

Abstract
Atherosclerosis is a chronic inflammatory disease in which initial vascular damage leads to extensive macrophage and lymphocyte infiltration. Although acutely glucocorticoids suppress inflammation, chronic glucocorticoid excess worsens atherosclerosis, possibly by exacerbating systemic cardiovascular risk factors. However, glucocorticoid action within the lesion may reduce neointimal proliferation and inflammation. Glucocorticoid levels within cells do not necessarily reflect circulating levels due to pre-receptor metabolism by 11β-hydroxysteroid dehydrogenases (11β-HSDs). 11β-HSD2 converts active glucocorticoids into inert 11-keto forms. 11β-HSD1 catalyses the reverse reaction, regenerating active glucocorticoids. 11β-HSD2-deficiency/ inhibition causes hypertension, whereas deficiency/ inhibition of 11β-HSD1 generates a cardioprotective lipid profile and improves glycemic control. Importantly, 11β-HSD1-deficiency/ inhibition is atheroprotective, whereas 11β-HSD2-deficiency accelerates atherosclerosis. These effects are largely independent of systemic risk factors, reflecting modulation of glucocorticoid action and inflammation within the vasculature. Here, we consider whether evidence linking the 11β-HSDs to vascular inflammation suggests these isozymes are potential therapeutic targets in vascular injury and atherosclerosis.
Keywords: Atherosclerosis, Neointima, Inflammation, Glucocorticoid, 11β-Hydroxysteroid dehydrogenase
Introduction
Atherosclerosis is widely acknowledged to be an inflammatory disease, with lesions resulting from an unchecked wound healing response to chronic arterial injury. Extensive inflammatory cell (macrophage and lymphocyte) invasion of the sub-endothelial space is key to lesion development [1–5]. Consequently, glucocorticoids, which are potent anti-inflammatory agents, might be expected to inhibit atherogenesis. In fact, glucocorticoid excess, whether endogenous (Cushing’s disease) or through pharmacotherapy, is associated with increased atherosclerosis and cardiovascular events [6–9], probably due, at least in part, to glucocorticoid-mediated exacerbation of systemic cardiovascular risk factors (including insulin resistance/type 2 diabetes, hypertension and dyslipidaemia). However, most atherosclerosis occurs independently of exogenous glucocorticoid administration, and plasma levels of glucocorticoids are not normally elevated in atherosclerosis. Nevertheless, recent evidence implicates cell-specific modulation of glucocorticoid action within the vasculature in this condition.
Glucocorticoid delivery and action within tissues can be modulated at a number of levels beyond the hypothalamic-pituitary-adrenal axis. Normally, the majority of plasma glucocorticoid (cortisol or corticosterone) is bound to the high affinity, but finite capacity, corticosterone binding globulin (CBG), leaving ~5 % being "free" and accessible to tissues. The mineralocorticoid aldosterone, and the intrinsically inactive 11-keto glucocorticoids cortisone and 11-dehydrocorticosterone, bind poorly to CBG, so that free concentrations are similar to free cortisol/corticosterone levels. However, CBG may not simply be an inert carrier; it can be cleaved by neutrophil elastase, releasing cortisol/corticosterone at sites of inflammation, thus potentially acting as a targeted glucocorticoid delivery mechanism [10]. Once inside cells, glucocorticoids can be actively removed by membrane pumps such as MDR1 [11], metabolised by 11β-hydroxysteroid dehydrogenase (11β-HSD; see below), or can bind to and activate cognate receptors, glucocorticoid receptor (GR) and, in cells which lack 11β-HSD2 (see below), the higher affinity mineralocorticoid receptor (MR). Activated receptors translocate to the nucleus to transcriptionally regulate specific gene networks.
Although all these mechanisms represent potential druggable targets in atherosclerosis, recent evidence points to the type 1 11β-HSD enzyme, which in vivo predominantly converts intrinsically inert glucocorticoids (cortisone, 11-dehydrocorticosterone) into corresponding active forms (cortisol, corticosterone), as a particularly attractive target [12]. Inhibition of 11β-HSD1 is atheroprotective, at least in animal models. Conversely, inactivation of renal 11β-HSD type 2 (11β-HSD2), which catalyses the opposite reaction, inactivating glucocorticoids in vivo, is well established as a cause of hypertension in humans. Recent data have shown that 11β-HSD2-deficiency worsens atherosclerosis independently of effects on hypertension [13•]. Here, we assess the potential of 11β-HSD1 as a therapeutic target in atherosclerosis by reviewing the actions of 11β-HSDs on vascular inflammation, considering local effects on the vasculature as well as effects on systemic cardiovascular risk factors.
Glucocorticoids and Cardiovascular Risk
To understand the potential benefits of 11β-HSD modulation in vascular inflammation, it is essential first to consider the actions of glucocorticoids. The complex relationship between glucocorticoids and cardiovascular disease is incompletely understood and may differ between humans and animal models (recently reviewed in [14–16]). Some of this complexity undoubtedly arises from the diverse actions of glucocorticoids: direct effects of glucocorticoids with the heart and vasculature may be largely beneficial, at least within certain cell types, whereas adverse effects may be mediated indirectly by changes in systemic risk factors (such as hypertension, lipids and insulin resistance/ diabetes).
The effect of glucocorticoids on dyslipidaemia and other systemic risk factors has been established for over 60 years [17–19]. Consistent with exacerbation of systemic risk factors, endogenous glucocorticoid excess or glucocorticoid pharmacotherapy in humans are associated with increased extent and severity of atherosclerosis, and predict cardiovascular morbidity and mortality [5, 20–28]. Discontinuation of glucocorticoid pharmacotherapy reduces cardiovascular risk [6, 29]. Similarly, normalisation of circulating glucocorticoids in Cushing’s disease largely reverses pathophysiological changes in vascular function and structure [9]. However, hyperglycaemia and dyslipidaemia are only modestly improved within the same time frame, suggesting distinct beneficial effects of reducing glucocorticoid action separate from effects on the classical systemic risk factors [9].
In contrast, animal studies suggest that glucocorticoids reduce atherosclerosis, despite causing hyperlipidaemia [30–40]. Furthermore, a recent report elegantly demonstrated that 'painting' dexamethasone onto atherosclerotic lesions improved markers of plaque stability (reducing macrophage content and increasing fibrous cap thickness) [41]. This discrepancy between the atherosclerosis-promoting effects of glucocorticoids in humans but not in animals remains unexplained, but may be related to the predominant use of dexamethasone as the glucocorticoid of choice in animals. Whereas endogenous glucocorticoids are agonists at both GR and MR, synthetic glucocorticoids, including dexamethasone, poorly activate MR. Chronically increased mineralocorticoid action, even at relatively modest levels, appears pro-inflammatory within the cardiovascular system, distinct from effects of MR activation upon blood pressure [42].
Glucocorticoids and the Acute Vascular Response to Injury
The introduction of percutaneous angioplasty for treatment of occluded arteries highlighted the fibroproliferative vascular response to acute mechanical injury. Indeed, re-occlusion (restenosis) of atherosclerotic arteries following angioplasty is a clinically significant complication of the technique. Acute arterial injury (eg by insertion of a wire or stent, by ligation, or with a laser) in animals provides a tool to elucidate the mechanisms underlying restenosis in atherosclerosis as well as exploring novel treatments. Mechanical arterial injury provokes an influx of inflammatory cells, recruited by factors such as monocyte-chemoattractant protein (MCP)-1 released from the injured vascular/endothelial cells [43], which stimulates proliferation and migration of vascular smooth muscle cells, forming the neointima [44]. Inhibition of this inflammatory response has been central to the use of glucocorticoids as potential inhibitors of neointimal lesion formation/restenosis [45–50].
Despite the largely positive data obtained in animals, clinical use of glucocorticoids (methylprednisolone, dexamethasone) for prevention of restenosis was initially disappointing [51–55]. However, recent trials are promising [56–59], reporting beneficial effects of prednisone [59, 60] or dexamethasone [55, 61]. It is not clear whether discrepancies between clinical and pre-clinical studies are due to animal models providing poor representations of human disease or reflect limitations in clinical trials (eg small group sizes, patient selection; route of administration). Alternatively, the discrepancies may result from a different balance in rodents and humans between the adverse effects of glucocorticoids upon systemic risk factors and beneficial effects on vascular inflammation, with the balance more in favour of the former in humans. Moreover, any systemic therapy will depress HPA axis activity, altering the balance of endogenous glucocorticoid ligands. Thus, identification of the cellular and molecular (eg MR or GR) targets of glucocorticoid action, including endogenous glucocorticoids, during vascular inflammation is crucial to development of novel, better targeted therapies.
Metabolism by 11β-HSDs Modulates Glucocorticoid Action
Glucocorticoid action within cells depends upon receptor density and ligand availability. The latter is potently influenced by the 11β-HSD isozymes. 11β-HSD1 is co-localised in the lumen of the endoplasmic reticulum with hexose-6-phosphate dehydrogenase which, by coupling glucose-6-phosphate oxidation to reduction of NADP+, generates the high NADPH/NADP+ ratio required to drive 11β-HSD1 reductase activity [62], converting inert cortisone and 11-dehydrocorticosterone into active cortisol and corticosterone, respectively. 11β-HSD2 is exclusively a dehydrogenase, inactivating glucocorticoids [63]. Some synthetic glucocorticoids, notably prednisolone/prednisone, are also substrates for the 11β-HSDs and are readily interconverted in vivo. Others, including dexamethasone, are poorly metabolised by these enzymes. Both isozymes are inhibited by the liquorice derivative, glycyrrhetinic acid or its synthetic analogue, carbenoxolone, which have contributed greatly to our current understanding of the function of 11β-HSDs, especially in humans. More recently, selective 11β-HSD1 inhibitors have been developed, allowing much greater discrimination of the roles of these important enzymes [64].
The 11β-HSDs are Expressed in the Cardiovascular System
11β-HSD1 is widely expressed, with highest expression in the liver and more modest expression elsewhere, typically in classical glucocorticoid target cells and tissues [63]. During pathogenesis, expression is increased at some sites, notably in adipose tissue in obesity and at sites of inflammation (see below). In contrast, 11β-HSD2 expression is restricted to mineralocorticoid target cells/tissues, including the distal nephron, and limited other sites such as the skin, lung and adrenal cortex [65]. Both isozymes are modestly expressed in the vasculature; 11β-HSD1 is probably restricted to vascular smooth muscle cells, with 11β-HSD2 expressed in the endothelium (reviewed in [15, 16]). Some studies have reported 11β-HSD2 expression in the vascular smooth muscle cells [66] and 11β-HSD1 in the endothelium [67•], though these studies depend on antibody specificity and others have not confirmed these findings [68]. Whilst co-expression cannot be excluded, normally 11β-HSD isozyme expression is mutually exclusive within individual cells.
The 11β-HSDs Modulate Cardiovascular Disease Risk Factors
Both 11β-HSD isozymes modulate systemic cardiovascular risk factors, with a well-established role for 11β-HSD2 in regulating mineralocorticoid effects, including blood pressure, and with 11β-HSD1 implicated in the pathogenesis of metabolic syndrome.
By inactivating glucocorticoids, 11β-HSD2 activity confers mineralocorticoid-specificity upon MR in cells in which they are co-expressed. Deficiency in, or inhibition of, 11β-HSD2 allows inappropriate activation of MR by glucocorticoids, causing the syndrome of Apparent Mineralocorticoid Excess, characterised by hypertension, hypokalaemia and suppression of the renin-angiotensin-aldosterone system [69].
A physiological role for 11β-HSD1 has been slower to emerge. However, over the last decade it has become apparent that amplification of intracellular glucocorticoid levels by 11β-HSD1, especially in adipose tissue and/or brain, contributes to obesity-associated metabolic disease and age-related cognitive decline (reviewed in [70]). 11β-HSD1 expression in adipose tissue is selectively elevated in obese humans and in rodents with monogenic obesity. Inhibition of 11β-HSD1 reduces levels of plasma glucose, glycated haemoglobin A1c (HbA1c) and cholesterol in patients with type 2 diabetes [71] and 11β-HSD1-deficiency or inhibition improves insulin sensitivity in animal models of diabetes and/or obesity [63]. Conversely, in mice, transgenic over-expression of 11β-HSD1 selectively in adipose tissue produces local, but not systemic, glucocorticoid excess and causes visceral obesity, hypertension, diabetes and dyslipidaemia [72, 73], whereas over-expression in liver causes hypertension and mild insulin resistance [74].
In a similar manner, age-related cognitive decline (itself associated with impaired glycaemic control [75]) is associated with higher 11β-HSD1 activity in humans [76] and in rodents [77], and 11β-HSD1-deficient mice are protected from age-related cognitive decline (reviewed in [78]). A causative role is supported by the phenotype of transgenic mice with selective over-expression of 11β-HSD1 in forebrain, which develops cognitive deficits at an earlier age than wild-type controls [77]. In humans, age-related cognitive decline, diabetes and metabolic syndrome, like atherosclerosis, are all associated with increased markers of inflammation, including in macrophages (or microglia in the brain). It is conceivable that altered inflammatory responses underlie at least part of the effects of 11β-HSD1 manipulation.
11β-HSDs and Inflammation
11β-HSD1 and 11β-HSD2 show opposite regulation at sites of inflammation, likely because of opposite regulation by the pro-inflammatory cytokines IL-1 and TNF-α, with up-regulation of 11β-HSD1 and down-regulation of 11β-HSD2 (reviewed in [12]), suggesting they modulate local glucocorticoid action during inflammation (Fig. 1a). 11β-HSD1 is expressed in immune cells, including macrophages and lymphocytes. Its expression, by-and-large, is increased following immune cell activation. In contrast, 11β-HSD2 is not normally expressed in immune cells, though it may become expressed in human macrophages and possibly other mononuclear cells in certain pathological situations [79], suggesting resistance to endogenous glucocorticoids under these circumstances.
Fig 1.
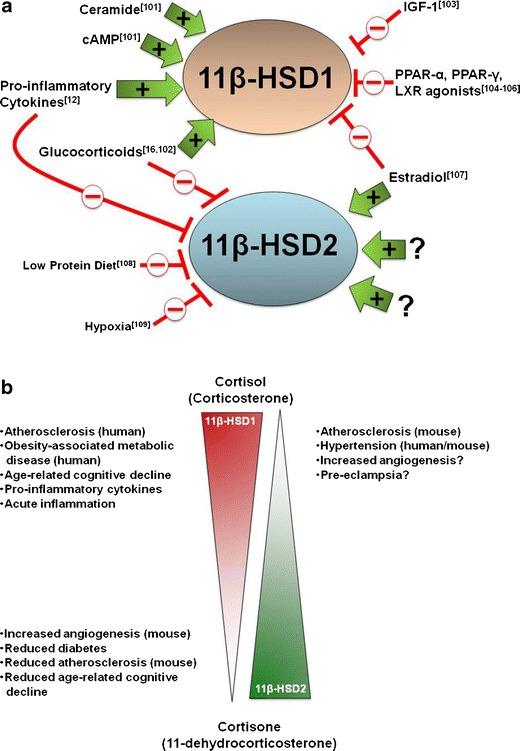
Regulation of 11β-hydroxysteroid dehydrogenase isozyme activity in health and disease. (a) The biochemical pathways that regulate 11β-HSDs. A number of factors have been identified that selectively increase (green arrows) or decrease (red lines) 11β-HSD isozyme expression [101–109]. Note: only some of the biochemical pathways that regulate 11β-HSD expression are shown; factors that may affect 11β-HSD activity (eg. glucose-6-phosphate availability, hexose-6-phosphate dehydrogenase activity [110] and insulin signaling) are not shown. Little is known about the pathways that regulate 11β-HSD2 activity in the vasculature. (b) The relationship between 11β-HSD isozyme activity and disease pathologies. High (top left) and low (bottom left) activity of 11β-HSD1, and low activity of 11β-HSD2 (top right) are all associated with disease pathologies
Unsurprisingly, given the well-known effects of glucocorticoids in limiting acute inflammation, 11β-HSD1-deficiency in mice increases the severity of acute inflammation. Inflammation is increased in an experimental model of arthritis, and more neutrophils and monocytes/macrophages are elicited in sterile peritonitis and pleurisy, consistent with a mechanism in which early induction of 11β-HSD1 during inflammation limits the severity of the response. 11β-HSD2-deficiency has no effect [80]. Similarly, following coronary artery ligation, more neutrophils and monocyte/macrophages are recruited in hearts of 11β-HSD1-deficient mice [81]. This contrasts with the attenuated inflammation that occurs in adipose tissue of 11β-HSD1-deficient mice during diet-induced obesity, with reduced macrophage and T cell infiltration of visceral adipose tissue [82].
Plausibly, an increased angiogenic response to hypoxia may be the common link in the beneficial effects of 11β-HSD1 inhibition or deficiency in metabolic disease (reviewed in [12]). Angiogenesis in vivo and in vitro is increased with 11β-HSD1-deficiency [83]. This results in better recovery of heart function following myocardial infarction in 11β-HSD1-deficient mice, despite (or possibly because of) greater inflammation acutely following coronary artery ligation [81, 83]. A similar increase in angiogenesis likely underlies the reduced hypoxia and inflammation within adipose tissue of high fat-fed 11β-HSD1-deficient mice [84]. Moreover, although it has not been tested in 11β-HSD1-deficient mice, angiogenesis is implicated in the pathogenesis of rheumatoid arthritis [85]. 11β-HSD2 may regulate angiogenesis in human endometrium, in the opposite manner. Higher endometrial 11β-HSD2 expression is associated with heavy menstrual bleeding, plausibly due to reduced thrombospondin-1-mediated inhibition of angiogenesis as a result of greater cortisol inactivation [86]. Importantly, these 11β-HSD actions are entirely consistent with the known actions of glucocorticoids, which limit acute inflammation whilst provoking adverse inflammatory cardiometabolic states. Whilst glucocorticoids promote adaption, including tissue remodelling when homeostasis is perturbed, they are also strongly anti-angiogenic both in vivo and in vitro [83].
11β-HSDs as a Therapeutic Target in Atherosclerosis
Levels of 11β-HSD1 mRNA are increased in human atherosclerotic vessels compared either with nearby intact (atheroma-free) vasculature or with vessels from control patients without coronary artery disease [87, 88]. This may reflect a pro-inflammatory peri-vascular environment in disease rather than systemic inflammation; mRNA levels of both 11β-HSD1 and CD68 (a macrophage marker) are increased in epicardial adipose tissue of patients with coronary artery disease compared with controls [87], consistent with inflammation within the peri-vascular adipose tissue associated with vascular inflammation. Pro-inflammatory cytokines increase 11β-HSD1 expression (Fig. 1a) in human and murine vascular smooth muscle cells in vitro [89, 90], but acute systemic inflammation has little or no effect on vascular 11β-HSD1 expression in mice [90]. This situation, with locally increased 11β-HSD1, is similar to that associated with inflammation in adipose tissue of metabolic syndrome patients. It suggests that dysregulated vascular 11β-HSD1, possibly initially induced to control local inflammation within the vasculature, might then drive local inflammation both within the vasculature and in the surrounding adipose tissue by restricting angiogenesis, thus promoting a hypoxic environment. This, of course, points to 11β-HSD1 inhibition as an attractive target in atherosclerosis. Dysregulation of 11β-HSD2 in human arterial disease has not been described (Fig. 1a), but the opposite regulation of 11β-HSD2 by pro-inflammatory cytokines is predicted to decrease levels within endothelial cells, allowing cortisol activation of MR and its pro-inflammatory consequences, given a setting of high levels of systemic risk factors (see below).
The potential for 11β-HSD1 inhibition as an effective pharmacotherapy for atherosclerosis was demonstrated by Hermanowski-Vosatka and colleagues from Merck [91]. Compound 544, a selective 11β-HSD1 inhibitor, modestly reduced circulating cholesterol levels and markedly reduced intra-aortic cholesterol in atherosclerosis-prone Apoe −/− mice fed a cholesterol-enriched 'western diet' [91]. More recently, this group has shown a similar reduction in intra-aortic cholesterol with a distinct inhibitor (Compound L-750), without effects on plasma lipids in Apoe −/− mice [67•]. However, another study in triple knock-out mice (lacking the LDL receptor, apolipoprotein-b and leptin) with a different selective inhibitor (Compound 2922), reported no effect on atherosclerosis and only slightly reduced plasma LDL levels, despite improved glucose homeostasis [92]. A previous study in Ldlr −/− mice with the non-selective inhibitor, carbenoxolone, only showed an effect on plasma lipids and atherosclerosis in severely obese mice (due to an additional mutation in the Agouti gene), though the drug treatment is likely to have made the mice hypertensive through inhibition of 11β-HSD2 (blood pressure was not reported) [93]. More recently, we have shown that generating either homozygous or heterozygous Hsd11b1 deletion and, thus, 11β-HSD1-deficiency (avoiding possible off-target drug effects) attenuates atherosclerosis in Apoe −/− mice without affecting plasma glucose or lipid levels. This suggests that atheroprotection in this background is not simply due to improvements in systemic risk factors, although it should be noted that the contribution of hemostatic factors has not been addressed. Reconstitution of lethally irradiated Apoe −/− mice with bone marrow from Hsd11b1 −/− Apoe −/− mice similarly reduced atherosclerosis compared to controls reconstituted with Apoe −/− donor bone marrow, implicating 11β-HSD1-deficiency in haematopoietic cells in the mechanism, and suggesting that any differences in plasma glucocorticoid turnover in 11β-HSD1-deficient mice [74, 94] are unlikely to be critical. The converse situation occurs with 11β-HSD2-deficiency, which accelerates atherosclerosis and worsens vascular inflammation in Apoe −/− mice independent of effects on blood pressure, even without the stimulus of western diet [13•]. Importantly, this suggests that it is intravascular glucocorticoid action that is the main culprit in 11β-HSD2-deficient Apoe −/− mice, rather than deficiency in the surrounding adipose tissue or circulating immune cells; 11β-HSD2 is not normally expressed either in immune cells or in adipose tissue, though inflammation within peri-vascular adipose tissue could, of course, contribute to pathology once initiated. A summary of experimental studies describing the effects of 11β-HSD inhibition or deficiency on atherosclerosis/ vascular inflammation is shown in Table 1. Interestingly, neither 11β-HSD1- nor 11β-HSD2-deficiency affected neointimal proliferation following femoral artery wire injury in lean healthy mice, though it was reduced by 11β-HSD1-deficiency in Apoe −/− mice (Hsd11b2 −/− Apoe −/− double knock-out mice were not tested) [95]. Thus, 11β-HSD1-inhibition is only likely to be effective in reducing neointimal proliferation when concurrent with exaggerated systemic risk factors.
Table 1.
Experimental studies describing the effects of 11β-HSD inhibition or deficiency on vascular inflammation
| 11β-HSD1 inhibition/deficiency | Species | Outcome | Ref |
|---|---|---|---|
| 11β-HSD1 inhibitor (Compound 544) | Apoe −/− mice | ↓ atherosclerosis | [91] |
| ↓ circulating cholesterol | |||
| ↓ circulating MCP-1 | |||
| 11β-HSD1 inhibitor (Compound L-750) | Apoe −/− mice | ↓ atherosclerosis | [67•] |
| ≠ circulating lipids | |||
| ↓ circulating MCP-1 and aortic MCP-1 mRNA expression | |||
| ↓ expression of: inflammatory, adhesive and coagulation factors in the vasculature | |||
| 11β-HSD inhibitor (Compound 2922) | Ldlr −/− Apob 100/100 Lep ob/ob mice | ≠ atherosclerosis | [92] |
| ↓ circulating LDL | |||
| improved glucose homeostasis | |||
| 11β-HSD inhibitor Carbenoxolone (inhibits both isozymes) | Ldlr −/− Agouti −/− mice | ↓ atherosclerosis in severely obese mice (additional agouti mutation) ↓ circulating lipids | [93] |
| 11β-HSD1 inhibitor (Compound 544) | Apoe −/− mice | ↓ atherosclerosis ≠ fasting plasma cholesterol, triglycerides or NEFA ↑ plaque stability | [96•] |
| 11β-HSD1 deficiency | Hsd11b1 −/− Apoe −/− mice | ↓ atherosclerosis | [96•] |
| ≠ circulating lipids | |||
| ↓ circulating MCP-1 | |||
| ↓ circulating Ly6Chi monocytes | |||
| ≠ macrophage density into lesions | |||
| ↓ T cell infiltration into lesions | |||
| ↓ aortic VCAM-1 mRNA expression | |||
| ↓ atherosclerosis in irradiated Apoe −/− mice receiving bone marrow cells from Hsd11b1 −/− Apoe −/− donor mice | |||
| 11β-HSD2 deficiency | Hsd11b2 −/− Apoe −/− mice | ↑ atherosclerosis | [13•] |
| ≠ blood pressure |
≠ no significant effect, ↓ reduced, ↑ increased, NEFA: non-esterified fatty acids
Mechanism of Action of 11β-HSD Manipulation
The mouse studies suggest at most a modest contribution of systemic risk factors to the effects of 11β-HSD deficiency or inhibition upon atherosclerosis, pointing instead to effects within both immune cells (11β-HSD1) and the vasculature (11β-HSD2). As stated above, immune cells are implicated as a target. Although macrophage density appears unaltered with 11β-HSD1-deficiency, T cell infiltration is reduced [96•] and 11β-HSD1 inhibition, either prophylactically or therapeutically in established atherosclerosis, reduces expression of pro-inflammatory and cell adhesion molecules in the vasculature of Apoe −/− mice [70, 96•]. Interestingly, the microarray studies on vasculature also highlight suppression of mRNAs encoding coagulation factors by 11β-HSD1, something that warrants further investigation. Experimental studies have implicated the coagulation system in the pathogenesis of atherosclerosis and atherothrombosis (reviewed in [97]) and megakaryocytes, the precursors to platelets, would be transferred in the bone marrow reconstitution experiments that implicate immune cells [96•].
Western diet-induced monocytosis is attenuated with 11β-HSD1-deficiency in Apoe −/− mice, attributable to the pro-inflammatory Ly6Chi monocyte subset. Instead, monocytes are retained in the bone marrow and spleen of Hsd11b1 −/− Apoe −/− mice [96•]. Lower circulating levels of MCP-1, the main macrophage chemoattractant in atherosclerosis [98], in Hsd11b1 −/− Apoe −/− mice are implicated in this difference [67•, 91, 96•], though whether the MCP-1 originates from the vasculature or adipose tissue is currently unclear [67•, 96•]. Either way, circulating MCP-1 is unlikely to be the sole or even the main target of 11β-HSD1-deficiency/inhibition.
Plausibly, the opposite effects of deficiency in 11β-HSD1 or 11β-HSD2 activity upon atherosclerosis reflect different but complementary mechanisms. 11β-HSD2 is not expressed in mouse immune cells, but it is expressed in vascular endothelial cells, where it restricts activation of MR by glucocorticoids. In mouse aortic endothelial cells in vitro, vascular cell adhesion molecule-1 was induced by corticosterone in an MR-dependent manner, but only following inhibition of 11β-HSD2. In vivo, antagonism of MR with eplerenone, at doses which had no effect on blood pressure, attenuated lesion development and inflammation, whilst also increasing collagen and smooth muscle content, markers of plaque stability [13•]. Together, these data clearly suggest that preservation of vascular endothelial cell 11β-HSD2 activity is important in controlling vascular injury and subsequent inflammation.
The target of 11β-HSD1-generated ligand is less obvious. Although GR has long been known to shape macrophage phenotype, recent evidence has also shown a crucial role for MR in governing macrophage polarisation, with MR-deficient macrophages skewing towards a "pro-resolution" alternatively activated (or M2) phenotype [99]. In vivo, MR deficiency in macrophages alone is sufficient to protect against the vascular damage and cardiac hypertrophy and fibrosis induced by L-NAME and angiogensin II [99], consistent with pro-inflammatory MR activation by glucocorticoids (11β-HSD2 is absent from macrophages). However, the high affinity MR is predicted to be activated even at basal plasma glucocorticoid levels, and these are normal in 11β-HSD1-deficient mice [94]. 11β-HSD1 may therefore amplify ligand availability to GR. But GR activation in macrophages would be expected to promote a pro-resolution phenotype. Exactly how intracellular glucocorticoid activation of MR versus GR is achieved in cells that express both without 11β-HSD2 is currently unclear [99], but could conceivably involve GR:MR heterodimers [100] and might be critical in pro- and anti-inflammatory actions of endogenous glucocorticoids. An alternative target is GR in megakaryocytes, but whether 11β-HSD1 is even expressed in these cells is unknown. Future dissection of the cellular and molecular targets will require selective receptor antagonism, as well as tissue-specific disruption of the 11β-HSD1 gene in mice, to elucidate specific roles in key processes during inflammation and angiogenesis.
Conclusions
There is clearly a counterbalance between the pro-atherogenic effects of 11β-HSD1 activity and the atheroprotective effects of 11β-HSD2 activity during vascular inflammation (Fig. 1b). Any therapy based on manipulation of glucocorticoid metabolism should inhibit 11β-HSD1 activity whilst preserving 11β-HSD2 activity. Future work should concentrate on elucidating whether the cellular and molecular targets are indeed complementary or may be overlapping. New drug delivery methods [41], targeting macrophages or indeed other cell types, open up exciting new therapeutic possibilities in vascular inflammation and atherosclerosis.
Acknowledgments
The authors are grateful to The Wellcome Trust, The British Heart Foundation and the BHF CoRE for supporting their research.
Conflicts of Interest
Patrick WF Hadoke declares no conflicts of interest.
Tiina Kipari declares no conflicts of interest.
Jonathan R. Seckl has been a consultant for several commercial drug development programmes for 11beta-HSD1 inhibitors.
Karen E. Chapman declares no conflicts of interest.
Abbreviations
- 11β-HSD
11β-Hydroxysteroid dehydrogenase
- GR
glucocorticoid receptor
- MR
mineralocorticoid receptor
Footnotes
This article is part of the Topical Collection on Vascular Biology
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
- 1.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6(7):508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 2.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12(3):204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 3.Taleb S, Tedgui A, Mallat Z. Adaptive T cell immune responses and atherogenesis. Curr Opin Pharmacol. 2010;10(2):197–202. doi: 10.1016/j.coph.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Packard RR, Lichtman AH, Libby P. Innate and adaptive immunity in atherosclerosis. Semin Immunopathol. 2009;31(1):5–22. doi: 10.1007/s00281-009-0153-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alevizaki M, Cimponeriu A, Lekakis J, Papamichael C, Chrousos GP. High anticipatory stress plasma cortisol levels and sensitivity to glucocorticoids predict severity of coronary artery disease in subjects undergoing coronary angiography. Metabolism. 2007;56(2):222–226. doi: 10.1016/j.metabol.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 7.Souverein PC, Berard A, Van Staa TP, Cooper C, Egberts AC, Leufkens HG, Walker BR. Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case–control study. Heart. 2004;90(8):859–865. doi: 10.1136/hrt.2003.020180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Etxabe J, Vazquez JA. Morbidity and mortality in Cushing's disease: an epidemiological approach. Clin Endocrinol (Oxf) 1994;40(4):479–484. doi: 10.1111/j.1365-2265.1994.tb02486.x. [DOI] [PubMed] [Google Scholar]
- 9.Faggiano A, Pivonello R, Spiezia S, De Martino MC, Filippella M, Di Somma C, Lombardi G, Colao A. Cardiovascular risk factors and common carotid artery caliber and stiffness in patients with Cushing's disease during active disease and 1 year after disease remission. J Clin Endocrinol Metab. 2003;88(6):2527–2533. doi: 10.1210/jc.2002-021558. [DOI] [PubMed] [Google Scholar]
- 10.Perogamvros I, Ray DW, Trainer PJ. Regulation of cortisol bioavailability – effects on hormone measurement and action. Nat Rev Endocrinol. 2012;8(12):717–727. doi: 10.1038/nrendo.2012.134. [DOI] [PubMed] [Google Scholar]
- 11.Gruol DJ, Bourgeois S. Expression of the mdr1 P-glycoprotein gene: a mechanism of escape from glucocorticoid-induced apoptosis. Biochem Cell Biol. 1994;72(11–12):561–571. doi: 10.1139/o94-075. [DOI] [PubMed] [Google Scholar]
- 12.Chapman KE, Coutinho AE, Gray M, Gilmour JS, Savill JS, Seckl JR. The role and regulation of 11beta-hydroxysteroid dehydrogenase type 1 in the inflammatory response. Mol Cell Endocrinol. 2009;301(1–2):123–131. doi: 10.1016/j.mce.2008.09.031. [DOI] [PubMed] [Google Scholar]
- 13.Deuchar GA, McLean D, Hadoke PWF, Brownstein D, Webb DJ, Mullins JJ, Chapman K, Seckl JR, Kotelevtsev YV. Loss of glucocorticoid inactivation by 11β-hydroxysteroid dehydrogenase type 2 causes pro inflammatory changes in the endothelium and exacerbates atherogenesis in Apoe−/− mice. Endocrinology. 2011;152:236–246. doi: 10.1210/en.2010-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker BR. Extra-adrenal regeneration of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase type 1: physiological regulator and pharmacological target for energy partitioning. Proc Nutr Soc. 2007;66:1–8. doi: 10.1017/S002966510700523X. [DOI] [PubMed] [Google Scholar]
- 15.Hadoke PWF, Macdonald L, Logie JJ, Small GR, Dover AR, Walker BR. Intra-vascular glucocorticoid metabolism as a modulator of vascular structure and function. Cell Mol Life Sci. 2006;63(5):565–578. doi: 10.1007/s00018-005-5427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hadoke PWF, Iqbal J, Walker BR. Therapeutic manipulation of glucocorticoid metabolism in cardiovascular disease. Brit J Pharmacol. 2009;156(5):689–712. doi: 10.1111/j.1476-5381.2008.00047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adlersberg D, Schaefer L, Drachman SR. Development of hypercholesteremia during cortisone and Acth therapy. JAMA J Am Med Assoc. 1950;144:909–914. doi: 10.1001/jama.1950.02920110021006. [DOI] [PubMed] [Google Scholar]
- 18.Adlersberg D, Schaefer LE, Dritch R. Studies on hormonal control of serum lipid partition in man. J Clin Endocrinol. 1950;10:814–815. [PubMed] [Google Scholar]
- 19.Etheridge EM, Hochligeti C. Lipid deposition in aortas in younger age groups following cortisone and adrenocorticotrophic hormone. Am J Pathol. 1952;28:315–319. [PMC free article] [PubMed] [Google Scholar]
- 20.Troxler RG, Sprague EA, Albanese RA, Fuchs R, Thompson AJ. Association of elevated plasma cortisol and early atherosclerosis as demonstrated by coronary angiography. Atherosclerosis. 1977;26:151–162. doi: 10.1016/0021-9150(77)90098-3. [DOI] [PubMed] [Google Scholar]
- 21.Colao A, Pivonello R, Spiezia S, Faggiano A, Ferone D, Filippella M, Marzullo P, Cerbone G, Siciliani M, Lombardi G. Persistence of increased cardiovascular risk in patients with Cushing's disease after five years of successful cure. J Clin Endocrinol Metab. 1999;84:2664–2672. doi: 10.1210/jc.84.8.2664. [DOI] [PubMed] [Google Scholar]
- 22.Wei L, MacDonald TM, Walker BR. Taking glucocorticoids by prescription is associated with subsequent cardiovascular disease. Ann Intern Med. 2004;141:764–770. doi: 10.7326/0003-4819-141-10-200411160-00007. [DOI] [PubMed] [Google Scholar]
- 23.Davis JM, Kremers HM, Crowson CS, Nicola PJ, Ballman KV, Therneau TM, Roger VL, Gabriel SE. Glucocorticoids and cardiovascular events in rheumatoid arthritis - a population-based cohort study. Arthritis Rheum. 2007;56:820–830. doi: 10.1002/art.22418. [DOI] [PubMed] [Google Scholar]
- 24.Sihvonen S, Korpela M, Mustonen J, Huhtala H, Karstila K, Pasternack A. Mortality in patients with rheumatoid arthritis treated with low-dose oral glucocorticoids. A population-based cohort study. J Rheumatol. 2006;33:1740–1746. [PubMed] [Google Scholar]
- 25.del Rincon I, O'Leary DH, Haas RW, Escalante A. Effect of glucocorticoids on the arteries in rheumatoid arthritis. Arthritis Rheum. 2004;50:3813–3822. doi: 10.1002/art.20661. [DOI] [PubMed] [Google Scholar]
- 26.Solomon DH, Avorn J, Katz JN, Weinblatt ME, Setoguchi S, Levin R, Schneeweiss S. Immunosuppressive medications and hospitalization for cardiovascular events in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54:3790–3798. doi: 10.1002/art.22255. [DOI] [PubMed] [Google Scholar]
- 27.Vlachoyiannopoulos PG, Kanellopoulos PG, Ioannidis JPA, Tektonidou MG, Mastorakou I, Moutsopoulos HM. Atherosclerosis in premenopausal women with antiphospholipid syndrome and systemic lupus erythematosus: a controlled study. Rheumatology. 2003;42:645–651. doi: 10.1093/rheumatology/keg182. [DOI] [PubMed] [Google Scholar]
- 28.Fischer-Betz R, Beer S, Schneider M. Considering systemic lupus erythematosus as example for accelerated atherosclerosis in rheumatoid diseases - what is the consequence? Z Rheumatol. 2005;64:229–238. doi: 10.1007/s00393-005-0733-5. [DOI] [PubMed] [Google Scholar]
- 29.Varas-Lorenzo C, Rodriguez LAG, Maguire A, Castellsague J, Perez-Gutthann S. Use of oral corticosteroids and the risk of acute myocardial infarction. Atherosclerosis. 2007;192:376–383. doi: 10.1016/j.atherosclerosis.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 30.Jain S, Pick R, Katz LN. Benefit from testosterone and hydrocortisone on coronary atherogenesis in cockerels on a low protein atherogenic diet. Circ Res. 1965;17:492. doi: 10.1161/01.RES.17.6.492. [DOI] [PubMed] [Google Scholar]
- 31.Bailey JM, Butler J. Anti-inflammatory drugs in experimental atherosclerosis.6. Combination therapy with steroid and non-steroid agents. Atherosclerosis. 1985;54:205–212. doi: 10.1016/0021-9150(85)90179-0. [DOI] [PubMed] [Google Scholar]
- 32.Naito M, Yasue M, Asai K, Yamada K, Hayashi T, Kuzuya M, Funaki C, Yoshimine N, Kuzuya F. Effects of dexamethasone on experimental atherosclerosis in cholesterol-fed rabbits. J Nutr Sci Vitaminol. 1992;38:255–264. doi: 10.3177/jnsv.38.255. [DOI] [PubMed] [Google Scholar]
- 33.Asai K, Funaki C, Hayashi T, Yamada K, Naito M, Kuzuya M, Yoshida F, Yoshimine N, Kuzuya F. Dexamethasone-induced suppression of aortic atherosclerosis in cholesterol-fed rabbits - possible mechanisms. Arterioscler Thromb. 1993;13:892–899. doi: 10.1161/01.ATV.13.6.892. [DOI] [PubMed] [Google Scholar]
- 34.Oppenheim E, Bruger M. Experimental cholesterol atherosclerosis.11. Studies with vitamin-A. Ama Arch Pathol. 1952;53:520–522. [PubMed] [Google Scholar]
- 35.Gordon D, Kobernick SD, Mcmillan GC, Duff GL. The effect of cortisone on the serum lipids and on the development of experimental cholesterol atherosclerosis in the rabbit. J Exp Med. 1954;99:371–386. doi: 10.1084/jem.99.4.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stumpf HH, Wilens SL. Inhibitory effect of cortisone hyperlipemia on arterial lipid deposition in cholesterol-fed rabbits. Proc Soc Exp Biol Med. 1954;86:219–223. doi: 10.3181/00379727-86-21055. [DOI] [PubMed] [Google Scholar]
- 37.Constantinides P, Hospes D, Gutmannauersperg N, Williams K. Estriol and prednisolone in rabbit atherosclerosis. Arch Pathol. 1962;73:277. [PubMed] [Google Scholar]
- 38.Cavallero C, Ditondo U, Mingazzini PL, Nicosia R, Pericoli MN, Sarti P, Spagnoli LG, Villaschi S. Cell-proliferation in atherosclerotic plaques of cholesterol-fed rabbits.3. Histological and autoradiographic observations on glucocorticoids-treated rabbits. Atherosclerosis. 1976;25:145–152. doi: 10.1016/0021-9150(76)90020-4. [DOI] [PubMed] [Google Scholar]
- 39.van der Sluis RJ, van Puijvelde GH, Van Berkel TJ, Hoekstra M. Adrenalectomy stimulates the formation of initial atherosclerotic lesions: reversal by adrenal transplantation. Atherosclerosis. 2012;221(1):76–83. doi: 10.1016/j.atherosclerosis.2011.12.022. [DOI] [PubMed] [Google Scholar]
- 40.Deitemeyer D, Yunker RL, Ashraf M, Subbiah MTR. Effect of glucocorticoid administration early in life on aortic prostaglandin synthesis and morphology in atherosclerosis-susceptible pigeons. Exp Clin Endocrinol. 1985;85:147–154. doi: 10.1055/s-0029-1210430. [DOI] [PubMed] [Google Scholar]
- 41.Kastrup CJ, Nahrendorf M, Figueiredo JL, Lee H, Kambhampati S, Lee T, Cho S-W, Gorbatov R, Iwamoto Y, Dang TT, Dutta P, Yeon JH, Cheng H, Pritchard CD, Vegas AJ, Siegel CD, MacDougall S, Okonkwo M, Thai A, Stone JR, Coury AJ, Weissleder R, Langer R, Anderson DG. Painting blood vessels and atherosclerotic plaques with an adhesive drug depot. Proc Nat Acad Sci (USA) 2012;109(52):21444–21449. doi: 10.1073/pnas.1217972110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galuppo P, Bauersachs J. Mineralocorticoid receptor activation in myocardial infarction and failure: recent advances. Eur J Clin Invest. 2012;42(10):1112–1120. doi: 10.1111/j.1365-2362.2012.02676.x. [DOI] [PubMed] [Google Scholar]
- 43.Ialenti A, Grassia G, Gordon P, Maddaluno M, Di Lauro MV, Baker AH, Guglielmotti A, Colombo A, Biondi G, Kennedy S, Maffia P. Inhibition of in-stent stenosis by oral administration of Bindarit in porcine coronary arteries. Arterioscler Thromb Vasc Biol. 2011;31:2448–2454. doi: 10.1161/ATVBAHA.111.230078. [DOI] [PubMed] [Google Scholar]
- 44.Wainwright CL, Miller AM, Wadsworth RM. Inflammation as a key event in the development of neointima formation following vascular balloon injury. Clin Exp Pharmacol Physiol. 2001;28:891–895. doi: 10.1046/j.1440-1681.2001.03543.x. [DOI] [PubMed] [Google Scholar]
- 45.Villa AE, Guzman LA, Chen W, Golomb G, Levy RJ, Topol EJ. Local delivery of dexamethasone for prevention of neointimal proliferation in a rat model of balloon angioplasty. J Clin Invest. 1994;93:1243–1249. doi: 10.1172/JCI117078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guzman LALV, Song CX, Jang YS, Lincoff AM, Levy R, Topol EJ. Local intraluminal infusion of biodegradable polymeric nanoparticles - a novel approach for prolonged drug delivery after balloon angioplasty. Circulation. 1996;94:1441–1448. doi: 10.1161/01.CIR.94.6.1441. [DOI] [PubMed] [Google Scholar]
- 47.Van Put DJM, Van Hove CE, DeMeyer GRY, Wuyts F, Herman AG, Bult H. Dexamethasone influences intimal thickening and vascular reactivity in the rabbit collared carotid artery. Eur J Pharmacol. 1995;294:753–761. doi: 10.1016/0014-2999(95)00635-4. [DOI] [PubMed] [Google Scholar]
- 48.Ribichini F, Joner M, Ferrero V, Finn AV, Crimins J, Nakazawa G, Acampado E, Kolodgie FD, Vassanelli C, Virmani R. Effects effects of oral prednisone after stenting in a rabbit model of established atherosclerosis. J Am Coll Cardiol. 2007;50:176–185. doi: 10.1016/j.jacc.2007.03.031. [DOI] [PubMed] [Google Scholar]
- 49.Poon M, Gertz SD, Fallon JT, Wiegman P, Berman JW, Sarembock IJ, Taubman MB. Dexamethasoneinhibits macrophage accumulation after balloon arterial injury in cholesterol-fed rabbits. Atherosclerosis. 2001;155:371–380. doi: 10.1016/S0021-9150(00)00605-5. [DOI] [PubMed] [Google Scholar]
- 50.Strecker EP, Gabelmann A, Boos I, Lucas CXZY, Haberstroh J, Freudenberg N, Stricker H, Langer M, Betz E. Effect on intimal hyperplasia of dexamethasone released from coated metal stents compared with non-coated stents in canine femoral arteries. Cardiovasc Inter Radiol. 1998;21:487–496. doi: 10.1007/s002709900309. [DOI] [PubMed] [Google Scholar]
- 51.Pepine CJ, Hirshfeld JW, Macdonald RG, Henderson MA, Bass TA, Goldberg S, Savage MP, Vetrovec G, Cowley M, Taussig AS, Whitworth HB, Margolis JR, Hill JA, Bove AA, Jugo R. A controlled trial of corticosteroids to prevent restenosis after coronary angioplasty. Circulation. 1990;81:1753–1761. doi: 10.1161/01.CIR.81.6.1753. [DOI] [PubMed] [Google Scholar]
- 52.Reimers B, Moussa I, Akiyama T, Kobayashi Y, Albiero R, Di Francesco L, Di Mario C, Colombo A. Persistent high restenosis after local intrawall delivery of long-acting steroids before coronary stent implantation. J Invasive Cardiol. 1998;10:323–331. [PubMed] [Google Scholar]
- 53.Hoffmann R, Langenberg R, Radke P, Franke A, Blindt R, Ortlepp J, Popma JJ, Weber C, Hanrath P. Evaluation of a high-dose dexamethasone-eluting stent. Am J Cardiol. 2004;94:193–195. doi: 10.1016/j.amjcard.2004.03.061. [DOI] [PubMed] [Google Scholar]
- 54.Hoffmann R, Radke P, Weber C, Ortlepp J, Blindt R, Haager P, Hanrath P. Failure of a high-dose dexamethasone-eluting stent to inhibit neointimal hyperplasia and restenosis. Eur Heart J. 2004;25:525–526. [Google Scholar]
- 55.Ribichini F, Tomai F, Paloscia L, Di Sciascio G, Carosio G, Romano M, Verna E, Galli M, Tamburino C, De Cesare N, Pirisi R, Piscione F, Lanteri G, Ferrero V, Vassanelli C, DESIRE i Steroid-eluting stents in patients with acute coronary syndrome: the Dexamethasone Eluting Stent Italian REgistry. Heart. 2007;93:598–600. doi: 10.1136/hrt.2006.098467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu X, De Scheerder I, Desmet W. Dexamethasone-eluting stent: an anti-inflammatory approach to inhibit coronary restenosis. Expert Rev Cardiovasc Ther. 2004;2:653–660. doi: 10.1586/14779072.2.5.653. [DOI] [PubMed] [Google Scholar]
- 57.Radke PW, Weber C, Kaiser A, Schober A, Hoffmann R. Dexamethasone and restenosis after coronary stent implantation: new indication for an old drug? Curr Pharm Des. 2004;10:349–355. doi: 10.2174/1381612043453324. [DOI] [PubMed] [Google Scholar]
- 58.Ferrero V, Ribichini F, Pesarini G, Brunelleschi S, Vassanelli C. Glucocorticoids in the prevention of restenosis after coronary angioplasty - therapeutic potential. Drugs. 2007;67:1243–1255. doi: 10.2165/00003495-200767090-00001. [DOI] [PubMed] [Google Scholar]
- 59.Versaci F, Gaspardone A, Tomai F, Ribichini F, Russo P, Proietti I, Ghini AS, Ferrero V, Chiariello L, Gioffre PA, Romeo F, Crea F. Immunosuppressive therapy for the prevention of restenosis after coronary artery stent implantation (IMPRESS Study) J Am Coll Cardiol. 2002;40:1935–1942. doi: 10.1016/S0735-1097(02)02562-7. [DOI] [PubMed] [Google Scholar]
- 60.Ferrero V, Ribichini F, Rognoni A, Marino P, Brunelleschi S, Vassanelli C. Comparison of efficacy and safety of lower-dose to higher-dose oral prednisone after percutaneous coronary interventions (the IMPRESS-LD Study) Am J Cardiol. 2007;99:1082–1086. doi: 10.1016/j.amjcard.2006.11.064. [DOI] [PubMed] [Google Scholar]
- 61.Liu XS, Huang YM, Hanet C, Vandormael M, Legrand V, Dens J, Vandenbossche JL, Missault L, Vrints C, De Scheerder I. Study of antirestenosis with the biodivysio dexamethasone-eluting stent (STRIDE): A First-in-Human Multicenter Pilot Trial. Catheter Cardiovasc Interv. 2003;60:172–178. doi: 10.1002/ccd.10636. [DOI] [PubMed] [Google Scholar]
- 62.Dzyakanchuk AA, Balazs Z, Nashev LG, Amrein KE, Odermatt A. 11beta-Hydroxysteroid dehydrogenase 1 reductase activity is dependent on a high ratio of NADPH/NADP(+) and is stimulated by extracellular glucose. Mol Cell Endocrinol. 2009;301:137–141. doi: 10.1016/j.mce.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 63.Seckl JR. 11β-hydroxysteroid dehydrogenases: changing glucocorticoid action. Curr Opin Pharmacol. 2004;4:597–602. doi: 10.1016/j.coph.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 64.Webster SP, Pallin TD. 11 beta-hydroxysteroid dehydrogenase type 1 inhibitors as therapeutic agents. Expert Opin Ther Patents. 2007;17:1407–1422. doi: 10.1517/13543776.17.12.1407. [DOI] [Google Scholar]
- 65.Draper N, Stewart P. 11β-Hydroxysteroid dehydrogenase and the pre-receptor regulation of corticosteroid hormone action. J Endocrinol. 2005;186:251–271. doi: 10.1677/joe.1.06019. [DOI] [PubMed] [Google Scholar]
- 66.Smith RE, Maguire JA, Stein Oakley AN, Sasano H, Takahashi K, Fukushima K, Krozowski ZS. Localization of 11 beta-hydroxysteroid dehydrogenase type II in human epithelial tissues. J Clin Endocrinol Metab. 1996;81:3244–3248. doi: 10.1210/jc.81.9.3244. [DOI] [PubMed] [Google Scholar]
- 67.Luo MJ, Thieringer R, Springer MS, Wright SD, Hermanowski-Vosatka A, Plump A, Balkovec JM, Cheng K, Ding GJ, Kawka DW, Koo GC, Le Grand CB, Luo Q, Maletic MM, Malkowitz L, Shah L, Singer I, Waddell ST, Wu KK, Yuan J, Zhu J, Stepaniants S, Yang X, Lum PY, Wang IM. 11β-HSD1 inhibition reduces atherosclerosis in mice by altering proinflammatory gene expression in the vasculature. Physiol Genomics. 2013;45:47–57. doi: 10.1152/physiolgenomics.00109.2012. [DOI] [PubMed] [Google Scholar]
- 68.Christy C, Hadoke PWF, Paterson JM, Mullins JJ, Seckl JR, Walker BR. Glucocorticoid action in mouse aorta; localisation of 11beta hydroxysteroid dehydrogenase type 2 and effects on response to glucocorticoid in vitro. Hypertension. 2003;42:580–587. doi: 10.1161/01.HYP.0000088855.06598.5B. [DOI] [PubMed] [Google Scholar]
- 69.Hammer F, Stewart PM. Cortisol metabolism in hypertension. Best Pract Res Clin Endocrinol Metab. 2006;20:337–353. doi: 10.1016/j.beem.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 70.Chapman KE, Seckl JR. 11beta-HSD1, inflammation, metabolic disease and age-related cognitive (dys)function. Neurochem Res. 2008;33:624–636. doi: 10.1007/s11064-007-9504-9. [DOI] [PubMed] [Google Scholar]
- 71.Rosenstock J, Banarer S, Fonseca VA, Inzucchi SE, Sun W, Yao W, Hollis G, Flores R, Levy R, Williams WV, Seckl JR, Huber R. The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care. 2010;33:1516–1522. doi: 10.2337/dc09-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. doi: 10.1126/science.1066285. [DOI] [PubMed] [Google Scholar]
- 73.Masuzaki H, Yamamoto H, Kenyon CJ, Elmquist JK, Morton NM, Paterson JM, Shinyama H, Sharp MG, Fleming S, Mullins JJ, Seckl JR, Flier JS. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest. 2003;112:83–90. doi: 10.1172/JCI17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Paterson JM, Holmes MC, Kenyon CJ, Carter R, Mullins JJ, Seckl JR. Liver-selective transgene rescue of hypothalamic-pituitary-adrenal axis dysfunction in 11beta-hydroxysteroid dehydrogenase type 1-deficient mice. Endocrinology. 2007;148(3):961–966. doi: 10.1210/en.2006-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61:661–666. doi: 10.1001/archneur.61.5.661. [DOI] [PubMed] [Google Scholar]
- 76.MacLullich AMJ, Ferguson KJ, Louise RM, Deary IJ, Starr JM, Wardlaw JM, Walker BR, Andrew R, Seckl JR. 11ß-Hydroxysteroid dehydrogenase type 1, brain atrophy and cognitive decline. Neurobiol Aging. 2012;33(1):207. doi: 10.1016/j.neurobiolaging.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Holmes MC, Carter RN, Noble J, Chitnis S, Dutia A, Paterson JM, Mullins JJ, Seckl JR, Yau JLW. 11β-hydroxysteroid dehydrogenase type 1 expression is increased in the aged mouse hippocampus and parietal cortex and causes memory impairments. J Neurosci. 2010;30:6916–6920. doi: 10.1523/JNEUROSCI.0731-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wyrwoll CS, Holmes MC, Seckl JR. 11β-Hydroxysteroid dehydrogenases and the brain: from zero to hero a decade of progress. Front Neuroendocrinol. 2011;32(3):265–286. doi: 10.1016/j.yfrne.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schmidt M, Weidler C, Naumann H, Anders S, Scholmerich J, Straub RH. Reduced capacity for the reactivation of glucocorticoids in rheumatoid arthritis synovial cells: possible role of the sympathetic nervous system? Arthritis Rheum. 2005;52:1711–1720. doi: 10.1002/art.21091. [DOI] [PubMed] [Google Scholar]
- 80.Coutinho AE, Gray M, Brownstein DG, Salter DM, Sawatzky DA, Clay S, et al. 11β-Hydroxysteroid dehydrogenase type 1, but not type 2, deficiency worsens acute inflammation and experimental arthritis in mice. Endocrinology. 2012;153(1):234–240. doi: 10.1210/en.2011-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McSweeney SJ, Kozak AM, Khaled H, Hadoke PWF, Walker BR, Gray GA. Sustained enhancement of cardiac function follows increased recruitment of pro-angiogenic macrophages to healing infarcts of 11β hydroxysteroid dehydrogenase type 1 deficient mice. Cardiovasc Res. 2010;88(1):159–167. doi: 10.1093/cvr/cvq149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wamil M, Battle JH, Turban S, Kipari T, Seguret D, de Peixoto RS, et al. Novel fat depot-specific mechanisms underlie resistance to visceral obesity and inflammation in 11{beta}-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes. 2011;60(4):1158–1167. doi: 10.2337/db10-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Small GR, Hadoke PWF, Sharif I, Dover AR, Armour D, Kenyon CJ, Gray GA, Walker BR. Preventing local regeneration of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase type 1 enhances angiogenesis. Proc Natl Acad Sci U S A. 2005;102:12165–12170. doi: 10.1073/pnas.0500641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Michailidou Z, Turban S, Miller E, Xiantong Z, Schrade J, Ratcliffe PJ, Hadoke PWF, Iredale J, Morton NM, Seckl JR. Increased angiogenesis protects against hypoxia and fibrosis in adipose tissue of metabolic disease-resistant 11β-hydroxysteroid dehydrogenase type 1 deficient mice. J Biol Chem. 2012;287(6):4188–4197. doi: 10.1074/jbc.M111.259325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maes C, Carmeliet G, Schipani E. Hypoxia-driven pathways in bone development, regeneration and disease. Nat Rev Rheumatol. 2012;8(6):358–366. doi: 10.1038/nrrheum.2012.36. [DOI] [PubMed] [Google Scholar]
- 86.Rae M, Mohamad A, Price D, Hadoke PWF, Walker BR, Mason JI, Hillier S, Critchley H. Cortisol inactivation by 11β-hydroxysteroid dehydrogenase-2 may enhance endometrial angiogenesis via reduced thrombospondin-1 in heavy menstruation. J Clin Endo Metab. 2009;94(4):1443–1450. doi: 10.1210/jc.2008-1879. [DOI] [PubMed] [Google Scholar]
- 87.Atalar F, Vural B, Ciftci C, Demirkan A, Akan G, Susleyici-Duman B, Gunay D, Akpinar B, Sagbas E, Ozbek U, Buyukdevrim AS. 11β-hydroxysteroid dehydrogenase type 1 gene expression is increased in ascending aorta tissue of metabolic syndrome patients with coronary artery disease. Genet Mol Res. 2012;11(3):3122–3132. doi: 10.4238/2012.August.31.10. [DOI] [PubMed] [Google Scholar]
- 88.Ayari H, Legedz L, Lantelme P, Feugier P, Randon J, Cerutti C, et al. Auto-amplification of cortisol actions in human carotid atheroma is linked to arterial remodeling and stroke. Fundam Clin Pharmacol. 2012. doi:10.1111/j.1472-8206.2012.01064.x. [Epub ahead of print]. PMID:23025717. [DOI] [PubMed]
- 89.Cai TQ, Wong BM, Mundt SS, Thieringer R, Wright SD, Hermanowski-Vosatka A. Induction of 11beta-hydroxysteroid dehydrogenase type 1 but not type 2 in human aortic smooth muscle cells by inflammatory stimuli. J Steroid Biochem. 2001;77:117–122. doi: 10.1016/S0960-0760(01)00041-3. [DOI] [PubMed] [Google Scholar]
- 90.Dover AR, Hadoke PWF, Macdonald LJ, Miller E, Newby DE, Walker BR. Intravascular glucocorticoid metabolism during inflammation and injury in mice. Endocrinology. 2007;148:166–172. doi: 10.1210/en.2006-0996. [DOI] [PubMed] [Google Scholar]
- 91.Hermanowski-Vosatka A, Balkovec JM, Cheng K, Chen HY, Hernandez M, Koo GC, Le Grand CB, Li ZH, Metzger JM, Mundt SS, Noonan H, Nunes CN, Olson SH, Pikounis B, Ren N, Robertson N, Schaeffer JM, Shah K, Springer MS, Strack AM, Strowski M, Wu K, Wu TJ, Xiao JY, Zhang BB, Wright SD, Thieringer R. 11 beta-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J Exp Med. 2005;202:517–527. doi: 10.1084/jem.20050119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lloyd DJ, Helmering J, Cordover D, Bowsman M, Chen M, Hale C, Fordstrom P, Zhou M, Wang M, Kaufman SA, Veniant MM. Antidiabetic effects of 11beta-HSD1 inhibition in a mouse model of combined diabetes, dyslipidaemia and atherosclerosis. Diabetes Obes Metab. 2009;11:688–699. doi: 10.1111/j.1463-1326.2009.01034.x. [DOI] [PubMed] [Google Scholar]
- 93.Nuotio-Antar AM, Hachey DL, Hasty AH. Carbenoxolone treatment attenuates symptoms of metabolic syndrome and atherogenesis in obese, hyperlipidemic mice. Am J Physiol Endocrinol Metab. 2007;293:E1517–E1528. doi: 10.1152/ajpendo.00522.2007. [DOI] [PubMed] [Google Scholar]
- 94.Carter RN, Paterson JM, Tworowska U, Stenvers DJ, Mullins JJ, Seckl JR, Holmes MC. Hypothalamic-pituitary-adrenal axis abnormalities in response to deletion of 11β-HSD1 is strain-dependent. J Neuroendocrinol. 2009;21(11):879–887. doi: 10.1111/j.1365-2826.2009.01899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Iqbal J, Macdonald LJ, Low L, Yau CW, Hadoke PWF, Walker BR. Contribution of endogenous glucocorticoids and their intra-vascular metabolism by 11β-HSDs to post-angioplasty neointimal proliferation in mice. Endocrinology. 2012;153(12):5896–5905. doi: 10.1210/en.2012-1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.• Kipari, T, Hadoke PWF, Iqbal J, Man T-Y, Miller E, Coutinho A, Sullivan KM, Mitic T, Livingstone DEW, Kenyon CJ, Samuel K, White CI, Sheraz SY, Blom J, Bouhlel MA, Chinetti-Gbaguidi G, Staels B, Andrew R, Walker BR, Savill JS, Chapman KE, Seckl JR. 11β-hydroxysteroid dehydrogenase type 1 deficiency reduces atherosclerosis and plaque inflammation. FASEB J. In Press. First study to demonstrate reduced lesion formation in atherosclerosis prone mice with transgenic deletion of 11β-HSD1; implicating a role for 11β-HSD1 deficiencyin haematopoietic cells in the mechanism of reduced lesion formation.
- 97.Borissoff JI, Spronk HMH, ten Cate H. Mechanisms of disease: the hemostatic system as a modulator of atherosclerosis. N Engl J Med. 2011;364:1746–1760. doi: 10.1056/NEJMra1011670. [DOI] [PubMed] [Google Scholar]
- 98.Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95:858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- 99.Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schütz G, Lumeng CN, Mortensen RM. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. 2010;120(9):3350–3364. doi: 10.1172/JCI41080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Trapp T, Holsboer F. Heterodimerization between mineralocorticoid and glucocorticoid receptor increases the functional diversity of corticosteroid action. Trends Pharmacol Sci. 1996;17:145–149. doi: 10.1016/0165-6147(96)81590-2. [DOI] [PubMed] [Google Scholar]
- 101.Arai N, Masuzaki H, Tanaka T, Ishii T, Yasue S, Kobayashi N, Tomita T, Noguchi M, Kusakabe T, Fujikura J, Ebihara K, Hirata M, Hosoda K, Hayashi T, Sawai H, Minokoshi Y, Nakao K. Ceramide and adenosine 5'-monophosphate-activated protein kinase are two novel regulators of 11β-hydroxysteroid dehydrogenase type 1 expression and activity in cultured preadipocytes. Endocrinology. 2007;148:5268–5277. doi: 10.1210/en.2007-0349. [DOI] [PubMed] [Google Scholar]
- 102.Clarke KA, Ward JW, Forhead AJ, Giussani DA, Fowden AL. Regulation of 11 beta-hydroxysteroid dehydrogenase type 2 activity in ovine placenta by fetal cortisol. J Endocrinol. 2002;172:527–534. doi: 10.1677/joe.0.1720527. [DOI] [PubMed] [Google Scholar]
- 103.Liu Y-J, Nakagawa Y, Nasuda K, Saegusa H, Igarashi Y. Effect of growth hormone, insulin and dexamethasone on 11ß-hydroxysteroid dehydrogenase activity on a primary culture of rat hepatocytes. Life Sciences. 1996;59:227–234. doi: 10.1016/0024-3205(96)00288-3. [DOI] [PubMed] [Google Scholar]
- 104.Berger J, Tanen M, Elbrecht A, Hermanowski-Vosatka A, Moller DE, Wright SD, Thieringer R. Peroxisome proliferator-activated receptor-gamma ligands inhibit adipocyte 11beta -hydroxysteroid dehydrogenase type 1 expression and activity. J Biol Chem. 2001;276:12629–12635. doi: 10.1074/jbc.M003592200. [DOI] [PubMed] [Google Scholar]
- 105.Hermanowski-Vosatka A, Gerhold D, Mundt SS, Loving VA, Lu M, Chen Y, Elbrecht A, Wu M, Doebber T, Kelly L, Milot D, Guo Q, Wang PR, Ippolito M, Chao YS, Wright SD, Thieringer R. PPARalpha agonists reduce 11beta-hydroxysteroid dehydrogenase type 1 in the liver. Biochem Biophys Res Commun. 2000;279:330–336. doi: 10.1006/bbrc.2000.3966. [DOI] [PubMed] [Google Scholar]
- 106.Stulnig TM, Oppermann U, Steffensen KR, Schuster GU, Gustafsson JA. Liver X receptors downregulate 11beta-hydroxysteroid dehydrogenase type 1 expression and activity. Diabetes. 2002;51:2426–2433. doi: 10.2337/diabetes.51.8.2426. [DOI] [PubMed] [Google Scholar]
- 107.Low SC, Assaad SN, Rajan V, Chapman KE, Edwards CR, Seckl JR. Regulation of 11 beta-hydroxysteroid dehydrogenase by sex steroids in vivo: further evidence for the existence of a second dehydrogenase in rat kidney. J Endocrinol. 1993;139:27–35. doi: 10.1677/joe.0.1390027. [DOI] [PubMed] [Google Scholar]
- 108.Langley-Evans SC, Philips G, Benediktsson R, Gardner D, Edwards CRW, Jackson AA, Seckl JR. Maternal dietary protein restriction, placental glucocorticoid metabolism and the programming of hypertension. Placenta. 1996;17:169–172. doi: 10.1016/S0143-4004(96)80010-5. [DOI] [PubMed] [Google Scholar]
- 109.Heiniger CD, Kostadinova RM, Rochat MK, Serra A, Ferrari P, Dick B, Frey BM, Frey FJ. Hypoxia causes down-regulation of 11 beta-hydroxysteroid dehydrogenase type 2 by induction of Egr-1. Faseb J. 2003;17:917–919. doi: 10.1096/fj.02-0582fje. [DOI] [PubMed] [Google Scholar]
- 110.Zielinska AE, Walker EA, Stewart PM, Lavery GG. Biochemistry and physiology of hexose-6-phosphate knockout mice. Mol Cell Endocrinol. 2011;336(1–2):213–8. doi: 10.1016/j.mce.2010.12.004. [DOI] [PubMed] [Google Scholar]