

Abstract
Crouzon syndrome is a debilitating congenital disorder involving abnormal craniofacial skeletal development caused by mutations in Fibroblast Growth Factor Receptor-2 (FGFR2). Phenotypic expression in humans exhibits an autosomal dominant pattern that commonly involves premature fusion of the coronal suture (craniosynostosis) and severe midface hypoplasia. To further investigate biologic mechanisms by which the Crouzon syndrome associated FGFR2C342Y mutation leads to abnormal craniofacial skeletal development we created congenic BALB/c FGFR2C342Y/+ mice. Here we show that BALB/c FGFR2C342Y/+ mice have a consistent craniofacial phenotype including partial fusion of the coronal and lambdoid sutures, intersphenoidal synchondrosis and multiple facial bones, with minimal fusion of other craniofacial sutures. This phenotype is similar to the classic and less severe form of Crouzon syndrome that involves significant midface hypoplasia with limited craniosynostosis. Linear and morphometric analyses demonstrate that FGFR2C342Y/+ mice on the BALB/c genetic background differ significantly in form and shape from their wild type littermates, and that in this genetic background the FGFR2C342Y mutation preferentially effects some craniofacial bones and sutures over others. Analysis of cranial bone cells indicates that the FGFR2C342Y mutation promotes aberrant osteoblast differentiation and increased apoptosis that is more severe in frontal than parietal bone cells. Additionally, FGFR2C342Y/+ frontal but not parietal bones exhibit significantly diminished bone volume and density compared to wild type mice. These results confirm that FGFR2-associated craniosynostosis occurs in association with diminished cranial bone tissue and may provide a potential biologic explanation for the clinical finding of phenotype consistency that exists between many Crouzon syndrome patients.
Keywords: craniofacial, fibroblast growth factor receptor, craniosynostosis, osteoblast, bone
Introduction
Craniosynostosis is a debilitating condition in which one or more of the sutures between growing cranial bones becomes prematurely fused. This fusion results in increased intracranial pressure as a result of limited growth at fused sutures [1,2], and abnormal skull and facial shapes presumably resulting from limited growth at fused craniofacial sutures with compensating overgrowth at non-fused cranial sutures [3,4]. Severity of the condition and the need for surgical reconstruction depends upon the timing of synostosis and on the number of affected cranial sutures [5]. Untreated craniosyostosis can lead to blindness, seizures and death [6–9]. Craniosynostosis has a relatively high incidence of approximately 1 in 2500 live births and current treatment options are limited to surgery, genetic counseling, dental, medical and social support [10]. Notably, even with an appropriately early and accurate diagnosis, craniosynostosis can carry high morbidity, with some patients requiring multiple surgeries throughout childhood for maintenance of an adequate airway, relief of high intracranial pressure, treatment of recurring craniosynostosis, achievement of ideal tooth relationships, and normalization of skull and facial shapes [11–14].
Craniosynostosis is known to occur in isolation or as part of a genetic syndrome [15,16]. While the molecular pathogenesis of craniosynostosis has yet to be fully elucidated, significant progress has been made regarding the genetics and biology underlying the phenotype of syndromic craniosynostosis. Activating mutations in fibroblast growth factor receptors (FGFR’s) are associated with several of the more common craniosynostosis syndromes and these mutations result in highly specific phenotypes including the premature fusion of specific cranial sutures, facial skeletal deficiencies, ocular abnormalities and the existence (or lack) of associated digit abnormalities [17–22]. Crouzon syndrome is one of the most common forms of syndromic craniosynostosis [23]. Typical features of Crouzon syndrome in humans include coronal suture synostosis with occasional pansynotosis, hypertelorism, ocular proptosis, hypoplastic maxilla and relative mandibular prognathism [12,24] Mice carrying the classic Crouzon syndrome associated FGFR2C342Y mutation were initially reported to have characteristics similar to those of Crouzon syndrome patients including a dome shaped skull, wide set and proptotic eyes, premature fusion of coronal and lambdoid sutures, a shortened maxilla and an apparently normal appendicular and axial skeleton [21]. Comprehensive craniofacial analysis of FGFR2C342Y/+ mice on a mixed genetic background subsequently revealed a rather severe phenotype that included obliteration (complete fusion) of the left and right coronal sutures in 90% of mice, obliteration of the sagittal suture in 70% of mice, and obliteration of the left and right lambdoid sutures in 30–40% of mice [25]. The intersphenoidal synchondrosis of the cranial base was also reported as fused in 100% of the mice with complete obliteration in most cases. These mice appear to reflect the more severe form of Crouzon syndrome that is reported to occur in some patients [12,26].
To further investigate biologic mechanisms that lead to craniosynostosis and abnormal craniofacial skeletal development, we created congenic BALB/c FGFR2C342Y/+ mice. Here report that the FGFR2C342Y/+ mutation in the BALB/c genetic background leads to a consistent and relatively mild craniofacial phenotype including partial fusion of the coronal and lambdoid sutures, intersphenoidal synchondrosis and multiple facial bones, with rare fusion of other craniofacial sutures. This phenotype appears to reflect the less severe and more typical form of Crouzon syndrome that involves significant midface hypoplasia with limited craniosynostosis. Despite this milder craniofacial bone fusion phenotype, linear and morphometric analyses demonstrate that FGFR2C342Y/+ mice on the BALB/c genetic background differ significantly in form and shape from wild type mice and that the FGFR2C342Y/+ mutation effects bones of the face, cranium and cranial base in a similar yet milder manner than that which was previously reported [25]. Because it is unknown why the FGFRC342Y mutation preferentially affects certain craniofacial bones and sutures over others, and because previous reports have demonstrated that activating mutations in FGF receptors lead to abnormal osteoblast differentiation and bone formation [21,22,27–34], we compared effects of the FGFRC342Y mutation on frontal and parietal bone cells and tissues. Our analyses show that the FGFR2C342Y/+ mutation promotes abnormal osteoblast differentiation, increased apoptosis and diminished cranial bone volume and density that is more severe in the frontal than the parietal bones. These results may provide a potential biologic explanation for the clinical finding of relative phenotype consistency that exists among many Crouzon syndrome patients.
Materials and Methods
FGFR2C342/+Y Mice
FGFR2C342Y/+mice on a mixed genetic background were generously provided by Dr. David Ornitz (Washington University School of Medicine, St. Louis, MO). Genotyping was performed as previously described [21]. Briefly, DNA from tail digests was amplified by polymerase chain reaction using 5′-gagtaccatgctgactgcatgc-3′ and 5′-ggagaggcatctctgtttcaagacc-3′ primers. Reaction product was resolved by gel electrophoresis to yield a 200 base pair band for wild type FGFR2 and a 300 base pair band for FGFR2C342Y. BALB/c mice were obtained from Charles River Laboratories International, Inc. (Wilmington, MA). Analyses of FGFR2C342Y/+ BALB/c craniofacial phenotype were performed on mice that had been backcrossed for at least eight generations. All animal procedures were performed according to University of Michigan’s University Committee on Use and Care of Animals.
Skeletal Staining
Whole skeletons from four week old mice were dissected, fixed in 95% ethanol and stained with Alcian blue (.015% Alcian Blue with 20% acetic acid in 95% ethanol). The skeletons were then washed with 95% ethanol, treated with 2% KOH and stained with Alizarin Red (.005% Alizarin Red in 1% KOH). Excess stain was removed by clearing in 0.2% KOH. Skeletons were imaged with an Olympus DP70 digital microscope camera mounted on an Olympus SZX9 stereo microscope.
Craniofacial Landmark Placement
Four week-old FGFR2C342Y/+ mice (n = 28; 12 female and 16 male) and wild type skulls (n = 28; 15 female and 13 male) were dissected and fixed in 100% ethanol overnight. Each specimen was embedded in 1% agarose, placed in a 48 mm diameter tube and scanned over the entire length of the skull using the Scanco μCT100 micro-computed tomography imaging system system (Scanco Medical, Bassersdorf, Switzerland). Scan settings were set at voxel size equal to 18 μm, operating voltage of 70 kVp and 114 μA current, using a 0.5 mm AL filter and an integration time of 500 ms. Scans were calibrated to a hydroxyapatite phantom and dicom files were exported for analysis.
Thirty-three craniofacial landmarks were used to quantifiably assess differences in craniofacial form and shape. These landmarks included twenty-seven landmarks originally published to quantify differences in craniofacial form by Richtsmeier et al. [35], plus an additional six landmarks previously utilized for three dimensional assessment of the Crouzon mouse craniofacial phenotype on a mixed genetic background [25] ( Fig. 1). Landmarks were placed via simultaneous viewing on two-dimensional slices of skulls using Dolphin Imaging 11.0 software (Dolphin Imaging and Management Solutions, Chatsworth, CA). This software is able to display the skulls in the axial, sagittal, and coronal slices with the three-dimensional surface reconstruction for landmark verification in all views simultaneously. Reliability of landmark placement was verified by inter-operator reliability statistics by calculating intraclass correlation coefficients (ICC) for each dimension. Inter-operator reliability of landmark placement was assessed by having a second investigator place all landmarks on the same fourteen mice. The ICC for inter-rater reliability was 0.999 for the x-coordinates, .999 for the y-coordinates, and .991 for the z-coordinates, p ≤ .05. These results demonstrate high reliability for landmark placement accuracy.
Fig. 1. Three-dimensional craniofacial skeletal landmarks.

Location and description of thirty-three craniofacial skeletal landmarks placed on micro CT scans of mouse skulls are indicated.
Linear Craniofacial Measurements
Fourteen linear craniofacial measurements between landmarks were calculated using Dolphin Imaging software. These measurements include six standard measurements currently in use by the Craniofacial Mutant Mouse Resource of Jackson Laboratory; which are nasal bone length (landmark 1 to 2), nose length (landmark 1 to 3), inner canthal distance (landmark 14 to 15), skull width (landmark 26 to 27) and upper jaw length (landmark 1 to 22, 23). The Jackson lab skull height measurement was substituted with a cranial height measurement measured between pari (landmark 4) and the inferior portion of the spheno-occipital synchondrosis (landmark 30), due to the omission of the mandible in our study. Linear measurements were also calculated for frontal bone length (landmark 2 to 3), parietal bone length (landmark 3 to 4), zygomatic arch length (landmarks 12,13 to 24,25), anterior cranial base length (landmark 2 to 30) and posterior cranial base length (landmark 30 to 32). Linear distances of bilateral structures (upper jaw length and zygomatic arch length) were averaged from right and left measurements for each mouse. Data is presented as means +/− standard deviations. Statistical significance between measurements was established by the student’s t test.
Morphometric Analysis
Landmark coordinate data was imported into WinEDMA 1.0.1 software (Theodore Cole, Department of Basic Medical Science, School of Medicine, University of Missouri, Kansas City, MO; http://www.getahead.psu.edu/comment/edma.asp). This software uses three dimensional X,Y,Z coordinate data from each sample to quantify and compare forms between two sample populations by Euclidean Distance Matrix Analysis (EDMA), which allows for an invariant statistical comparison of forms. The analysis also allows for an overall statistical comparison of shapes, if size is estimated to be significantly different between the two sample populations. Briefly, EDMA is a morphometric analysis that uses landmark coordinate data without using a fixed coordinate axis [36]. The analysis calculates all the linear distances between all possible pairs of landmarks in each individual and compares these distances as ratios between groups. This method has been previously used in human and non-human [25,35,36] studies, and is a widely accepted method for morphometric comparisons.
Cranial Bone Quality Analysis
Four week old FGFR2C342Y/+ (n = 21; 11 female and 10 male) and wild type (n = 21; 10 female and 11 male) whole dissected calvaria were scanned with the Scanco μCT100 micro-computed tomography imaging system, as described above at an 18 um isotropic voxel resolution. Regions of interest (ROI’s) for parietal and frontal bones were established as 1 mm in length, 1 mm in width, depth equivalent to thickness of bone and position starting at a 0.75 mm distance from sagittal and coronal sutures. Density measurements were calibrated to the manufacturer’s hydroxyapatite phantom. Analysis was performed using the manufacturer’s evaluation software, and a fixed global threshold of 28% (280 on a grayscale of 0–000) was used to segment bone from non-bone. Student’s t-tests comparing quantitative results were performed to establish statistically significant differences between genotypes and bones. Micro CT bone data was analyzed and is reported in accordance with the recommendations of Bouxsein et al. 2010 [37].
Suture Fusion Assessment
Four week old FGFR2C342Y/+ (n = 21; 11 female and 10 male) and wild type (n = 21; 10 female and 11 male) whole dissected calvaria were scanned in water at an 18 um isotropic voxel resolution using the eXplore Locus SP micro-computed tomography imaging system (GE Healthcare Pre-Clinical Imaging, London, ON, Canada) for suture fusion assessment. Measurements were taken at an operating voltage of 80 kV and 80 mA of current, with an exposure time of 1600 ms using the Parker method scan technique, which rotates the sample 180 degrees plus a fan angle of 20 degrees. Scans were calibrated to a hydroxyapatite phantom and imaged at an effective voxel size of 18 μm3. Patency or fusion of cranial and facial sutures was then assessed using Microview version 2.2 software (GE Healthcare Pre-Clinical Imaging, London, ON). Sutures were viewed using the 2D micro CT slices in orthogonal views across the entire length of the suture in question, in a manner similar to that previously described for analysis of FGFR2C342Y/+ mice on a mixed genetic background [25]. Fisher’s exact test based upon the number of fused vs. patent sutures was performed to establish statistical significance between genotypes within gender, and to establish statistical significance between genders within genotype. McNemar’s test was performed to establish statistical significance between the different craniofacial sutures.
Calvarial Cell Isolation and Analysis
Primary calvarial cells were isolated from dissected frontal and parietal bones by collagenase digestion, as previously described [38–41]. Briefly, bones were rinsed with media then serially digested in a solution containing 2 mg/ml collagenase P and 1 mg/ml trypsin. Each digestion was followed by centrifugation for cell isolation. Cells from the third digestion were utilized for experiments. Cells were induced to differentiate by culture in αMEM containing 50 μg/ml ascorbate, 10% fetal bovine serum (FBS) and 10,000 μg/ml penicillin/streptomycin (P/S). RNA was isolated using Trizol reagent (Invitrogen) following manufacturer protocols. mRNA levels were assayed by reverse transcription and real time PCR. Real time PCR was performed utilizing the murine hypoxanthine guanine phosphoribosyl transferase (Hprt1) primer/probe set Mm01545399_m1, the murine osteocalcin (OCN) Mm03413826_mH primer/probe set, the murine bone sialoprotein (BSP) primer/probe set Mm00492555_m1, the murine tissue non-specific alkaline phosphatase (TNAP) primer/probe set Mm00475834_m1, the murine Runx2 primer/probe set Mm00501578_m1 and Taqman Universal PCR Master Mix (Applied Biosystems). Real-time PCR was performed on a GeneAmp 7700 thermocyler (Applied Biosystems) and quantified by comparison to a standard curve. mRNA levels are reported after normalization to Hprt1 mRNA levels. Cells were induced to form mineral by addition of 10 mM β-glycerophosphate. Mineralized nodules were stained by Von Kossa. Briefly, cells were rinsed with phosphate-buffered saline, fixed with 100% ethanol and rehydrated in a graded ethanol series. Cells were then incubated in 5% AgNO3, rinsed with dH2O and exposed to light for 1 hour. Von Kossa staining was quantified by densitometry. Tissue non-specific alkaline phosphatase (TNAP) enzyme activity was assayed using the colorimetric substrate, NBT/ BCIP (Sigma). Cells were fixed in 70% ethanol for 10 minutes at room temperature, air dried and incubated with substrate for 1 hour at 37C. Cells were then rinsed with dH2O, air dried, and visualized macroscopically for evidence of staining. For quantification, wells were scanned and densitometry was measured using NIH Image software. To assay cellular apoptosis, a Cell Death Detection kit (Roche) was utilized according to the manufacturer’s instructions. This assay uses antibodies directed against DNA and histones, to quantify mono- and oligonucleosomes that are released into the cytoplasm of cells that die from apoptosis. Briefly, 10,000 cells were seeded into 96-well plates in quadruplicate and grown in media containing 10% or 0.5% fetal bovine serum for 48 hours. Cell lysate was utilized to quantify apoptosis by a colorimetric reaction and absorbance was measured at 405 nm (reference wavelength of 490nm). To assay cellular proliferation, cells were seeded at 150,000 cells per 6-well plate and grown in media containing 10% fetal bovine serum for six days. Cells were stained with trypan blue and counted in sextuplet for each time point.
Results
Qualitative Craniofacial Phenotype of FGFR2C342Y/+ Mice in BALB/c Genetic Background
Photographs of four-week old (Fig. 2A,C) and eight-week old (Fig. 2B,C) BALB/c FGFR2C342Y/+ mice and wild type littermates reveal a mild somatic growth deficiency and severe midface hypoplasia. Notably, unlike the previously reported phenotype of mixed genetic background FGFR2C342Y/+ mice [25], anterior cross bite dental malocclusion (mandibular incisor teeth positioned anteriorly to maxillary incisor teeth) requiring tooth filing does not occur in these mice. Stained calvaria of four week BALB/c FGFR2C342Y/+ mice and wild type littermates reveal a mutation associated phenotype that appears primarily restricted to the anterior cranial base (ACB) and midface (MF) in that the posterior cranial base (PCB) appears fairly normal in size and shape while both the anterior cranial base and midface appear shortened in FGFR2C342Y/+ mice, as compared to wild type littermates (Fig. 3A). Significant overlap of frontal and parietal bones is evident in FGFR2C342Y/+ mice compared to FGFR2+/+ mice, although much of the coronal suture still appears patent in FGFR2C342Y/+ mice (Fig. 3B,C). Close inspection of the palatomaxillary suture (PMS) reveals fusion of the maxillary to palatine bones in FGFR2C342Y/+ but not FGFR2+/+ mice (Fig. 3D,E). Additionally, points of fusion are apparent between the frontal and nasal bones of FGFR2C342Y/+ mice and the frontonasal suture lacks interdigitation compared to FGFR2+/+ mice, and (Fig. 3F,G).
Fig. 2. BALB/c FGFR2C342Y/+ mice exhibit somatic growth deficiency and midface hypoplasia.

(A,C) At four weeks of age, FGFR2C342Y/+ mice are slightly smaller than their wild type littermates and show evidence of developing midface hypoplasia. (B,D) By eight weeks of age, FGFR2C342Y/+ mice are still slightly smaller than their wild type littermates and exhibit severe midface hypoplasia.
Fig. 3. Skeletal stains of BALB/c FGFR2C342Y/+ and FGFR2+/+ calvaria.

(A) Stained whole calvaria from four week old FGFR2C342Y/+ mice and wild type littermates demonstrates that the anterior cranial base (ACB) and midface (MF) appear shorter in FGFR2C342Y/+ mice, while the posterior cranial base (PCB) appears relatively unaffected. (B,C) Visualization of the coronal sutures at 32x magnification shows increased frontal to parietal bone overlap with potential partial fusions in FGFR2C342Y/+ mice (b) compared to wild type mice (c). (D,E) Visualization of the inferior aspect of the midface at 6.3x magnification shows apparent fusion of the palatomaxillary suture (PMS) in FGFR2C342Y/+ mice (d) but not in FGFR2+/+ mice (e). (F,G) Visualization of the nasofrontal suture at 32x magnification shows a lack of interdigitation with points of apparent fusion between the frontal and nasal bones in FGFR2C342Y/+ mice (f) but not in FGFR2+/+ mice (g).
Craniofacial Suture Fusions in BALB/c FGFR2C342Y/+ Mice
To establish fusion between adjacent craniofacial bones we next analyzed the mice by visualization of micro CT scans. While the data is presented by genotype and gender, no statistically significant differences were found for fusion of any of the analyzed sutures by gender, regardless of genotype. Results of this analysis confirm that the abnormal craniofacial bone fusion phenotype of BALB/c FGFR2C342Y/+ mice is consistent and occurs in a statistically significant fashion when compared to wild type littermates (Figs. 4,5). The spheno-occipital synchondrosis (SOS, located between basisphenoid and basioccipital bones of the posterior cranial base) was partially fused in a low percentage of FGFR2C342Y/+ mice and was not fused in any of the wild type mice (Fig. 4A,B). In contrast, the intersphenoidal synchondrosis (ISS, located between presphenoid and basisphenoid bones of the anterior cranial base) was partially fused in 100% of FGFR2C342Y/+ mice but not fused in any of the FGFR2+/+ mice (Fig. 4A,B). Partial fusion of the coronal suture (between the frontal and parietal bones) was evident in 91% of FGFR2C342Y/+ female and 78% of FGFR2C342Y/+ male mice (Fig. 4G,H), and was most commonly located close to the sagittal midline. The sagittal suture (between paired parietal bones) was not fused in any of the mice, regardless of genotype. Finally, the lambdoid suture was partially fused in in 91% of FGFR2C342Y/+ female and 89% of FGFR2C342Y/+ male mice, and in one of the wild type mice. Interestingly, the lambdoid suture was not fused along the top of the skull (Fig. 4G,H) where the interparietal bone lies adjacent to the parietal bones but was fused at the far lateral edges of that suture (Fig. 4I,J), where the occipital bone lies adjacent to the squamosal bones. Statistical analysis of suture fusion (Fig. 5) demonstrates that the SOS and sagittal sutures were not fused to a significant extent, while the ISS, coronal and lambdoid, sutures were fused to a significant extent in FGFR2C342Y/+ mice when compared to wild type littermates (p<.005). Statistical analysis also demonstrates that the ISS is fused in FGFR2C342Y/+ mice to a significantly greater extent than the SOS, and that the coronal and lambdoid sutures are fused in FGFR2C342Y/+ mice to a significantly greater extent than the sagittal suture (p<.0001). Of note and in contrast to the small areas of partial fusion seen in BALB/c FGFR2C342Y/+ coronal and lamdoid sutures; the coronal, lamdoid and sagittal sutures were previously reported to be fully fused/obliterated in FGFR2C342Y/+ mice on a mixed genetic background in 90%, 70% and 40% of mice, respectively [25]. Together, these findings indicate that a difference in severity of the Crouzon craniosynostosis phenotype is dependent upon genetic background. Of note, the craniofacial phenotype of the FGFR3P244R mouse model of Muenke craniosynostosis syndrome was also previously reported to be genetic strain dependent and significantly less severe on the BALB/c genetic background [34].
Fig. 4. Craniofacial bone fusions in BALB/c FGFR2C342Y/+ mice involve both cranial and facial bones.

Micro computed tomographic images of calvaria from four week old mice were viewed in axial, sagittal and coronal planes scanning the entire image to establish fusion or patency between adjacent bones. Images show premature fusion of the presphenoid and basisphenoid bones of the anterior cranial base, premature fusion of the frontal and nasal bones in FGFR2C342Y/+ (B) but not FGFR2+/+ (A) mice, and no fusion of the basisphenoid and basioccipital bones of the posterior cranial base in FGFR2C342Y/+ or FGFR2+/+ mice. Fusion between the maxillary and palatine bones, and of the bones of the zygomatic arch was also evident in FGFR2C342Y/+ (D,F) but not in FGFR2+/+ (C,E) mice. Fusion between the parietal and intraparietal bones was not found in either FGFR2C342Y/+ (H) or FGFR2+/+ (G) mice while fusion between the occipital and squamosal bones was evident in FGFR2C342Y/+ mice (J) but not in FGFR2+/+ mice (I). Fusion between the parietal and frontal bones was also evident in FGFR2C342Y/+ (H) but not FGFR2+/+ (G) mice. Fusion of paired parietal bones was not apparent in any of the mice (not shown). SOS: Spheno-occipital synchondrosis suture (located between basisphenoid and basioccipital bones); ISS: Intersphenoidal synchondrosis suture (located between presphenoid and basisphenoid bones); FN: frontonasal suture (located between frontal and nasal bones); PalMx: palatomaxillary suture (located between palatine and maxillary bones); Zyg: zygomatic arch sutures (located between squamosal, zygomatic and maxillary bones); Lam: lambdoid suture (located between intraparietal and parietal bones along top of cranium, located between occipital and squamosal bones along inferior edges of suture); Cor: coronal suture (located between frontal and parietal bones).
Fig. 5. Consistent pattern of premature craniofacial bone fusions in BALB/c FGFR2C342Y/+ mice.
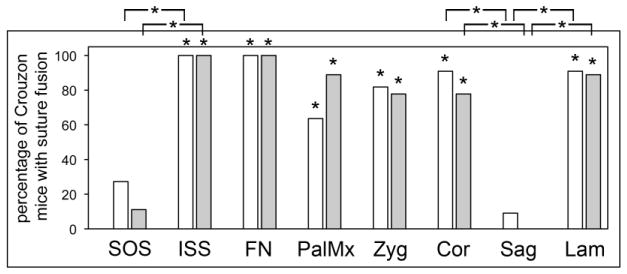
Percentage of BALB/c FGFR2C342Y/+ mice with fusion of indicated sutures is shown. Statistical analysis on the number of identified fused and patent sutures was performed to establish statistical significance between genotypes and genders, and to establish significance between sutures. No statistically significant differences were found for fusion of any of the analyzed sutures by gender, regardless of genotype. Results demonstrate a consistent pattern of bone fusion in BALB/c FGFR2C342Y/+ mice that primarily involves sutures involving bones of the neural crest lineage. The spheno-occipital synchondrosis and the sagittal sutures were not fused to a significant extent in the FGFR2C342Y/+ mice, while the intersphenoidal synchondrosis, coronal and lambdoid sutures were fused to a significant extent in the FGFR2C342Y/+ mice, when compared to wild type littermates. The nasofrontal suture, the palatomaxillary suture and bones of the zygomatic arch were also fused to a significant extent in FGFR2C342Y/+ compared to wild type mice. *p<.005 vs. percentage of fused FGFR2+/+ sutures, or between indicated sutures.
Here we also report that multiple facial bones are fused in BALB/c FGFR2C342Y/+ mice. The frontonasal suture (between frontal and nasal bones) was fused in all FGFR2C342Y/+ mice but in no FGFR2+/+ mice (Fig. 4A,B). The palatomaxillary suture (between palatine and maxillary bones) was fused in 64% of FGFR2C342Y/+ female and 89% of FGFR2C342Y/+ male mice and in no FGFR2+/+ mice (Fig. 4C,D). The zygomatic arch was fused in 82% of FGFR2C342Y/+ female and 78% of FGFR2C342Y/+ male mice and in none of the FGFR2+/+ mice (Fig. 4E,F). Fusion of these facial sutures occurred to a significantly greater extent in FGFR2C342Y/+ compared to FGFR2+/+ mice (Fig. 5). These findings are consistent with previously published reports of facial bone fusions in mouse models of both Apert and Pfeiffer syndromes [42,43] and indicate that the fusion of facial bones is common to multiple forms of FGFR-associated syndromic craniosynostosis.
Craniofacial Skeletal Linear Analysis
To initially quantify form abnormalities in the FGFR2C342Y/+ craniofacial skeleton, we utilized three-dimensional coordinate data generated from micro CT files to conduct a linear measurement comparison of FGFR2C342Y/+ and FGFR2+/+ mice, by gender. Measurements demonstrate that the craniofacial skeletal abnormalities of FGFR2C342Y/+ mice in the Balb/C genetic background are consistent, and are similar in pattern to that which was previously reported for FGFR2C342Y/+ mice on a mixed genetic background [25] (Fig. 6A). Cranial height and width are significantly increased in FGFR2C342Y/+ mice while cranial length is diminished, which is consistent with the previously reported overall dome-like shape of the Crouzon mouse cranium [21]. Upper jaw length and nose length are also significantly smaller in FGFR2C342Y/+ mice, indicative of the midface hypoplasia that has been previously reported for mice carrying the FGFR2C342Y/+ mutation on a mixed genetic background [21,25]. Zygomatic arch length was also significantly shorter in FGFR2C342Y/+ mice than FGFR2+/+ mice, indicating that fusion exists between zygomatic and maxillary bones, and/or between zygomatic and squamosal bones; and/or that lack of growth of other structures is inhibiting the growth of the bones that comprise the zygomatic arch. Notably, nasal and frontal bone lengths are smaller, while parietal bones lengths are longer in FGFR2C342Y/+ than wild type mice, indicating that anterior-posterior growth of the parietal bone may compensate for diminished anterior-posterior growth of other craniofacial bones in these FGFR2C342Y/+ mice. Also of note, anterior cranial base length but not posterior cranial base length is significantly shorter in FGFR2C342Y/+ than wild type mice, confirming our qualitative interpretation of the phenotype as being restricted to the anterior cranial base and face. Additionally, while the overall pattern for male and female FGFR2C342Y/+ mice is similar, some differences between the two genders are apparent. Both nose length and frontal bone length are significantly larger in male than female FGFR2C342Y/+ mice. All other measures, including skull length are not significantly different between the two genders. Together, these results indicate that the nose length and frontal bone length gender differences are not necessarily due to an overall difference in skull size.
Fig. 6. Linear and morphometric analysis of craniofacial forms demonstrate consistent and craniofacial bone specific skeletal abnormalities in BALB/c FGFR2C342Y/+ mice.

(A) Three-dimensional coordinate data generated from landmarks placed micro CT scans of FGFR2C342Y/+ and FGFR2+/+ mice were used to generate linear measurements (Fig. 1). Measurements of craniofacial bones and cranial vault dimensions demonstrate significantly diminished skull length, upper jaw length, nasal bone length, frontal bone length and zygomatic arch length with significantly increased cranial height, cranial width, inner canthal distance and parietal bone length in FGFR2C342Y/+ as compared to wild type mice, regardless of gender. Nose length and frontal bone length were significantly larger in male FGFR2C342Y/+ as compared to female FGFR2C342Y/+ mice but no other gender differences were found. Data is presented as means +/− standard deviations. Statistical significance was established by the student’s t test. *p < .001 vs. wild type or between indicated groups. White = FGFR2+/+ female, light grey = FGFR2+/+ male, dark grey = FGFR2C342Y/+ female, black = FGFR2C342Y/+ male. (B) A representative subset of landmark distance EDMA mean ratios is shown on a sagittal micro CT section of an FGFR2C342Y/+ mouse. Blue lines indicate those distances that are significantly smaller in FGFR2C342Y/+ as compared to FGFR2+/+ mice. Red lines indicate those distances that are significantly larger in FGFR2C342Y/+ as compared to FGFR2+/+ mice. All distances are for midline landmarks other than those for landmarks 8,9 and 10,11. Distance ratios for bilateral landmarks are represented as a single line and were significant for both right and left sides. Note that larger distances in the FGFR2C342Y/+ mouse are primarily restricted to the parietal bones, interparietal bones and posterior cranial base dimensions.
Comparison of Craniofacial Form and Shape
A statistical comparison of forms by EDMA analysis reveals that the overall craniofacial form of FGFR2C342Y/+ mice is significantly different from that of wild type mice (T = 2.16, p <.001). Because differences in form can be due to differences in shape and/or size and because the body weight and length of the mice were significantly larger for the wild type mice than for the Crouzon mice regardless of gender (13.7 +/− 0.9 vs. 11.0 +/− 1.9 g weight, p <.001 and 76.9 +/− 4.0 vs. 69.5 +/−4.6 mm body length, p<.001), we next used EDMA to determine if FGFR2C342Y/+ and FGFR2+/+ skulls are statistically different in shape. Estimation of the confidence interval for a difference in the geometric mean (mean distance between landmarks) between the two groups demonstrated a significant difference in size between FGFR2C342Y/+ and FGFR2+/+ skulls. EDMA shape analysis using geometric mean as a scaling factor confirmed that FGFR2C342Y/+ and FGFR2+/+ skulls are statistically different in shape (Z = −0.313, a = 0.01). Because an EDMA analysis of forms by gender indicated that neither wild type nor mutant male skulls were significantly different in form than their female counterparts, genders were combined for the EDMA analyses of form, size and shape; as well as for localized confidence intervals.
Localized form differences generated by the EDMA analysis were consistent with results of our linear measurement analysis (Fig. 6B). Confidence intervals indicated that skull length distance ratios were significantly smaller and that multiple landmark distance ratios were smaller in the face and anterior cranial base when FGFR2C342Y/+ mice were compared to wild type littermates. Also similar to our linear cranial measurement results, posterior cranial base length ratios (measured from SOS to opisthion or ISS to opisthion) were not significantly different between FGFR2C342Y/+ and wild type mice. Cranial height and cranial width ratios were significantly larger in FGFR2C342Y/+ as compared to wild type mice. Also of significance, ratios involving parietal and interparietal bones were larger but ratios involving frontal and occipital bones were smaller in FGFR2C342Y/+ as compared to wild type mice. Given that bony fusions were found in the far lateral lamdoid suture and the coronal suture, but not in the sagittal suture or more medial portion of the lamdoid suture, these landmark distance ratio results suggest that compensating overgrowth in parietal and interparietal bones occurs to compensate for diminished growth of frontal and occipital bones in the cranium of BALB/c FGFR2C342Y/+ mice.
Diminished Bone Volume and Density in BALB/c FGFR2C342Y/+ Cranial Bones
To determine if the coronal suture fusion seen in FGFR2C342Y/+ mice occurs in association with differences in the surrounding cranial bones, we utilized micro computed tomography to analyze parameters of bone quality and quantity in frontal and parietal bones of FGFR2C342Y/+ and FGFR2+/+ mice. No significant differences were seen between genders, so genders were combined for genotype analysis. Results demonstrate that FGFR2C342Y/+ mice have significantly diminished bone volume, bone mineral density and tissue mineral density in frontal bones but not in parietal bones (Fig. 7A,C,D), when compared to FGFR2+/+ mice. Additionally, both wild type and mutant frontal bones have diminished bone volume, bone volume/total volume, bone mineral content and tissue mineral content compared to their respective parietal bones (Fig. 7A,B,C,D). Together, these results demonstrate that the FGFR2C342Y/+ mutation causes craniosynostosis in association with decreased cranial bone volume and density, and that frontal bones may be more susceptible to FGFR2C342Y/+ induced abnormalities than parietal bones.
Fig. 7. Diminished bone volume and density in frontal bones of BALB/c FGFR2C342Y/+ mice.

(A,B,C,D) Micro CT analyses demonstrate significantly diminished bone volume, bone mineral density and tissue mineral density in frontal bones but not parietal bones of FGFR2C342Y/+ (Cz) mice as compared to wild type (WT) mice. Bone volume, bone volume/total volume, bone mineral density and tissue mineral density are also significantly diminished in frontal compared to parietal bones of both FGFR2C342Y/+ and wild type mice. *p<.05. (E,F) Wild type (E) and FGFR2C342Y/+ (F) sagittal sections of parietal (left) and frontal (right) bones surrounding the coronal suture.
BALB/c FGFR2C342Y/+ Cells Exhibit Cranial Bone Specific Changes in Osteoblastic Gene Expression
Because the frontal bones of wild type mice showed diminished parameters of bone volume and density compared to the parietal bones of these same mice, and because these measures were even lower in the frontal bones of FGFR2C342Y/+ mice, we next sought to determine if cells isolated from the mutant and wild type cranial bones differed in their tendency to osteoblast differentiate and form mineralized nodules. Results demonstrate that cells isolated from parietal bones have significantly diminished expression of osteoblastic marker genes than cells isolated from frontal bones. Runx2, tissue non-specific alkaline phosphatase (TNAP), bone sialoprotein (BSP) and osteocalcin (OCN) mRNA expression are all significantly decreased in undifferentiated, differentiating and differentiated parietal bone cells when compared to frontal bone cells, regardless of genotype (Fig. 8).
Fig. 8. FGFR2C342Y/+ mutation induces cranial bone specific changes in osteoblastic gene expression.

(A–H) Primary cells were isolated from FGFR2C342Y/+ and wild type, frontal (A,C,E,G) and parietal (B,D,F,H) bones then cultured with or without ascorbate to induce osteoblast differentiation. RNA was isolated at indicated time points. Osteocalcin (OCN), bone sialoprotein (BSP), tissue non-specific alkaline phosphatase (TNAP) and Runx2 mRNA levels were measured by real time PCR. Results are presented as normalized to Hprt1. *p<.05 vs. WT. Note: all parietal cell mRNA levels are significantly lower than frontal cell mRNA levels, regardless of genotype (p<.05). Grey lines indicate cells cultured without ascorbate; black lines indicate cells cultured with ascorbate; dashed lines indicate FGFR2C342Y/+, solid lines indicate FGFR2+/+.
When comparing the expression of osteoblastic marker genes by genotype, a slightly different pattern emerges, dependent upon the bone of cell origin. For cells isolated from parietal bones, FGFR2C342Y/+ cells appear to show a fairly similar overall pattern of mRNA expression to that of wild type cells, albeit to a lower extent (Fig. 8B,D,F,H). Parietal FGFR2C342Y/+ cells express similar levels of Runx2 by 18 days of differentiation, but Runx2 expression is significantly diminished in FGFR2C342Y/+ cells at multiple earlier time points when compared to wild type cells. Similarly, while parietal FGFR2C342Y/+ cells express significantly more TNAP mRNA upon 1 day of culture, TNAP expression is significantly diminished at multiple later time points in differentiating cells. Additionally, both BSP and OCN expression are similar in FGFR2C342Y/+ and wild type parietal cells upon 1 day of culture, but expression of these genes is significantly diminished at multiple later time points in differentiating cells.
For cells isolated from frontal bones, FGFR2C342Y/+ cells show a different overall pattern of mRNA expression than that of wild type cells (Fig. 8A,C,E,G). Runx2 mRNA expression increases upon differentiation in wild type frontal bone cells and this increased expression is maintained for up to 18 days of culture in differentiation media. In contrast, Runx2 mRNA expression also increases upon differentiation in FGFR2C342Y/+ frontal bone cells, but the high expression level of Runx2 is not maintained and actually decreases at 12 and 18 days of culture in differentiation media. Similarly, while TNAP and BSP mRNA expression increase in both wild type and mutant frontal bone cells at early differentiation time points, expression increases to a significantly greater extent in wild type cells than FGFR2C342Y/+ cells at later time points of differentiation. While these results suggest that FGFR2C342Y/+ frontal bone cells appear to successfully initiate osteoblastic differentiation but not sustain it, the results also show that FGFR2C342Y/+ frontal bone cells do express significantly higher levels of both BSP and OCN mRNA at 6 days of differentiation. Overall it appears that the expression of osteoblastic genes may be normal or even enhanced in early differentiating FGFR2C342Y/+ frontal bone cells but this expression becomes diminished with continued differentiation, when compared to wild type cells.
Consistent with our gene expression data showing lower TNAP mRNA expression in parietal than frontal bone cells, staining with a colorimetric substrate demonstrates significantly lower TNAP enzyme activity in parietal than frontal bone cells. Concordantly, parietal bone cells form significantly less mineral than frontal bone cells. Also consistent with our gene expression data, when comparing by genotype results show that FGFR2C342Y/+ frontal bone cells express higher levels of TNAP enzyme activity in undifferentiated cells but lower levels of TNAP enzyme activity after induction of differentiation by ascorbate (Fig. 9A,B), and that the FGFR2C342Y/+ mutation decreases mineralized nodule formation by frontal bone cells (Fig. 9C,D). Together these results demonstrate that while the FGFR2C342Y/+ may enhance expression of some osteoblastic genes in undifferentiated and early differentiating frontal bone cells (including TNAP), the mutation also appears to inhibit the ability of frontal bone cells to fully differentiate into mature osteoblasts when compared to wild type cells. The findings also indicate that the FGFR2C342Y/+ mutation induces a cell autonomous defect in calvarial osteoblast differentiation and mineralization.
Fig. 9. FGFR2C342Y/+ mutation induces cranial bone specific changes in mineralization, apoptosis and proliferation.

(A,B) Primary cells were cultured with or without ascorbate to induce osteoblast differentiation. TNAP enzyme activity was visualized by incubation of cells with a colorimetric substrate. Enzyme activity was quantified by densitometry. Results are shown as means +/− standard deviations of triplicate experiments. (C,D) Cells were cultured with ascorbate and b-glycerophosphate to induce mineralized nodule formation. Mineralized nodules were stained by Von Kossa and quantified by densitometry. *p<.05 vs. WT. (E) Cells were cultured with 0.5% serum to induce apoptosis. Generation of apoptotic changes in DNA was assayed by a colorimetric reaction. Results are shown as means +/− standard deviations of quadruplicate experiments. (F) Cells were cultured without ascorbate for up to six days. Cells were stained with trypan blue and counted at indicated time points. Results are shown as means +/− standard deviations of sextuplet experiments.
Primary Cells Isolated from BALB/c FGFR2C342Y/+ Frontal Bones Exhibit Increased Apoptosis and no Change in Proliferation
Because FGFR-associated craniosynostosis may result from changes in cranial cell behavior other than or in addition to changes in osteoblastic differentiation, we next examined primary cells isolated from FGFR2C342Y/+ and wild type cranial bones for their tendency to undergo apoptosis and to proliferate. Apoptosis assays show that a greater percentage of cells isolated from frontal bones undergo apoptosis when deprived of serum than cells isolated from parietal bones, regardless of genotype. Apoptosis results also show that the FGFR2C342Y/+ mutation increases the tendency for apoptosis in frontal bone cells. In contrast to these findings, proliferation assays reveal that the FGFR2C342Y/+ mutation decreases proliferation of parietal bone cells upon confluency (at the 6 day time point) but has no effect on the proliferation of frontal bone cells.
Discussion
Each craniosynostosis syndrome involves the premature fusion of specific cranial sutures with resultant characteristic skull and facial shapes [3,4,12]. Here we report that FGFR2C342Y/+ mice on a BALB/c congenic background have a consistent and relatively mild phenotype, reflecting the classic form of human Crouzon syndrome involving limited craniosynostosis and severe midface hypoplasia [12]. While the craniofacial form of BALB/c FGFR2C342Y/+ mice is similar, the severity of craniosynososis is significantly diminished when compared to that which was previously reported for FGFR2C342Y/+ mice on a mixed genetic background [25]. This more moderate BALB/c based phenotype allows us to determine which craniofacial tissues are more susceptible to FGFR2C342Y/+ induced abnormalities and begin to explain how this specificity of effect contributes to the predictable Crouzon craniofacial form. BALB/c FGFR2C342Y/+ mice have a phenotype involving high rates of fusion of multiple facial bones, the frontal bones and bones of the anterior cranial base (intersphenoidal synchondrosis); but rare fusions between the parietal bones, between the parietal and intraparietal bones or between bones of the posterior cranial base (spheno-occipital synchondrosis). The anterior cranial base but not the posterior cranial base is significantly shorter, and parietal and intraparietal bone lengths are longer while frontal and nasal bone lengths are shorter in the mutant mice, when compared to wild type mice. Is has been previously suggested that the predictable abnormal craniofacial shapes associated with each of the FGFR-associated craniosynostosis syndromes is due to the diminished growth of bones predictably involved in fusions, in combination with the overgrowth of bones not involved in fusions [3]. Results of our suture fusion assessment, in combination with our linear and morphometric craniofacial analyses, demonstrate that this is indeed the case for Crouzon syndrome.
BALB/c FGFR2C342Y/+ mice have a phenotype involving decreased frontal bone length but increased parietal bone length, as well as fusions in the coronal and nasofrontal but not in the sagittal or medial lambdoid sutures. These results, combined with our data showing that the FGFR2C342Y/+ mutation decreases frontal but not parietal bone volume and density when compared to wild type bones, suggests that frontal bone tissue may be more susceptible to effects of the FGFR2C342Y/+ mutation than parietal bone tissue. Additionally, our results show that the FGFR2C342Y/+ mutation alters frontal bone cell gene expression in a different manner than parietal cell gene expression and that the FGFR2C342Y/+ mutation increases apoptosis and inhibits mineralization in frontal but not in parietal bone cells. These results indicate that frontal bone tissues may be more affected by the FGFR2C342Y/+ mutation because frontal bone cells are more susceptible to effects of the FGFR2C342Y/+ mutation than parietal bone cells. This idea is supported by previous reports demonstrating significant differences between frontal and parietal bone cell behavior. Frontal bone cells were previously shown to exhibit higher levels of osteoblastic gene expression and TNAP enzyme activity, and to have greater healing capacity than parietal bone cells in a murine calvarial defect model [44,45]. Frontal bone cells were also previously shown to have higher expression levels of FGF’s and FGFR’s when compared to parietal bone cells [45,46]. Together, these findings suggest that the FGFR2C342Y/+ mutation may be more likely to lead to abnormalities of clinical significance in frontal bone tissues than parietal bone tissues due to innate differences between frontal and parietal bone cells. Future studies are required to determine if the differences are due to lower expression levels of mutant FGFR2 in the less effected tissues, or to other modifying factors.
FGFR2 is expressed in osteoprogenitor cells and FGFR1 is expressed in differentiating cells within the osteogenic fronts of growing cranial bones, indicating that FGFR activity could control cranial bone and suture development by altering precursor cell behavior [47,48]. Multiple previous reports have demonstrated that the cranial cells and tissues of Apert and Crouzon mouse models of craniosynostosis exhibit aberrant osteoblast differentiation. Previous analyses of the FGFR2P253R/+ and FGFR2S252W/+ mouse models of Apert syndrome revealed increased osteoblastic gene expression around the coronal suture [22,27]. Analysis of FGFR2C342Y/+ mice in a mixed genetic background were also previously shown to have increased osteoblast numbers and Runx2 levels around the coronal suture [21]. Here we show that FGFR2C342Y/+ mutant frontal bone cells exhibit higher expression levels of TNAP, BSP and OCN at early differentiation time points (day 1, day 6 and day 6; respectively) and lower expression levels of TNAP, BSP and OCN at later differentiation time points (day 18, days 12 and 18, and day 12; respectively). In addition, our results show that while Runx2 mRNA levels rise during early differentiation of FGFR2C342Y/+ mutant frontal bone cells, this increase in expression is not maintained at later stages of differentiation, and that TNAP enzyme activity is increased in undifferentiated but decreased in differentiated FGFR2C342Y/+ frontal bone cells when compared to wild type. Taken together, these results suggest that the FGFR2C342Y/+ mutation may stimulate early osteoblast differentiation while inhibiting later stages of differentiation.
As was previously proposed for Apert craniosynostosis [27,29], our results suggest that the FGFR2C342Y mutation stimulates the early osteoblastic differentiation of precursor cells. This effect could lead to a larger pool of cranial osteoblasts along the osteogenic front of growing cranial bones thereby promoting craniosynostosis. Our results also indicate that the FGFR2C342Y mutation inhibits later stages of osteoblast differentiation and inhibits mineralization in vitro. These results suggest that FGFR2C342Y/+ frontal bone cells are inhibited in their ability to differentiate into fully functional osteoblasts and is in accordance with previous studies showing that S252W, C342Y, P253R mutations in FGFR2 can inhibit osteoblast differentiation [30,31]. In combination, expression of the FGFR2C342Y/+ mutation could lead to a larger pool of osteoblastic cells that are less fully functional than wild type cells thereby promoting craniosynostosis in the context of diminished cranial bone volume and density. Importantly, our results are in accordance with reports of other mouse models of FGFR-associated craniosynostosis. FGFR2S250W/+ mice and FGFR3P244R/+ mice were also previously shown to exhibit craniosynostosis in association with diminished cranial bone volume and/or formation when compared to wild type littermates [32,34].
Notably, we also find that FGFR2C342Y/+ frontal bone cells show an increased tendency to undergo apoptosis when compared to FGFR2+/+ frontal bone cells. These results are similar to previously published reports which showed that FGFR2S252W/+ Apert and FGF2-overexpressor cranial cells exhibit increased levels of apoptosis [31,49]. Apoptosis has been proposed to clear suture cells, allowing for continued cranial bone growth into the suture area. Via this mechanism, higher levels of apoptosis could promote craniosynostosis [49].
Taken together, our results indicate that the Crouzon FGFR2C342Y mutation promotes early osteoblastic differentiation while inhibiting the full maturation of those cells, and increases apoptosis. These effects are consistent with that which has been previously reported for the Apert FGFR2S252W/+ mutation [49]. This phenomenon could explain the consistent finding of craniosynostosis in association with diminished cranial bone seen in mouse models of FGFR-associated craniosynostosis, although future studies are required to definitively determine if FGFR2C342Y/+ cranial bone cells form less bone in vivo. Importantly, our results also show for the first time that the FGFR2C342Y mutation influences frontal bone cells and tissues to a greater extent than parietal bone cells and tissues, which likely contributes to the relative consistency in craniofacial shape of Crouzon syndrome patients [12].
Acknowledgments
We would like to thank Dr. David Ornitz for providing the FGFR2C342Y/+ mice. This work was supported by NIH/NIDCR grant R03DE021082.
Footnotes
The authors have stated that they have no conflict of interest.
Disclosures: None
References
- 1.Renier D, Lajeunie E, Arnaud E, Marchac D. Management of craniosynostoses. Childs Nerv Syst. 2000;16(10–11):645–58. doi: 10.1007/s003810000320. [DOI] [PubMed] [Google Scholar]
- 2.Seruya M, Oh AK, Boyajian MJ, Posnick JC, Keating RF. Treatment for delayed presentation of sagittal synostosis: challenges pertaining to occult intracranial hypertension. J Neurosurg Pediatr. 2011;8(1):40–8. doi: 10.3171/2011.4.PEDS1160. [DOI] [PubMed] [Google Scholar]
- 3.Morriss-Kay GM, Wilkie AO. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat. 2005;207(5):637–53. doi: 10.1111/j.1469-7580.2005.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kreiborg S. Craniofacial growth in plagiocephaly and Crouzon syndrome. Scand J Plast Reconstr Surg. 1981;15(3):187–97. doi: 10.3109/02844318109103433. [DOI] [PubMed] [Google Scholar]
- 5.Cohen MM., Jr Sutural biology and the correlates of craniosynostosis. Am J Med Genet. 1993;47:581–616. doi: 10.1002/ajmg.1320470507. [DOI] [PubMed] [Google Scholar]
- 6.Okajima K, Robinson LK, Hart MA, Abuelo DN, Cowan LS, Hasegawa T, Maumenee IH, Jabs EW. Ocular anterior chamber dysgenesis in craniosynostosis syndromes with a fibroblast growth factor receptor 2 mutation. Am J Med Genet. 1999;85(2):160–70. doi: 10.1002/(sici)1096-8628(19990716)85:2<160::aid-ajmg11>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 7.Stavrou P, Sgouros S, Willshaw HE, Goldin JH, Hockley AD, Wake MJ. Visual failure caused by raised intracranial pressure in craniosynostosis. Childs Nerv Syst. 1997;13(2):64–7. doi: 10.1007/s003810050043. [DOI] [PubMed] [Google Scholar]
- 8.Abe H, Ikota T, Akino M, Kitami K, Tsuru M. Functional prognosis of surgical treatment of craniosynostosis. Childs Nerv Syst. 1985;1(1):53–61. doi: 10.1007/BF00706732. [DOI] [PubMed] [Google Scholar]
- 9.Shah PS, Siriwardena K, Taylor G, Steele L, Ray P, Blaser S, Chitayat D. Sudden infant death in a patient with FGFR3 P250R mutation. Am J Med Genet A. 2006;140(24):2794–6. doi: 10.1002/ajmg.a.31517. [DOI] [PubMed] [Google Scholar]
- 10.Rasmussen SA, Yazdy MM, Frías JL, Honein MA. Priorities for public health research on craniosynostosis: summary and recommendations from a Centers for Disease Control and Prevention-sponsored meeting. Am J Med Genet A. 2008;146A(2):149–58. doi: 10.1002/ajmg.a.32106. [DOI] [PubMed] [Google Scholar]
- 11.Cohen MM, Jr, Kreiborg S. Upper and lower airway compromise in the Apert syndrome. Am J Med Genet. 1992;44(1):90–3. doi: 10.1002/ajmg.1320440121. [DOI] [PubMed] [Google Scholar]
- 12.Cunningham ML, Seto ML, Ratisoontorn C, Heike CL, Hing AV. Syndromic craniosynostosis: from history to hydrogen bonds. Orthod Craniofac Res. 2007;10(2):67–81. doi: 10.1111/j.1601-6343.2007.00389.x. [DOI] [PubMed] [Google Scholar]
- 13.Flapper WJ, Anderson PJ, Roberts RM, David DJ. Intellectual outcomes following protocol management in Crouzon, Pfeiffer, and Muenke syndromes. J Craniofac Surg. 2009;20(4):1252–5. doi: 10.1097/SCS.0b013e3181acdf9a. [DOI] [PubMed] [Google Scholar]
- 14.Baird LC, Gonda D, Cohen SR, Evers LH, Lefloch N, Levy ML, Meltzer HS. Craniofacial reconstruction as a treatment for elevated intracranial pressure. Childs Nerv Syst. 2011;28(3):411–8. doi: 10.1007/s00381-011-1615-6. [DOI] [PubMed] [Google Scholar]
- 15.Boyadjiev SA. International Craniosynostosis Consortium. Genetic analysis of non-syndromic craniosynostosis. Orthod Craniofac Res. 2007;10(3):129–37. doi: 10.1111/j.1601-6343.2007.00393.x. [DOI] [PubMed] [Google Scholar]
- 16.Wilkie AO, Byren JC, Hurst JA, Jayamohan J, Johnson D, Knight SJ, Lester T, Richards PG, Twigg SR, Wall SA. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics. 2010;126(2):e391–400. doi: 10.1542/peds.2009-3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet. 1994;8:98–103. doi: 10.1038/ng0994-98. [DOI] [PubMed] [Google Scholar]
- 18.Schell U, Hehr A, Feldman GJ, Robin NH, Zackai EH, de Die-Smulders C, Viskochil DH, Stewart JM, Wolff G, Ohashi H, et al. Mutations in FGFR1 and FGFR2 cause familial and sporadic Pfeiffer syndrome. Hum Mol Genet. 1995;4:323–28. doi: 10.1093/hmg/4.3.323. [DOI] [PubMed] [Google Scholar]
- 19.Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9:165–72. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- 20.Ibrahimi OA, Zhang F, Eliseenkova AV, Linhardt RJ, Mohammadi M. Proline to arginine mutations in FGF receptors 1 and 3 result in Pfeiffer and Muenke craniosynostosis syndromes through enhancement of FGF binding affinity. Hum Mol Genet. 2004;13:69–78. doi: 10.1093/hmg/ddh011. [DOI] [PubMed] [Google Scholar]
- 21.Eswarakumar VP, Horowitz MC, Locklin R, Morriss-Kay GM, Lonai P. A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proc Natl Acad Sci USA. 2004;101:12555–60. doi: 10.1073/pnas.0405031101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin L, Du X, Li C, Xu X, Chen Z, Su N, Zhao L, Qi H, Li F, Xue J, Yang J, Jin M, Deng C, Chen L. A Pro253Arg mutation in fibroblast growth factor receptor 2(Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone. 2008;42:631–643. doi: 10.1016/j.bone.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 23.Cohen MM, Jr, Kreiborg S. Birth prevalence studies of the Crouzon syndrome: comparison of direct and indirect methods. Clin Genet. 1992;41(1):12–5. doi: 10.1111/j.1399-0004.1992.tb03620.x. [DOI] [PubMed] [Google Scholar]
- 24.Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University; Baltimore, MD: MIM Number: 123500: last edited 01/31/2011. World Wide Web URL: http://omim.org/entry/123500. [Google Scholar]
- 25.Perlyn CA, DeLeon VB, Babbs C, Govier D, Burell L, Darvann T, Kreiborg S, Morriss-Kay G. The craniofacial phenotype of the Crouzon mouse: analysis of a model for syndromic craniosynostosis using three-dimensional MicroCT. Cleft Palate Craniofac J. 2006;43(6):740–8. doi: 10.1597/05-212. [DOI] [PubMed] [Google Scholar]
- 26.Flippen JH., Jr Cranio-facial dysostosis of Crouzon; report of a case in which the malformation occurred in four generations. Pediatrics. 1950;5(1):90–6. [PubMed] [Google Scholar]
- 27.Holmes G, Rothschild G, Roy UB, Deng C, Mansukhani A, Basilico C. Early onset of craniosynostosis in an Apert mouse model reveals critical features of this pathology. DevBio. 2009;328:273–84. doi: 10.1016/j.ydbio.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou YX, Xu X, Chen L, Li C, Brodie SG, Deng CX. A Pro250Arg substitution in mouse Fgfr1 causes increased expression of Cbfa1 and premature fusion of calvarial sutures. Hum Mol Genet. 2000;9:2001–8. doi: 10.1093/hmg/9.13.2001. [DOI] [PubMed] [Google Scholar]
- 29.Lomri A, Lemonnier J, Hott M, de Parseval N, Lajeunie E, Munnich A, Renier D, Marie PJ. Increased calvaria cell differentiation and bone matrix formation induced by fibroblast growth factor receptor 2 mutations in Apert syndrome. J Clin Invest. 1998;101:1310–17. [PMC free article] [PubMed] [Google Scholar]
- 30.Fragale A, Tartaglia M, Bernardini S, Di Stasi AM, Di Rocco C, Velardi F, Teti A, Battaglia PA, Migliaccio S. Decreased proliferation and altered differentiation in osteoblasts from genetically and clinically distinct craniosynostotic disorders. Am J Pathol. 1999;154:1465–77. doi: 10.1016/S0002-9440(10)65401-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mansukhani A, Bellosta P, Sahni M, Basilico C. Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J Bone Min Res. 2000;149:1297–308. doi: 10.1083/jcb.149.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Li D, Li C, Engel A, Deng CX. A Ser252Trp [corrected] substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone. 2003;33:169–17. doi: 10.1016/s8756-3282(03)00222-9. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Sun M, Uhlhorn VL, Zhou X, Peter I, Martinez-Abadias N, Hill CA, Percival CJ, Richtsmeier JT, Huso DL, Jabs EW. Activation of p38 MAPK pathway in the skull abnormalities of Apert syndrome Fgfr2(+P253R) mice. BMC Dev Biol. 2010;10:22. doi: 10.1186/1471-213X-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Twigg SR, Healy C, Babbs C, Sharpe JA, Wood WG, Sharpe PT, Morriss-Kay GM, Wilkie AO. Skeletal analysis of the Fgfr3(P244R) mouse, a genetic model for the Muenke craniosynostosis syndrome. Dev Dyn. 2009;238(2):331–42. doi: 10.1002/dvdy.21790. [DOI] [PubMed] [Google Scholar]
- 35.Richtsmeier JT, Baxter LL, Reeves RH. Parallels of craniofacial maldevelopment in Down syndrome and Ts65Dn mice. Dev Dyn. 2000;217(2):137–45. doi: 10.1002/(SICI)1097-0177(200002)217:2<137::AID-DVDY1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 36.Lele S, Richtsmeier JT. An invariant approach to statistical analysis of shapes. Chapman & Hall/CRC; Boca Raton, FL: 2001. [Google Scholar]
- 37.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25(7):1468–86. doi: 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- 38.Hatch N, Li Y, Franceschi RT. FGF2 stimulated expression of the pyrophosphate generating enzyme, PC-1, is mediated by Runx2. J Bone Min Res. 2009;24(4):652–62. doi: 10.1359/JBMR.081213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCarthy TL, Centrella M, Canalis E. Further biochemical and molecular characterization of primary rat parietal bone cell cultures. J Bone Miner Res. 1988 Aug;3(4):401–8. doi: 10.1002/jbmr.5650030406. [DOI] [PubMed] [Google Scholar]
- 40.Harris SE, Sabatini M, Harris MA, Feng JQ, Wozney J, Mundy GR. Expression of bone morphogenetic protein messenger RNA in prolonged cultures of fetal rat calvarial cells. J Bone Miner Res. 1994 Mar;9(3):389–94. doi: 10.1002/jbmr.5650090314. [DOI] [PubMed] [Google Scholar]
- 41.Ducy P, Karsenty G. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol Cell Biol. 1995 Apr;15(4):1858–69. doi: 10.1128/mcb.15.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martínez-Abadías N, Heuzé Y, Wang Y, Jabs EW, Aldridge K, Richtsmeier JT. FGF/FGFR signaling coordinates skull development by modulating magnitude of morphological integration: evidence from Apert syndrome mouse models. PLoS One. 2011;6(10):e26425. doi: 10.1371/journal.pone.0026425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holmes G, Basilico C. Mesodermal expression of Fgfr2(S252W) is necessary and sufficient to induce craniosynostosis in a mouse model of Apert syndrome. Dev Biol. 2012 doi: 10.1016/j.ydbio.2012.05.026. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.LI S, Quarto N, Longaker MT. Activation of FGF signaling mediates proliferative and osteogenic differences between neural crest derived frontal and mesoderm parietal derived bone. PLoS One. 2010;5(11):e14033. doi: 10.1371/journal.pone.0014033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Behr B, Panetta NJ, Longaker MT, Quarto N. Different endogenous threshold levels of Fibroblast Growth Factor-ligands determine the healing potential of frontal and parietal bones. Bone. 2010;47(2):281–94. doi: 10.1016/j.bone.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 46.Quarto N, Behr B, Li S, Longaker MT. Differential FGF ligands and FGF receptors expression pattern in frontal and parietal calvarial bones. Cells Tissues Organs. 2009;190(3):158–69. doi: 10.1159/000202789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iseki S, Wilkie AO, Heath JK, Ishimaru T, Eto K, Morriss-Kay GM. Fgfr2 and osteopontin domains in the developing skull vault are mutually exclusive and can be altered by locally applied FGF2. Development. 1997;124:3375–84. doi: 10.1242/dev.124.17.3375. [DOI] [PubMed] [Google Scholar]
- 48.Iseki S, Wilkie AO, Morriss-Kay GM. Fgfr1 and Fgfr2 have distinct differentiation- and proliferation-related roles in the developing mouse skull vault. Development. 1999;126:5611–20. doi: 10.1242/dev.126.24.5611. [DOI] [PubMed] [Google Scholar]
- 49.Lemonnier J, Haÿ E, Delannoy P, Fromigué O, Lomri A, Modrowski D, Marie PJ. Increased osteoblast apoptosis in apert craniosynostosis: role of protein kinase C and interleukin-1. Am J Pathol. 2001;158(5):1833–42. doi: 10.1016/S0002-9440(10)64139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]