammation and damage.
FIGURE 1.

A2BR expression in the lungs of ADA−/− mice. Transcript levels for the A2BR were measured in whole-lung RNA extracts from postnatal day 18 ADA-containing (ADA+) and ADA-deficient (ADA−/−) mice using quantitative RT-PCR. Data are presented as mean percentage of β-actin transcripts ± SEM; n = 4 for each. *, p ≤ 0.05 compared with ADA+.
ADA/A2BR double-knockout mice die precociously and exhibit enhanced pulmonary inflammation
ADA/A2BR double-knockout mice were generated to assess the impact of genetically removing this receptor on the pulmonary phenotypes seen in ADA−/− mice. ADA−/− mice develop progressive pulmonary inflammation and die between postnatal days 18 and 21 (12). ADA/A2BR double-knockout mice did not survive past postnatal day 11 or 12 (data not shown), demonstrating that the genetic removal of the A2BR from ADA−/− mice leads to precocious death. The loss of animals at this stage made detailed assessment of pulmonary phenotypes difficult. Therefore, ADA−/− mice and ADA/A2BR double-knockout mice were identified at birth and maintained on ADA enzyme therapy until postnatal day 21 to prevent defects in alveolarization (19). ADA enzyme therapy was then halted to allow adenosine levels to elevate and pulmonary phenotypes to develop. ADA−/− mice treated in this manner exhibit signs of respiratory distress ~16 days after the cessation of ADA enzyme therapy. In contrast, ADA/A2BR double-knockout mice began to show signs of respiratory distress as early as day 11 after the cessation of enzyme therapy, and succumbed by 16 days following the removal of ADA enzyme therapy. Therefore, detailed analysis of pulmonary phenotypes was conducted 14 days after the cessation of ADA enzyme therapy, which was postnatal day 35. At this stage, ADA−/− mice had diffuse monocytic inflammation relative to that seen in wild-type mice (compare Fig. 2, A and C). There was no evidence of inflammation in ADA+ mice lacking the A2BR (Fig. 2B), whereas ADA/A2BR double-knockout mice exhibited pronounced pulmonary inflammation and thickening of the alveolar septae (Fig. 2D).
FIGURE 2.

Lung histopathology. Lungs were collected on postnatal day 35 and prepared for sectioning and H&E staining. A, Lung section from an ADA+ A2BR+/+ mouse. B, Lung section from an ADA+ A2BR−/− mouse. C, Lung section from an ADA−/−A2BR+/+ mouse. D, Lung section from an ADA−/−A2BR−/− mouse. Sections are representative of eight mice from each genotype. Bars, 100 μm.
To quantify the level of inflammation in ADA/A2BR double-knockout mice, BAL was performed, and recovered cells were counted. There was a significant increase in the number of inflammatory cells recovered from ADA/A2BR double-knockout mice compared with ADA−/− mice containing the A2BR (Fig. 3A). Differential staining of airway cells demonstrated a significant increase in neutrophils, lymphocytes, and alveolar macrophages (Fig. 3, B and C). To better visualize increases in lung tissue neutrophils, lung sections were incubated with an Ab against mouse neutrophils. There were robust increases in tissue neutrophils in the lung parenchyma of ADA/A2BR double-knockout mice relative to the lungs of ADA−/− mice (Fig. 3, D and E). Flow cytometric analysis of whole lung cellular digest confirmed selective increases in neutrophils within the lungs of ADA/A2BR double-knockout mice (data not shown). Together, these data demonstrate that there is enhanced pulmonary neutrophilia in the lungs of ADA−/− mice lacking the A2BR.
FIGURE 3.

Pulmonary inflammation. A, BAL fluid was collected on postnatal day 35, and total cell numbers were determined. B, BAL cells were cytospun and stained with Diff-Quick, allowing for determination of cellular differentials. Data are mean cell counts ± SEM. *, p ≤ 0.05 vs ADA+ mice; #, p ≤ 0.05 vs ADA−/− A2BR+/+ mice; n = 6 (ADA+ A2BR+/+ and ADA+ A2BR−/−), n = 8 (ADA−/−A2BR+/+ and ADA−/−A2BR−/−). C, Cytospun BAL cells stained with Diff-Quick. Bar, 10 μm. D, Lung sections from an ADA−/−A2BR−/− mouse and an ADA−/−A2BR−/− mouse (E) were stained with an Ab against neutrophils to visualize infiltrated tissue neutrophils (brown). Bars, 100 μm.
Inflammatory cytokine and chemokine production is enhanced in ADA/A2BR double-knockout mice
To better understand the mechanisms involved in the regulation of inflammatory responses in ADA/A2BR double-knockout mice, cytokine and chemokine transcript levels were monitored in whole-lung RNA extracts. There were no differences in the levels of cytokines or chemokines in the lungs of ADA+ mice with or with-transcripts were out the A2BR (Fig. 4 and data not shown). TNF-α elevated in the lungs of ADA−/− mice, and levels were increased in the absence of the A2BR (Fig. 4A). IL-6 transcripts were elevated in the lungs of ADA−/− mice but were significantly decreased with the removal of the A2BR (Fig. 4B). Among the chemokines examined, levels of CXCL-1 and MCP-1 were markedly increased in the lungs of ADA−/− mice, and levels of CXCL-1 were further increased after the genetic removal of the A2BR (Fig. 4, C and D). These data demonstrate that removal of the A 2BR results in exaggerated production of TNF-α and CXCL-1 in the lungs of ADA−/− mice.
FIGURE 4.

Production of cytokines and chemokines. Transcript levels of various proinflammatory cytokines and chemokines were measured in whole-lung extracts from postnatal day 35 mice using quantitative RT-PCR. Shown are levels of TNF-α (A), IL-6 (B), CXCL1 (C), and MCP-1 (D). Results are presented as mean fold increase compared with controls or percent of 18S RNA ± SEM. *, p ≤ 0.05 vs ADA+ mice; #, p ≤ 0.05 vs ADA−/−A2BR+/+ mice. n = 4 (ADA+ A2BR+/+ and ADA+A2BR−/−), n = 8 (ADA−/− A2BR+/+ and ADA−/− A2BR−/−).
Cytokine and chemokine protein levels were determined in plasma and BAL fluid using Milliplex mouse cytokine/chemokine panels (Table I). CXCL-1 and MCP-1 protein levels were increased in the plasma of ADA−/− mice, and levels were increased further after the genetic removal of the A2BR. Levels for IL-1α and IL-1β were not elevated in ADA−/− mice containing the A2BR; however, they were significantly elevated in ADA/A2BR double-knockout mice. MIG protein levels were decreased in the plasma of ADA−/− mice and were further decreased in mice lacking the A2BR. Levels of CXCL-1 and LIF-1 were elevated in the BAL fluid of ADA−/− mice, and levels were significantly elevated after the genetic removal of the A2BR. RANTES protein levels were not elevated in the BAL fluid of ADA−/− mice containing the A2BR; however, they were significantly elevated in ADA/A2BR double-knockout mice. MCP-1 protein levels were not altered after the genetic removal of the A2BR although they were increased in ADA−/− mice compared with wild-type mice. These data demonstrate that theA2BR differentially regulates cytokine and chemokine production in this setting of elevated adenosine levels.
Table I.
Cytokine and chemokine protein levels in plasma and BALa fluid
| Plasma (pg/ml)
|
BALF (pg/ml)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| ADA+ A2BR+/+ | ADA+ A2BR−/− | ADA−/− A2BR+/+ | ADA−/− A2BR −/− | ADA+ A2BR+/+ | ADA+ A2BR−/− | ADA−/− A2BR+/+ | ADA−/− A2BR −/− | |
| CXCL-1 | 111 ± 10 | 155 ± 18 | 493 ± 121* | 1086 ± 191# | 3 ± 1 | 5 ± 1 | 50 ± 24* | 116 ± 29# |
| MCP-1 | 7 ± 1 | 11 ± 4 | 27 ± 8* | 190 ± 98 # | 18 ± 16 | 26 ± 14 | 153 ± 83* | 245 ± 85 |
| LIF | ND | ND | ND | ND | 0.3 ± 0.2 | 0.1 ± 0.1 | 9 ± 4* | 26 ± 8# |
| RANTES | 18 ± 4 | 25 ± 4 | 18 ± 6 | 20 ± 4 | 0.4 ± 0.3 | 0.2 ± 0.1 | 0.6 ± 0.2 | 2 ± 1# |
| MIG | 143 ± 21 | 123 ± 8 | 41 ± 14* | 19 ± 3 | ND | ND | ND | ND |
| IL-1α | 89 ± 19 | 52 ± 6 | 68 ± 37 | 522 ± 218# | 4 ± 1 | 5 ± 2 | 2 ± 2 | 3 ± 2 |
| IL-1β | 17 ± 4 | 26 ± 4 | 33 ± 7 | 81 ± 29 # | 5 ± 2 | 4 ± 1 | 5 ± 1 | 5 ± 1 |
Levels of inflammatory mediators in plasma and BAL fluid. Plasma and BAL fluid were collected from postnatal day 35 mice. Protein levels of inflammatory cytokines and chemokines were determined by Luminex assay. Data are presented as mean ± SEM.
, p ≤ 0.05 vs ADA+ mice;
p ≤ 0.05 vs ADA−/− A2BR+/+ mice. n = 4 (ADA+), 6 (ADA−/−).
ND, not detected.
Expression of cell adhesion molecules in the absence of the A2BR
The genetic removal of the A2BR is associated with up-regulation of adhesion molecules such as E-selectin, P-selectin, and ICAM-1 in association with NF-κB activation (27). These observations together with the enhanced pulmonary neutrophilia seen in ADA/A2BR double-knockout mice prompted the examination of the expression levels of adhesion molecules and IκB-α as an indicator of NF-κB activation. Whole-lung extracts from postnatal day 35 mice were subjected to Western blot analysis to determine the protein levels of E-selectin, ICAM-1, and IκB-α. These experiments were analyzed in parallel with lungs from postnatal day 22, a stage preceding inflammation and elevated adenosine levels (19). The expression of E-selectin, ICAM-1 and IκB-α were not altered in ADA−/− or A2BR−/− mice on day 22 (Fig. 5). On day 35 when pulmonary inflammation was robust, the expression of E-selectin and ICAM-1 were elevated in the lungs of ADA−/− mice; however, levels were not elevated further in the lungs of ADA/A2BR double-knockout mice (Fig. 5). In addition, the expression of IκB-α was not altered in any of the genotypes (Fig. 5). These data demonstrate that there is no baseline increase in key cell adhesion molecules in the lung in the absence of the A2BR. Furthermore, the absence of increases in ADA/A2BR double-knockout mice compared with that in ADA−/− mice suggest that these pathways do not mediate the enhanced pulmonary neutrophilia seen.
FIGURE 5.
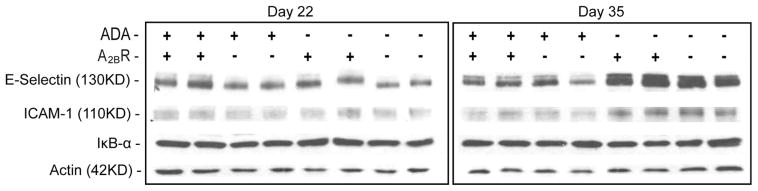
Western blot analysis of E-selectin, ICAM-1, and IκB-α in whole-lung extracts. Samples from postnatal day 22 and 35 mice were subjected to Western blotting with the indicated Abs. Antiactin was used as a loading control.
Enhanced vascular permeability in ADA/A2BR double-knockout mice
Previous studies have suggested that A2BR signaling serves an important role in the preservation of endothelial barrier function during hypoxia and lung injury (17). Thus, we hypothesized that A2BR deficiency would enhance vascular leakage in ADA−/− mice. To address this, BAL fluid was collected from postnatal day 35 mice, and total protein levels were determined. Total protein content was elevated in the lungs of ADA−/− mice and was markedly increased in the absence of the A2BR (Fig. 6A). Lung water content was determined by measuring lung wet-dry weight ratios. ADA deficiency was associated with a significant degree of lung water increase, but this increase was not affected by the genetic removal of the A2BR (Fig. 6B). However, comparative analysis of vascular permeability as assessed by Evans blue dye extravasation, revealed that vascular leakage was significantly increased in lungs of ADA/A2BR double-knockout mice compared with that in ADA−/− mice (Fig. 6, C and D). These findings identify signaling through the A2BR as a critical control point for pulmonary endothelial barrier functions associated with elevated levels of adenosine in ADA−/− mice.
FIGURE 6.

Vascular permeability analysis. A, BAL fluid was collected from postnatal day 35 mice, and total BAL fluid protein levels were determined by Bradford assay. Data are mean total protein content ± SEM. *, p ≤ 0.05 vs ADA+ mice; #, p ≤ 0.05 vs ADA−/− A2BR+/+ mice. n = 6 (ADA+), 8 (ADA−/−). B, Lungs from postnatal day 35 mice were collected, and lung water content was determined by lung wet-dry weight ratio. Data are mean ratios ± SEM. *, p ≤ 0.05 vs ADA+ mice; #, p ≤ 0.05 vs ADA−/− A2BR+/+ mice. n = 4 (ADA+ A2BR+/+ and ADA+A2BR−/−), n = 6 (ADA−/−A2BR+/+ and ADA−/−A2BR−/−). Mice were given i.p. Evans blue dye (0.2 ml of 0.5% in PBS) and sacrificed 4 h later, and the hearts and lungs were harvested. C, Representative images of lungs. Evans blue dye concentrations were quantified in lung (D). Data are mean dye content in micrograms per gram of lung tissue ± SEM. *, p ≤ 0.05 vs ADA+ mice; #, p ≤ 0.05 vs ADA−/− A2BR+/+ mice. n = 4 (ADA+ A2BR+/+ and ADA+ A2BR−/−), n = 8 (ADA−/− A2BR+/+ and ADA−/−A2BR−/−).
Enhanced mucous cell metaplasia in the lungs of ADA/A2BR double-knockout mice
Mucous cell metaplasia is a prominent feature seen in the bronchial airways of ADA−/− mice (12). To determine the impact of genetically removing the A2BR on mucous cell metaplasia, tissue sections were stained with PAS, and morphometry was used to quantify mucous cell metaplasia. A2BR−/− mice exhibited increased numbers of PAS-positive cells in the bronchial airways (compare Fig. 7, A, B, and E). As expected, a large number of mucin-containing cells were seen in the bronchial airways of ADA−/− mice (Fig. 7, C and E). Moreover, there was increased PAS staining in the bronchial airways of ADA/A2BR double-knockout mice (Fig. 7, D and E). These results demonstrate that there is mucus accumulation in A2BR−/− mice at baseline and that this feature is enhanced in ADA/A2BR double-knockout mice.
FIGURE 7.

Mucus metaplasia. Lungs were collected on postnatal day 35 and prepared for sectioning and PAS staining. A, Lung section from an ADA+ A2BR+/+ mouse. B, Lung section from an ADA+ A2BR−/− mouse. C, Lung section from an ADA−/− A2BR+/+ mouse. D, Lung section from an ADA−/− A2BR−/− mouse. Sections are representative of eight different mice from each genotype. Bars, 100 μm. E, A mucus index was determined, and data are presented as mean mucus index ± SEM. *, p ≤ 0.05 vs ADA+ mice; #, p ≤ 0.05 vs ADA−/− A2BR+/+ mice. n = 4 (ADA+ A2BR+/+ and ADA+ A2BR−/−), n = 8 (ADA−/− A2BR+/+ and ADA−/− A2BR−/−).
Enhanced alveolar destruction in the lungs of ADA/A2BR double-knockout mice
ADA−/− mice develop features of alveolar airspace enlargement that are characteristic of emphysema (19). To determine the consequences of genetically removing A2BR on airway enlargement, alveolar destruction was analyzed in ADA/A2BR double-knockout mice. As expected, ADA−/− mice exhibited a significant increase in alveolar size (Fig. 8, A and E). Furthermore, the degree of air-way enlargement was increased in the lungs of ADA/A2BR double-knockout mice (Fig. 8, B and E). These findings were associated with increased apoptosis in the alveolar airways of ADA/A2BR double-knockout mice (Fig. 8, C, D, and F). Together, these findings demonstrated enhanced alveolar destruction in the lungs of ADA−/− mice lacking the A2BR.
FIGURE 8.

Alveolar airway enlargement. Lungs from postnatal day 35 mice were infused with fixative under constant pressure (25 cm H2O) and processed for H&E or TUNEL staining. A, H&E-stained lung section from an ADA−/− A2BR+/+ mouse. B, H&E-stained lung section from an ADA−/−A2BR+/+ mouse. C, TUNEL-stained lung section from an ADA−/− A2BR+/+ mouse. D, TUNEL-stained lung section from an ADA−/− A2BR−/− mouse. Images are representative of eight animals from each genotype. E, Alveolar airspace size was calculated using ImagePro analysis software; data are mean chord length ± SEM. F, TUNEL-positive cells were counted, and data are presented as mean TUNEL scores ± SEM. *, p ≤ 0.05 vs ADA+ mice; #, p ≤ 0.05 vs ADA−/−A2BR+/+ mice. n = 4 (ADA+ A2BR+/+ and ADA+ A2BR−/−), n = 8 (ADA−/− A2BR+/+ and ADA−/− A2BR−/−). Bars, 100 μm.
Increased collagen production in the lungs of ADA/A2BR double-knockout mice
Enhanced collagen deposition and production are other features noted in the lungs of ADA−/− mice (13, 19). Examination of α1 procollagen transcript (Fig. 9A) and soluble collagen levels in BAL fluid (Fig. 9B) revealed enhanced collagen production in the lungs of ADA/A2BR double-knockout mice.
FIGURE 9.

Collagen levels. A, Transcript levels for α1-procollagen were measured in lung extracts from postnatal day 35 mice using quantitative RT-PCR, and results are presented as mean fold increase compared with controls or mean pg transcript/μg RNA ± SEM. B, Collagen protein levels in BAL fluid from postnatal day 35 mice were quantified using the Sircol assay. Data are presented as mean collagen levels ± SEM. *, p ≤ 0.05 vs ADA+ mice; #, p ≤ 0.05 vs ADA−/− A2BR+/+ mice. n = 4 (ADA+ A2BR+/+ and ADA+ A2BR−/−), n = 8 (ADA−/− A2BR+/+ and ADA−/− A2BR−/−).
Enhanced tracheal vascularity in ADA/A2BR double-knockout mice
ADA−/− mice develop increased tracheal vascularity in an adenosine-dependent manner, which is thought to contribute to the progression of pulmonary fibrosis seen in this model (26). To determine the effect of genetically removing the A2BR on this end-point, whole-mount immunohistochemistry on tracheas was conducted to assess vessel numbers (Fig. 10). Consistent with previous findings, there was a significant increase in vessel numbers in the tracheas of ADA−/− mice. Furthermore, ADA/A2BR double-knockout mice exhibited increased tracheal vascularity compared with ADA−/− mice possessing the A2BR (Fig. 10). These results indicate that the genetic removal of the A2BR leads to enhanced tracheal angiogenesis in ADA−/− mice.
FIGURE 10.

Tracheal angiogenesis. A, Tracheas were removed from postnatal day 35 mice and analyzed by whole-mount CD31 immunostaining for the visualization of vessels. Results are representative of four mice from each genotype. Bars, 100 μm. B, Tracheal vascularity was quantified by counting the number of vessels intersecting a line down the length of the cartilage ring. At least 12 cartilage rings were analyzed per sample. Data are represented as mean vessels (in millimeters) ± SEM; n = 4. *, p ≤ 0.05 vs ADA+ mice; #, p ≤ 0.05 vs ADA−/− A2BR+/+ mice.
Discussion
Adenosine is rapidly generated in response to cellular injury and plays important roles in orchestrating the balance between tissue protection and repair, and the progression to pathological tissue remodeling (28). Evidence for this is provided by studies in the ADA-deficient mouse model demonstrating that adenosine elevations are closely associated with the activation of pathways that increase airway inflammation and remodeling (12, 13). Furthermore, pharmacological studies suggest the A2BR contributes to the adenosine-dependent phenotypes seen in the ADA-deficient model (19) and thus may represent an important therapeutic target for regulating pathological tissue remodeling characteristic of chronic lung disease. The hypothesis of the current study was that inactivation of the A2BR in ADA-deficient mice would result in attenuation of the observed pulmonary phenotypes. However, the genetic removal of the A2BR from ADA-deficient mice led to enhanced pulmonary inflammation and airway destruction, revealing an important protective role for A2BR signaling during acute stages of lung disease in this model.
These findings are consistent with the enhanced pulmonary inflammation seen in A2BR knockout mice subjected to either hypoxia (17) or ventilator-induced lung injury (18). In these acute lung injury models, A2BR knockout mice exhibited enhanced loss of pulmonary barrier function together with enhanced pulmonary neutrophilia. Further-more, treatment of wild-type mice with an A2BR agonist protected from the loss of barrier function and neutrophilic inflammation (17, 18). These studies clearly indicate that A2BR engagement is critical for regulating barrier function following acute lung injury. Consistent with this, we now demonstrate that ADA/A2BR double-knockout mice exhibit enhanced loss of pulmonary barrier function and a selective increase in pulmonary neutrophils. These findings further support the notion that adenosine generation and the promotion of barrier function through A2BR engagement are prominent features of acute lung injury, and that targeting this pathway could provide therapeutic benefits for the treatment of various pulmonary disorders (18). The mechanisms by which A2BR engagement regulates barrier function are not clear, but they may involve protein kinase A-induced phosphorylation of vasodilator-stimulated phosphoprotein, which can influence the geometry and distribution of junctional proteins, thereby affecting the characteristics of the junctional complex and promoting increases in barrier function (29 –31).
We propose that the enhanced loss of barrier function in ADA/A2BR double-knockout mice is a major mechanism accounting for the increased number of neutrophils found in the lungs of these mice. However, it is also possible that increased expression of inflammatory mediators also influence the neutrophilia seen. In particular, TNF-α levels were markedly increased in the lungs of ADA/A2BR double-knockout mice, which could activate pathways that influence the trafficking of neutrophils in the lung. This would be consistent with previous studies demonstrating enhanced systemic TNF-α levels in A2BR knockout mice at baseline and following LPS exposure (27). In that study, increased production of TNF-α in A2BR knockout mice was associated with enhanced expression of leukocyte adhesion molecules and increased leukocyte adhesion and rolling in mesenteric vessels (27). However, levels of leukocyte adhesion molecules were not found to be increased in the lungs of ADA/A2BR double-knockout mice, suggesting enhanced leukocyte adhesion or rolling in the lungs does not contribute to the increased airway neutrophilia seen. This may be due to the absence of IκB-α activation and subsequent NF-κB activation, which has been suggested to account for the increases in the mesenteric vessels of A2BR knockout mice (27). These differences could be attributed to the different tissues examined or to the nature of the inflammatory insults in the different models.
Another interesting distinction between these two studies was in the regulation of IL-6. Previous studies demonstrated increased IL-6 levels in A2BR knockout mice at baseline and after LPS exposure (27). In contrast, we found that IL-6 was not elevated at baseline in the lungs of A2BR knockout mice compared with wild-type mice and that levels of IL-6 were reduced in the lungs of ADA/A2BR double-knockout mice compared with ADA knockout mice with the A2BR. Consistent with this, treatment of ADA-deficient mice with an A2BR antagonist results in diminished IL-6 production in the lungs of ADA-deficient mice (19). These findings suggest that the A2BR regulates IL-6 production in the lungs of ADA-deficient mice, a feature consistent with those seen in isolated pulmonary cell types such as bronchial smooth muscle (32), pulmonary fibroblast (33), bronchial epithelial cells (34), and alveolar macrophages (M. Pedroza and M. R. Blackburn, unpublished observations). Current efforts are focused on understanding the mechanisms and function of A2BR-mediated IL-6 production in ADA-deficient mice. The regulation of this fibrogenic cytokine may represent an important distinguishing feature underlying the protective effects of A2BR signaling, namely the promotion of barrier function, and tissue remodeling responses of this receptor, such as the promotion of fibrosis.
In addition to increases in the number of pulmonary neutrophils, ADA/A2BR double-knockout mice exhibited increased pulmonary tissue damage and remodeling. This included enhanced destruction of the alveolar airways, mucous cell metaplasia, airway angiogenesis, and collagen production. Mediators generated by activated neutrophils have been shown to contribute to these features of chronic lung disease (35–37), and it is possible that the enhancement of the pulmonary phenotypes noted in ADA/A2BR knockout mice is attributed to the increases in pulmonary neutrophils seen. For example, neutrophils produce degradative enzymes such as Matrix metalloproteinase-9 (38) and neutrophil elastase (39), which can contribute to alveolar airway enlargement and could account for the enhanced airway enlargement seen in ADA/A2BR double-knockout mice exhibiting increases in airway neutrophils. Similarly, oxidative burst from neutrophils in the airways might contribute to apoptosis of alveolar airway epithelial cells also contributing to enhanced airway enlargement (40) or to the enhanced mucous cell metaplasia seen (41). Thus, the enhancement of many of the pulmonary phenotypes noted in the lungs of ADA/A2BR double-knockout mice are likely related to the increased number of neutrophils found in the airways of these animals.
These findings differ from observations following the pharmacological blockade of the A2BR in the ADA-deficient model (19), in which treatment with an A2BR antagonist beginning 4 days after the cessation of ADA enzyme therapy resulted in diminished airway inflammation, neutrophilia, alveolar airway enlargement, and pulmonary fibrosis (19). Concentrations of the A2BR antagonists used in that study did not reach effective concentrations until ~10 days after the cessation of ADA enzyme therapy, which suggests that A2BR antagonism was capable of attenuating active lung disease in this model. These findings, together with those in the current study, suggest that A2BR signaling has important anti-inflammatory activities during early stages of lung disease that were revealed by the global genetic removal of the A2BR, whereas A2BR engagement during active disease may contribute to disease progression. These findings also suggest that the A2BR is not necessary for the development of many of the pulmonary phenotypes seen in the ADA-deficient model but likely contributes to the progression of chronic remodeling features including alveolar airway destruction and fibrosis. Moreover, it is likely that treatment of mice with an A2BR antagonist during active stages of disease when A2BR levels are increased does not lead to a complete inactivation of this receptor, a feature that may be important for allowing the protective effects of the A2BR to be maintained in this model despite antagonistic treatments. Hence, the stage of disease and the degree of A2BR antagonism or agonism will be critical variables to assess when using A2BR based therapeutics in the treatment of lung diseases.
Differential effects of A2BR signaling in and among models are not without precedence, which underscores the important role this signaling pathway has on distinct cell populations at different disease stages. In acute injury models, the A2BR plays an anti-inflammatory role in that tissue damage and injury is amplified in the absence of A2BR signaling (17, 18, 42). In addition, the ablation of the A2BR causes augmentation of proinflammatory cytokines, such as TNF-α, and a consequent activation of NF-κB signaling pathway (27), and the loss of this Gs-coupled receptor on mouse bone marrow-derived mast cells results in decreased levels of cAMP and enhanced mast cell activation (43). Furthermore, A2BR engagement protects the kidney (42) and heart (44) from ischemia reperfusion injury. Finally, a recent study has demonstrated that the neuronal guidance molecule netrin-1 can interact with the A2BR on neutrophils and attenuate hypoxia-induced inflammation (45). This provides a novel mechanism for A2BR-mediated anti-inflammatory responses.
In contrast to the protective functions of A2BR signaling, evidence exists to support a role for A2BR signaling in disease progression. A2BR engagement can promote the differentiation of pulmonary fibroblasts into myofibroblasts, which may contribute to increases in the deposition of collagen and fibrosis (33). In addition, A2BR activation mediates the direct expression of proinflammatory cytokines such as IL-8, IL-4, and IL-13 from human HMC-1 cells (46 – 48), and up-regulates IL-6 production from mouse macrophages (49) and IL-13 and VEGF secretion from mouse bone marrow mast cells (46). In keeping with these findings, A2BR antagonism is beneficial in a ragweed model of allergic lung inflammation (50). Similarly, blockade or deletion of the A2BR in a mouse model of colitis showed reduced clinical symptoms, histological scores, and IL-6 and keratinocyte-derived chemokine levels (51, 52). Interestingly, although colonic inflammation was attenuated in A2BR-deficient mice compared with their wild-type counterparts, A2BR-deficient mice actually showed increased susceptibility to systemic Salmonella infection (51). As with the current study, this observation highlights the emerging theme that A2BR signaling serves diverse functions in regulating inflammatory responses.
It will be important to define the specific mechanisms by which A2BR signaling contributes to tissue-protective or tissue-destructive processes in specific diseases. In this regard, the ADA-deficient model will be useful in examining stage specific effects in adenosine-mediated lung injury. Current and future efforts will use this model to examine the stage-specific effects of A2BR antagonists or agonists on features of acute and chronic lung disease. In addition, approaches to conditionally remove the A2BR from specific cell types in the lung will prove informative in defining mechanisms on specific cellular processes. In addition, this model will prove useful in examining the role of novel A2BR interactions with netrin-1, which could provide a novel distinction between the anti-inflammatory and proinflammatory actions of the A2BR.
In conclusion, we demonstrate here that the genetic deletion of the A2BR from ADA-deficient mice causes enhanced pulmonary inflammation and injury. Marked loss of pulmonary barrier function leading to excessive airway neutrophilia is thought to be the underlying mechanism for the enhanced tissue damage observed. These findings are particularly striking in light of the previous observations that A2BR antagonism during active disease can attenuate disease progression in the same model (19). These findings suggest that engagement of the A2BR plays important protective roles in acute injury processes such as barrier function, but may be detrimental in processes associated with chronic tissue remodeling. Determining the mechanisms underlying the protective and destructive components of A2BR signaling will be critical for the development of A2BR-based therapies for the treatment of different diseases.
Acknowledgments
We thank Dr. Daniel J. Schneider for his helpful comments on the manuscript.
Footnotes
This work was supported by National Institutes of Health Grants AI43572 and HL70952 to M.R.B. W.M. was supported by an Odyssey Program Fellowship and The Cockrell Foundation Award for Scientific Achievement at M. D. Anderson Cancer Center.
Abbreviations used in this paper: ADA, adenosine deaminase; AR, adenosine receptor; PEG-ADA, polyethelene glycol-ADA; BAL, bronchial alveolar lavage; PAS, periodic acid-Schiff.
Disclosures
Dr. M. R. Blackburn is a paid consultant for CV Therapeutics, which develops AR-based therapeutics.
References
- 1.Vestbo J, Prescott E. Update on the “Dutch hypothesis” for chronic respiratory disease. Thorax. 1998;53(Suppl 2):S15–S19. doi: 10.1136/thx.53.2008.s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thannickal VJ, Toews GB, White ES, Lynch JP, 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med. 2004;55:395–417. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- 3.Sime PJ, O’Reilly KM. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol. 2001;99:308–319. doi: 10.1006/clim.2001.5008. [DOI] [PubMed] [Google Scholar]
- 4.Tillie-Leblond I, Pugin J, Marquette CH, Lamblin C, Saulnier F, Brichet A, Wallaert B, Tonnel AB, Gosset P. Balance between proinflammatory cytokines and their inhibitors in bronchial lavage from patients with status asthmaticus. Am J Respir Crit Care Med. 1999;159:487–494. doi: 10.1164/ajrccm.159.2.9805115. [DOI] [PubMed] [Google Scholar]
- 5.Sheppard D. Pulmonary fibrosis: a cellular overreaction or a failure of communication? J Clin Invest. 2001;107:1501–1502. doi: 10.1172/JCI13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valls MD, Cronstein BN, Montesinos MC. Adenosine receptor agonists for promotion of dermal wound healing. Biochem Pharmacol. 2009;77:1117–1124. doi: 10.1016/j.bcp.2008.11.002. Epub November 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Driver AG, Kukoly CA, Ali S, Mustafa SJ. Adenosine in bronchoalveolar lavage fluid in asthma. Am Rev Respir Dis. 1993;148:91–97. doi: 10.1164/ajrccm/148.1.91. [DOI] [PubMed] [Google Scholar]
- 8.Huszar E, Vass G, Vizi E, Csoma Z, Barat E, Molnar Vilagos G, Herjavecz I, Horvath I. Adenosine in exhaled breath condensate in healthy volunteers and in patients with asthma. Eur Respir J. 2002;20:1393–1398. doi: 10.1183/09031936.02.00005002. [DOI] [PubMed] [Google Scholar]
- 9.Blackburn MR, Lee CG, Young HW, Zhu Z, Chunn JL, Kang MJ, Banerjee SK, Elias JA. Adenosine mediates IL-13-induced inflammation and remodeling in the lung and interacts in an IL-13-adenosine amplification pathway. J Clin Invest. 2003;112:332–344. doi: 10.1172/JCI16815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma B, Blackburn MR, Lee CG, Homer RJ, Liu W, Flavell RA, Boyden L, Lifton RP, Sun CX, Young HW, Elias JA. Adenosine metabolism and murine strain-specific IL-4-induced inflammation, emphysema, and fibrosis. J Clin Invest. 2006;116:1274–1283. doi: 10.1172/JCI26372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Volmer JB, Thompson LF, Blackburn MR. Ecto-5′-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J Immunol. 2006;176:4449–4458. doi: 10.4049/jimmunol.176.7.4449. [DOI] [PubMed] [Google Scholar]
- 12.Blackburn MR, Volmer JB, Thrasher JL, Zhong H, Crosby JR, Lee JJ, Kellems RE. Metabolic consequences of adenosine deaminase deficiency in mice are associated with defects in alveogenesis, pulmonary inflammation, and airway obstruction. J Exp Med. 2000;192:159–170. doi: 10.1084/jem.192.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chunn JL, Molina JG, Mi T, Xia Y, Kellems RE, Blackburn MR. Adenosine-dependent pulmonary fibrosis in adenosine deaminase-deficient mice. J Immunol. 2005;175:1937–1946. doi: 10.4049/jimmunol.175.3.1937. [DOI] [PubMed] [Google Scholar]
- 14.Chunn JL, Mohsenin A, Young HW, Lee CG, Elias JA, Kellems RE, Blackburn MR. Partially adenosine deaminase-deficient mice develop pulmonary fibrosis in association with adenosine elevations. Am J Physiol Lung Cell Mol Physiol. 2006;290:L579–L587. doi: 10.1152/ajplung.00258.2005. [DOI] [PubMed] [Google Scholar]
- 15.Fredholm BB, APIJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 16.Mohsenin A, Blackburn MR. Adenosine signaling in asthma and chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2006;12:54–59. doi: 10.1097/01.mcp.0000199002.46038.cb. [DOI] [PubMed] [Google Scholar]
- 17.Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. 2008;111:2024–2035. doi: 10.1182/blood-2007-10-117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest. 2008;118:3301–3315. doi: 10.1172/JCI34203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun CX, Zhong H, Mohsenin A, Morschl E, Chunn JL, Molina JG, Belardinelli L, Zeng D, Blackburn MR. Role of A2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Invest. 2006;116:2173–2182. doi: 10.1172/JCI27303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blackburn MR, Datta SK, Kellems RE. Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J Biol Chem. 1998;273:5093–5100. doi: 10.1074/jbc.273.9.5093. [DOI] [PubMed] [Google Scholar]
- 21.Young HW, Molina JG, Dimina D, Zhong H, Jacobson M, Chan LN, Chan TS, Lee JJ, Blackburn MR. A3 adenosine receptor signaling contributes to airway inflammation and mucus production in adenosine deaminase-deficient mice. J Immunol. 2004;173:1380–1389. doi: 10.4049/jimmunol.173.2.1380. [DOI] [PubMed] [Google Scholar]
- 22.Sun CX, Young HW, Molina JG, Volmer JB, Schnermann J, Blackburn MR. A protective role for the A1 adenosine receptor in adenosine-dependent pulmonary injury. J Clin Invest. 2005;115:35–43. doi: 10.1172/JCI22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohsenin A, Mi T, Xia Y, Kellems RE, Chen JF, Blackburn MR. Genetic removal of the A2A adenosine receptor enhances pulmonary inflammation, mucin production and angiogenesis in adenosine deaminase deficient mice. Am J Physiol Lung Cell Mol Physiol. 2007;293:L753–L763. doi: 10.1152/ajplung.00187.2007. [DOI] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−δ δCt method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Burnette WN. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem. 1981;112:195–203. doi: 10.1016/0003-2697(81)90281-5. [DOI] [PubMed] [Google Scholar]
- 26.Mohsenin A, Burdick MD, Molina JG, Keane MP, Blackburn MR. Enhanced CXCL1 production and angiogenesis in adenosine-mediated lung disease. FASEB J. 2007;21:1026–1036. doi: 10.1096/fj.06-7301com. [DOI] [PubMed] [Google Scholar]
- 27.Yang D, Zhang Y, Nguyen HG, Koupenova M, Chauhan AK, Makitalo M, Jones MR, St Hilaire C, Seldin DC, Toselli P, et al. The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Invest. 2006;116:1913–1923. doi: 10.1172/JCI27933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blackburn MR. Too much of a good thing: adenosine overload in adenosine-deaminase-deficient mice. Trends Pharmacol Sci. 2003;24:66–70. doi: 10.1016/S0165-6147(02)00045-7. [DOI] [PubMed] [Google Scholar]
- 29.Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Comerford KM, Lawrence DW, Synnestvedt K, Levi BP, Colgan SP. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. FASEB J. 2002;16:583–585. doi: 10.1096/fj.01-0739fje. [DOI] [PubMed] [Google Scholar]
- 31.Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, Gertler FB. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–521. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- 32.Zhong H, Belardinelli L, Maa T, Feoktistov I, Biaggioni I, Zeng D. A2B adenosine receptors increase cytokine release by bronchial smooth muscle cells. Am J Respir Cell Mol Biol. 2004;30:118–125. doi: 10.1165/rcmb.2003-0118OC. [DOI] [PubMed] [Google Scholar]
- 33.Zhong H, Belardinelli L, Maa T, Zeng D. Synergy between A2B adenosine receptors and hypoxia in activating human lung fibroblasts. Am J Respir Cell Mol Biol. 2005;32:2–8. doi: 10.1165/rcmb.2004-0103OC. [DOI] [PubMed] [Google Scholar]
- 34.Sun Y, Wu F, Sun F, Huang P. Adenosine promotes IL-6 release in airway epithelia. J Immunol. 2008;180:4173–4181. doi: 10.4049/jimmunol.180.6.4173. [DOI] [PubMed] [Google Scholar]
- 35.Chua F, Laurent GJ. Neutrophil elastase: mediator of extracellular matrix destruction and accumulation. Proc Am Thorac Soc. 2006;3:424–427. doi: 10.1513/pats.200603-078AW. [DOI] [PubMed] [Google Scholar]
- 36.Shao MX, Nadel JA. Neutrophil elastase induces MUC5AC mucin production in human airway epithelial cells via a cascade involving protein kinase C, reactive oxygen species, and TNF-α-converting enzyme. J Immunol. 2005;175:4009–4016. doi: 10.4049/jimmunol.175.6.4009. [DOI] [PubMed] [Google Scholar]
- 37.Foronjy R, Nkyimbeng T, Wallace A, Thankachen J, Okada Y, Lemaitre V, D’Armiento J. Transgenic expression of matrix metalloproteinase-9 causes adult-onset emphysema in mice associated with the loss of alveolar elastin. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1149–L1157. doi: 10.1152/ajplung.00481.2007. [DOI] [PubMed] [Google Scholar]
- 38.Atkinson JJ, Senior RM. Matrix metalloproteinase-9 in lung remodeling. Am J Respir Cell Mol Biol. 2003;28:12–24. doi: 10.1165/rcmb.2002-0166TR. [DOI] [PubMed] [Google Scholar]
- 39.Snider GL, Ciccolella DE, Morris SM, Stone PJ, Lucey EC. Putative role of neutrophil elastase in the pathogenesis of emphysema. Ann NY Acad Sci. 1991;624:45–59. doi: 10.1111/j.1749-6632.1991.tb17005.x. [DOI] [PubMed] [Google Scholar]
- 40.Quint JK, Wedzicha JA. The neutrophil in chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2007;119:1065–1071. doi: 10.1016/j.jaci.2006.12.640. [DOI] [PubMed] [Google Scholar]
- 41.Takeyama K, Dabbagh K, Jeong Shim J, Dao-Pick T, Ueki IF, Nadel JA. Oxidative stress causes mucin synthesis via trans activation of epidermal growth factor receptor: role of neutrophils. J Immunol. 2000;164:1546–1552. doi: 10.4049/jimmunol.164.3.1546. [DOI] [PubMed] [Google Scholar]
- 42.Grenz A, Osswald H, Eckle T, Yang D, Zhang H, Tran ZV, Klingel K, Ravid K, Eltzschig HK. The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med. 2008;5:e137. doi: 10.1371/journal.pmed.0050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hua X, Kovarova M, Chason KD, Nguyen M, Koller BH, Tilley SL. Enhanced mast cell activation in mice deficient in the A2b adenosine receptor. J Exp Med. 2007;204:117–128. doi: 10.1084/jem.20061372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eckle T, Krahn T, Grenz A, Kohler D, Mittelbronn M, Ledent C, Jacobson MA, Osswald H, Thompson LF, Unertl K, Eltzschig HK. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–1590. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- 45.Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, Unertl K, Eltzschig HK. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol. 2009;10:195–202. doi: 10.1038/ni.1683. [DOI] [PubMed] [Google Scholar]
- 46.Ryzhov S, Zaynagetdinov R, Goldstein AE, Novitskiy SV, Dikov MM, Blackburn MR, Biaggioni I, Feoktistov I. Effect of A2B adenosine receptor gene ablation on proinflammatory adenosine signaling in mast cells. J Immunol. 2008;180:7212–7220. doi: 10.4049/jimmunol.180.11.7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ryzhov S, Goldstein AE, Biaggioni I, Feoktistov I. Cross-talk between G (s)- and G (q)-coupled pathways in regulation of interleukin-4 by A2B adenosine receptors in human mast cells. Mol Pharmacol. 2006;70:727–735. doi: 10.1124/mol.106.022780. [DOI] [PubMed] [Google Scholar]
- 48.Feoktistov I, Biaggioni I. Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells: an enprofylline-sensitive mechanism with implications for asthma. J Clin Invest. 1995;96:1979–1986. doi: 10.1172/JCI118245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryzhov S, Zaynagetdinov R, Goldstein AE, Novitskiy SV, Blackburn MR, Biaggioni I, Feoktistov I. Effect of A2B adenosine receptor gene ablation on adenosine-dependent regulation of proinflammatory cytokines. J Pharmacol Exp Ther. 2008;324:694–700. doi: 10.1124/jpet.107.131540. [DOI] [PubMed] [Google Scholar]
- 50.Mustafa SJ, Nadeem A, Fan M, Zhong H, Belardinelli L, Zeng D. Effect of a specific and selective A2B adenosine receptor antagonist on adenosine agonist AMP and allergen-induced airway responsiveness and cellular influx in a mouse model of asthma. J Pharmacol Exp Ther. 2007;320:1246–1251. doi: 10.1124/jpet.106.112250. [DOI] [PubMed] [Google Scholar]
- 51.Kolachala VL, Vijay-Kumar M, Dalmasso G, Yang D, Linden J, Wang L, Gewirtz A, Ravid K, Merlin D, Sitaraman SV. A2B adenosine receptor gene deletion attenuates murine colitis. Gastroenterology. 2008;135:861–870. doi: 10.1053/j.gastro.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kolachala VL, Ruble BK, Vijay-Kumar M, Wang L, Mwangi S, Figler HE, Figler RA, Srinivasan S, Gewirtz AT, Linden J, Merlin D, Sitaraman SV. Blockade of adenosine A2B receptors ameliorates murine colitis. Br J Pharmacol. 2008;155:127–137. doi: 10.1038/bjp.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]