

Abstract
The human thioredoxin (TRX)-interacting protein is found in multiple subcellular compartments and plays a major role in redox homeostasis, particularly in the context of metabolism (e.g., lipidemia and glycemia) and apoptosis. A molecular approach to the protein's modus operandi is still needed because some aspects of the TRX-interacting protein-mediated regulation of TRX are not clearly understood. To this end, His-tagged TRX-interacting proteins were over-expressed in Escherichia coli. Because the protein is expressed mainly in inclusion bodies, it was denatured in high concentrations of guanidium hydrochloride, centrifuged, and purified by Ni-NTA affinity chromatography. His-TRX-interacting protein was then refolded by dialysis and its restructuring monitored by circular dichroism spectrometry. This preparation resulted in the formation of a covalent complex with recombinant human TRX, demonstrating that association occurs without the intervention of other partner proteins. Multiple cysteine-to-serine mutants of TRX-interacting protein were produced and purified. These mutations were efficient in limiting the formation of disulfide-linked homo-oligomers in an oxidizing environment. The mutants were also used to gain functional insight into the formation of the TRX-interacting protein-TRX complexes. These complexes were able to form in the absence of internal disulfide bridges. A mutant with all but one cysteine changed to serine (Cys247) also showed an enhanced capacity to form complexes with TRX demonstrating, in a pure molecular system, that this particular cysteine is likely responsible for the disulfide bridge between TRX-interacting protein and TRX.
Keywords: thioredoxin-interacting protein, thioredoxin, heterodimers, denaturation conditions, refolding, mutagenic analysis, regulation
Introduction
Thioredoxin-interacting protein (Txnip, also known as Vitamin D3 up-regulated protein, VDUP-1) is a 43-kDa multifunctional protein involved in various cellular responses.1 Txnip was originally detected as an up-regulated protein in HL60 cells stimulated with 1,25-(OH)2 vitamin D3,2 but it was identified later as a thioredoxin (TRX) partner in yeast two-hybrid screening experiments.3, 4 TRX is a major component of the thiol-reducing system and implicated in the regulation of various cellular processes, including proliferation, apoptosis, and gene expression. The effects of Txnip were suggested initially to be due to its negative regulation of TRX function and expression,5 but this protein might also have other functions in inflammation, and possibly cancer,6 through TRX-independent mechanisms.7 The interaction between Txnip and TRX appears to be a key redox-controlled regulatory process implicated in hepatic glucose production,8 and more recently in response to nutrient signals, in the regulation of metabolism and glucose homeostasis,9 as well as inflammation in endothelial cells.10 The interaction between Txnip and TRX was recently proposed to act as a regulatory process and may impede the proteasomal degradation of Txnip.11, 12
A model has been described for the formation of Txnip-TRX complexes based on experiments with transfected HEK293 cells over-expressing Txnip bearing a single cysteine-to-serine mutation.13 Initially, reduced TRX was suggested to interact with Txnip residue Cys247 through the recognition of an intra-chain Cys247-Cys63 disulfide bond.13 Subsequent results indicated that Cys63 is not involved in the formation of the mixed disulfide,14 raising questions about the molecular reactions resulting in the formation of a disulfide-linked complex.
This covalent complex formation is quite unusual because TRX-regulated proteins are generally subjected to a disulfide swap promoted by the reducing activity of TRX. This disulfide exchange results in TRX oxidation and subsequent release from its partner protein.15 Current knowledge about the Txnip-TRX interaction is limited to whole-cell analyses,13 which need to be extended to molecular interactions demonstrate directly the formation of a stable, disulfide-linked complex without the implication of any other protein(s).
To this end, human Txnip was produced, purified, and characterized in this study and successfully assayed for in vitro complex formation with TRX. A series of Txnip mutants was created to provide functional insight into the formation of this covalent complex, an important preliminary step for an assay aiming to discover antagonists of this protein-protein interaction. The results indicate that Txnip Cys247 is both compulsory and sufficient to promote the formation of a mixed disulfide bridge with TRX.
Modulation of the formation of this protein-protein interaction by small molecules may be an interesting way to treat metabolic pathologies, and some cancer processes. In addition, this particular behavior toward TRX may have specific features permitting inhibition specificity.
Results
Expression and purification of recombinant Txnip
The expression of human Txnip was studied in three different expression systems: Escherichia coli, baculovirus-insect cells, and mammalian cells. The protein was expressed in E. coli using two different constructs: Txnip with an N-terminal 6xHis tag (His-Txnip) and Txnip with an N-terminal fusion to maltose-binding protein (MBP-Txnip). Expression conditions were studied in small-scale using different protocols to maximize the solubility of the recombinant protein.16 Soluble expression was assayed by SDS-PAGE and anti-Txnip immunoblotting (data not shown). Afterward, His-Txnip or MBP-Txnip was expressed under conditions permitting the best yield of soluble protein and then affinity purified. His-Txnip was also expressed in insect cells and affinity purified. The coding region of human Txnip cDNA, supplemented at the amino terminal end with a DYKDDDDK tag (M2-Txnip), was expressed in HEK293 cells and affinity purified. Eluted material from different affinity purification trials were analyzed by SDS-PAGE, followed by Coomassie blue staining and anti-Txnip immunoblotting [Fig. 1(A)].
Figure 1.
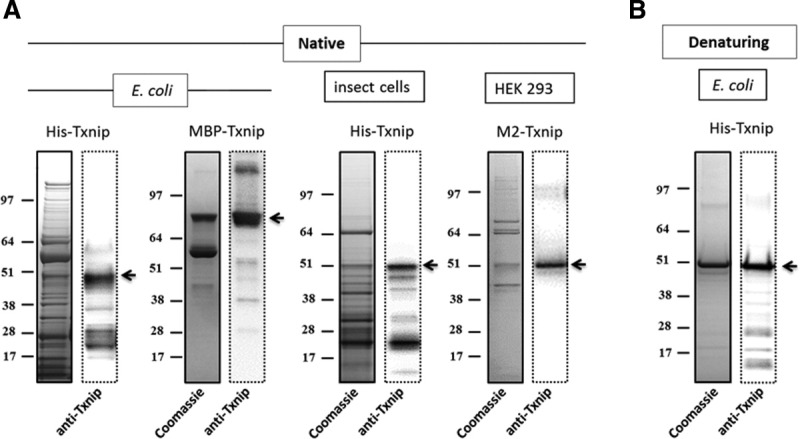
SDS-PAGE analysis of recombinant Txnip preparations. (A) Affinity purification under native conditions. Eluates were subjected to SDS-PAGE followed by Coomassie blue staining (left) and anti-Txnip immunoblotting (right). The arrow indicates His-Txnip, MBP-Txnip, or M2-Txnip. (B) Affinity purification under denaturing conditions. His-Txnip was expressed in E. coli. Eluates were subjected to SDS-PAGE followed by Coomassie blue staining (left) and anti-Txnip immunoblotting (right). The arrow indicates His-Txnip.
In all eluates, Txnip was highly contaminated by host proteins, with purity ranging from 5 to 30% as judged by SDS-PAGE and Coomassie blue staining. Several chemicals, including imidazole, maltose, detergents, salts, and reducing agents, were used to reduce non-specific binding during the purification process. All these chemicals were inefficient at improving the purity of the eluted material. Subsequent purification of affinity-purified material resulted in >80% loss of the proteins in chromatographic media with no improvement in the quality of the recombinant preparations. The proteins co-eluting with Txnip expressed by E. coli or HEK were identified by MALDI-TOF mass spectrometry as being primarily chaperone proteins: E. coli 60 kDa chaperonin 1 (A1AJ51) and DnaK (A7ZHA4) and human HSP70 (P08107) and protein disulfide isomerase (P07237). Purification of His-Txnip under denaturing conditions resulted in >70% purity as assessed by SDS-PAGE and Coomassie blue staining [Fig. 1(B)]. Cysteine mutants of Txnip (see Fig. 2 for details of the mutants) were expressed in E. coli under conditions similar to those used for wild-type (wt) Txnip. The expression of soluble protein was slightly increased for mutants B, C, D, and E. The soluble material was purified under conditions similar to those used for wt Txnip, and comparable purity was obtained (data not shown). We then decided to purify these mutants using the same denaturation/renaturation conditions described for wt Txnip.
Figure 2.

Schematic representation of the wt Txnip and cysteine-to-serine mutants used in this study. Cysteine residues are represented by white squares and numbered on the Txnip sequence. Substitutions with serine are indicated by black dots.
Refolding of wt His-Txnip from E. coli
The refolding procedure was set-up based on the Pierce Protein refolding kit with slight modifications.17 First, the yield of soluble protein recovery was monitored based on SDS-PAGE under reducing conditions and Coomassie blue staining after high-speed centrifugation. As seen in Figure 3(A), a combination of Tris and l-arginine was the most efficient additive for refolding His-Txnip. An important consideration was the possibility that one or more intra-chain and extra-chain disulfide bridges would form among the 11 cysteine residues in Txnip;13 thus, different redox conditions were tested during refolding. Analyses of the soluble material after refolding was performed using reducing and non-reducing SDS-PAGE, followed by anti-Txnip immunoblotting [Fig. 3(B)]. Initial trials were conducted without pre-treating the sample with a reducing agent, resulting in highly disulfide-linked material (i.e., homo-oligomers, data not shown). Trials were then conducted by pre-treating the sample with the reducing agent before refolding dialysis. The analysis under non-reducing conditions or with the shuffling agent, glutathione, revealed a high heterogeneity of the preparation as seen by the supra-molecular disulfide-linked species (doublet arrows) and smeared band at 40–50 kDa, which is likely intra-chain disulfide species (discontinuous arrows), in Figure 3(B, right panel). Therefore, we decided to perform refolding under reducing conditions (5 mM DTT) to improve the homogeneity of the Txnip preparation.
Figure 3.
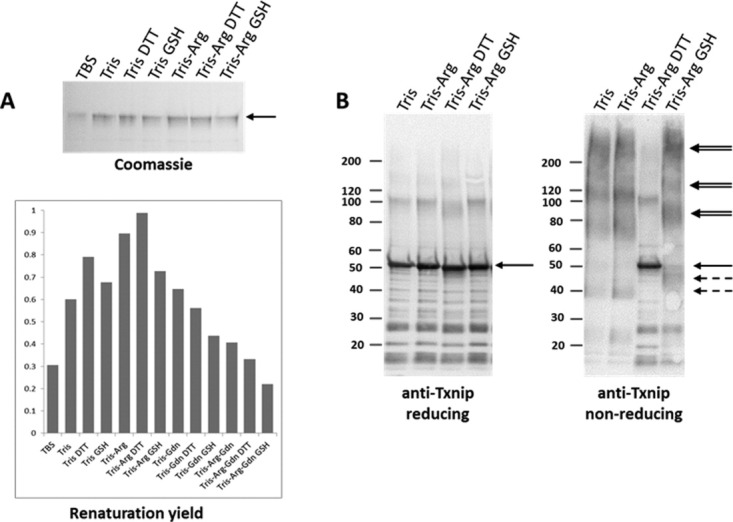
The setup for Txnip refolding. His-Txnip was purified under denaturing conditions and refolding performed by dialysis against various buffers: Tris–HCl 50 mM, NaCl 50 mM, EDTA 1 mM, pH 7.8 (TBS); Tris–HCl 0.5 M, NaCl 50 mM, EDTA 1 mM, pH 7.8 (Tris); Tris buffer + DTT 5 mM (Tris DTT); Tris buffer + oxidized glutathione 0.4 mM and reduced glutathione 4 mM (Tris GSH); Tris buffer + l-arginine-HCl 0.8 M (Tris-Arg); Tris-Arg buffer + DTT 5 mM (Tris-Arg DTT); Tris-Arg buffer + oxidized glutathione 0.4 mM and reduced glutathione 4 mM (Tris-Arg GSH). (A) Solubility screening of Txnip refolding. After overnight dialysis, the samples were centrifuged at 20,000 × g for 20 min and analyzed by SDS-PAGE under reducing conditions followed by Coomassie blue staining. The relative renaturation yield was then estimated by densitometry analysis. (B) Redox screening of Txnip refolding. Before dialysis, the denatured Txnip was supplemented with 50 mM DTT and heated at 37°C for 1 hour. After overnight dialysis, the samples were centrifuged at 20,000 × g for 20 min and analyzed by SDS-PAGE under reducing (left) and non-reducing conditions (right), followed by anti-Txnip immunoblotting. The solid arrows indicate reduced Txnip, the doublet arrows indicate Txnip homo-multimers, and the discontinuous arrows indicate suggested intra-chain disulfide species.
Analysis of refolded human Txnip
We then analyzed the refolded Txnip preparations by circular dichroism (CD) analysis to obtain information on secondary structures. The CD analyses of wt and Txnip mutant C showed similar spectra with a positive peak at 196 nm and negative peak at 216 nm (Fig. 4), both of which are characteristic of proteins with a primarily β-sheet structure. The three-dimensional structure of Txnip is unknown but, from computational studies and homologous structures, appears to be essentially composed of β-sheets.18 Comparing this model with our experimental data suggests that the recombinant preparations folded correctly. We also noticed an absence of major changes in the secondary structure between the wt and mutant C in which all the cysteine residues were mutated to serine residues except Cys247 (see Fig. 2 for details).
Figure 4.
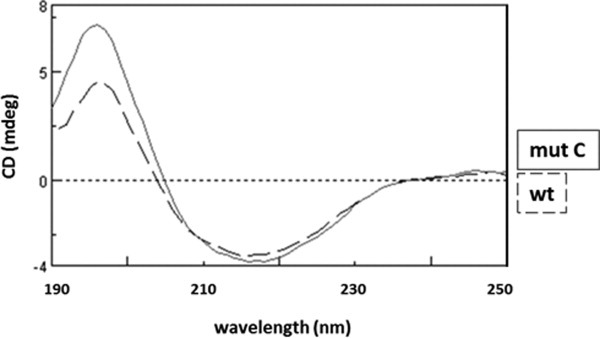
CD spectra of refolded wild-type Txnip and Txnip mutant C. The wt protein and mutant C (mut C) were refolded, extensively dialyzed against a buffer containing 20 mM Tris–sulfuric acid and 0.5 M sodium fluoride (pH 7.5), and then concentrated to 0.5–1 mg/ml using an Amicon ultra-4 concentrator with a 10,000 molecular weight cut-off. Spectra were recorded between 190 and 260 nm with a 1 nm pitch at a 50 nm/min scanning speed using a JASCO J-815 CD spectrometer. The discontinuous line is the spectra for wt Txnip and the solid line is the spectra for mut C.
The recombinant wt and Txnip mutant C proteins were also subjected to analytical gel filtration. Significant differences were observed in the homogeneity of these proteins (Fig. 5). The first peak, which is representative of aggregated species, was significantly diminished for mutant C compared with the wt protein. The second peak corresponds to a calculated molecular weight of ∼59 kDa for mutant C and ∼67 kDa for the wt based on the relative elution volumes of globular protein markers. These species were consistent with a mixture of dimers (86 kDa) and monomers (43 kDa). The shape of Txnip, which is supposed to be extended,18 may also have a modified elution position, suggesting that this species, especially in the case of mutant C, is the Txnip monomer. The later eluting population (peak 3, calculated mass 20–30 kDa) likely consists of contaminant proteins, as detected by SDS-PAGE and Coomassie blue staining [Fig. 5 (inset)].
Figure 5.
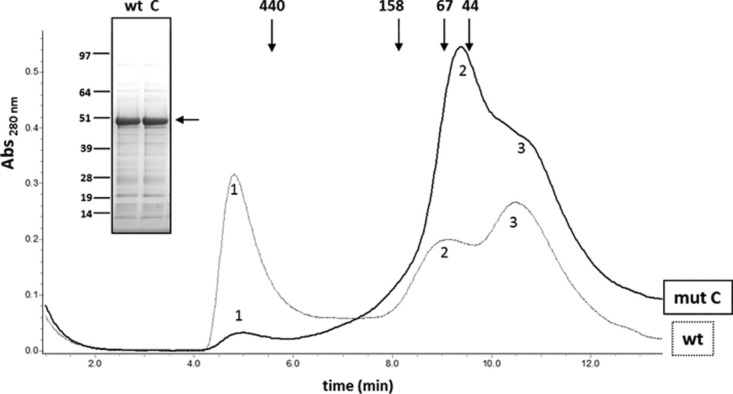
Analytical size exclusion chromatography of refolded wild-type Txnip and Txnip mutant C. Proteins were refolded and dialyzed overnight in a buffer containing 50 mM Tris–HCl, 500 mM NaCl, and 5 mM DTT (pH 7.5). The resulting proteins were concentrated to 0.5 mg/ml using an Amicon ultra-4 concentrator with a 10,000 molecular weight cut-off and centrifuged at 40,000 × g for 20 min. Concentrated preparations were then loaded on a Superdex 200 5/150 GL column (GE healthcare) equilibrated with 50 mM Tris–HCl and 500 mM NaCl (pH 7.5) at a flow rate of 0.2 ml/min and the absorbance recorded at 280 nm. The thin line represents wt Txnip and the bold line represents mutant C. Molecular weight was estimated based on the elution volumes of globular proteins using the HMW column calibration kit (GE life sciences) and linear regression. Inset: SDS-PAGE and Coomassie blue staining of wt Txnip and Txnip mutant C preparations.
Functional characterization of Txnip-TRX complex formation
The ability of the recombinant wt His-Txnip to form a covalent complex with recombinant TRX was checked. Initial trials failed to form detectable amounts of a Txnip-TRX complex, possibly because of traces of DTT (data not shown). Next, Txnip and TRX were mixed and extensive dialysis performed against a DTT-free buffer. A band appeared at 60 kDa that was recognized by both anti-Txnip and anti-TRX antibodies and likely a covalent 1:1 Txnip-TRX complex (Fig. 6). In addition, the apparent molecular weight of wt Txnip increased on non-reducing SDS-PAGE after extensive dialysis against a DTT-free buffer. Before dialysis, Txnip migrated at the expected 50 kDa [Fig. 6(A)], and after dialysis in the absence of TRX most of the material was blocked at the bottom of the well [Fig. 6(B)], suggesting the formation of high molecular weight (HMW) disulfide-linked Txnip aggregates.
Figure 6.
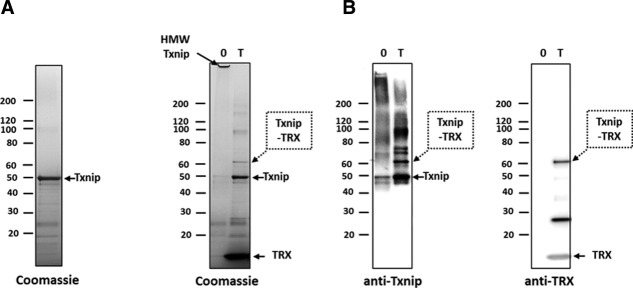
Recombinant Txnip and TRX form a covalent complex in vitro. (A) Refolded His-Txnip was incubated with recombinant TRX (T) or TRX buffer (0) at a 1:10 Txnip:TRX molar ratio overnight at 4°C. Samples were then alkylated and subsequently analyzed by non-reducing SDS-PAGE and stained with Coomassie blue or analyzed by Western blot using anti-Txnip or anti-TRX antibodies. Dotted arrows indicate the 60 kDa species corresponding to the Txnip-TRX disulfide-linked complex. Arrows with a continuous line indicate free Txnip and free TRX. HMW Txnip: HMW, disulfide-linked Txnip homo-oligomers.
The first effects of cysteine-to-serine mutations in Txnip on the formation of complexes between the Txnip mutants and TRX were seen after overnight incubation without TRX. Wild-type Txnip underwent aggregation, as most of the material did not migrate at the expected ∼50 kDa molecular weight, but remained on top of the gel (Fig. 7). The single Cys247Ser mutant (mutant A) had no effect on oligomerization, but multiple cysteine mutations (mutants B, C, and D) significantly reduced aggregation and oligomerization, which were completely absent with the cysteine-less mutant (mutant E). Mutated Txnip lacking Cys247 (mutants A, D, and E) did not form any complexes with TRX, reinforcing the current hypothesis that this residue is the site of a mixed disulfide bridge with TRX.13, 14 Intriguingly, mutants B and C both exhibited increased Txnip-TRX complex formation compared with wt Txnip. The most striking result was that mutant C, which contains only one cysteine (Cys247), efficiently complexed with TRX and to a similar extent as mutant B, which contains both Cys247 and Cys63. This result indicates that Cys247 is both necessary and sufficient to form a covalent complex with TRX.
Figure 7.
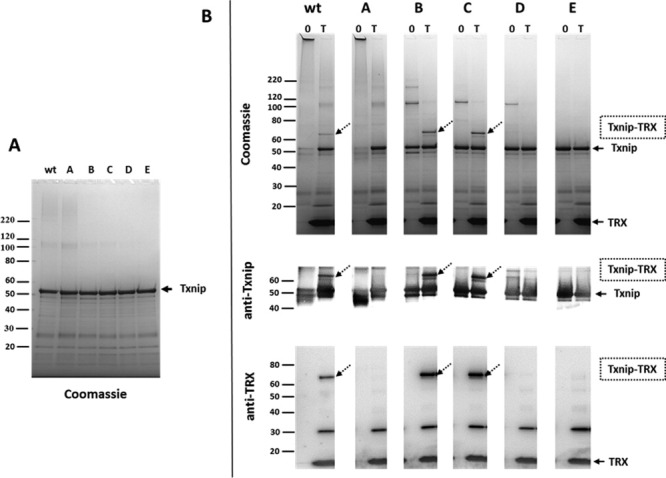
Functional characterization of Txnip-TRX complex formation. (A) Non-reducing SDS-PAGE analysis of refolded wt Txnip and Txnip mutants A, B, C, D, and E. (B) Non-reducing SDS-PAGE analysis of refolded wt Txnip and Txnip mutants A, B, C, D, and E after overnight incubation with TRX (T) or control buffer (0). Dotted arrows indicate the 60 kDa species corresponding to the Txnip-TRX disulfide-linked complex. Arrows with a continuous line indicate free Txnip and free TRX.
Incubation of Txnip and TRX had multiple effects on Txnip oligomerization and aggregation compared to control (buffer) (Fig. 7) and whether TRX prevented or reverted the formation of these higher molecular weight species is unclear. Wild-type Txnip and mutants A, B, and C, as well as TRX, were extensively dialyzed to remove DTT and mixed with TRX. Non-reducing SDS-PAGE analysis (Fig. 8) showed two effects of TRX: the reduction of aggregates and oligomeric forms of Txnip that appeared to be non-specific and the formation of a covalent complex that was highly specific to Cys247. The results were similar to previous complex formation experiments, but this co-incubation was performed for 20 minutes compared with overnight incubation in the previous protocol. In this context, mutant C appeared as a simplified model for studying the Txnip-TRX interaction and could be an interesting tool for designing a screening assay.
Figure 8.
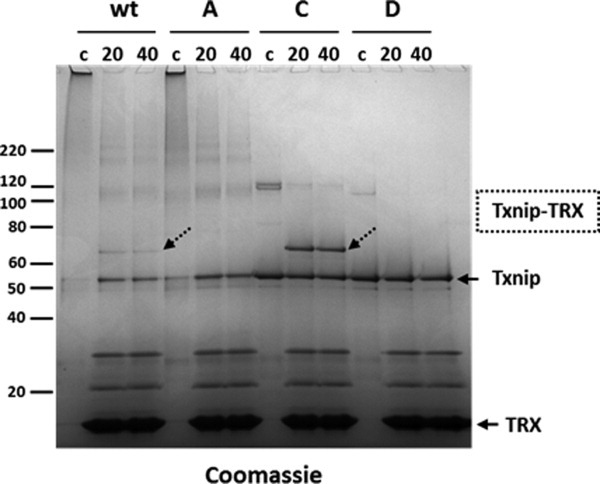
Effect of TRX on wild-type Txnip and Txnip mutants A, C, and D. First, wt and mutant Txnip were purified and refolded. TRX and Txnip preparations were then extensively dialyzed separately against a buffer containing 50 mM Tris–HCl and 500 mM NaCl (pH 7.5). After dialysis, the TRX and Txnip preparations were centrifuged at 40,000 × g for 20 min. The dialyzed wt Txnip and Txnip mutants were mixed with dialyzed TRX at a 1:10 Txnip:TRX ratio and incubated at 25°C for 20 min (20) or 40 min (40) before alkylation and non-reducing SDS-PAGE. Alternatively, control experiments (0) were performed using Txnip mixed with dialysis buffer before alkylation and non-reducing SDS-PAGE. Samples were analyzed by SDS-PAGE under non-reducing conditions and stained with Coomassie blue. Dotted arrows indicate the Txnip/TRX complexes. Free Txnip and TRX are indicated by an arrow with a continuous line.
Discussion
To produce a functionally active recombinant human His-Txnip protein from insoluble material expressed in E. coli, a denaturation/renaturation strategy was applied, yielding ∼70% purity. In all expression systems and conditions tested, affinity purification of the soluble portion of the expressed material resulted in highly contaminated preparations, with difficulties separating contaminants from Txnip under non-denaturing conditions. This problem may be the result of incomplete folding of the over-expressed protein, as well as interactions with chaperone proteins via the hydrophobic regions of Txnip.19 The contamination could be related to intrinsic scaffolding properties of Txnip; the protein is described as an α-arrestin,20 and some of the functions of Txnip could specifically favor the transient association of two proteins in a cascade process, such as in receptor signaling kinase/kinase pathways. After the expression and purification trials, the study pursued refolding the insoluble His-Txnip preparation from E. coli. The choice was based on the yield, purity, and stability of the preparations, as well as the ease of creating mutants for a functional purpose with this approach.
The correct folding of the refolded His-Txnip preparation was validated by CD, which indicated a mostly β-sheet structure, which is in agreement with the known structures of homologous β- and visual arrestins.18 Furthermore, the formation of a covalent complex with TRX validated the functionality of the refolded preparation. Taken together, these data strongly suggest correct folding of the protein in this preparation, validating its use for functional characterization.
The protein partners of TRX usually form transient mixed disulfide complexes, resulting in reduction of the target,15 implicating the formation of an intra-protein SS bridge. In contrast, the interaction between TRX and Txnip promotes the formation of a covalent mixed disulfide complex. This behavior was demonstrated by Western blot analysis of whole-cell lysates over-expressing Txnip.14 This methodology was limited by the complexity of the medium, which can interfere with protein-protein recognition or favor complex formation. Working with purified proteins, Txnip and TRX were demonstrated to form a stable covalent complex in the absence of other proteins for the first time.
Cysteine-to-serine mutants were designed to solve some of the expression problems but did not succeed, leading us to purify these mutants under the same conditions as wt Txnip. Nevertheless, the results obtained with these mutant proteins were useful, especially in the case of multiple cysteine-to-serine mutants B, C, and D, which exhibited enhanced resistance to oxidative conditions, an important context in the formation of the Txnip-TRX complex. In fact, mutants B (bearing only Cys247 and Cys63) and C (bearing only Cys247) also exhibited an enhanced complex formation capacity towards Txnip, which could represent an interesting gain in sensitivity for the development of an assay aimed at drug discovery. The explanation for this enhanced capacity is not clear but could be linked to the diminished anomalous disulfide linkage and aggregation, which may cause diminished accessibility to TRX. Another possibility is that an unidentified missing cysteine residue participates in disulfide shuffling on Txnip as described for phosphoribulokinase.21
These cysteine-to-serine substitutions were also useful for gaining insight into Txnip-TRX complex formation, because Cys247 was both compulsory and sufficient to promote efficient complex formation with TRX. Consequently, no internal disulfide bridge was required for formation of the complex, which contradicts the current hypothesis.13 Nevertheless, we could not exclude the possibility of the recognition of an extra-chain disulfide, possibly between two Cys247 of a disulfide-linked Txnip dimer.
This study provides a rapid and versatile method for producing recombinant Txnip. The creation of cysteine-to-serine substitutions yielded functional insight into Txnip-TRX complex formation, which surprisingly requires only Cys247 out of the 11 cysteines of Txnip. Thus, mutant C exhibited an enhanced capacity to form a complex with TRX and less aggregation compared with wt Txnip. This mutant appeared to be a simplified model for studying this particular type of protein-protein interaction and will be used as a tool in drug discovery and to gain further insight into the functional and structural characterization of Txnip. Indeed, a peptide from Txnip Cys247 is already being derived, with the challenge of finding a peptide long enough to fold properly and associate with TRX. We intend to label this peptide with a fluorophore to perform a series of assessments similar to those we performed in the past for other targets in fluorescence polarization assays.22, 23 However, such a screening for inhibitors of the Txnip/TRX interaction would be strongly limited by the relatively high instability of the recombinant Txnip protein, as well as its low capability of complex formation with TRX. Thus, another goal of this study was to assess the background necessary to study this interaction.
Materials and Methods
Reagents and antibodies
All reagents were purchased from Sigma-Aldrich (Saint Louis, MO) unless otherwise indicated. Plasmids were purchased from GTP Technology (Labège, France). The His-Trap FF affinity column was purchased from GE Healthcare (Orsay, France). Proteins were quantified using Bio-Rad protein assay and densitometry on SDS-PAGE stained with Coomassie blue. Anti-Txnip monoclonal antibody (JY-2) was purchased from MBL Intl (Woburn, MA) and anti-TRX polyclonal antibody (SC-20146) from Santa Cruz Biotechnology (Santa Cruz, CA). Complete EDTA-free protease inhibitor was purchased from Roche Diagnostics (Meylan, France).
Construction of expression plasmids
The cDNA coding for human TRX was synthesized with codon optimization for expression in E. coli (Geneart, Regensburg, Germany) and subsequently inserted into pGTPc301, a pET14b derivative (Novagen, Merck4Biosciences, Darmstadt, Germany) with a modified multiple cloning site. The cDNA for human Txnip was synthesized without codon optimization for constructs used in E. coli and baculovirus-insect cells. For the E. coli expression plasmid, cDNA was digested by NcoI and XhoI and subsequently inserted into pGTPc301. For E. coli expression of a fusion maltose-binding protein (MBP), cDNA was digested by EcoRI and SacI and subsequently inserted into pMAL-c5X (New England Biolabs). For expression in the baculovirus-insect cell system, synthesized cDNA was digested by NcoI and XhoI and subsequently inserted into pGTPb302, a pFastbac derivative (Novagen, Merck4Biosciences, Darmstadt, Germany) with a modified multiple cloning site. All constructs were characterized by restriction mapping and checked by double-stranded DNA sequencing.
Expression plasmid modifications
Cysteine-to-serine mutant DNAs were obtained by gene synthesis, cloned in the same vectors used for wt constructs and subsequently checked by double-stranded DNA sequencing.
Protein expression and purification
Human TRX
The pGTPc301/TRX wt or mutants were integrated into the BL21 (DE3) E. coli host strain (Novagen, Merck4Biosciences, Darmstadt, Germany). Cultures were grown in 1 L of LB medium to an absorbance of 0.6–0.8 at 600 nm. Protein production was induced by the addition of 5 mM isopropyl 1-thio-β-D-galactopyranoside and the culture incubated for 3 hours at 37°C. Cells were isolated by centrifugation and stored at −20°C. TRX was purified using a previously described method (e.g., as shown in Ref. 24) with slight modifications. Purification was performed at 4°C in the presence of 5 mM DTT. The first steps consisted of two successive anion exchange chromatography purifications (DEAE sepharose fast-flow, GE Healthcare, Orsay, France). TRX was then concentrated to 1 mg/ml using an Amicon filter with a molecular weight cut-off (MWCO) of 5000 and subjected to gel filtration chromatography using a HiLoad 26/60 Superdex column. TRX was eluted under a single peak and fractions containing TRX were collected and concentrated to ∼1 mg/ml. Glycerol was added to 10% and purified TRX aliquots snap-frozen in liquid nitrogen.
Human His-Txnip in E. coli
The pGTPc301/His-Txnip wt or mutants were integrated into the DL21 (DE3) E. coli host strain (Novagen, Merck4Biosciences, Darmstadt, Germany). Cultures were grown in 1 L Luria-Broth medium to an absorbance of 0.6–0.8 at 600 nm. Protein production was induced by the addition of 1 mM isopropyl 1-thio-β-D-galactopyranoside and the culture incubated for 4 hours at 37°C. Cells were isolated by centrifugation and stored at −80°C. His-Txnip wt and mutants were purified under denaturing conditions using the same protocol. Cells were then suspended in 60 ml of 50 mM Tris–HCl, 25 mM NaCl, 1 mM DTT (pH 7.5), and complete EDTA-free protease inhibitor cocktail (Roche Diagnostics France, Aulnay-sous-Bois, France). Lysis was performed using a cell disrupter (CellD, Sauveterre, France) at 1200 bars. The lysate was centrifuged at 40,000 × g for 40 min at 4°C. The pellets containing insoluble Txnip were suspended in 60 ml solubilization buffer containing 50 mM Tris–HCl, 6 M guanidinium hydrochloride, and 4 mM DTT (pH 8) and incubated for 2 hours at 37°C on a rocking shaker. Guanidinium-solubilized proteins were clarified by 100,000 × g centrifugation for 40 min. The supernatant was loaded on a 1 ml Ni-NTA superflow column (QIAgen) equilibrated with solubilization buffer. After extensive washes with loading buffer, Txnip was eluted using loading buffer containing 250 mM imidazole. The denatured preparation was then snap-frozen in liquid nitrogen and stored at −80°C.
Human MBP-Txnip in E. coli
The pMAL-c5X-Tev-TXNIP plasmid was integrated into the Arctic Express (DE3) host strain. Cultures were grown in 1 L Luria-Broth medium to an absorbance of 0.6–0.8 at 600 nm. Protein production was induced by the addition of 0.1 mM isopropyl 1-thio-β-D-galactopyranoside and the culture incubated for 20 hours at 20°C. Cells were isolated by centrifugation and stored at −80°C. The fusion protein was then affinity purified using amylose resin (New England Biolabs) according to the manufacturer's instructions.
Insect cells
The pGTPb302-His-Txnip plasmid was integrated into DH10bac (Invitrogen) to generate the recombinant bacmid. The bacmid was transfected into Sf9 cells (Invitrogen) to produce recombinant baculovirus. The Sf9 cells were then infected by the baculovirus at a multiplicity of infection of 1, and cells were harvested 72 hours post-infection. The cells were isolated by centrifugation and stored at –80°C. The His-tagged protein was then affinity purified using 1 ml His-Trap HP (GE healthcare) according to the manufacturer's instructions.
His-Txnip refolding
After thawing, the purified His-Txnip was diluted to 75 μg/ml in a buffer containing 50 mM Tris–HCl, 6 M guanidinium hydrochloride, 50 mM DTT, and 1 mM EDTA (pH 8) and incubated at 37°C for 1 hour. Next, the protein was refolded by dialysis for 4 hours at 4°C against a buffer containing 0.5 M Tris–HCl, 0.8 M l-arginine, and 5 mM DTT (pH 8).
M2-Txnip: HEK 293
The coding region cDNA of human Txnip (NM_006472) was supplemented at the amino terminal end with a DYKDDDDK tag and synthesized by Genecust (Genecust Europe, Dudelange, Luxembourg). This cDNA was inserted between the NheI and NotI restriction sites of pIMLP1A.CMV vector (Cellectis, Romainville, France). The cGPS HEK-293/flag-Txnip(h) cell line was established using the protocol described in the cGPS HEK-293 full kit purchased from Cellectis (Romainville, France). Positive cell clones were selected by immunofluorescence using anti-Flag antibodies (Sigma, F-3165) and anti-mouse IgG Alexa 488 antibodies (Invitrogen, A-11029). cGPS HEK-293/flagTxnip(h) cells were grown in DMEM Glutamax medium (Invitrogen 61965) supplemented with 10% v:v bovine fetal serum, 1% v:v sodium pyruvate, 400 μg/ml neomycin, and 1 μg/ml puromycin. Cells were recovered by centrifugation and frozen at –80°C in liquid nitrogen. One billion cells were resuspended in 25 ml hypotonic buffer containing 50 mM Tris–HCl, 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT (pH 7.5), and complete EDTA-free protease inhibitor (Roche). After 10 min incubation on ice, cell lysates were homogenized with 20 strokes of a 40 ml tight dounce potter. Lysates were then centrifuged at 230 × g for 5 min to pellet the nuclei and cell fragments. The supernatant was recovered and 5 M NaCl added to adjust salt concentration to 150 mM, and then centrifuged for 40 min at 70,000 × g. Clarified lysate was then loaded at a flow rate of 0.5 ml/min onto a 1 ml anti-M2 flag agarose (Sigma) column pre-equilibrated with a buffer containing 50 mM Tris–HCl and 150 mM NaCl (pH 7.5). After sample loading, the column was washed with 60 column volumes of equilibration buffer, M2-Txnip was then eluted using 100 μg/ml M2 peptide (Sigma) in the same buffer.
Electrophoresis and western blot analysis
SDS-PAGE analysis was performed using Nu-PAGE 4–12% in MOPS buffer (Invitrogen). For sample preparation under reducing conditions, samples were boiled for 5 min at 95°C with LDS sample buffer (Invitrogen) and 50 mM DTT. For non-reducing SDS-PAGE analysis, samples were first treated with 30 mM iodoacetamide for 20 min at 20°C and subsequently incubated for 45 min at 37°C in LDS sample buffer. Electrophoretic transfer was performed using an iBlot apparatus (Invitrogen) and 0.45 μm nitrocellulose membranes according to the manufacturer's protocol. Anti-Txnip monoclonal antibody (JY-2) was used at 1:1000 dilution, and anti-TRX polyclonal antibody (sc-20146) at 1:400 dilution in a TBS Buffer (Sigma), 0.05% Tween-20, and 5% (w/v) ECL advance blocking agent (GE Healthcare, Orsay, France). Protein bands were detected using ECL Advance (GE Healthcare, Orsay, France). Densitometry analysis was performed using a Chemi Doc XRS camera and Quantity One software (Bio-Rad).
CD
Refolded Txnip proteins were extensively dialyzed against a buffer containing 20 mM Tris–sulfuric acid and 0.5 M sodium fluoride (pH 7.5), concentrated to ∼0.7 mg/ml using an Amicon ultra-4 MWCO 10,000 concentrator, and centrifuged at 20,000 × g for 20 min. Spectra were acquired at 20°C with a 0.1 cm path length using a JASCO J-815 CD spectrometer (JASCO UK, Great Dunmow, Essex, England). Scans were performed between 260 and 190 nm with a 1 nm pitch at a 50 nm/min scanning speed.
Analytical size-exclusion chromatography
For size-exclusion chromatography, samples were concentrated to ∼0.5 mg/ml using an Amicon Ultra-4 concentrator with an MWCO of 10,000. The recombinant Txnip preparations were analyzed on a Superdex 200 5/150 GL column (GE Healthcare, Orsay, France) equilibrated with 50 mM Tris–HCl and 500 mM NaCl (pH 7.5) at a flow rate of 0.2 ml/min, and the absorbance was recorded at 280 nm. Molecular weight was estimated based on the elution volumes of globular proteins using the HMW column calibration kit (GE Life Sciences, Orsay, France) and linear regression.
Formation of mixed disulfide linked TRX-Txnip complexes
Recombinant Txnip was mixed with recombinant human TRX at a 1:10 molar ratio. Proteins were then dialyzed to remove DTT using a 7000 MWCO Slide-A-Lyzer MINI dialysis unit or G2 dialysis cassettes (Pierce) against a buffer containing 20 mM Tris–HCl and 200 mM NaCl (pH 8) for 2 hours. The dialysis buffer was exchanged and dialysis proceeded overnight at 4°C. The samples were then alkylated by the addition of 30 mM iodoacetamide and incubated for 20 min at room temperature. When Txnip and TRX were dialyzed separately, the buffer contained 50 mM Tris–HCl and 500 mM NaCl (pH 7.5) to prevent aggregation. After dialysis, protein samples were centrifuged at 58,000 × g for 20 min, subsequently mixed at a 1:10 molar ratio, and incubated at 25°C under mild agitation. Disulfide exchange was stopped by the addition of 30 mM iodoacetamide and subsequent incubation at 20°C for 20 min.
Mass spectrometric analysis of contaminant proteins
Mass spectrometric analyses were performed by Proteodynamics (Saint Beauzire). Briefly, protein samples were exchanged in a buffer containing 50 mM ammonium bicarbonate and concentrated using a vivaspin 500. Afterward, 5 μg of protein was reduced and digested with sequencing grade modified trypsin (Promega) according to the provider's instructions. Peptides were then analyzed by a nanoLC-Ultimate 3000 (Dionex) and hybrid mass spectrometer (LTQ-Orbitrap-XL, ThermoElectron). Raw data analysis was performed using Proteome Discover 1.2 software (Thermo scientific) and the databanks SEQUEST (Thermo scientific) and Mascot (Matrix sciences).
Acknowledgments
The authors thank Grégory Leclerc and Jérôme Dupuis for identifying contaminant proteins by mass spectrometry. They also thank Manfred Theisen (Proteodynamics, Saint-Beauzire, France) for helpful discussions. The present manuscript was edited by San Francisco Edit.
References
- 1.Kim SY, Suh HW, Chung JW, Yoon SR, Choi I. Diverse functions of VDUP1 in cell proliferation, differentiation, and diseases. Cell Mol Immunol. 2007;4:345–351. [PubMed] [Google Scholar]
- 2.Chen KS, DeLuca HF. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochim Biophys Acta. 1994;1219:26–32. doi: 10.1016/0167-4781(94)90242-9. [DOI] [PubMed] [Google Scholar]
- 3.Yamanaka H, Maehira F, Oshiro M, Asato T, Yanagawa Y, Takei H, Nakashima Y. A possible interaction of thioredoxin with VDUP1 in HeLa cells detected in a yeast two-hybrid system. Biochem Biophys Res Commun. 2000;271:796–800. doi: 10.1006/bbrc.2000.2699. [DOI] [PubMed] [Google Scholar]
- 4.Junn E, Han SH, Im JY, Yang Y, Cho EW, Um HD, Kim DK, Lee KW, Han PL, Rhee SG, Choi I. Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J Immunol. 2000;164:6287–6295. doi: 10.4049/jimmunol.164.12.6287. [DOI] [PubMed] [Google Scholar]
- 5.Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, Takagi Y, Sono H, Gon Y, Yodoi J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem. 2000;274:21645–21650. doi: 10.1074/jbc.274.31.21645. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe R, Nakamura H, Masutani H, Yodoi J. Anti-oxidative, anti-cancer and anti-inflammatory actions by thioredoxin 1 and thioredoxin-binding protein-2. Pharmacol Ther. 2010;127:261–270. doi: 10.1016/j.pharmthera.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Kaimul AM, Nakamura H, Masutani H, Yodoi J. Thioredoxin and thioredoxin-binding protein-2 in cancer and metabolic syndrome. Free Radic Biol Med. 2007;43:861–868. doi: 10.1016/j.freeradbiomed.2007.05.032. [DOI] [PubMed] [Google Scholar]
- 8.Chutkow WA, Patwari P, Yoshioka J, Lee RT. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J Biol Chem. 2008;283:2397–2406. doi: 10.1074/jbc.M708169200. [DOI] [PubMed] [Google Scholar]
- 9.Blouet C, Schwartz GJ. Nutrient-sensing hypothalamic TXNIP links nutrient excess to energy imbalance in mice. J Neurosci. 2011;31:6019–6027. doi: 10.1523/JNEUROSCI.6498-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.World C, Spindel ON, Berk BC. Thioredoxin-interacting protein mediates TRX1 translocation to the plasma membrane in response to tumor necrosis factor-{alpha}: a key mechanism for vascular endothelial growth factor receptor-2 transactivation by reactive oxygen species. Arterioscler Thromb Vasc Biol. 2011;31:1890–1897. doi: 10.1161/ATVBAHA.111.226340. [DOI] [PubMed] [Google Scholar]
- 11.Chutkow WA, Lee RT. Thioredoxin regulates adipogenesis through thioredoxin interacting protein (Txnip) protein stability. J Biol Chem. 2011;286:29139–29145. doi: 10.1074/jbc.M111.267666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang P, Wang C, Gao K, Wang D, Mao J, An J, Xu C, Wu D, Yu H, Liu JO, Yu L. The ubiquitin ligase itch regulates apoptosis by targeting thioredoxin-interacting protein for ubiquitin-dependent degradation. J Biol Chem. 2010;85:8869–8879. doi: 10.1074/jbc.M109.063321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patwari P, Higgins LJ, Chutkow WA, Yoshioka J, Lee RT. Evidence for formation of a mixed disulfide by disulfide exchange. J Biol Chem. 2006;81:21884–21891. doi: 10.1074/jbc.M600427200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patwari P, Chutkow WA, Cummings K, Verstraeten VL, Lammerding J, Schreiter ER, Lee RT. Thioredoxin-independent regulation of metabolism by the alpha-arrestin proteins. J Biol Chem. 2009;284:24996–25003. doi: 10.1074/jbc.M109.018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holmgren A. Thioredoxin structure and mechanism: conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure. 1995;3:239–243. doi: 10.1016/s0969-2126(01)00153-8. [DOI] [PubMed] [Google Scholar]
- 16.Sorensen HP, Mortensen KK. Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microb Cell Fact. 2005;4:1. doi: 10.1186/1475-2859-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vallejo LF, Rinas U. Strategies for the recovery of active proteins through refolding of bacterial inclusion body proteins. Microb Cell Fact. 2004;3:11. doi: 10.1186/1475-2859-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aubry L, Guetta D, Klein G. The arrestin fold: variations on a theme. Curr Genomics. 2009;10:133–142. doi: 10.2174/138920209787847014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grallert H, Buchner J. Review: a structural view of the GroE chaperone cycle. J Struct Biol. 2001;135:95–103. doi: 10.1006/jsbi.2001.4387. [DOI] [PubMed] [Google Scholar]
- 20.Alvarez CE. On the origins of arrestin and rhodopsin. BMC Evol Biol. 2008;8:222. doi: 10.1186/1471-2148-8-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brandes HK, Larimer FW, Hartman FC. The molecular pathway for the regulation of phosphoribulokinase by thioredoxin f. J Biol Chem. 1996;271:3333–3335. doi: 10.1074/jbc.271.7.3333. [DOI] [PubMed] [Google Scholar]
- 22.Nosjean O, Souchaud S, Deniau C, Geneste O, Cauquil N, Boutin JA. A simple theoretical model for fluorescence polarization binding assay development. J Biomol Screen. 2006;11:949–958. doi: 10.1177/1087057106294841. [DOI] [PubMed] [Google Scholar]
- 23.Furger C, Derick S, Boutin JA, Nosjean O. Image-free assessment of protein translocation in live cells. Curr Opin Pharmacol. 2009;9:650–656. doi: 10.1016/j.coph.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Oblong JE, Berggren M, Gasdaska PY, Powis G. Site-directed mutagenesis of active site cysteines in human thioredoxin produces competitive inhibitors of human thioredoxin reductase and elimination of mitogenic properties of thioredoxin. J Biol Chem. 1994;269:11714–11720. [PubMed] [Google Scholar]