

Abstract
The photodissociation of cyanide from ferric myoglobin (MbCN) and horseradish peroxidase (HRPCN) has been definitively observed. This has implications for the interpretation of ultrafast IR (Helbing et al. Biophys. J. 2004, 87, 1881–1891) and optical (Gruia et al. Biophys. J. 2008, 94, 2252–2268) studies that had previously suggested the Fe-CN bond was photostable in MbCN. The photolysis of ferric MbCN takes place with a quantum yield of ~75% and the resonance Raman spectrum of the photoproduct observed in steady-state experiments as a function of laser power and sample spinning rate is identical to that of ferric Mb (metMb). The data are quantitatively analyzed using a simple model where cyanide is photodissociated and, although geminate rebinding with a rate kBA ≈ (3.6 ps)−1 is the dominant process, some CN− exits from the distal heme pocket and is replaced by water. Using independently determined values for the CN− association rate, we find that the CN− escape rate from the ferric myoglobin pocket to the solution at 293 K is kout ≈ 1–2 × 107 s−1. This value is very similar to, but slightly larger than, the histidine gated escape rate of CO from Mb (1.1×107 s−1) under the same conditions. The analysis leads to an escape probability kout/(kout+kBA) ~ 10−4, which is unobservable in most time domain kinetic measurements. However, the photolysis is surprisingly easy to detect in Mb using cw resonance Raman measurements. This is due to the anomalously slow CN− bimolecular association rate (170 M−1s−1), which arises from the need for water to exchange at the ferric heme binding site of Mb. In contrast, ferric HRP does not have a heme bound water molecule and its CN− bimolecular association rate is larger by ~103 making the CN− photolysis more difficult to observe.
Keywords: Myoglobin CN, heme cyanide, photolysis, resonance Raman
Introduction
Ferric heme systems can be ligated by CN−, a strong field ligand, which places the heme into a low-spin state that is generally thought to be very stable and that does not undergo photodissociation1. The CN− adducts of many heme systems have been studied using a variety of physical methods including EPR 2–4, NMR 5–8, Mössbauer 9, X-ray crystallography10, XANES11 and resonance Raman spectroscopy 12–21. More recently, the met-myoglobin complex with CN− (MbCN) has been studied using ultrafast laser pump-probe spectroscopy 22–24.
Helbing et al. 22 used the CN oscillator as a probe of the system dynamics using infrared (IR) spectroscopy, while Gruia et al. 23 studied the system using vibrational coherence spectroscopy (VCS) and broad band continuum probes in the near ultraviolet (Soret band) region. Very recently, Consani et al. 24 used MbCN in studies that probe tryptophan-to-heme electron transfer and FRET relaxation pathways where the photochemical stability of MbCN was implied. Although Helbing et al. 22 acknowledged the possibility of CN− photodissociation, they favored a model where the CN remained bound to the heme following photoexcitation and attributed the observed infrared vibrational dynamics of CN to excited d-electron states that appeared on the 3.6 ps relaxation path. On the other hand, Gruia et al. 23 observed the formation of a 5-coordinate ferric state that relaxed with a ~4 ps time constant and suggested that one of the heme ligands undergoes photodissociation with a quantum yield of 75%. Both the time constants and the quantum yields found by Gruia et al. 23 are in excellent agreement with those found by Helbing et al. 22 using independent methods.
Because the continuum studies, along with VCS, strongly indicated that ligand photolysis was involved, it was proposed that proximal histidine was photolabile in the metMbCN system 23. Many of the ultrafast IR observations 22, such as the similarity of the signals between metMb and hemoglobin I (from Lucina pectinata) and the time-independent anisotropy of the CN oscillator, are explained by the hypothesis of proximal histidine photolysis. Thus, although there was no definitive spectroscopic evidence, the combination of the IR observations, various chemical considerations 23, and the likelihood that the CN in metMbCN is stabilized by an H-bond 25 suggested that the proximal histidine ligand, rather than the bound CN ligand might be photolabile. There was also a precedent for photo-dissociation of a heme amino acid axial ligand. This had been observed previously in cytochrome c where methionine photolysis and ~ 6 ps rebinding has been observed 26.
Given this background, we were surprised to find, during more extensive VCS studies of the metMbCN complex, that there is clear and compelling evidence for CN− photolysis. These studies involved the creation and probing of photostationary states of metMbCN using cw laser based resonance Raman spectroscopy. Because the inherent geminate rebinding rate of the photolyzed ligand is kBA = (3.6 ps)−1 = 2.78×1011 s−1, it initially appears that a cw laser does not have the capacity to generate a photo-excitation rate, kγ = JσA, that is competitive with the rebinding rate. The photon flux, J, is typically of order J ~ 1021 s−1cm−2 for ~ 1 mW of blue light focused to a diameter of 20 μm. Since the absorption cross-section for the Soret band is typically σA ~ 4×10−16 cm2, this leads to a photo-excitation rate of only kγ ~ 4×105 s−1, which is much smaller than kBA. Thus, only a vanishing ratio (kγ/kBA ~ 10−6 to 10−4 for 1 to 100 mW excitation) of photo-product might be expected to be observed in a photo-stationary state situation where the resonance Raman spectrum is being recorded. However, as will be seen below, the presence of the protein means that we must also consider the small, but finite, escape of the CN− ligand from the heme pocket. This, along with the relatively slow bimolecular entry rate 27–28 of CN− back into the protein pocket (170 M−1s−1), leads to the build-up of photoproduct population that can easily be detected using resonance Raman spectroscopy. Because the geminate rebinding rate and the bimolecular entry rates are known, and the photo-excitation rate can be controlled experimentally, we can perform experiments at different laser power and ligand concentration in order to extract the escape rate of CN− from the protein matrix and compare it to other ligands such as CO. Thus, the main goals of the paper are to first demonstrate that CN− definitely photo-dissociates from the heme in metMb (contrary to the assumption in several recent ultrafast experimental studies) and then to extract the rate of its exit from the protein matrix, even though there is very rapid (~3×1011 s−1) geminate rebinding taking place.
Materials and Methods
Sample preparation
Horse heart myoglobin, horseradish peroxidase (HRP), and potassium cyanide (KCN) were purchased from Sigma (St. Louis, MO) and used without further purification. The myoglobin was dissolved in potassium phosphate buffer solution (0.1 M, pH 7.4) and the concentration was 0.05 mM. An aliquot of KCN (4 μL of either 25 mM or 50 mM) in deionized water was added to 200 μL myoglobin solution to form the ferric MbCN (metMbCN) complex. The CN− concentrations (0.5 mM or 1 mM) correspond to either a 10 fold or 20 fold excess of KCN. A similar protocol was followed for the HRP samples, with the exception that the final HRP and CN− concentrations were 0.12 mM and 1.2 mM, respectively. UV-Vis absorption spectra were recorded with a spectrophotometer (Hitachi, model U4100, Tokyo, Japan) to confirm the complete formation of the cyanide-bound complex before the Raman measurements and to ensure sample integrity after the Raman measurements.
Resonance Raman measurements
To obtain Raman spectra, the MbCN or HRPCN solution was transferred to a cylindrical quartz cell (NSG Precision Cells, Type 76, Farmingdale, NY) which was mounted to a home-built electric motor assembly that spins the sample at a known frequency; in this case 1500 rpm. The sample was excited using the 413.1 nm line from a krypton laser (Innova 300, Coherent). The laser beam interacted with the solution through the flat bottom of the quartz cell and the scattering signals were collected in a 90 degree configuration. The focal spot size of laser beam was 20 μm. To minimize the absorption of the Raman signal, the laser beam was focused on the sample as close to the outer wall of the quartz cell as possible. Laser light and Rayleigh scattering were extinguished by a notch filter (Kaiser Optical Systems, Ann Arbor, MI). The polarized scattered light was scrambled by passing through a depolarizer. The Raman signal was detected by a liquid-nitrogen-cooled CCD after being dispersed by a 0.5 m triple grating monochromator (model SP-2500i, Princeton Instruments, Acton, MA). For each cyanide concentration, spectra with different excitation powers were acquired by varying the laser power, and without changing any other settings. The Raman spectra were calibrated using fenchone29.
Kinetic Analysis
Kinetic model and rates
Ferric myoglobin (metMb) has a water molecule as its distal ligand and a detailed kinetic analysis involving NO photolysis and water binding has been carried out previously 30. One important result is that water enters through the distal histidine gate and binds to the heme of metMb very rapidly (~6×106 s−1), ultimately being stabilized by a distal histidine H-bond so that the effective ratio of water-bound to water-unbound heme in ferric metMb is30 ~103. This, along with the need for the charged CN− ligand to enter hydrophobic regions of the protein, leads to a very slow (170 M−1s−1) CN− association rate for metMb 27–28. For comparison, if we examine the case of ferric horseradish peroxidase (HRP), which does not have a heme-bound water 31, we find the bimolecular rate for CN− binding is 9×104 M−1s−1 32. The ratio of the bimolecular CN− rates for metMb and ferric HRP (1.9×10−3) is on the same order as the probability (1.1×10−3) that the heme water site is vacant in metMb 30. Thus, the difference between CN− binding to HRP and Mb is primarily due to the fact that the heme-bound water dissociation is rate limiting for CN− binding in metMb but is not rate limiting for HRP.
The much faster time-scale of the water binding and dissociation rate compared to CN− binding means that there are only two observed states in the cw resonance Raman experiments reported here. The MbCN bound state (A) and the water bound metMb state (C). They are connected kinetically by the CN− dissociated state with CN− still in the distal heme pocket (B-state). The B-state has vanishingly small population under the photostationary conditions of the present work because of the rapid CN− rebinding or escape to solution. The rapid binding of water whenever the CN− exits to solution means that only A or C will be observed under the present experimental conditions. The overall kinetic scheme can be expressed as:
Scheme I.
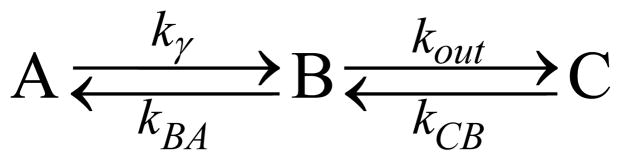
Three-state kinetic model.
In this scheme, kγ = YJσA represents the photo-dissociation rate in terms of the photo-excitation rate (JσA) and the quantum yield (Y) for dissociation in the excited electronic state. The quantum yield will be taken as Y = 0.75 as observed independently by Helbing et al. 22 and Gruia et al. 23. The photo-excitation rate is given by the product of the laser flux, J, and the absorption cross-section, σA. In our experiment, for a 1 mW laser (at 413.1 nm) focusing on a 20 μm diameter spot, J = 6.62×1020 s−1cm−2. The absorption cross section at 413.1 nm for ferric MbCN is σA = 3.67×10−16 cm2. The decay rate of the photoexcited state B has been determined by previous studies 22–23, kBA=(3.6 ps)−1=2.78×1011 s−1, and this is much larger than the rate of escape to solution, kout, which will be determined in the following analysis. The CN− entry into a vacant heme pocket is rate limiting for association so that kCB = 170 M−1s−1, or kCB = 0.17 s−1 under the condition of 1 mM CN−. In terms of the two observable populations (A=MbCN and C= metMb) we can re-cast Scheme I to read:
Scheme II.
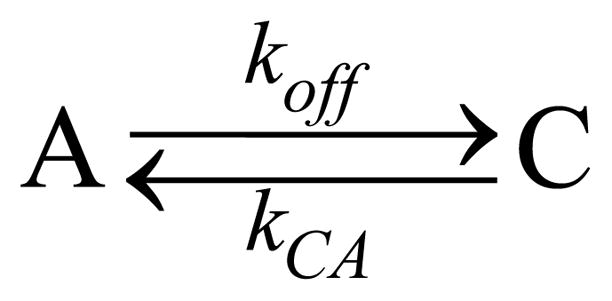
Two-state kinetic model.
where koff = kγkout/(kBA+kout) ≈ kγkout/kBA and kCA = 170 M−1 s−1×[CN−].
Photostationary state populations
In a spinning Raman cell, the material is subject to alternating periods of light and dark as it passes through the focused laser beam where light scattering takes place. The two observable material states (A and C) are differentiated by their Raman spectra so that their relative populations can be determined as a function of laser power, CN− concentration, or spinning rate.
To be more specific, we use an experimental spinning rate of 1500 rpm. This corresponds to a spinning period of τ = 0.04 s. With this angular frequency, and a cell radius of 0.5 cm, the protein solution resides in the 20 μm diameter laser focal area for T = 25 μs. The Raman scattering of the laser beam by the sample in the focal spot consists of ferric MbCN (A-state) and metMb (C-state) and yields their relative population ratio as determined below.
When the protein solution is in the focal spot, during the time (0 ≤ t ≤ T = 25 μs), both photolysis and rebinding of the cyanide ligand can take place during what we call the “light phase”. In contrast, during the “dark phase”, when the photolyzed volume element is no longer in the focal spot (T ≤ t ≤ τ) there is no photolysis, and during this phase bimolecular binding (and possibly diffusion) replenishes the MbCN population. Both the rebinding of CN− and the diffusion of MbCN into the volume element are slow. (Please see the Supporting Information section for a full discussion of the relative roles of diffusion and rebinding.) Thus, during the dark phase, the system may not completely reset to 100% MbCN population in the laser scattering volume element. The evolution of the population of the A and C states can be described by the following differential equations for the light phase (0 ≤ t ≤ T).
| (1) |
Suppose at t = 0, when the protein solution just enters the laser focal spot, the populations of A and C are A(0)=A0 and C(0)=1−A0. The solution to the population of A and C during the light phase (0 ≤ t ≤ T) is
| (2) |
At t = T, A(T) and C(T) act as the initial conditions for the differential equations during the dark phase (T ≤ t ≤ τ).
| (3) |
The dark phase lasts for a time τ = 40 ms, which is large compared to T. This means that a solution for A0 can be found by using 1−A0 = C (τ) with
| (4) |
Expanding the exponentials in Eq. (4) (koffT ≪ 1, kCAτ ≪ 1) and use of T/τ ≪ 1 leads to an expression for the photostationary state population of MbCN in the laser beam:
| (5) |
This is the population of ferric MbCN that contributes to the Raman signal. The population of photolyzed product (metMb) is given by 1−A0.
With a fixed CN− concentration and laser beam area, AB, the laser excitation power can be varied in an experiment that probes dependence of <A> =A0 on the laser power P. Since koff depends on the excitation power, we can write the above equation for A0 as
| (6) |
where
| (7) |
with [CN−] as the cyanide concentration in mM and P is the laser power in mW and h is Planck’s constant and νL is the laser frequency. By measuring the population ratio A/C=A0/(1−A0) as a function of laser power and fitting A0(P) vs P, we can obtain the parameter b so that kout can be evaluated using the above expression.
It is important to note that the fitting parameter b, as written, assumes that MbCN recovery is due to CN− rebinding from solution rather than from MbCN diffusion into the laser focal volume. This assumption is discussed in more detail in the Supporting Information. A simple experimental test of this assumption involves the bimolecular nature of kCA and the fact that b should depend inversely upon the CN− concentration when bimolecular rebinding is the main channel for MbCN population recovery within the focal volume.
Population ratio determination
In order to determine the A-state and C-state populations as a function of laser power using the Raman spectra, we must deconvolve the ferric MbCN (A state) and metMb (C state) spectra. The major difficulty in doing this precisely is determining the basis spectra for pure MbCN and metMb, obtained under exactly the same excitation configuration (reabsorption corrections, etc.) as the spectra in the mixture. We approach this problem by taking, as a first approximation, the spectra excited by the lowest and highest power as fitting basis spectra. This guarantees the same excitation configurations for the basis spectra and the spectra in the mixture.
We denote the basis spectrum with the lowest power as B1 ( P = 2.4 mW) and the basis spectrum with the highest power as B2 ( P = 180 mW) as shown in Fig. 1. It is apparent that these basis spectra will still include a small degree of admixture of ferric MbCN and metMb. We introduce two parameters α and β to express the fractions of MbCN and metMb in B1 and B2:
| (8) |
Figure 1.

Resonance Raman spectra of ferric MbCN in 0.1M, pH 7.4 potassium phosphate buffer, taken with 413.1 nm excitation. The concentrations of horse myoglobin and potassium cyanide are 0.05 mM and 1 mM, respectively. UV-Vis absorption indicated complete formation of ferric MbCN (see Fig. S2). To obtain the Raman spectra, the protein solution was spun at 1500 rpm in a quartz cell (10 mm inner diameter). The spectrum for static sample (2.4 mW) is labeled C and shown in red. Independent spectra of ferric MbCN (0.07 mM with 50 fold KCN added, 3.4 mW, spin) and metMb (0.07 mM, 24 mW, spin) are also shown (in blue, A and D, respectively) as reference spectra. The power was varied as indicated to the right of each spectrum. All spectra are normalized by dividing the spectral intensity by the product of excitation power and accumulation time.
α and β represent the fraction of ferric MbCN in the basis spectra B1 and B2, respectively and IMbCN and ImetMb are the pure spectra of A and C states (if the B1 and B2 spectra were composed of pure A and C states, respectively, the condition α = 1 and β = 0 would apply). The spectra S(P) obtained at the intermediate powers will be a mixture of B1 and B2 and they can be fit using B1 and B2 in the following way,
| (9) |
where f is the fraction of B1 contained in the mixture as determined using the least-squares fitting method. Putting the expressions for B1 and B2 into the above equation,
| (10) |
so the fraction of MbCN actually in the mixture is
| (11) |
Equating Eq. (11) to Eq. (6), we can be express f (P) as determined using the fits with B1 and B2.
| (12) |
We then fit f (P) using the expression
| (13) |
with the fitting parameters ã = α−β, b̃ = (α−β)b and c̃ = β/(α−β), the parameters α and β can be found using α = ã + c̃ · ã and β = c̃ · ã. The fraction of ferric MbCN as a function of laser power, A0(P), can thus be determined using Eq. (11) and when fit using Eq. (6) the value of b in Eq (7) can be found, leading to the value of kout.
Results
Spectral observations
Figure 1 shows the Resonance Raman spectra in the 420 to 470 cm−1 and 1350 to 1660 cm−1 regions of ferric MbCN with 20 fold excess (1 mM) of KCN added. The spectra (A) and (D) are pure ferric MbCN and metMb, respectively, acting as reference spectra in the figure. Experimental conditions for these two spectra are listed in the caption. Excitation powers were varied from 2.4 mW (B1) to 180 mW (B2) for the spinning ferric MbCN sample (only a selection of spectra at different powers are shown in order to minimize congestion in the figure). It can be readily seen from the figure that, as the power increases, the low-spin ferric MbCN features decrease while the high-spin ferric metMb features increase. The ν4 band downshifts from 1375 cm−1 towards 1372 cm−1; the ν3 band increases the high-spin component at 1482 cm−1 while decreasing the low-spin component at 1509 cm−1; the ν2 band at 1563 cm−1 emerges and becomes a prominent feature, signaling the high-spin ferric myoglobin complex. Finally, the ν10 band at 1644 cm−1 and the Fe-CN stretching mode19 at 454 cm−1, which are signatures of the low-spin ferric MbCN complex are decreasing as the power increases.
The spectrum labeled “static” in Fig. 1 (denoted C) was acquired with an excitation power of 2.4 mW after stopping the spinning cell. The spectral features of ferric MbCN in this spectrum nearly disappear and the resultant spectrum resembles that of pure metMb (Fig. 1, D). Another set of experiments was carried out using only a 10 fold excess of KCN (0.5 mM) in order to reduce the bimolecular component of the rate kCA. Very similar results are obtained and the Raman spectra are displayed as Fig. S1 in the Supporting Information. Additional studies with ferric HRP are also presented in the Supporting Information. It is shown that CN− photolysis is also taking place in HRP, but with a kCA for CN− that is ~103 larger than for metMb. Because of the enhanced bimolecular association rate (due to the absence of a heme bound water molecule), the HRPCN photolysis must be studied in a stationary cell where quantitative measurements are more difficult due to heating and convection.
Fitting results
The fraction (f) of the basis spectrum B1 was obtained by a global least squares fit of each Raman spectrum according to Eq. (9). Figure 2 shows an example of the fitting for the spectrum with excitation power of 19.4 mW and 1 mM KCN concentration. The fitting results lead to f = 0.6 and A0(P = 19.4 mW) = 0.63, demonstrating that the effect of using the basis spectra B1 and B2 compared to the “pure” spectra of metMb and MbCN is only about 3%. The results for all values of f (P) are shown in Fig. 3 for the case of 1 mM cyanide (upper panel) and 0.5 mM cyanide (lower panel). The fit of f (P) according to Eq. (13) leads to α = 0.932, β = 0.169 for the case with 1 mM cyanide, and to α = 0.831, β = 0.063 for the case with 0.5 mM cyanide. The precise fraction of ferric MbCN, A0(P), can be calculated using Eq. (11) and this is plotted for all powers in Fig. 4. The fits of the data points in Fig. 4, using Eq. (6) gives b = 0.02966 and kout = 1.2×107 s−1 for 1 mM cyanide. For 0.05 mM cyanide we find b = 0.07412 and kout = 1.5×107 s−1. The inverse dependence of b on [CN−], along with the very similar values for kout, demonstrates that rebinding, rather than diffusion, is of primary importance in the MbCN population recovery during the dark phase of the spinning cell.
Figure 2.

The measured Raman spectrum (A, open circle) was fitted in the range of 1350 ~ 1660 cm−1 with the basis spectra (B1 and B2 in Fig. 1) to determine the fraction of each basis component. Because the basis spectra are not necessarily pure ferric MbCN or metMb, two parameters, α and β, are introduced to account for small amounts of metMb in B1 and MbCN in B2 (see Eq. (8) for details). For the fit at 19.4 mW, shown in this figure, the spectrum contains 60% contribution from B1 and 40% contribution from B2. Since we determine that α = 0.932 and β = 0.169 (see Fig. 3 for details), the actual contribution of ferric MbCN is calculated to be 63%, while that of metMb is 37%.
Figure 3.

Open circles represents the fraction of basis spectrum 1 (top panel = B1 in Fig. 1; bottom panel = B1 in Fig. S1) in the Raman spectra for the case of [Mb] = 0.05 mM, [CN] = 1 mM (top panel) and [Mb] = 0.05 mM, [CN] = 0.5 mM (bottom panel). These fractions were obtained by fitting the Raman spectra with the lowest and highest power spectra, which were taken as “basis spectra”. The solid line is the fitted relationship between the fraction of B1 and excitation power according to Eq. (13) in the main text. The fitting results in the top panel are ã = 0.763, b̃ = 0.0226 and c̃ = 0.221 so that α = 0.932 and β = 0.169. For the bottom panel we have ã = 0.768, b̃ = 0.0569 and c̃ = 0.0819 so that α = 0.831 and β = 0.063.
Figure 4.

The faction of ferric MbCN, A0(P), calculated by using the fitting results from Fig. 3 and Eq. (11). The open triangles are for the case of 1 mM cyanide (20 fold of that of myoglobin) and open circles are for the case of 0.5 mM cyanide (10 fold of that of myoglobin). Solid curves represent the fitting results for the dependence of A0(P) on excitation power P according to Eq. (6). For the case with 1 mM cyanide, b = 0.02966, which yields kout = 1.2×107 s−1; for the case with 0.5 mM cyanide, b = 0.07412, which yields kout = 1.5×107 s−1.
Discussion
The results presented in the prior section demonstrate unequivocally that CN− is a photolabile heme ligand and, based on other recent observations 22–23, the results are fully consistent with a quantum yield of photodissociation that is 0.75. Because the geminate recombination rate is ~ 2–3×1011 s−1 and the escape rate is on the order of 107 s−1, very little (~10−4) of the dissociated CN− escapes from the heme pocket into solution. As a result, it is very difficult for time domain pump-probe experiments to unambiguously detect the CN− photodissociation and escape process. The inability to directly observe CN− photolysis in visible-infrared pump-probe experiments has led to the hypothesis that the observed ~ 4 ps infrared probe response might be related to heme internal electronic processes that are affecting the CN vibrational frequency 22. On the other hand, another recent study acknowledged the likelihood of ligand photolysis, but suggested that histidine, rather than CN−, was the photodissociable ligand 23. Clearly, both of the prior studies must be reconsidered in the context of the present results. Another very recent study involving MbCN proposes transient electron transfer between tryptophan and heme as an alternate to the normal FRET non-radiative pathway 24. However, these studies are probably immune to the CN− photolysis reported here because the time-scales for the electron transfer and FRET process are so much slower than the ~4 ps geminate recombination following CN− photolysis.
The quantitative analysis of the power dependence of CN− dissociation using cw Raman spectroscopy has allowed us to independently determine the CN− escape rate and we find it to be in very good agreement with the CO escape rate. The CO escape from Mb has been studied in detail and found to be controlled by the opening and closing of the distal histidine “gate” 33–39. The fact that the independently determined CN− escape rate is so similar to the gate fluctuation 34 and CO escape 35 rates (~107 s−1 in aqueous buffer solution at 293 K), strongly suggests that the escape of CN− in metMb is also rate-limited by the opening of the histidine gate. On the other hand, the binding of CN− to metMb is significantly retarded by the fact that a water molecule is bound to the heme 99.9% of time 30. This reduces the bimolecular rate for CN− binding to metMb (170 M−1s−1) 27 by a factor of ~103 compared to ferric HRP (9×104 M−1s−1) 32, which does not have a water-bound heme.
The fact that changing the CN− concentration by a factor of two increases the fitting constant b by approximately the same amount is a strong indication that bimolecular rebinding is, in fact, the major process affecting the recovery of the MbCN population. The possibility of MbCN diffusion into the laser beam focal volume during the dark phase of the spinning cell rotation is explored in more depth in the Supporting Information section. It is shown that, under optimal conditions, diffusion along the radial direction in the cell might compete with the unusually slow bimolecular rebinding of metMbCN. However, the diffusion contribution to MbCN population recovery cannot be the dominant process and, based on conservative assumptions, diffusive repopulation is shown to be (at most) on the same order as repopulation due to bimolecular binding. In fact, the experimental measurements in Fig. 4 that monitor the dependence of the population ratios on the CN− concentration are the best test of the relative contributions of rebinding and diffusion. In the limit of very high power, Eq. (6), which assumes no diffusive contribution to MbCN population recovery, predicts that the observed value of A0 should be a factor of two larger for the 1 mM CN− solution than for the 0.5 mM solution. Since this is very close to the ratio observed in the high power limit in Fig. 4, we conclude that diffusion effects can be neglected in these experiments.
We have also developed and implemented a simple methodology for extracting the populations of different species within a mixture that is being driven into a photostationary state. This was done by using low-power and high-power resonance Raman “basis spectra” that are not necessarily the resonance Raman spectra of the “pure” compounds composing the system. In the present analysis, the basis spectra were shown to be very similar to the “pure” spectra. However, in other applications this could be a useful methodology for separating out the actual populations within a mixture without having to be concerned about differential resonance Raman enhancements, corrections due to sample geometry, or the inability to isolate the pure compounds for independent analysis.
Finally, we conclude that CN− is a photolabile heme ligand and this must be taken into account when studying CN− bound heme systems on ultrafast timescales. This work also vividly demonstrates that water binding to the heme can reduce the bimolecular binding rate of exogenous ligands by three orders of magnitude. These studies also demonstrate that the charged CN− ligand must undergo a very similar escape trajectory compared to the neutral CO ligand and both trajectories are regulated by distal histidine gate fluctuations.
Supplementary Material
Acknowledgments
This work was supported by NSF MCB-0744738 and NIH DK35090. We thank Professor J. Timothy Sage for helpful discussions.
Footnotes
Supporting Information Available
Supporting figures and diffusion analysis are presented. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Danielsson J, Meuwly M. Energetics and Dynamics in MbCN: CN−-Vibrational Relaxation from Molecular Dynamics Simulations. J Phys Chem B. 2007;111:218–226. doi: 10.1021/jp0662698. [DOI] [PubMed] [Google Scholar]
- 2.Blumberg WE, Peisach J, Wittenberg BA, Wittenberg JB. The Electronic Structure of Protoheme Proteins. I. An Electron Paramagnetic Resonance and Optical Study of Horseradish Peroxidase and Its Derivatives. J Biol Chem. 1968;243:1854–1862. [PubMed] [Google Scholar]
- 3.Wittenberg BA, Kampa L, Wittenberg JB, Blumberg WE, Peisach J. The Electronic Structure of Protoheme Proteins. II. An Electron Paramagnetic Resonance and Optical Study of Cytochrome C Peroxidase and Its Derivatives. J Biol Chem. 1968;243:1863–1870. [PubMed] [Google Scholar]
- 4.Peisach J, Blumberg WE, Wittenberg BA, Wittenberg JB. The Electronic Structure of Protoheme Proteins. III. Configuration of the Heme and Its Ligands. J Biol Chem. 1968;243:1871–1880. [PubMed] [Google Scholar]
- 5.La Mar GN, Budd DL, Viscio DB, Smith KM, Langry KC. Proton Nuclear Magnetic Resonance Characterization of Heme Disorder in Hemoproteins. Proc Natl Acad Sci USA. 1978;75:5755–5759. doi: 10.1073/pnas.75.12.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rajarathnam K, La Mar GN, Chiu ML, Sligar SG. Determination of the Orientation of the Magnetic Axes of the Cyano-Metmb Complexes of Point Mutants of Myoglobin by Solution 1h NMR: Influence of His E7 → Gly and Arg Cd3 → Gly Substitutions. J Am Chem Soc. 1992;114:9048–9058. [Google Scholar]
- 7.Rajarathnam K, Qin J, La Mar GN, Chiu ML, Sligar SG. Solution Structure Determination of the Heme Cavity in the E7 His → Val Cyano-Met Myoglobin Point Mutant Based on the Proton NMR Detected Dipolar Field of the Iron: Evidence for Contraction of the Heme Pocket. Biochemistry. 1993;32:5670–5680. doi: 10.1021/bi00072a024. [DOI] [PubMed] [Google Scholar]
- 8.La Mar GN, Asokan A, Espiritu B, Yeh DC, Auclair K, Ortiz de Montellano PR. Solution 1h NMR of the Active Site of Substrate-Bound, Cyanide-Inhibited Human Heme Oxygenase: Comparison to the Crystal Structure of the Water-Ligated Form. J Biol Chem. 2001;276:15676–15687. doi: 10.1074/jbc.M009974200. [DOI] [PubMed] [Google Scholar]
- 9.Lang G, Marshali W. Mössbauer Effect in Some Haemoglobin Compounds. Proc Phys Soc. 1966;87:3–34. doi: 10.1016/s0022-2836(66)80032-3. [DOI] [PubMed] [Google Scholar]
- 10.Bolognesi M, Rosano C, Losso R, Borassi A, Rizzi M, Wittenberg JB, Boffi A, Ascenzi P. Cyanide Binding to Lucina pectinata Hemoglobin I and to Sperm Whale Myoglobin: an X-ray Crystallographic Study. Biophys J. 1999;77:1093–1099. doi: 10.1016/S0006-3495(99)76959-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arcovito A, Benfatto M, Cianci M, Hasnain SS, Nienhaus K, Nienhaus GU, Savino C, Strange RW, Vallone B, Della Longa S. X-ray Structure Analysis of a Metalloprotein with Enhanced Active-site Resolution Using in situ X-ray Absorption Near Edge Structure Spectroscopy. Proc Natl Acad Sci USA. 2007;104:6211–6216. doi: 10.1073/pnas.0608411104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu NT, Benko B, Kerr EA, Gersonde K. Iron-Carbon Bond Lengths in Carbonmonoxy and Cyanomet Complexes of the Monomeric Hemoglobin III from Chironomus Thummi Thummi: A Critical Comparison between Resonance Raman and X-Ray Diffraction Studies. Proc Natl Acad Sci USA. 1984;81:5106–5110. doi: 10.1073/pnas.81.16.5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henry ER, Rousseau DL, Hopfield JJ, Noble RW, Simon SR. Spectroscopic Studies of Protein-Heme Interactions Accompanying the Allosteric Transition in Methemoglobins. Biochemistry. 1985;24:5907–5918. doi: 10.1021/bi00342a033. [DOI] [PubMed] [Google Scholar]
- 14.Sitter AJ, Reczek CM, Terner J. Observation of the FeIV=O Stretching Vibration of Ferryl Myoglobin by Resonance Raman Spectroscopy. Biochim Biophys Acta (BBA) - Protein Structure and Molecular Enzymology. 1985;828:229–235. doi: 10.1016/0167-4838(85)90301-2. [DOI] [PubMed] [Google Scholar]
- 15.López-Garriga JJ, Oertling WA, Kean RT, Hoogland H, Wever R, Babcock GT. Metal-Ligand Vibrations of Cyanoferric Myeloperoxidase and Cyanoferric Horseradish Peroxidase: Evidence for a Constrained Heme Pocket in Myeloperoxidase. Biochemistry. 1990;29:9387–9395. doi: 10.1021/bi00492a012. [DOI] [PubMed] [Google Scholar]
- 16.Hu S, Treat RW, Kincaid JR. Distinct Heme Active-Site Structure in Lactoperoxidase Revealed by Resonance Raman Spectroscopy. Biochemistry. 1993;32:10125–10130. doi: 10.1021/bi00089a031. [DOI] [PubMed] [Google Scholar]
- 17.Al-Mustafa J, Kincaid JR. Resonance Raman Study of Cyanide-Ligated Horseradish Peroxidase. Detection of Two Binding Geometries and Direct Evidence for the “Push-Pull” Effect. Biochemistry. 1994;33:2191–2197. doi: 10.1021/bi00174a028. [DOI] [PubMed] [Google Scholar]
- 18.Simianu MC, Kincaid JR. Resonance Raman Spectroscopic Detection of Both Linear and Bent Fe-CN Fragments for the Cyanide Adducts of Cytochrome P-450 Camphor and Its Substrate-Bound Forms. Relevance to the “Charge Relay” Mechanism. J Am Chem Soc. 1995;117:4628–4636. [Google Scholar]
- 19.Hirota S, Ogura T, ShinzawaItoh K, Yoshikawa S, Kitagawa T. Observation of Multiple CN-Isotope-Sensitive Raman Bands for CN− Adducts of Hemoglobin, Myoglobin, and Cytochrome C Oxidase: Evidence for Vibrational Coupling between the Fe-C-N Bending and Porphyrin in-Plane Modes. J Phys Chem. 1996;100:15274–15279. [Google Scholar]
- 20.Das TK, Couture M, Guertin M, Rousseau DL. Distal Interactions in the Cyanide Complex of Ferric Chlamydomonas Hemoglobin. J Phys Chem B. 2000;104:10750–10756. [Google Scholar]
- 21.Pinakoulaki E, Vamvouka M, Varotsis C. The Active Site Structure of Heme a33+-C≡N-CuB2+ of Cytochrome aa3 Oxidase as Revealed from Resonance Raman Scattering. J Phys Chem B. 2003;107:9865–9868. [Google Scholar]
- 22.Helbing J, Bonacina L, Pietri R, Bredenbeck J, Hamm P, van Mourik F, Chaussard F, Gonzalez-Gonzalez A, Chergui M, Ramos-Alvarez C, Ruiz C, López-Garriga J. Time-Resolved Visible and Infrared Study of the Cyano Complexes of Myoglobin and of Hemoglobin I from Lucina pectinata. Biophys J. 2004;87:1881–1891. doi: 10.1529/biophysj.103.036236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gruia F, Kubo M, Ye X, Champion PM. Investigations of Vibrational Coherence in the Low-Frequency Region of Ferric Heme Proteins. Biophys J. 2008;94:2252–2268. doi: 10.1529/biophysj.107.122119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Consani C, Auböck G, van Mourik F, Chergui M. Ultrafast Tryptophan-to-heme Electron Transfer in Myoglobins Revealed by UV 2D Spectroscopy. Science. 2013;339:000–000. doi: 10.1126/science.1230758. [DOI] [PubMed] [Google Scholar]
- 25.Yoshikawa S, O’Keeffe DH, Caughey WS. Investigations of Cyanide as an Infrared Probe of Hemeprotein Ligand Binding Sites. J Biol Chem. 1985;260:3518–3528. [PubMed] [Google Scholar]
- 26.Wang W, Ye X, Demidov AA, Rosca F, Sjodin T, Cao WX, Sheeran M, Champion PM. Femtosecond Multicolor Pump-Probe Spectroscopy of Ferrous Cytochrome C. J Phys Chem B. 2000;104:10789–10801. [Google Scholar]
- 27.Antonini E, Brunori M. Hemoglobin and Myoglobin in Their Reactions with Ligands. North-Holland: Amsterdam: 1971. [Google Scholar]
- 28.Blanck J, Graf W, Scheler UW. Kinetische Untersuchungen über die Bildung von Metmyoglobin-komplexen. Acta Biol Med Germ. 1961;7:323–326. [PubMed] [Google Scholar]
- 29.Yu NT, Srivastava RB. Resonance Raman Spectroscopy of Heme Proteins with Intensified Vidicon Detectors: Studies of Low Frequency Modes and Excitation Profiles in Cytochrome c and Hemoglobin. J Raman Spectrosc. 1980;9:166–171. [Google Scholar]
- 30.Cao WX, Christian JF, Champion PM, Rosca F, Sage JT. Water Penetration and Binding to Ferric Myoglobin. Biochemistry. 2001;40:5728–5737. doi: 10.1021/bi010067e. [DOI] [PubMed] [Google Scholar]
- 31.Gajhede M, Schuller DJ, Henriksen A, Smith AT, Poulos TL. Crystal Structure of Horseradish Peroxidase C at 2. 15 Å Resolution. Nat Struct Mol Biol. 1997;4:1032–1038. doi: 10.1038/nsb1297-1032. [DOI] [PubMed] [Google Scholar]
- 32.Ellis WD, Dunford HB. The Kinetics of Cyanide and Fluoride Binding by Ferric Horseradish Peroxidase. Biochemistry. 1968;7:2054–2062. doi: 10.1021/bi00846a006. [DOI] [PubMed] [Google Scholar]
- 33.Tian WD, Sage JT, Champion PM. Investigations of Ligand Association and Dissociation Rates in the “Open” and “Closed” States of Myoglobin. J Mol Biol. 1993;233:155–166. doi: 10.1006/jmbi.1993.1491. [DOI] [PubMed] [Google Scholar]
- 34.Tian WD, Sage JT, Champion PM, Chien E, Sligar SG. Probing Heme Protein Conformational Equilibration Rates with Kinetic Selection. Biochemistry. 1996;35:3487–3502. doi: 10.1021/bi952474u. [DOI] [PubMed] [Google Scholar]
- 35.Cao W, Ye X, Sjodin T, Christian JF, Demidov AA, Berezhna S, Wang W, Barrick D, Sage JT, Champion PM. Investigations of Photolysis and Rebinding Kinetics in Myoglobin Using Proximal Ligand Replacements. Biochemistry. 2004;43:11109–11117. doi: 10.1021/bi049077g. [DOI] [PubMed] [Google Scholar]
- 36.Perutz MF, Mathews FS. An X-ray Study of Azide Methaemoglobin. J Mol Biol. 1966;21:199–202. doi: 10.1016/0022-2836(66)90088-x. [DOI] [PubMed] [Google Scholar]
- 37.Case DA, Karplus M. Dynamics of Ligand Binding to Heme Proteins. J Mol Biol. 1979;132:343–368. doi: 10.1016/0022-2836(79)90265-1. [DOI] [PubMed] [Google Scholar]
- 38.Ringe D, Petsko GA, Kerr DE, Ortiz de Montellano PR. Reaction of Myoglobin with Phenylhydrazine: A Molecular Doorstop. Biochemistry. 1984;23:2–4. doi: 10.1021/bi00296a001. [DOI] [PubMed] [Google Scholar]
- 39.Morikis D, Champion PM, Springer BA, Sligar SG. Resonance Raman Investigations of Site-directed Mutants of Myoglobin: Effects of Distal Histidine Replacement. Biochemistry. 1989;28:4791–4800. doi: 10.1021/bi00437a041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.