

Abstract
Genetic studies in the mouse have demonstrated multiple roles for β-catenin in the skeleton. In the embryo, β-catenin is critical for the early stages of osteoblast differentiation. Postnatally, β-catenin in mature osteoblasts and osteocytes indirectly suppresses osteoclast differentiation. However, a direct role for β-catenin in regulating osteoblast number and/or function specifically in the postnatal life has not been demonstrated. Addressing this knowledge gap is important because LRP5, a co-receptor for WNT signaling proposed to function through β-catenin, controls osteoblast number and function in postnatal mice or humans. To overcome the neonatal lethality caused by embryonic deletion of β-catenin in early-stage osteoblast-lineage cells, we utilize Osx-CreERT2 to remove β-catenin in Osx-expressing cells by administering tamoxifen (TM) temporarily to postnatal mice. Lineage-tracing experiments in the long bones demonstrate that Osx-CreERT2 targets predominantly osteoblast-lineage cells on the bone surface, but also transient progenitors that contribute to bone marrow stromal cells and adipocytes. Deletion of β-catenin by this strategy greatly reduces the bone formation activity of the targeted osteoblasts. However, the targeted osteoblasts rapidly turn over and are replaced by an excessive number of non-targeted osteoblasts, causing an unexpected increase in bone formation, but an even greater increase in osteoclast number and activity produces a net effect of severe osteopenia. With time, the mutant mice also exhibit a marked increase in bone marrow adiposity. Thus, β-catenin in postnatal Osx-lineage cells critically regulates bone homeostasis by promoting osteoblast activity and suppressing osteoblast turnover, while restraining osteoclast and marrow fat formation.
Introduction
The WNT molecules belong to a family of secreted proteins consisting of 19 members in mammals and play important roles in embryonic development and tissue homeostasis (1). WNT proteins activate multiple downstream signaling cascades depending on the cell context, one of which is the β-catenin-dependent pathway. In this pathway, binding of WNT to a Frizzled (Fz) receptor and a LRP5/6 co-receptor leads to stabilization of β-catenin, which subsequently translocates to the nucleus where it interacts with members of the TCF/LEF transcription factors to activate transcription of downstream target genes (2).
Genetic studies by conditionally deleting β-catenin at different stages of osteoblast development during embryogenesis have implicated WNT/β-catenin signaling in regulating multiple steps of osteoblast differentiation. Specifically, deletion of both LRP5 and LRP6, or β-catenin in mesenchymal progenitors results in a failure to initiate or maintain Osterix-positive preosteoblasts (3-6). Deletion of β-catenin in Osterix-positive cells abolishes subsequent differentiation to mature osteoblasts (7). Interestingly, deletion of β-catenin in mature osteoblasts or osteocytes indirectly increases osteoclast number and activity without a notable effect on osteoblasts (8-10). Thus, WNT/β-catenin signaling plays multiple and stage-specific roles in the osteoblast lineage.
Other genetic studies have implicated WNT signaling in promoting postnatal bone formation. In humans, loss-of-function mutations in LRP5 cause osteoporosis-pseudoglioma syndrome, a form of early-onset osteoporosis (11), whereas gain-of-function point mutations in LRP5 result in high bone mass (12,13). Moreover, deficiency in either expression or function of SOST, a secreted protein that inhibits the binding of WNT molecules to LRP5/6, results in Van Buchem disease or sclerosteosis, both exhibiting high bone mass (14,15). Mouse genetic studies have demonstrated that LRP5 promotes postnatal bone mass accrual by increasing osteoblast number and function (16,17), with LRP6 performing a redundant role (18). In addition, mice lacking SOST or missing one DKK1 allele exhibit higher bone mass predominantly due to increased bone formation (19,20). Thus, multiple lines of genetic evidence support an important role for WNT-LRP5/6 signaling in stimulating postnatal bone formation, but an alternative mechanism for LRP5 function has also been proposed (21). Because embryonic deletion of β-catenin in early-stage osteoblast-lineage cells leads to perinatal lethality whereas deletion in the more mature cells did not affect osteoblasts per se, it remains an open question whether WNT-LRP5/6 signaling regulates postnatal osteoblast number or function in a β-catenin-dependent manner.
Here we employ the Osx-CreERT2 mouse that expresses tamoxifen (TM)-inducible Cre from the regulatory sequences of Osterix (Osx, official symbol Sp7), both to examine the contribution of postnatal Osx-expressing cells, and to determine the role of β-catenin in these cells. Osx is a zinc-finger transcription factor whose expression initiates in preosteoblasts and is indispensable for osteoblast differentiation (22,23). During endochondral bone development in the mouse embryo, Osx-expressing cells migrate to the nascent bone marrow cavity and produce trabecular osteoblasts, osteocytes and stromal cells (24). Here we report that in the postnatal mice, Osx-expressing cells include both osteoblast-lineage cells and transient progenitors that give rise to bone marrow stromal cells and adipocytes. We further provide genetic evidence that β-catenin in Osx-expressing cells is necessary for osteoblast function in postnatal bones.
Materials and Methods
Mouse strains
β-catenin conditional mouse (Ctnnb1c/c), R26-mT/mG reporter mice, and Osx-CreERT2 mice are reported previously (24-26). The Animal Studies Committee at Washington University has reviewed and approved all mouse procedures used in this study.
Tamoxifen administration
Two-month-old Osx-CreERT2; R26-mT/mG, Osx-CreERT2; Ctnnb1c/c or Ctnnb1c/c mice were subjected to either corn oil (vehicle) or tamoxifen administration (80 μg per gram of body weight) by oral gavage once daily for five consecutive days. The mice were harvested at 8, 21, or 49 days after the first treatment for analyses. All data presented were derived from male mice.
Cryojane Section and Immunofluorescence
Mice were perfused with 4% PFA according to a standard protocol. After perfusion, mouse femurs were isolated and then fixed in 4%PFA overnight. After 3 days of decalcification by 14% EDTA, femurs were snap-frozen in OCT embedding medium and then sectioned at 8 μm by using a Leica cryostat equipped with Cryojane (Leica, IL). For detection of GFP, perilipin or osteoclacin, immunostaining was performed on frozen sections using a chicken polyclonal GFP antibody (Abcam, Cambridge, MA), or rabbit monoclonal perilipin antibody (Cell Signaling Technology, Danvers, MA), or rabbit polyclonal osteocalcin antibody (Santa Cruz Biotechnology).
X-ray Radiography and Microcomputerized Tomography (μCT)
Radiographic images of either whole body or the hindlimbs were acquired using a Faxitron X-ray system (Faxitron X-ray Corp) at 25 kv for 20 seconds. The femurs were scanned by a microcomputed tomography system (μCT 40, Scanco Medical AG) following the recommendations by American Society for Bone and Mineral Research (27). For quantifying trabecular bone parameters, 100 μCT slices (1.6 mm total) immediately below the growth plate of femurs were analyzed (threshold set at 300). For quantifying cortical bone parameters, 50 μCT slices (0.8 mm total) starting from 6.8 mm below the articular surface of femurs were analyzed.
Histology and Histomorphometry
For histological analysis, femurs were isolated from mice after perfusion with 4%PFA, and fixed in 10% buffered formalin overnight at room temperature, followed by decalcification in 14% EDTA for 2 weeks. After decalcification, femurs were processed for paraffin embedding and then sectioned at 6μm thickness. H&E, alcian blue/picro-sirius red staining, and TRAP staining were performed on paraffin sections following the standard protocols. For dynamic histomorphometry, calcein (Sigma, St. Louis, MO) was injected intraperitoneally at 20 mg/kg on days 7 and 2 prior to sacrifice, and bones were fixed in 70% ethanol and embedded in methyl-methacrylate for sectioning. Both static and dynamic bone histomophometry were performed with the computer software Bioquant II.
RNA Extraction and qPCR
For RNA extraction, femurs and tibias were cleanly dissected. After removal of the epiphysis and the bone marrow (by centrifugation), the bones were rinsed twice in the cold PBS, and cut into small pieces. Then 1 ml Trizol (Invitrogen) was added to extract RNA. 1 μg RNA was used to synthesize cDNA using the iScript™ cDNA Synthesis Kit (BIO-RAD). qPCR was performed with SYBR-Green supermix (BIO-RAD). The primers used were: Ctnnb1 (5′-CCTCCCAAGTCCTTTATGAATGG-3′, and 5′-CCGTCAATATCAGCTACTTGCTCTT-3′); OPG (5′-CCGAGGACCACAATGAACAAGT-3′, and 5′-CTGGGTTGTCCATTCAATGATG-3′); Rankl (5′-CTGGGCCAAGATCTCTAACATGA-3′, and 5′-GGTACGCTTCCCGATGTTTC-3′); and 18S (5′-CGGCTACCACATCCAAGGAA-3′, and 5′-GCTGGAATTACCGCGGCT-3′).
Serum CTX and PINP Assays
For serum CTX and PINP assays, serum was collected from mice after 6-hour fasting. CTX and PINP assays were performed with the RatLaps ELISA kit and the Rat/Mouse PINP EIA kit (both from Immunodiagnostic Systems, Ltd.), respectively.
Results
Postnatal Osx-expressing cells include transient progenitors for bone marrow stromal cells and adipocytes
We first examined what cells are targeted by Osx-CreERT2 upon tamoxifen (TM) administration in two-month-old mice. To this end, we generated Osx-CreERT2; R26-mT/mG animals so that GFP expression can be activated by TM only in Osx-expressing cells and their progeny. The mice were administered either vehicle or TM once daily for five consecutive days, and then harvested at different time points for GFP detection (Fig. 1A). To detect the cells that were initially targeted by this strategy, we harvested the mice at eight days after the first treatment (D8). As expected, bones from the vehicle-treated animals did not show any GFP (Fig. 1B). In contrast, bones from the TM-treated mice exhibited many GFP-positive cells (Fig. 1C). Most positive cells were detected at the chondro-osseous junction of the primary ossification center (Fig. 1C1), the trabecular bone surface (Fig. 1C2) and the endocortical surface (Fig. 1C3). Quantitative analyses revealed that GFP-positive cells covered approximately 65% of the trabecular bone surface and 85% of the endocortical surface in the femur. GFP was also detected at the secondary ossification center albeit at a lower intensity (Fig. 1C, asterisk), but none within the growth plate (Fig. 1C, bracket) or at the periosteum (Fig. 1C3, red arrow). Relatively few osteocytes expressed GFP (Fig. 1C3, white arrow), and no GFP was detected in the bone marrow (Fig. 1I). Double immunostaining for GFP and osteocalcin (OC) confirmed that a majority of the GFP-positive cells on the bone surface express OC (Fig. 1D). Thus, Osx-CreERT2 initially targets mainly osteoblast-lineage cells on the bone surface.
Figure 1.

Osx-expressing cells in postnatal mice include osteoblast-lineage cells and transient progenitors for bone marrow stromal cells. (A) Experimental design. Two-month-old mice injected TM or corn oil once daily for five consecutive days, and harvested at D8, D21 or D49. (B-C) Immunostaining for GFP on longitudinal sections of femur from mice with the genotype of Osx-CreERT2;R26-mT/mG injected with either corn oil (B) or TM (C) and harvested at D8. *: secondary ossification center. Bracket denotes growth plate. (C1-C3) Higher magnification views of boxed regions in C representing chondro-osseous junction (C1), trabecular bone (C2) and cortical bone (C3). White arrow: osteocytes; red arrow: periosteum. (D) Immunostaining for GFP (green) and osteocalcin (magenta) on trabecular bone surface in Osx-CreERT2;R26-mT/mG mice harvested at D8. M: marrow; T: trabecular bone. (E-G) GFP immunostaining on trabecular bone region from Osx-CreERT2;R26-mT/mG mice harvested at D8 (E), D21(F) or D49 (G). (H) Quantification of trabecular bone surface covered by GFP-positive cells. (I-K) GFP detection at a higher magnification in the bone marrow at D8 (I), D21 (J) or D49 (K). T: trabecular bone; arrow: GFP+ stromal cell clusters. (L) Quantification of GFP+ stromal cell clusters. *: p<0.05, results from 4 sections each from three mice at each time point.
To track the fate of the cells initially targeted by Osx-CreERT2, we analyzed GFP expression in animals harvested at 21 days after the first TM treatment (D21). Here, 55% of the trabecular bone surface remained GFP-positive, slightly but statistically significantly reduced from the coverage at D8 (Fig. 1F, H). However, many GFP-positive cells appeared within the bone marrow (Fig. 1F). These cells exhibited a typical reticular morphology of stromal cells, and were often present in clusters (Fig. 1J, arrow). Because they were not detected at D8 (Fig. 1I), the GFP-positive bone marrow stromal cells were most likely derived from Osx-expressing progenitors initially targeted by Osx-CreERT2.
To examine the fate of the targeted cells for a longer term, we analyzed the mice at D49. GFP-positive cells were still detected on both trabecular and endocortical bone surfaces, but their coverage of the total surfaces was greatly reduced (down to 25% for trabecular bone) (Fig. 1G, H, and data not shown). Similarly, the number of GFP-positive stromal cell clusters and the size of each cluster were dramatically reduced when compared to D21 (Fig. 1K, L). Thus, both bone-surface osteoblast-lineage cells, and bone marrow stromal cells experienced appreciable turnover between D21 and D49, and the Osx-expressing progenitors had only a limited capacity to replenish these populations.
Because adipocytes are an important constituent of the bone marrow in adult mice, we next asked whether Osx-expressing cells normally contribute to adipocytes. We immunostained the bone sections from the Osx-CreERT2; R26-mT/mG mice for both GFP and the adipocyte-specific marker perilipin. At D8, the TM-treated mice, like the vehicle-treated controls, exhibited no co-localization of GFP and perilipin in the 261 adipocytes counted, indicating that adipocytes were not directly targeted by Osx-CreERT2 (Fig. 2A, B). However, at D21, GFP and perilipin were found to co-localize in 3% of the 436 adipocytes counted (Fig. 2C). The co-localization was specific to bone marrow adipocytes, as it was not found in other adipose tissue (data not shown). At D49, co-localization was observed in 1% of 637 adipoctyes counted (Fig. 2D). Thus, an Osx-expressing progenitor normally contributes to adipocytes in the bone marrow of postnatal mice at a low rate.
Figure 2.
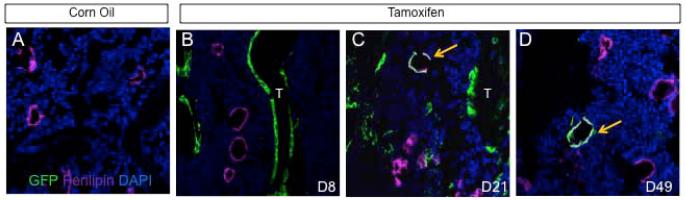
Osx-expressing cells in postnatal mice include transient progenitors for bone marrow adipocytes. Double immunostaining for GFP and perilipin on longitudinal sections of femur from mice with the genotype of Osx-CreERT2;R26-mT/mG. (A) Mice injected with corn oil (A) and harvested at D8. (B-C) Mice injected with TM and harvested at D8 (B), D21 (C) or D49 (D). Arrows denote co-localization of GFP and perilipin. T: trabecular bone. Green: GFP. Magenta: perilipin. Blue: DAPI.
Deletion of β-catenin in postnatal Osx-lineage cells results in high-turnover osteopenia and increased bone marrow adiposity
Having established the cell populations targeted by Osx-CreERT2, we proceeded with using this approach to delete β-catenin (Ctnnb1) in the postnatal skeleton. We administered TM to two-month-old Osx-CreERT2; Ctnnb1c/c mice and their littermate controls (i.e., Ctnnb1c/+ or Ctnnb1c/c) once daily for five consecutive days. These mice were harvested at 8, 21 or 49 days after the first TM administration (hereafter KO-D8, -D21, and -D49, respectively). X-ray radiography detected no obvious change in the KO-D8 mice, but severe osteopenia throughout the skeleton in the KO-D21 and -D49 animals (Fig. 3A, B, data not shown). Because a similar phenotype was observed with both male and female mice, we have focused all subsequent analyses on males. μCT analyses of the trabecular bone in the femur confirmed that bone mass (BV/TV) in the KO-D8 mice was relatively normal, but that in the KO-D21 and −49 mice was progressively reduced when compared to their respective littermate controls (Fig. 3C-E). The decrease in bone mass in the KO-D21 and −49 mice was associated with reduced trabecular number and increased trabecular spacing, with little change in trabecular thickness (Fig. 3D, E). In keeping with the μCT results, picro-sirius red staining of the femur revealed progressive worsening of the trabecular bone loss in the KO-D21 and -D49 mice, although the KO-D8 mice were relatively normal (Fig. 3F). This staining also showed a loss of the cortical bone at the metaphyseal region in the KO-D49 mice (Fig. 3F, arrows). Moreover, μCT analyses of the diaphyseal cortical bone showed a smaller diameter and reduced cortical thickness in the KO-49, but not the KO-21 mice when compared to their respective littermate controls (Fig.). Overall, TM-induced deletion of β-catenin by Osx-CreERT2 in two-month-old mice results in progressive general osteopenia.
Figure 3.

Deletion of β-catenin in postnatal Osx-lineage cells causes severe osteopenia. (A-B) X-ray radiography of Ctnnb1c/c (A) or Osx-CreERT2;Ctnnb1c/c mice (B) harvested at D49. (A1-A3, B1-B3) Higher magnification of boxed areas in A and B, respectively. Arrows denote osteopenic skull (B1), rib (B2) and vertebra (B3). (C-E) Parameters from μCT analyses of the distal femur at D8 (C), D21 (E) or D49 (E). *: p<0.05, n=5 for D8, 4 for D21, 5 for D49. (F) Picro-sirius staining of the distal femur. Arrows denote cortical bone loss. (G) Representative images of μCT scan of diaphyseal cortical bone. (H, I) Quantification of diaphyseal cortical bone by μCT at D21 (H) and D49 (I). Tt. Ar: total cross-sectional area; Ct.Ar: cortical bone area; Ct. Th: average cortical thickness. *: p<0.05, n=4 for D21, 5 for D49. C: control (Ctnnb1c/c); M: mutant (Osx-CreERT2;Ctnnb1c/c).
We next sought to understand the cellular basis for the osteopenia caused by β-catenin deletion. We assessed total bone resorption activity in the body by measuring serum CTX levels. There was no change in the KO-D8 mice, but CTX was increased by 118% and 149% over the controls in the KO-D21 and -D49 mice, respectively (Fig. 4A, E, I). Tartrate-resistant acid phosphatase (TRAP) staining on femur sections revealed an increase in both osteoclast number per bone perimeter (No. OC/BS) and the spreading of individual osteoclasts (μm/OC) in the KO-D21 and -D49 mice, which together led to a significant increase in the percentage of bone surface covered by osteoclasts (OC. S./BS) (Fig. 4F-H, J-L). In contrast, these osteoclast parameters were not altered in the KO-D8 mice (Fig. 4B-D). To probe the molecular basis for the enhanced osteogenesis, we measured RANKL and OPG levels in the serum. However, these assays did not detect any significant difference between any of the mutant mice and their respective controls (Fig. 4 M-R). On the other hand, qPCR experiments with RNA extracted from the bone surface cells detected a decrease in Opg but no change in Rankl mRNA in the KO-D8 versus control mice (Fig. 4S, T). These experiments also revealed that β-catenin (official name Ctnnb1) mRNA was reduced by ~40% in the KO-D8 mice (Fig. 4U). Whether or not the observed changes in Opg can account for the increased osteoclastogenesis remains to be tested. Overall, deletion of β-catenin in Osx-expressing cells induces excessive osteoclastogenesis and bone resorption.
Figure 4.

Deletion of β-catenin leads to high turnover osteopenia. (A-L) Analyses of osteoclast parameters at D8 (A-D), D21 (E-H) or D49 (I-L). (M-O) Serum RANKL levels at D8 (M), D21 (N) or D49 (O). (P-R) Serum OPG levels at D8 (P), D21 (Q) or D49 (R). (S-U) Expression analyses of mRNA by qPCR in bone surface cells at D8. Data normalized to 18S rRNA. Expression level in control designated 1. (V-X) Serum PINP levels at D8 (V), D21 (W) or D49 (X). C: control; M: mutant. *: p<0.05, n=6 for D8, 4 for D21, 5 for D49. #: p<0.05, n=3.
We next examined total bone formation activity by measuring serum amino-terminal propeptide of type I procollagen (PINP). Although the KO-D8 mice did not show an overall reduction in bone mass, they exhibited a marked reduction in PINP (Fig. 4V). In contrast, the KO-21 and -49 mice that were severely osteopenic showed a much higher level of PINP than their control littermates (Fig. 4W, X). Thus, β-catenin deletion in the postnatal Osx-expressing cells initially decreases bone formation activity without overtly reducing bone mass, but subsequently exuberant bone resorption causes high turnover osteopenia.
In analyzing the bone phenotype, we observed a marked increase in bone marrow adiposity in the KO-D49 mice (Fig. 5A, B). The increase was localized specifically to the metaphyseal regions of the long bones (Fig. 5A1-A3, B1-B3). On H&E sections of the femur, we found a 14-fold increase in the number of adipocytes in the KO-D49 mice over their littermate controls (Fig. 5C), and that the average size of the adipocytes in the mutant mice also increased (Fig. 5A1, B1). To investigate the possibility that Osx-positive progenitors might differentiate preferentially to adipocytes upon β-catenin deletion, we generated mice with the genotype of Osx-CreERT2;R26-mT/mG;Ctnnb1c/c (KO-GFP), and compared their percentage of GFP-positive adipocytes among total adipocytes with that in the Osx-CreERT2; R26-mT/mG mice at D21. Although we observed an increase in one out of three KO-GFP mice, a similar change was not seen in the other two (Fig. 5D). We conclude that fate switch of the Osx-positive progenitors may not be the driving force for the excessive adipogenesis in the KO-D49 mice.
Figure 5.

Deletion of β-catenin increases bone marrow adiposity. (A-B) H&E staining of longitudinal sections through the distal femur of control (A) or mutant (B) mice at D49. (A1-A3, B1-B3) Higher magnification views of boxed areas in A and B. (C) Quantification of adipocyte numbers in metaphyseal regions of Ctnnb1c/c (C) or Osx-CreERT2; Ctnnb1c/c (M) mice at D49. *: p<0.05, n=3 mice per genotype with 3 or 4 sections per mouse. (D) Percentage of GFP-positive adipocytes (perilipin-positive) among total adipocytes in Osx-CreERT2;R26-mT/mG (Ctrl) versus Osx-CreERT2;Ctnnb1 c/c; R26-mT/mG (CKO-GFP) mice at D21. Each dot represents one mouse, and the line indicates the average. More than 200 adipocytes were counted in each mouse.
Loss of β-catenin impairs osteoblast activity and increases osteoblast turnover
We next sought to determine the basis for the impaired bone formation in the KO-D8 mice. Because Osx-CreERT2 mainly targets bone-surface osteoblast-lineage cells at D8, we reasoned that deletion of β-catenin might directly affect osteoblast number and/or function. Histomorphometry showed that osteoblast number normalized to bone surface was not changed in the KO-D8 mice, although slightly increased in the KO-D21 and -D49 mice (Fig.6A-C). However, mineral apposition rate (MAR), an indicator of osteoblast function, was greatly reduced in the KO-D8 mice (Fig. 6D, E). Moreover, both the double-labeled surface normalized to total bone surface (Dls/BS) and the mineralized surface normalized total bone surface (MS/BS) were markedly decreased in the KO-D8 mice (Fig. 6F and data not shown). Consequently, the bone formation rate (BFR) was much reduced in these mice (Fig. 6G). Because of the extremely low bone mass, we could not obtain reliable data for osteoblast activity in the KO-21 or -49 mice. Thus, β-catenin deletion in osteoblast-lineage cells greatly suppresses osteoblast activity in postnatal mice.
Figure 6.

Deletion of β-catenin suppresses osteoblast activity. (A-C) Quantification of osteoblast numbers by histomorphometry at D8 (A), D21 (B) or D49 (C). (D) Representative micrographs of calcein double labeling at D8. Box areas shown at a higher magnification to the right. (E-G) Quantification of bone formation parameters at D8. C: control; M: mutant. *: p<0.05, n=4.
We next sought to explain the unexpected observation that osteoblast numbers and bone formation activity were increased in the KO-D21 and −49 mice. We hypothesized that osteoblasts initially targeted for β-catenin deletion might be replaced by newly produced wild-type osteoblasts by D21. To investigate this possibility, we monitored the presence of the β-catenin-deficient and GFP-positive osteoblasts in the KO-GFP mice at D21. Indeed, there were notably fewer GFP-positive osteoblast-lineage cells associated with bone surfaces in the KO-GFP mice than the Osx-CreERT2;R26-mT/mG control mice at D21 (Fig. 7A,B). Quantification revealed that only 28% of the trabecular bone surfaces was covered by GFP-positive cells in the KO-GFP mice at D21, compared to the Osx-CreERT2;R26-mT/mG mice with 55% at D21 and 25% at D49. Thus, loss of β-catenin greatly accelerates the turnover of osteoblast-lineage cells at the bone surface.
Figure 7.

Deletion of β-catenin accelerates osteoblast turnover. Immunostaining for GFP on longitudinal sections through the trabecular bone region of the femur in control (A) or mutant (B) mice at D21. Quantification of GFP+ bone surface shown below the images was obtained from multiple sections from three mice for each genotype.
Discussion
We have investigated the fate of postnatal Osx-expressing cells, and the role of β-catenin in the Osx-lineage cells in postnatal bone homeostasis. We find that Osx-expressing cells in postnatal mice include not only osteoblast-lineage cells, but also transient progenitors that contribute to stromal cells and adipocytes in the bone marrow. Deletion of β-catenin in the Osx-lineage cells initially greatly reduces bone formation, but later markedly increases bone resorption and marrow adiposity. These results provide to our knowledge, the first genetic evidence that β-catenin critically regulates osteoblast behavior in postnatal bones.
This study provides new insight about the turnover of osteoblast-linage cells. In a normal mouse, the trabecular bone surface covered by labeled osteoblast-lineage cells decreased from 55% to 25% within 4 weeks (D21 to D49). Assuming no major replenishment of the labeled cells, we estimate that approximately half of the cells turn over within this time, and that the average life span of bone surface osteoblast-lineage cells is about two months. This estimation is in agreement with a previous report about calvarial osteoblasts (28). The apparent turnover rate between D8 and D21 was lower than expected (from 65% to 55%), but this could be explained if a labeled progenitor population transiently replenished the bone-surface cells during this time. Alternatively, this could simply reflect the time delay required for sufficient removal of β-catenin from the targeted cells. Importantly, deletion of β-catenin in the Osx-lineage cells markedly accelerated the apparent turnover rate of bone-surface cells by D21. Because the high apparent turnover rate exceeded what could be expected from a normal osteoblast life span even if the replenishment rate was reduced to zero, we conclude that loss of β-catenin shortens the life span of osteoblasts. It is not clear at present whether the role of β-catenin in osteoblast life span reflects a similar role for LRP5. Studies to date have only reported on osteoblast apoptosis in the calvaria of LRP5 mouse models. Whereas one study showed that overexpression of a high-bone-mass variant of LRP5 reduced apoptosis (29), another study detected no change in osteoblast apoptosis in the LRP5−/− mice (16).
Following the initial suppression of osteoblast activity, deletion of β-catenin in postnatal Osx-expressing cells subsequently caused excessive osteoclastogenesis. The increased osteoclastogenesis is consistent with previous reports about deletion of β-catenin in mature osteoblasts or osteocytes (8-10). These previous studies have proposed downregulation of OPG expression by osteoblasts or osteocytes as the primary mechanism for the increased osteoclastogenesis. Although we detected a modest decrease in OPG mRNA in bone surface cells in the KO-D8 mice (3 days after last TM administration), we did not observe a change in OPG protein concentrations in the serum at any time point. In addition, we did not detect any changes in the serum RANKL protein level in our mice, even though it was reported to be higher than normal when β-catenin was deleted specifically in osteocytes (10). It is possible that a change in the local RANKL/OPG ratio within the bone microenvironment was responsible for the drastic increase in osteoclastogenesis, but this possibility needs to be further examined in the future. Finally, because Osx-CreERT2 also targeted a subset of osteocytes in response to TM, deletion of β-catenin in this population might have contributed to the increased osteoclastogenesis, especially at the later time points when the targeted osteoblasts have been replaced largely by normal osteoblasts.
Concurrent with the increased bone resorption, bone formation also increased in the KO-D21 and -D49 mice. The increased bone formation correlated with an increased number of osteoblasts per bone surface area. This increase in osteoblast number and total bone formation activity is unexpected, but is explained by the finding that a large majority of the osteoblasts at the later time points following TM treatment were not targeted by our gene deletion strategy. The mechanism for the large increase in wild-type osteoblasts is not understood at present, but may be secondary to the excessive bone resorption, which may stimulate osteoblastogenesis through the release of growth factors from the bone matrix (30,31).
Through lineage-tracing experiments, we show that in postnatal mice, there are Osx-expressing cells that give rise to bone marrow stromal cells and adipocytes. This result extends the previous finding that during embryogenesis, Osx-expressing progenitors within the perichondrium contribute to bone marrow stromal cells (24). Because the postnatal Osx-expressing cells cannot sustain a stable population of stromal cells or adipocytes, they are most likely transient progenitors, not self-renewing stem cells. It is not known at present whether stromal cells, adipocytes and osteoblasts share the same progenitor, or simply the same marker Osx among the different lineage-restricted progenitors.
Finally, it is not fully understood at present what caused the marked increase in bone marrow adiposity following β-catenin deletion in postnatal Osx-expressing cells. While this paper was in preparation, others reported a similar observation when β-catenin was deleted with the Tet-off Osx-Cre by keeping the mice off Dox for two to four months beginning at two months of age (32). Through both in vitro cell culture and in vivo lineage tracing experiments, the authors concluded that loss of β-catenin cell-autonomously shifted the differentiation potential of Osx-positive preosteoblasts to adipocytes. Here we show that Osx-positive progenitors normally contribute to a small percentage of bone marrow adipocytes in our experimental setting, but deletion of β-catenin did not cause a consistent increase in the contribution at D21. It remains possible that a shift in differentiation to adipocytes occurred at a later stage to cause the marked increase in bone marrow adiposity. On the other hand, because the increase in adipocyte number was not obvious until long after TM treatment (D49), loss of β-catenin in the Osx-positive progenitors may not be the sole driver for the excessive adipogenesis. Instead, changes in the bone marrow microenvironment caused by excessive bone resorption may be an important factor. Overall, the present work demonstrates that β-catenin in postnatal Osx-expressing cells plays multiple roles in maintaining bone homeostasis. In particular, the role of β-catenin in regulating postnatal osteoblast function and life span lends support to the view that WNT/LRP5 signaling may regulate postnatal bone accrual in part through β-catenin.
Acknowledgement
We thank Dr. Henry M. Kronenberg (Massachusetts General Hospital, Harvard Medical School) for his generous donation of the Osx-CreERT2 mouse strain. This work is supported by NIH grants AR060456 and AR055923 (FL), and P30 AR057235 (Washington University Musculoskeletal Research Center).
Footnotes
Disclosure
All authors state that there are no conflicts of interest.
References
- 1.van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136(19):3205–14. doi: 10.1242/dev.033910. [DOI] [PubMed] [Google Scholar]
- 2.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127(3):469–80. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 3.Joeng KS, Schumacher CA, Zylstra-Diegel CR, Long F, Williams BO. Lrp5 and Lrp6 redundantly control skeletal development in the mouse embryo. Developmental biology. 2011;359(2):222–9. doi: 10.1016/j.ydbio.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu H, Hilton MJ, Tu X, Yu K, Ornitz DM, Long F. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development. 2005;132(1):49–60. doi: 10.1242/dev.01564. [DOI] [PubMed] [Google Scholar]
- 5.Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8(5):739–50. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 6.Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8(5):727–38. doi: 10.1016/j.devcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133(16):3231–44. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 8.Glass DA, 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8(5):751–64. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 9.Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere MC, Bouxsein ML, Deng L, Clemens TL, Williams BO. Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem. 2005;280(22):21162–8. doi: 10.1074/jbc.M501900200. [DOI] [PubMed] [Google Scholar]
- 10.Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, Feng JQ, Bonewald LF, Kneissel M. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Molecular and cellular biology. 2010;30(12):3071–85. doi: 10.1128/MCB.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, De Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Juppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, Van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, Warman ML. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107(4):513–23. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 12.Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, Manning SP, Swain PM, Zhao SC, Eustace B, Lappe MM, Spitzer L, Zweier S, Braunschweiger K, Benchekroun Y, Hu X, Adair R, Chee L, FitzGerald MG, Tulig C, Caruso A, Tzellas N, Bawa A, Franklin B, McGuire S, Nogues X, Gong G, Allen KM, Anisowicz A, Morales AJ, Lomedico PT, Recker SM, Van Eerdewegh P, Recker RR, Johnson ML. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70(1):11–9. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346(20):1513–21. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 14.Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001;10(5):537–43. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 15.Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39(2):91–7. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kato M, Patel MS, Levasseur R, Lobov I, Chang BH, Glass DA, 2nd, Hartmann C, Li L, Hwang TH, Brayton CF, Lang RA, Karsenty G, Chan L. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157(2):303–14. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui Y, Niziolek PJ, Macdonald BT, Zylstra CR, Alenina N, Robinson DR, Zhong Z, Matthes S, Jacobsen CM, Conlon RA, Brommage R, Liu Q, Mseeh F, Powell DR, Yang QM, Zambrowicz B, Gerrits H, Gossen JA, He X, Bader M, Williams BO, Warman ML, Robling AG. Lrp5 functions in bone to regulate bone mass. Nature medicine. 2011;17(6):684–91. doi: 10.1038/nm.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holmen SL, Giambernardi TA, Zylstra CR, Buckner-Berghuis BD, Resau JH, Hess JF, Glatt V, Bouxsein ML, Ai M, Warman ML, Williams BO. Decreased BMD and limb deformities in mice carrying mutations in both Lrp5 and Lrp6. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2004;19(12):2033–40. doi: 10.1359/JBMR.040907. [DOI] [PubMed] [Google Scholar]
- 19.Morvan F, Boulukos K, Clement-Lacroix P, Roman Roman S, Suc-Royer I, Vayssiere B, Ammann P, Martin P, Pinho S, Pognonec P, Mollat P, Niehrs C, Baron R, Rawadi G. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2006;21(6):934–45. doi: 10.1359/jbmr.060311. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2008;23(6):860–9. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 21.Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schutz G, Glorieux FH, Chiang CY, Zajac JD, Insogna KL, Mann JJ, Hen R, Ducy P, Karsenty G. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135(5):825–37. doi: 10.1016/j.cell.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 23.Long F. Building strong bones: molecular regulation of the osteoblast lineage. Nature reviews. Molecular cell biology. 2012;13(1):27–38. doi: 10.1038/nrm3254. [DOI] [PubMed] [Google Scholar]
- 24.Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, Carmeliet G, Kronenberg HM. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Developmental cell. 2010;19(2):329–44. doi: 10.1016/j.devcel.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miao D, He B, Jiang Y, Kobayashi T, Soroceanu MA, Zhao J, Su H, Tong X, Amizuka N, Gupta A, Genant HK, Kronenberg HM, Goltzman D, Karaplis AC. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1-34. J Clin Invest. 2005;115(9):2402–11. doi: 10.1172/JCI24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45(9):593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 27.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2010;25(7):1468–86. doi: 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- 28.Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL, Lin CP, Kronenberg HM, Scadden DT. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell stem cell. 2012;10(3):259–72. doi: 10.1016/j.stem.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Babij P, Zhao W, Small C, Kharode Y, Yaworsky PJ, Bouxsein ML, Reddy PS, Bodine PV, Robinson JA, Bhat B, Marzolf J, Moran RA, Bex F. High bone mass in mice expressing a mutant LRP5 gene. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2003;18(6):960–74. doi: 10.1359/jbmr.2003.18.6.960. [DOI] [PubMed] [Google Scholar]
- 30.Xian L, Wu X, Pang L, Lou M, Rosen CJ, Qiu T, Crane J, Frassica F, Zhang L, Rodriguez JP, Jia X, Yakar S, Xuan S, Efstratiadis A, Wan M, Cao X. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nature medicine. 2012 doi: 10.1038/nm.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, Zhao L, Nagy TR, Peng X, Hu J, Feng X, Van Hul W, Wan M, Cao X. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nature medicine. 2009;15(7):757–65. doi: 10.1038/nm.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song L, Liu M, Ono N, Bringhurst FR, Kronenberg HM, Guo J. Loss of wnt/beta-catenin signaling causes cell fate shift of preosteoblasts from osteoblasts to adipocytes. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2012;27(11):2344–58. doi: 10.1002/jbmr.1694. [DOI] [PMC free article] [PubMed] [Google Scholar]