

Abstract
1,3,5-Tri-N-alkylcarbamylphloroglucinols (1–4) are synthesized as conformationally constrained analogs of triacylglycerols (TGs) to probe Jenck's proximity effect in the cholesterol esterase inhibition. For the cholesterol esterase inhibition, inhibitors 1–4 are 220–760-fold more potent than 1,2,3-tri-N-alkylcarbamylglycerols (13–15) that are substrate analogs of TG. Comparison of tridentate inhibitors 1–4, bidentate inhibitors 3,5-di-N-n-alkylcarbamyloxyphenols (5–8) and monodentate inhibitors 5-N-n-alkylcarbamyloxyresorcinols (9–12) indicates that inhibitory potencies are as followed: tridentate inhibitor > bidentate inhibitor > monodentate inhibitor. The log ki and pKi values of tridentate inhibitors, bidentate inhibitors, and monodentate inhibitors are linearly correlated with the alkyl chain length indicating a common mechanism in each inhibition. Also, positive slopes of these correlations indicate that the longer chain inhibitors bind more tightly to the enzyme than the shorter ones. Molecular dockings of tridentate 1, bidentate 5, and monodentate 9 into the X-ray crystal structure of cholesterol esterase suggest that one carbamyl group in the cis form of the inhibitor binds to the acyl chain-binding site of the enzyme. The second carbamyl groups in the trans forms of inhibitors 1 and 5 bind to the second acyl chain-binding site of the enzyme. The third carbamyl group in the trans form of inhibitor 1 binds to the third acyl chain-binding site of the enzyme. Moreover, the configuration of the inhibitor in the enzyme-inhibitor complex is the (1,3,5)-(cis, trans, trans)-tricarbamate form that mimics the (+gauche, −gauche)-conformation of TG.
Keywords: cholesterol esterase, inhibitors, carbamates, molecular docking
Introduction
Cholesterol esterase (CEase, EC 3.1.1.13) also known as bile salt-activated lipase, carboxyl ester lipase, lysophospholipase, and nonspecific lipase, is responsible for the hydrolysis of dietary cholesterol esters, fat soluble vitamin esters, phosphatidylcholines, and triacylglycerols (TGs).1–4 Like other α/β hydrolases, CEase possesses a catalytic triad, Ser194–His435–Asp320, that serves as a nucleophilic and general acid-base catalytic entity.5–9
CEase has also been demonstrated that it is involved directly in lipoprotein metabolism, in that the enzyme catalyzes the conversion of large low-density lipoprotein (LDL) to smaller, denser, more cholesterol ester-rich lipoproteins and that the enzyme may regulate serum cholesterol levels.4 Therefore, the ability of CEase to convert large LDL to smaller LDL subspecies, and the relationship between plasma CEase and LDL levels, suggest that high-plasma CEase levels may constitute a potential risk factor for atherosclerosis. Thus, CEase inhibitors may be suitable for the treatment of combined lipoprotein disorders characterized by elevation of cholesterol.10, 11
In the presence of substrate, carbamates serve as the pseudo or alternate substrates inhibitors of CEase (Fig. 1).12–21 The mechanism for CEase-catalyzed hydrolysis of substrate involves the formation of the noncovalent enzyme-substrate Michaelis complex, followed by nucleophilic attack of the active site serine on the substrate, which leads to the acyl enzyme intermediate; hydrolysis of the acylenzyme completes the catalytic cycle.
Figure 1.
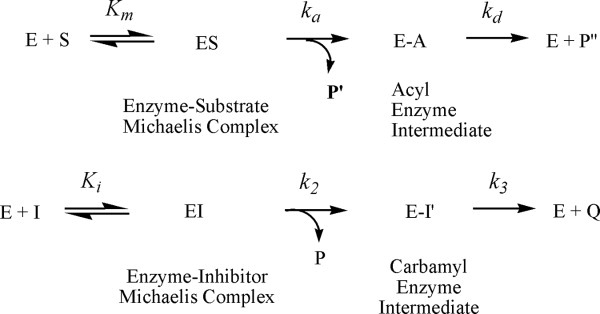
Kinetic scheme for the pseudo-substrate inhibition of inhibitors in the presence of substrate. E, enzyme (CEase); S, substrate (PNPB); ES, acylenzyme intermediate; I, inhibitors 1–17; EI, enzyme-inhibitor tetrahedral intermediate; EI′, carbamyl enzyme intermediate; P, the product from substrate reaction (4-nitrophenol); P′, the product from pseudo substrate reaction (alcohols); Q, the second product (carbamic acids).
The first step for this pseudo-substrate inhibition is reversible formation of the noncovalent enzyme-inhibitor complex, with inhibition constant Ki. Subsequent attack of serine residue of the enzyme on the carbamate carbonyl carbon of the inhibitor forms the carbamylenzyme, with rate constant k2. The carbamate group of the inhibitor must bind to the acyl group-binding site of the active site,5, 6 which is located deeply inside the enzyme. The third step is decarbamylation, governed by k3.
Although different bile salt-activation mechanisms were proposed, the active site of CEase from two different X-ray crystal structures was similar to that of lipases (Fig. 2).5, 6 Because TGs are also substrates of CEase, the active sites of CEase also contain three acyl group-binding sites like all other lipases (Fig. 2).15, 22 The active site of CEase may contain the following six binding sites. (i) The catalytic triad comprises Ser194–His435–Asp320, which is located at the bottom of the gorge.5, 6 (ii) The oxyanion hole, Gly107, Ala108, and Ala195, is also located at the bottom of the gorge and at the opposite direction of the catalytic triad and stabilizes the tetrahedral intermediate. (iii) The first acyl chain-binding site (ACS) is deeply buried in the enzyme and binds the acyl group of cholesterol ester5, 6 or the sn-1 (or sn-3) acyl chain of TGs.15 (iv) The leaving group-binding site (LGS) is located extending from the entrance (mouth) to the bottom of the active site gorge that may bind to the cholesterol moiety of cholesterol ester. (v) The second acyl chain-binding site (SACS) is located at the top of oxyanion hole and is about 90° to LGS and may bind the sn-2 acyl chain of TGs.15 (vi) The third acyl chain-binding site (TACS) is located at the opposite direction of SACS, is also about 90° to LGS, and may bind the sn-3 (or sn-1) acyl chain of TGs.
Figure 2.
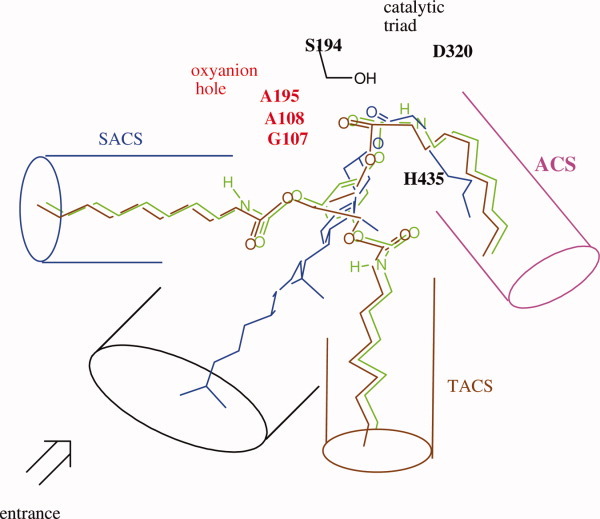
Superimposition of tridentate inhibitor 1,3,5-tri-N-n-octylcarbamyl-phloroglucinol (1), cholesterol ester, and TG into the active sites of CEase that contains the catalytic triad, an oxyanion hole, ACS, SACS, TACS, and LGS based on the X-ray crystal structures.5, 6 One of octylcarbamate moiety of inhibitor 1, the acyl chain of cholesterol ester, and the sn-1 or sn-3 acyl chain of TG are orientated to fit into ACS of the enzyme. The carbonyl oxygen atoms of these ACS-bound acyl or carbamyl groups of substrates or inhibitor are orientated to fit into the oxyanion hole of the enzyme, and the carbonyl carbon atoms of those are in the correct position for the attack by the Ser 194 of the enzyme. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Among five conformers for the glycerol backbones of TGs (Figs. 3 and 4), CEase may stereospecifically bind few of them. Therefore, the substrate analogs with constrained conformations23–32 should increase the reactivity according to William Jencks' proximity effect.33, 34 We then synthesized 1,3,5-tri-N-alkylcarbamylphloroglucinols (1–4) (Fig. 5) as conformationally constrained glycerol backbone analogs of TGs that might tightly and simultaneously bind to ACS, SACS, and TACS of CEase and completely block all entrances of the enzyme. For comparison, we also synthesized analogs of TGs, 1,3,5-tri-N-substituted carbamylglycerols (13–17) (Fig. 5) that might freely rotate around their glycerol backbones, adapt many stable conformations (Fig. 4), and should have less inhibitory potency for CEase than inhibitors 1–4.
Figure 3.
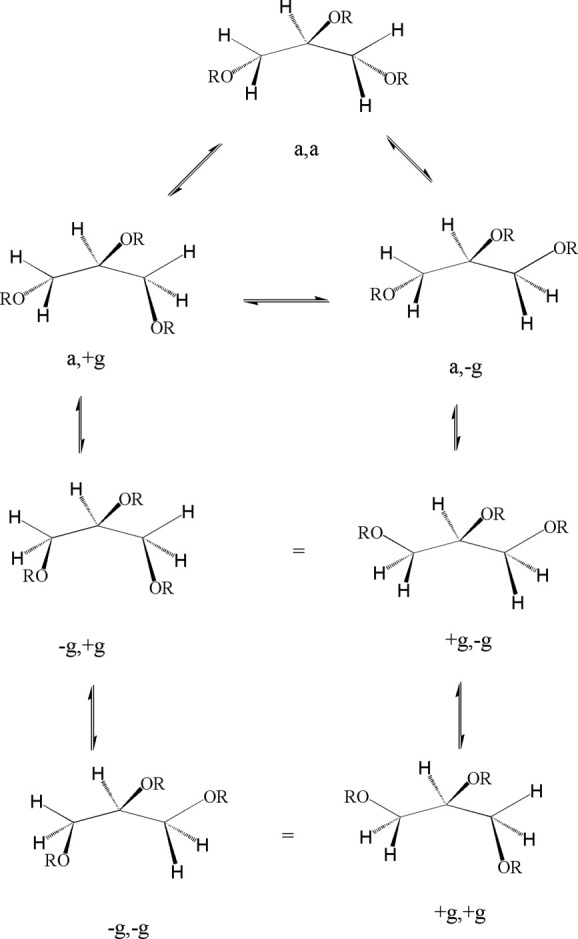
Five stable conformations for the glycerol backbone of TG.
Figure 4.
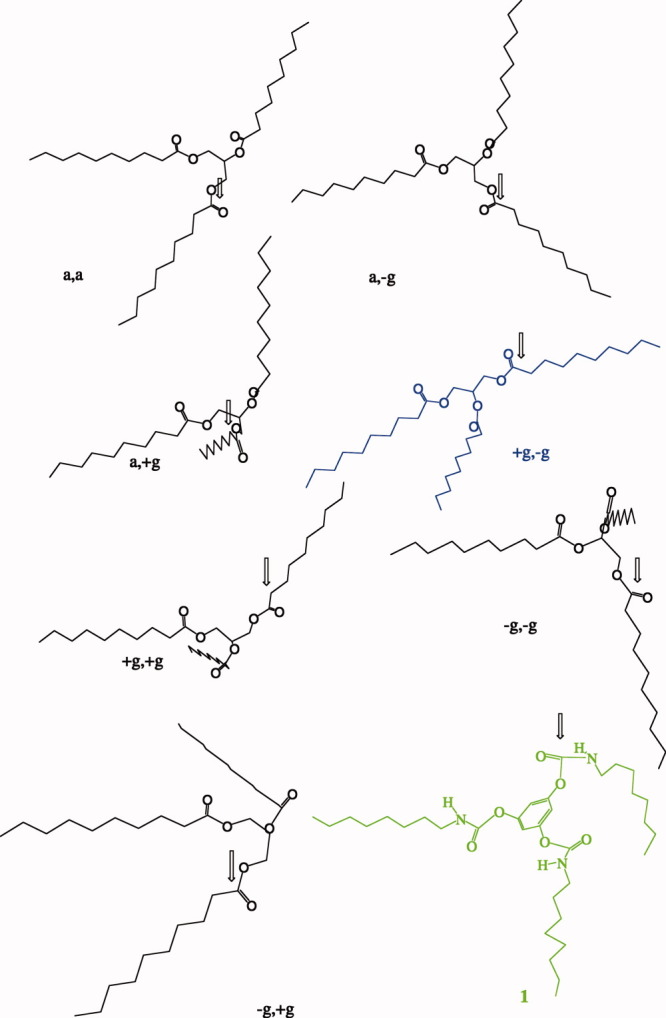
Five stable conformations for the glycerol backbone of 1,2,3-tridecanoylglycerol and structure of tridentate inhibitor 1. The structure of tridentate inhibitor 1 mimics (+g, −g)-form. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 5.
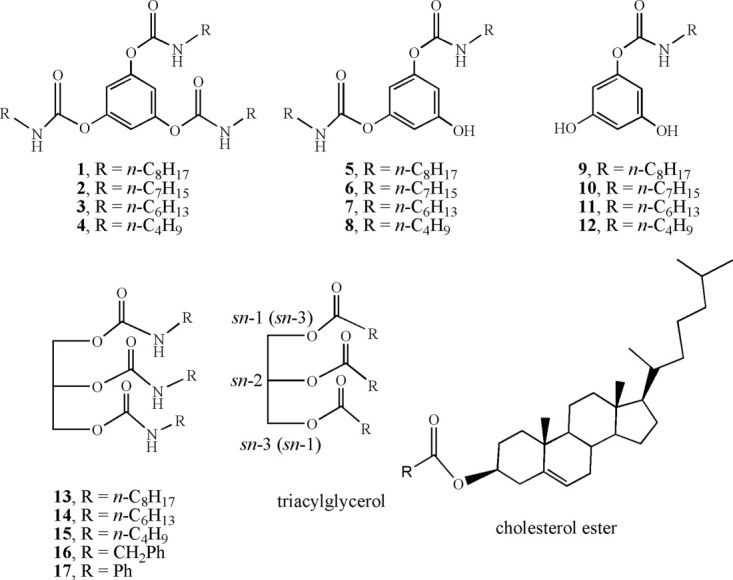
Chemical structures of inhibitors 1–17.
Quantitative structure activity relationship (QSAR) or linear-free energy relationship plays an important role in the development of a new drug and in the elucidation of a reaction mechanism.35–38 The relationships between variations of the alkyl group chain lengths or the substituent and the logarithmic values of inhibition constants of tridentate inhibitors 1–4, bidentate inhibitors 5–8, monodentate inhibitors 9–12, and inhibitors 13–17 for CEase were also studied in this work in order to explore the reaction mechanism of CEase in more detail.
Results
Synthesis of 1,3,5-tri-N-n-alkylcarbamylphloroglucinols (1–4), 3,5-di- N-n-alkylcarbamyloxyphenols (5–8), and 5-N-n-alkylcarbamyloxyresorcinols (9–12)
1,3,5-Tri-N-n-alkylcarbamylphloroglucocinols (1–4), 3,5-di-N-n-alkylcarbamyloxyphenols (5–8), and 5-N-n-alkylcarbamyloxyresorcinols (9–12; Fig. 5) were synthesized from one-pot reaction of 1,3,5-benzene-triol with Eq. (2) of the corresponding n-alkylisocyanate and Eq. (2) of triethylamine in tetrahydrofuran at 25°C for 5 h. Three products were easily separated by liquid chromatography. Under this optimal condition, the product ratio for tridentate inhibitors 1–4: bidentate inhibitors 5–8: monodentate inhibitors 9–12 was about 2:1:1.5. When Eq. (3) of n-alkylisocyanate and Eq. (3) of triethylamine were used, the reactions yielded mainly tridentate inhibitors 1–4. When Eq. (1) of n-alkylisocyanate and Eq. (1) of triethylamine were used, the reaction yield is too low.
1–17 were potent inhibitors of cholesterol esterase
In the presence of substrate, carbamates serve as the pseudo or alternate substrates inhibitors of CEase.12–19 Similarly, inhibitors 1–17 were characterized as potent inhibitors of CEase (Tables I and II; Fig. 6). Thus, inhibitors 1–17 might bind to the enzyme through the nucleophilic attack of the catalytic serine 194 of the enzyme to one carbamyl carbon of inhibitors 1–17. Therefore, one n-alkylcarbamyl moieties of inhibitors 1–17 might bind to ACS of the enzyme (Fig. 2). Furthermore, the inhibitory potencies were as follows: tridentate inhibitors 1–4 > bidentate inhibitors 5–8 > monodentate inhibitors 9–12 (Table I). Conformationally constrained analogs tridentate inhibitors 1–4 were 220–760-fold more potent inhibitors than TG analogs inhibitors 13–17 (Tables I and II).
Table I.
Pseudo-Substrate Inhibition of CEase by 1,3,5-Tri-N-Alkylcarbamyl-Phloroglucinols (1–12)
| Inhibitors | k2 (10−3 s−1) | Ki (nM) | ki (103 M−1 s−1)a |
|---|---|---|---|
| 1 | 2.2 ± 0.1b | 10 ± 2 | 220 ± 40 |
| 2 | 3.4 ± 0.3 | 25 ± 6 | 140 ± 30 |
| 3 | 2.27 ± 0.07 | 14 ± 2 | 160 ± 20 |
| 4 | 2.19 ± 0.02 | 11 ± 3 | 200 ± 60 |
| 5 | 1.8 ± 0.1 | 20 ± 4 | 90 ± 20 |
| 6 | 1.46 ± 0.06 | 22 ± 5 | 70 ± 20 |
| 7 | 1.39 ± 0.05 | 10 ± 2 | 140 ± 30 |
| 8 | 1.49 ± 0.07 | 12 ± 2 | 120 ± 20 |
| 9 | 1.00 ± 0.02 | 20 ± 4 | 50 ± 10 |
| 10 | 1.01 ± 0.06 | 23 ± 7 | 40 ± 10 |
| 11 | 1.11 ± 0.05 | 9 ± 2 | 120 ± 30 |
| 12 | 0.89 ± 0.02 | 12 ± 2 | 70 ± 10 |
The ki values were calculated from k2/Ki and uncertainty in ki values = {(uncertainty of k2)2 + (uncertainty of Ki)2}1/2.
Data followed by ± signs were standard deviations obtained from least-squares curve fittings with n (number of separate experiments) > 9.
Table II.
Pseudo-Substrate Inhibition of CEase by 1,3,5-Tri-N-Substituted Carbamylglycerols (13–17)
| Inhibitors | σ* | k2 (10−4 s−1) | Ki (μM) | ki (M−1 s−1)a |
|---|---|---|---|---|
| 13 | −0.13 | 1.4 ± 0.1b | 0.14 ± 0.01 | 1000 ± 100 |
| 14 | −0.15 | 1.5 ± 0.1 | 0.72 ± 0.04 | 210 ± 20 |
| 15 | −0.13 | 1.4 ± 0.1 | 0.39 ± 0.01 | 360 ± 20 |
| 16 | 0.22 | 2.8 ± 0.2 | 1.90 ± 0.06 | 150 ± 10 |
| 17 | 0.6 | 3.7 ± 0.2 | 22 ± 1 | 17 ± 1 |
The ki values were calculated from k2/Ki and uncertainty in ki values = {(uncertainty of k2)2 + (uncertainty of Ki)2}1/2.
Data followed by ± signs were standard deviations obtained from least-squares curve fittings with n (number of separate experiments) > 9.
Figure 6.
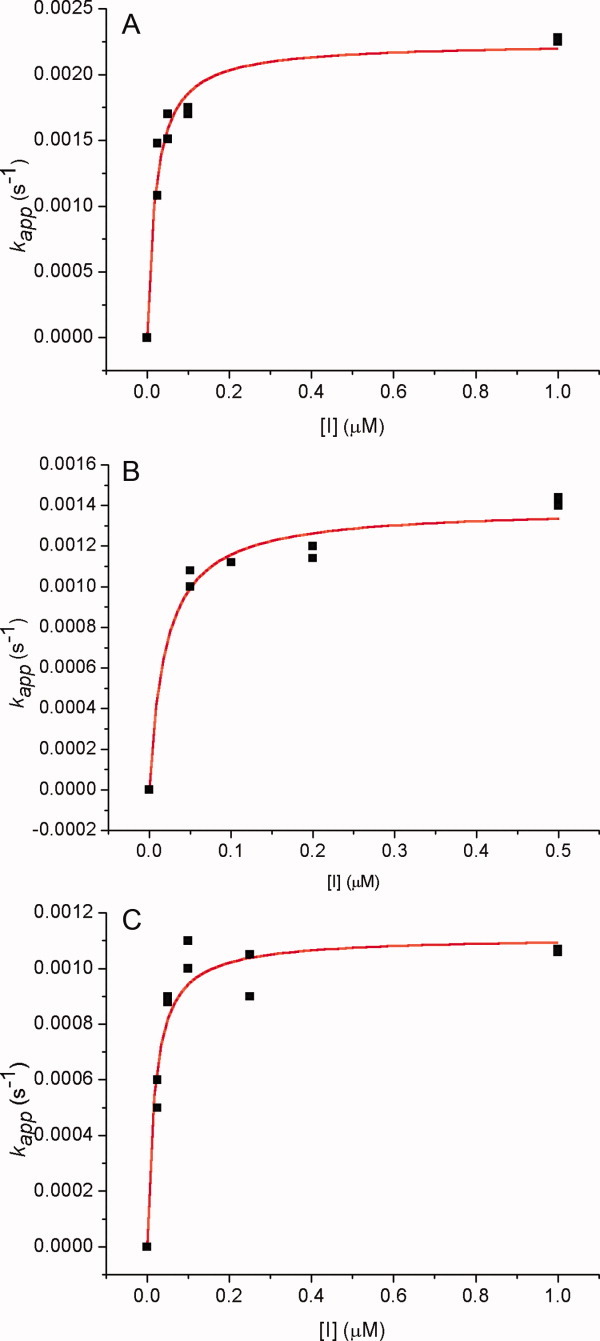
Nonlinear least-squares curve fittings of kapp versus inhibitor concentration ([I]) plots against Eq. (2) for the pseudo-substrate inhibition12–21 of CEase by (A) 1,3,5-tri-N-n-octylcarbamylphloroglucinol (1), (B) 3,5-di-N-n-hexylcarbamyl- oxyphenol (7), and (C) 5-N-n-heptylcarbamyloxyresorcinol (10). For (A), k2 = 0.0027 ± 0.0001 s−1 and Ki = 15 ± 3 nM (R2= 0.965). For (B), k2 = 0.0016 ± 0.0002 s−1 and Ki = 110 ± 40 nM (R2 = 0.913). For C, k2 = 0.0018 ± 0.0002 s−1 and Ki = 60 ± 20 nM (R2 = 0.926). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Discussion
Enhancement of inhibitory potencies by conformationally constrained tridentate inhibitors 1–4
Theoretically, there are at least five stable conformers at the glycerol backbone of TG (Fig. 3). After MM-2 energy minimization, the structures of these five conformers of TG were shown in Figure 4. These structures were all oriented for the sn-1 acyl chain to fit into ACS and for the carbonyl oxygen of the sn-1 acyl chain to fit into the oxyanion hole. Among them, (+g, −g)-TG was the best form to fit into the other parts (SACS and TACS) of the enzyme (Fig. 2). The structure of tridentate inhibitor 1 was similar to that of (+g, −g)-TG. On the other hand, inhibitors 13–17 mimicked TG with five convertible conformers. Therefore, tridentate inhibitors 1–4 were much more potent inhibitors than inhibitors 13–17 due to the fact that the conformations of tridentate inhibitors 1–4 were constrained (loss degree of freedom) that might increase the inhibitory potencies due to Jenck's proximity effect.33, 34
Inhibitory potencies for varied the chain lengths of n-alkylcarbamyl moieties of inhibitors 1–12
When different n-alkylcarbamyl substituents in tridentate inhibitors 1–12 were compared, the inhibitory potency increased with the chain length of the inhibitors (Fig. 7 and Table I). The possible reason was that the long alkyl chain inhibitor bound better than the short one in the ACS of CEase (Fig. 2).
Figure 7.
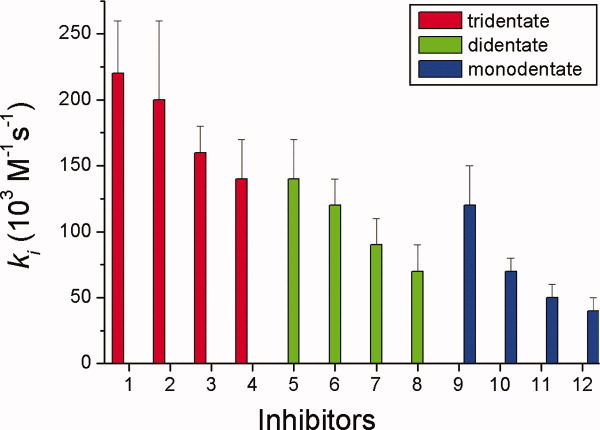
Comparisons of the ki values of pseudo-substrate inhibitions of CEase by tridentate inhibitors 1–4, bidentate inhibitors 5–8, and monodentate inhibitors 9–12. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
QSAR for the CEase by inhibitors 1–12
For log ki and pKi values of tridentate inhibitors 1–4, bidentate inhibitors 5–8, and monodentate inhibitors 9–12, there all existed in linear relationships with the alkyl group chain length of the carbamyl moiety of the inhibitors (n) or the hydrophobicity constant (π), which was defined as n/2 (Fig. 8).39, 40 Thus, one n-alkylcarbamyl group of inhibitors 1–12 might bind to ACS of the enzyme through a common mechanism, and the longer carbamyl groups of the inhibitors might bind better to ACS of the enzyme.
Figure 8.
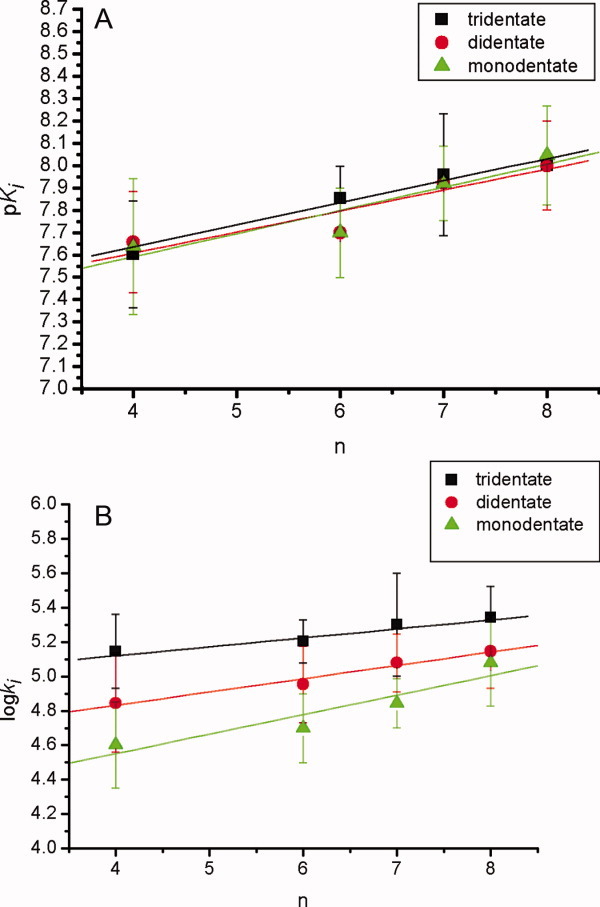
The (A) pKi and (B) log ki values versus carbamyl carbon numbers (n) of inhibitors 1–12 for the pseudo-substrate inhibition of CEase. In (A), linear correlations between the pKi and n values are observed (pKi = 7.24 ± 0.09 + (0.10 ± 0.01)n and R = 0.979 for tridentate inhibitors 1–4; pKi = 7.2 ± 0.2 + (0.10 ± 0.03)n and R = 0.933 for bidentate inhibitors 5–8; pKi = 7.2 ± 0.2 + (0.09 ± 0.32)n and R = 0.916 for monodentate inhibitors 9–12). In (B), linear correlations between the log ki and n values are observed (log ki = 4.91 ± 0.07 + (0.05 ± 0.01)n and R = 0.957 for tridentate inhibitors 1–4; log ki = 4.52 ± 0.06 + (0.077 ± 0.009)n and R = 0.986 for bidentate inhibitors 5–8; log ki = 4.1 ± 0.2 + (0.11 ± 0.03)n and R = 0.934 for monodentate inhibitors 9–12). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
QSAR for the CEase by inhibitors 13–17
For conformationally free analogs inhibitors 13–17, linear correlations between pKi as well as log ki values and σ* were observed, and the slopes of these two correlations were all negative (Fig. 9). The negative slope indicated that the electron-donating substituents accelerated the reaction.18 However, the Ki steps of these reactions were all approaching to negative transition states (noncolvent enzyme-inhibitor Michaelis complexes) that should resemble negative tetrahedral intermediates (enzyme-inhibitor tetrahedral intermediates) and therefore should have a positive ρ* value for the pKi − σ* correlation.18 Therefore, we suggested that the pre-equilibrium protonations of inhibitors 13–17 at the carbamate NHs would result in positive protonated inhibitors and a negative ρ* value (ρ* = −1.5).41 Overall, one carbamate group of inhibitors 13–17 might bind to ACS of CEase through a common mechanism.
Figure 9.
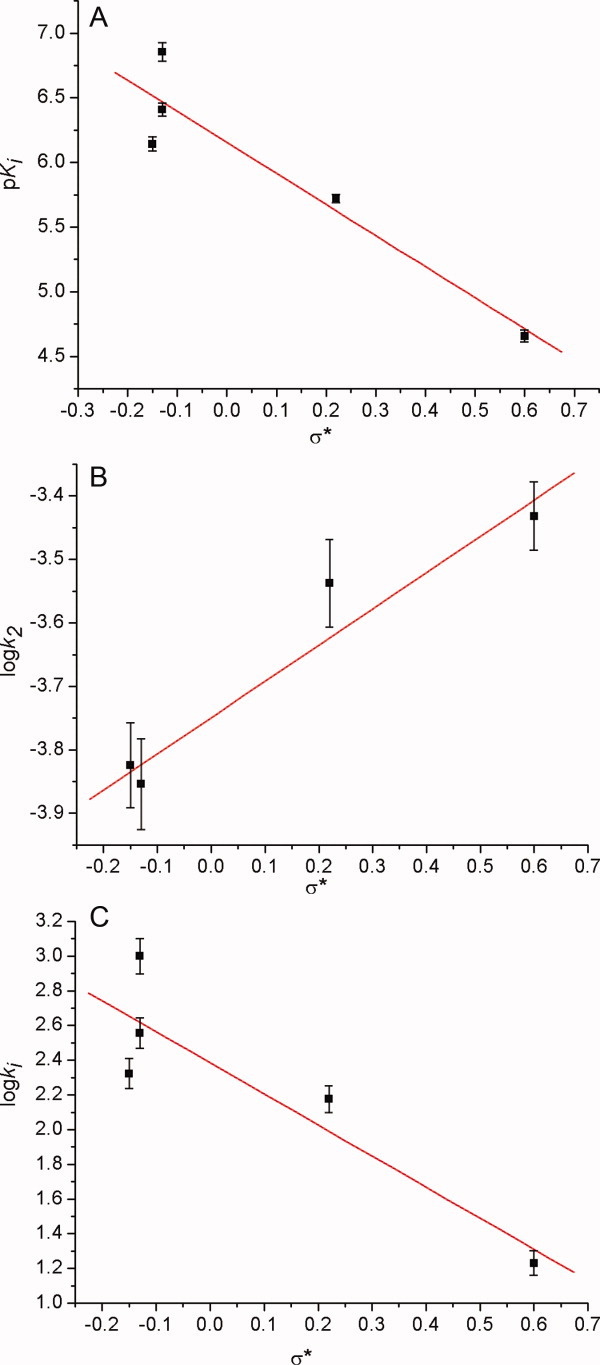
Linear correlations of the (A) pKi, (B) log k2, and (C) log ki values with the substituent constant (σ*) of 1,2,3-tri-N-substituted carbamylglycerols (13–17) for the pseudo-substrate inhibition of CEase. In (A), pKi = 6.1 ± 0.1 – (2.4 ± 0.5)σ* and R = 0.944. In (B), log k2 = 3.75 ± 0.03 + (0.57 ± 0.09)σ* and R = 0.974. In (C), log ki = 2.39 ± 0.04 – (1.8 ± 0.1)σ* and R = 0.929. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The log k2 values for CEase inhibition by inhibitors 12–17 also linearly correlated with σ* with a positive ρ* value of 0.57, indicating that an electron-withdrawing substituents accelerated the reaction, and all inhibitors 13–17 were through a common mechanism in the k2 step.
Conformational analysis of the carbamyl CN bonds of inhibitor 1 by quantum chemistry
Conformational analysis of the carbamyl CN bonds of inhibitor 1 by quantum chemical semiempirical method42 suggested that the most two stable rotamers were (1,3,5)-(trans, trans, trans)- and (1,3,5)-(cis, trans, trans)-tri-carbamate forms (Fig. 10). The energy barrier between these two rotamers was calculated to be 41 kJ/mol by variations of both bond angles and bond lengths from semiempirical method of GAUSSIAN 03.42 This value was relatively small; therefore, these two rotamers were easily convertible to each other at room temperature.43
Figure 10.
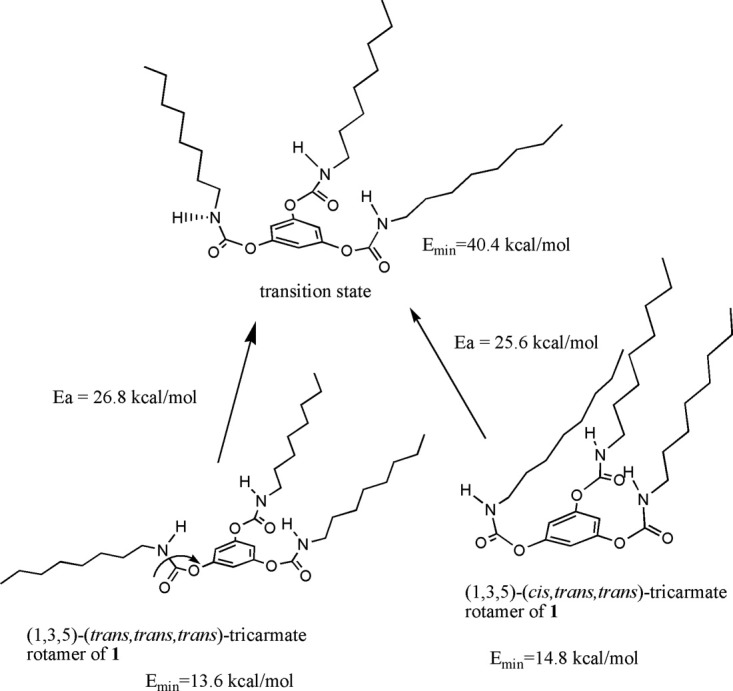
Internal changes between (1,3,5)-(trans, trans, trans)- and (1,3,5)-(cis, trans, trans)-tricarbamate rotamers of inhibitor 1.
Docking of inhibitors 1, 5, and 9 into the X-ray crystal structure of CEase
Molecular docking of inhibitors 1, 5, and 9 with the mode of free rotation around the carbamayl partial CN double bond of the inhibitor into the X-ray crystal structure of CEase6 indicated that the configurations of the inhibitors in ACS of the enzyme-inhibitor complexes were all in the cis-carbamate forms (Figs. 11 and 12). Therefore, it could be predicted that the configurations of the alkylcarbamyl moieties of the inhibitors 1–12 in ACSs of the enzyme-inhibitor complexes were all in the cis forms.
Figure 11.
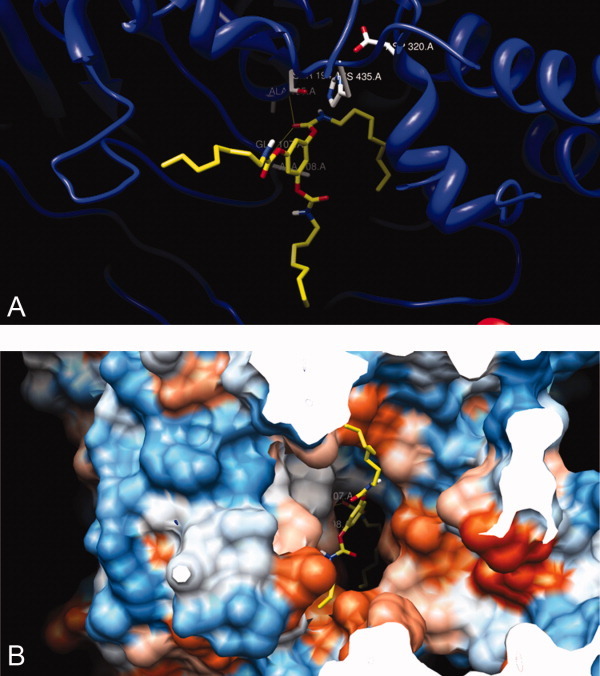
Molecular docking of tridentate inhibitor 1, with the mode of free rotation around the carbamyl CN partial double bond, into the active sites of X-ray crystal structure of CEase6: (A) the active site view and (B) the view from the entrance (mouth) of the enzyme. The configuration of the inhibitor after docked is the (1,3,5)-(cis, trans, trans)-tricarbamate form. The carbamyl carbonyl carbon atom of the inhibitor is close to S194 of the catalytic triad, and the carbamyl ester oxygen atom of the inhibitor is closed to H435 of the catalytic triad. The cis octylcarbamyl moiety of the inhibitor binds to ACS of the enzyme. The other two octylcarbamyl groups of the inhibitor, in the trans forms, bind to SACS and TACS of the enzyme. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 12.
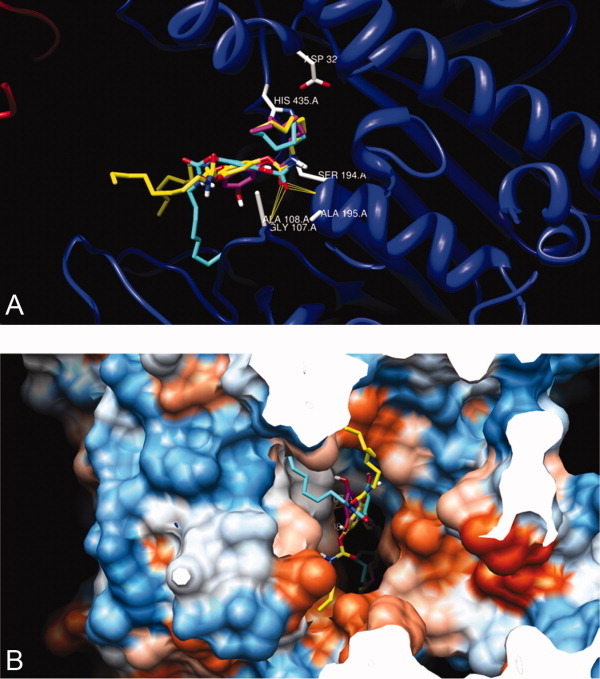
Superimpositions of structures of tridentate 1 (yellow), bidentate 5 (turquoise), and monodentate 9 (mangenta) that have been automatically docked into the X-ray crystal of CEase 1AQL6 by AutoDock program.41, 44–46 View from the active site (A) and from the entrance (mouth) (B) of the enzyme. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The other n-octylcarbamyl group of inhibitor 5 might bind to SACS of the enzyme (Fig. 12). Three carbamyl groups of tridentate inhibitor 1 might bind simultaneously to ACS, SACS, and TACS of the enzyme (Fig. 11). Therefore, tridentate inhibitor 1 might bind to the catalytic triad, oxyanion hole, ACS, SACS, and TACS of CEase and seemed to block completely all possible entrances of the enzyme (Fig. 11). On the other hand, inhibitors 5 and 9 might not completely block all entrances of the enzyme (Fig. 12); therefore, inhibitory potencies were as followed: tridentate inhibitor 1 > bidentate inhibitor 5 > monodentate inhibitor 9.
Conclusion
Tridentate inhibitors 1–4 acted as conformationally constrained analogs of TG and were 200–760-fold more potent inhibitors of CEase than inhibitors 13–17 that were not conformationally restricted at the glycerol backbone. It could therefore be concluded that Jenck's proximity effect played an important role in the CEase inhibition reaction.
Materials and Methods
Materials
Porcine pancreatic CEase (lyophilized powder, activity > 15,000 U/g), substrate p-nitrophenyl butyrate (PNPB), detergent triton-X 100 (TX), 1,3,5-benzene-triol, n-butyl isocyanate, n-hexyl isocyanate, n-heptyl isocyanate, n-octyl isocyanate, phenyl isocyanate, benzyl isocyanate, triethylamine, CDCl3, tetramethylsilane, glycerol, and pyridine were purchased from Sigma-Aldrich, St. Louis, USA. Silica gel and TLC plate were obtained from Merck, Darmstadt, Germany. Hexane, CH2Cl2, ethyl acetate, and tetrahydrofuran were obtained from TEDIA, Fairfield, USA. Sodium dihydrogen phosphate (NaH2PO4:2H2O), disodium hydrogen phosphate (Na2HPO4:12H2O), hydrogen chloride (HCl), sodium hydroxide (NaOH), potassium hydroxide (KOH), calcium chloride (CaCl2), and sodium chloride (NaCl) were purchased from UCW, Taipei, Taiwan. All chemicals and biochemicals were reagent grades (purity >99.9%) and were used directly without further purification.
Synthesis of inhibitors 1–17
1,3,5-Tri-N-alkylcarbamylphloroglucinols (1–4), 3,5-di-N-alkylcarbamyl-oxyphenols (5–8), and 5-N-alkylcarbamyloxyresorcinols (9–12; Fig. 2) were synthesized from the condensation of phloroglucinol with Eq. (2) of the corresponding n-alkylisocyanate and Eq. (2) of triethylamine in tetrahydrofuran at 25°C for 5 h. For example, condensation of phloroglucinol with Eq. (2) of octylisocyanate and Eq. (2) of triethylamine in tetrahydrofuran at 25°C for 5 h yielded 1,3,5-tri-N-n-octyl-carbamylphloroglucinol (1) (26%), 3,5-di-N-n-octylcarbamyloxyphenol (5) (12%), and 5-N-n-octylcarbamyloxyresorcinol (9; 19%) These three products were separated and purified by liquid chromatography (silica gel, hexane-ethyl acetate gradient) and characterized by 1H-, 13C-NMR, and high-resolution mass spectra. 1,2,3-Tri-N-n-octylcarbamylglycerol (13), 1,2,3-tri-N-n-hexylcarbamylglycerol (14), 1,2,3-tri-N-n-butylcarbamylglycerol (15), 1,2,3-tri-N-benzylcarbamylglycerol (16), and 1,2,3-tri-N-phenylcarbamylglycerol (17; Fig. 5) were synthesized from the condensation of glycerol with Eq. (4) of the corresponding isocyanate in pyridine at 25°C for 24 h (60–80% yield). These products were separated and purified by liquid chromatography (silica gel, hexane-ethyl acetate gradient) and characterized by 1H, 13C-NMR, and high-resolution mass spectra.
1,3,5-Tri-N-n-octylcarbamylphloroglucinol (1)
1H-NMR (CDCl3, 400 MHz) δ/ppm 0.88 (t, J = 7 Hz, 9H, ω-CH3), 1.28–1.35 (m, 30H, γ- to ω-1-CH2), 1.50 (m, 6H, β-CH2), 3.20 (dt, J = 6.4 and 13.6 Hz, 6H, α-CH2), 5.29 (t, J = 5.6 Hz, 3H, NH), and 6.82 (s, 3H, benzene-H); 13C-NMR (CDCl3, 100 MHz, assignment from DEPT experiments) δ/ppm 14.0 (ω-CH3), 22.5, 26.7, 29.1, 29.1, 29.6 (γ- to ω-1-CH2), 31.7 (β-CH2), 41.2 (α-CH2), 111.8 (C-2, C-4, and C-6 of benzene ring), 151.4 (C-1, C-3, and C-5 of benzene ring), and 153.8 (carbamate C=O); mass spectra, exact mass: calculated 591.4247, found 591.4242; elemental analysis: calculated for C33H57N3O6: C, 66.97; H, 9.71; N, 7.10, found C, 66.89; H, 9.80; N, 7.05.
1,3,5-Tri-N-n-heptylcarbamylphloroglucinol (2)
1H-NMR (CDCl3, 400 MHz) δ/ppm 0.85 (t, J = 7 Hz, 9H, ω-CH3), 1.28–1.35 (m, 24H, γ- to ω-1-CH2), 1.47 (m, 6H, β-CH2), 3.13 (dt, J = 6.6 and 13.4 Hz, 6H, α-CH2), 5.37 (t, J = 6 Hz, 3H, NH), and 6.77 (s, 3H, benzene-H); 13C-NMR (CDCl3, 100 MHz) δ/ppm 13.9 (ω-CH3), 22.5, 26.6, 28.8, 29.6 (γ- to ω-1-CH2), 31.6 (β-CH2), 41.1 (α-CH2), 111.8 (C-2, C-4, and C-6 of benzene ring), 151.3 (C-1, C-3, and C-5 of benzene ring), and 153.6 (carbamate C=O); mass spectra, exact mass: calculated 549.3778, found 549.3771; elemental analysis: calculated for C30H51N3O6: C, 65.54; H, 9.35; N, 7.64, found C, 65.50; H, 9.53; N, 7.52.
1,3,5-Tri-N-n-hexylcarbamylphloroglucinol (3)
1H-NMR (CDCl3, 400 MHz) δ/ppm 0.86 (t, J = 7 Hz, 9H, ω-CH3), 1.28–1.34 (m, 18H, γ- to ω-1-CH2), 1.48 (m, 6H, β-CH2), 3.42 (dt, J = 6.6 and 13.4 Hz, 6H, α-CH2), 5.30 (t, J = 6 Hz, 3H, NH), and 6.78 (s, 3H, benzene-H). 13C-NMR (CDCl3, 100 MHz) δ/ppm 13.9 (ω-CH3), 22.4, 26.3, 30.0 (γ- to ω-1-CH2), 31.3 (β-CH2), 41.2 (α-CH2), 111.8 (C-2, C-4, and C-6 of benzene ring), 151.4 (C-1, C-3, and C-5 of benzene ring), and 153.8 (carbamate C=O); mass spectra, exact mass: calculated 507.3308, found 507.3302; elemental analysis: calculated for C27H45N3O6: C, 63.88; H,8.93; N, 8.28, found C, 63.79; H, 9.01; N, 8.19.
1,3,5-Tri-N-n-butylcarbamylphloroglucinol (4)
1H-NMR (CDCl3, 400 MHz) δ/ppm 0.90 (t, J = 7 Hz, 9H, ω-CH3), 1.28 (sextet, J = 7 Hz, 6H, γ-CH2), 1.45 (quintet, J = 7 Hz, 6H, β-CH2), 3.18 (dt, J = 6.6 and 13.4 Hz, 6H, α-CH2), 5.19 (t, J = 6 Hz, 3H, NH), and 6.80 (s, 3H, benzene-H). 13C-NMR (CDCl3, 100 MHz) δ/ppm 13.9 (ω-CH3), 20.1 (γ-CH2), 32.0 (β-CH2), 41.1 (α-CH2), 112.1 (C-2, C-4, and C-6 of benzene ring), 151.7 (C-1, C-3, and C-5 of benzene ring), and 154.1 (carbamate C=O); mass spectra, exact mass: calculated 423.2369, found 424.2364; elemental analysis: calculated for C21H33N3O6: C, 59.56; H,7.85; N, 9.92, found C, 59.51; H, 7.90; N, 9.83.
3,5-Di-N-n-octylcarbamyloxyphenol (5)
1H-NMR (CDCl3, 400 MHz) δ/ppm 0.86 (t, J = 7 Hz, 6H, ω-CH3), 1.28–1.35 (m, 20H, γ- to ω-1-CH2), 1.50 (m, 4H, β-CH2), 3.18 (dt, J = 6.6 and 13.4 Hz, 4H, α-CH2), 5.44 (t, J = 6.0 Hz, 2H, NH), 6.26–6.40 (m, 3H, benzene-H), and 6.42 (s, 1H, 1-OH). 13C-NMR (CDCl3, 100 MHz, assignment from DEPT experiments) δ/ppm 14.0 (ω-CH3), 22.5, 26.7, 28.9, 29.1, 29.6 (γ- to ω-1-CH2), 31.7(β-CH2), 41.1 (α-CH2), 106.5 (C-2 and C-4 of benzene ring), 106.8 (C-6 of benzene ring), 151.2 (C-3 and C-5 of benzene ring), 154.8 (C-1 of benzene ring) and 157.7 (carbamate C=O); mass spectra, exact mass: calculated 436.2937, found 436.2932; elemental analysis: calculated for C24H40N2O5: C, 66.03; H, 9.23; N, 6.42, found C, 65.96; H, 9.57; N, 6.31.
3,5-Di-N-n-heptylcarbamyloxyphenol (6)
1H-NMR (CDCl3, 400 MHz) δ/ppm 0.86 (t, J = 7 Hz, 6H, ω-CH3), 1.28–1.34 (m, 16H, γ- to ω-1-CH2), 1.50 (m, 4H, β-CH2), 3.18 (dt, J = 6.6 and 13.4 Hz, 4H, α-CH2), 5.44 (t, J = 6.0 Hz, 2H, NH), 6.22–6.24 (m, 3H, benzene-H), and 6.34 (s, 1H, 1-OH). 13C-NMR (CDCl3, 100 MHz, assignment from DEPT experiments) δ/ppm 14.0 (ω-CH3), 22.5, 26.7, 28.9, 29.6 (γ- to ω-1-CH2), 31.7 (β-CH2), 41.1 (α-CH2), 106.5 (C-2 and C-4 of benzene ring), 106.8 (C-6 of benzene ring), 151.2 (C-3 and C-5 of benzene ring), 154.8 (C-1 of benzene ring), and 157.7 (carbamate C=O); mass spectra, exact mass: calculated 408.2624, found 408.2618; elemental analysis: calculated for C22H36N2O5: C, 64.68; H, 8.88; N, 6.86, found C, 64.60; H, 9.01; N, 6.53.
3,5-Di-N-n-hexylcarbamyloxyphenol (7)
1H-NMR ((CD3)2SO, 400 MHz) δ/ppm 0.86 (t, J = 7 Hz, 6H, ω-CH3), 1.28–1.34 (m, 12H, γ- to ω-1-CH2), 1.44 (m, 4H, β-CH2), 3.02 (dt, J = 7 and 13 Hz, 4H, α-CH2), 6.26–6.32 (m, 3H, benzene-H), and 7.68 (t, J = 5.6 Hz, 2H, NH), and 9.79 (s, 1H, 1-OH); 13C-NMR ((CD3)2SO, 100 MHz, assignment from DEPT experiments) δ/ppm 14.0 (ω-CH3), 22.5, 26.7, 28.9, 29.6 (γ- to ω-1-CH2), 31.7 (β-CH2), 41.1 (α-CH2), 106.5 (C-2 and C-4 of benzene ring), 106.8 (C-6 of benzene ring), 151.2 (C-3 and C-5 of benzene ring), 154.8 (C-1 of benzene ring), and 157.7 (carbamate C=O); mass spectra, exact mass: calculated 380.2311, found 380.2306; elemental analysis: calculated for C20H32N2O5: C, 63.13; H, 8.48; N, 7.36, found C, 63.02; H, 8.62; N, 7.31.
3,5-Di-N-n-butylcarbamyloxyphenol (8)
1H-NMR ((CD3)2SO, 400 MHz) δ/ppm 0.89 (t, J = 7 Hz, 6H, ω-CH3), 1.29 (sextet, J = 7 Hz, 4H, γ-CH2), 1.44 (quintet, J = 7 Hz, 4H, β-CH2), 3.08 (dt, J = 6.6 and 13.2 Hz, 4H, α-CH2), 6.27–6.33 (m, 3H, benzene-H), 7.70 (t, J = 5.6 Hz, 3H, NH), and 9.80 (s, 1H, 1-OH); 13C-NMR ((CD3)2SO, 100 MHz) δ/ppm 13.5 (ω-CH3), 19.4 (γ-CH2), 31.3 (β-CH2), 40.1 (α-CH2), 105.4 (C-2 and C-6 of benzene ring), 105.8 (C-4 of benzene ring), 152.1 (C-1 of benzene ring), 153.9 (C-3 and C-5 of benzene ring), and 158.1 (carbamate C=O); mass spectra, exact mass: calculated 324.1685, found 324.1680; elemental analysis: calculated for C16H24N2O5: C, 59.24; H, 7.46; N, 8.64, found C, 59.11; H, 7.58; N, 8.37.
5-N-n-Octylcarbamyloxyresorcinol (9)
1H-NMR ((CD3)2SO, 400 MHz) δ/ppm 0.90 (t, J = 7 Hz, 3H, ω-CH3), 1.28–1.35 (m, 10H, γ- to ω-1-CH2), 1.52 (m, 2H, β-CH2), 3.21 (t, J = 6.8 Hz, 2H, α-CH2), 4.63 (s, 1H, NH), 4.90 (s, 2H, 1-OH), 6.03–6.11 (m, 3H, benzene-H); 13C-NMR ((CD3)2SO, 100 MHz, assignment from DEPT experiments) δ/ppm 14.4 (ω-CH3), 23.6, 27.8, 30.1, 30.7, 31.8 (γ- to ω-1-CH2), 32.9 (β-CH2), 42.0 (α-CH2), 100.7 (C-2 of benzene ring), 101.6 (C-4 and C-6 of benzene ring), 154.1 (C-5 of benzene ring), and 157.2 (carbamate C=O), and 159.9 (C-3 and C-5 of benzene ring); mass spectra, exact mass: calculated 281.1627, found 281.1622; elemental analysis: calculated for C15H23NO4: C, 64.03; H, 8.24; N, 4.98, found C, 63.91; H, 8.37; N, 4.86.
5-N-n-Heptylcarbamyloxyresorcinol (10)
1H-NMR ((CD3)2SO, 400 MHz) δ/ppm 0.89 (t, J = 7 Hz, 3H, ω-CH3), 1.29–1.35 (m, 8H, γ- to ω-1-CH2), 1.52 (m, 2H, β-CH2), 3.20 (t, J = 6.8 Hz, 2H, α-CH2), 4.63 (s, 1H, NH), 4.89 (s, 2H, 1-OH), and 6.03–6.11 (m, 3H, benzene-H); 13C-NMR ((CD3)2SO, 100 MHz, assignment from DEPT experiments) δ/ppm 14.4 (ω-CH3), 23.6, 27.8, 30.7, 31.7 (γ- to ω-1-CH2), 32.9 (β-CH2), 42.0 (α-CH2), 100.7 (C-2 of benzene ring), 101.6 (C-4 and C-6 of benzene ring), 154.1 (C-5 of benzene ring), 157.2 (carbamate C=O), and 159.9 (C-3 and C-5 of benzene ring); mass spectra, exact mass: calculated 267.164, found 267.158; elemental analysis: calculated for C14H21NO4: C, 62.90; H, 7.92; N, 5.24, found C, 62.78; H, 8.02; N, 5.08.
5-N-n-Hexylcarbamyloxyresorcinol (11)
1H-NMR ((CD3)2SO, 400 MHz) δ/ppm 0.86 (t, J = 7 Hz, 3H, ω-CH3), 1.29–1.35 (m, 6H, γ- to ω-1-CH2), 1.43 (m, 2H, β-CH2), 3.00 (dt, J = 5.6 and 6.8 Hz, 2H, α-CH2), 5.92 (s, 2H, 4,6-benzene-H), 6.03 (s, 1H, 2-benzene-H), 7.56 (t, J = 5.2 Hz, 1H, NH), and 9.37 (s, 2H, 1,3-OH); 13C-NMR ((CD3)2SO, 100 MHz, assignment from DEPT experiments) δ/ppm 14.7 (ω-CH3), 22.7, 26.6, 29.9 (γ- to ω-1-CH2), 31.6 (β-CH2), 41.1 (α-CH2), 99.9 (C-2 of benzene ring), 100.7 (C-4 and C-6 of benzene ring), 153.3(carbamate C=O), 154.8 (C-5 of benzene ring), and 159.2 (C-3 and C-5 of benzene ring); mass spectra, exact mass: calculated 253.1314, found 253.1308; elemental analysis: calculated for C13H19NO4: C, 61.64; H, 7.56; N, 5.53, found C, 61.53; H, 7.64; N, 5.42.
5-N-n-Butylcarbamyloxyresorcinol (12)
1H-NMR ((CD3)2SO, 400 MHz) δ/ppm 0.90 (t, J = 7 Hz, 3H, ω-CH3), 1.29 (sextet, J = 7 Hz, 2H, γ-CH2), 1.44 (quintet, J = 7 Hz, 2H, β-CH2), 3.06 (dt, J = 6.4 and 6.8 Hz, 2H, α-CH2), 5.93 (s, 2H, 4,6-benzene-H), 6.04 (s, 1H, 2-benzene-H), 7.57 (t, J = 5.2 Hz, 1H, NH), and 9.36 (s, 2H, 1,3-OH); 13C-NMR ((CD3)2SO, 100 MHz, assignment from DEPT experiments) δ/ppm 13.551 (ω-CH3), 19.4 (γ-CH2), 31.3 (β-CH2), 40.3 (α-CH2), 105.0 (C-2 of benzene ring), 106.0 (C-4 and C-6 of benzene ring), 155.6 (carbamate C=O), 156.0 (C-5 of benzene ring), and 162.0 (C-3 and C-5 of benzene ring); mass spectra, exact mass: calculated 225.1001, found 225.0095; elemental analysis: calculated for C11H15NO4: C, 58.66; H, 6.71; N, 6.22, found C,58.58; H, 6.90; N, 6.13.
1,2,3-Tri-N-n-octylcarbamylglycerol (13)
1H-NMR (CDCl3, 400 MHz) δ/ppm 0.88 (t, J = 7 Hz, 9H, ω-CH3), 1.28–1.35 (m, 30H, γ- to ω-1-CH2), 1.48 (m, 6H, β-CH2), 3.14 (t, J = 7 Hz, 6H, α-CH2), 4.20 (d, J = 5.1 Hz, 4H, sn-1- and sn-3-CH2), 4.79 (m, 3H, NH), and 5.12 (m, 1H, sn-2-CH); 13C-NMR (CDCl3, 100 MHz, assignment from DEPT experiments) δ/ppm 14.0 (ω-CH3), 22.6, 26.9, 29.3, 29.8, 30.2 (γ- to ω-1-CH2), 31.7 (β-CH2), 40.5 (α-CH2), 63.0 (sn-1- and sn-3-CH2), 70.2 (sn-2-CH), and 158.6 (carbamate C=O); mass spectra, exact mass: calculated 557.4404, found 557.4411; elemental analysis: calculated for C30H59N3O6: C, 64.60; H, 10.66; N, 7.53, found C, 64.43; H, 10.81; N, 7.46.
1,2,3-Tri-N-n-hexylcarbamylglycerol (14)
1H-NMR (CDCl3, 400 MHz) δ/ppm 0.88 (t, J = 7 Hz, 9H, ω-CH3), 1.28–1.34 (m, 18H, γ- to ω-1-CH2), 1.48 (m, 6H, β-CH2), 3.15 (t, J = 7 Hz, 6H, α-CH2), 4.20 (d, J = 4.8 Hz, 4H, sn-1- and sn-3-CH2), 4.36 (m, 3H, NH), and 5.11 (m, 1H, sn-2-CH). 13C-NMR (CDCl3, 100 MHz) δ/ppm 13.9 (ω-CH3), 22.5, 26.3, 29.7 (γ- to ω-1-CH2), 31.5 (β-CH2), 40.4 (α-CH2), 63.0 (sn-1- and sn-3-CH2), 70.2 (sn-2-CH), and 158.7 (carbamate C=O); mass spectra, exact mass: calculated 473.3465, found 473.3453; elemental analysis: calculated for C24H47N3O6: C, 60.86; H,10.00; N, 8.87, found C, 60.79; H, 10.09; N, 8.77.
1,2,3-Tri-N-n-butylcarbamylglycerol (15)
1H-NMR (CDCl3, 400 MHz) δ/ppm 0.92 (t, J = 7 Hz, 9H, ω-CH3), 1.35 (sextet, J = 7 Hz, 6H, γ-CH2), 1.47 (quintet, J = 7 Hz, 6H, β-CH2), 3.16 (dt, J = 7 and 13 Hz, 6H, α-CH2), 4.20 (d, J = 4.8 Hz, 4H, sn-1- and sn-3-CH2), 4.76 (m, 3H, NH), and 5.12 (m, 1H, sn-2-CH). 13C-NMR (CDCl3, 100 MHz) δ/ppm 13.6 (ω-CH3), 19.997 (γ-CH2), 31.9 (β-CH2), 40.7 (α-CH2), 63.0 (sn-1- and sn-3-CH2), 70.3 (sn-2-CH), and 156.0 (carbamate C=O); mass spectra, exact mass: calculated 389.2526, found 389.2521; elemental analysis: calculated for C18H35N3O6: C, 55.51; H, 9.06; N, 10.79, found C, 55.42; H, 9.11; N, 10.70.
1,2,3-Tri-N-benzylcarbamylglycerol (16)
1H-NMR (CDCl3, 400 MHz) δ/ppm 3.96 (m, 6H, CH2 Ph), 4.19, 4.24 (d, J = 6 Hz, 4H, sn-1- and sn-3-CH2), 5.18 (d, J = 5 Hz, 1H, sn-2-CH), 6.46 (t, J = 5 Hz, 3H, NH), and 7.25–7.32 (m, C6H5, 15H). 13C-NMR (CDCl3, 100 MHz) δ/ppm 43.0 (CH2C6H5), 65.3 (sn-1- and sn-3-CH2), 67.0 (sn-2-CH), 126.7, 126.8, 128.3, 139.9, 141.0 (C6H5), and 158.2 (carbamate C=O); mass spectra, exact mass: calculated 491.2056, found 491.2050; elemental analysis: calculated for C27H29N3O6: C, 65.97; H, 5.95; N, 8.55, found C, 65.83; H, 6.01; N, 8.50.
1,2,3-Tri-N-phenylcarbamylglycerol (17)
1H-NMR (CDCl3, 400 MHz) δ/ppm 4.28 (m, 4H, sn-1- and sn-3-CH2), 4.50 (m, 1H, sn-2-CH), 5.12 (m, 3H, NH), and 7.25–7.32 (m, C6H5, 15H). 13C-NMR (CDCl3, 100 MHz) δ/ppm 62.5 (sn-1- and sn-3-CH2), 70.3 (sn-2-CH), 118.2, 122.7, 128.9, 139.0 (C6H5), and 153.2 (carbamate C=O); mass spectra, exact mass: calculated 449.1587, found 449.1593; elemental analysis: calculated for C24H23N3O6: C, 64.13; H, 5.16; N, 9.35, found C, 64.01; H, 5.22; N, 9.27.
Instrumental methods
All steady-state kinetic data were obtained from a UV–vis spectrophotometer (Agilent 8453) with a cell holder circulated with a water bath. 1H and 13C-NMR spectra were recorded in CDCl3 at 400 and 100 MHz, respectively, with an internal reference tetramethylsilane at 25°C on a Varian Gemini 400 spectrometer. Mass spectra (EI) were recorded at 71 eV in a mass spectrometer (Joel JMS-SX/SX 102A). Elemental analyses were performed on a Heraeus instrument.
Data reduction
Origin (version 6.0) was used for linear and nonlinear least-square curve fittings. Data followed by ± signs were standard deviations obtained from least-squares curve fittings with n (number of separate experiments) >9.
CEase inhibition
CEase inhibition reactions were determined as described by Hosie et al.12–14 CEase-catalyzed hydrolysis of PNPB in the presence of a carbamate inhibitor was followed continuously at 410 nm on the UV–vis spectrometer. The temperature was maintained at 25.0°C by a refrigerated circulating water bath. All reactions were performed in sodium phosphate buffer (1 mL, 0.1 M, pH 7.0) containing NaCl (0.1 M), CH3CN (2% by volume), detergent triton-X 100 (TX; 0.5% by weight), substrate PNPB (0.1 mM), and varying concentration of the inhibitors. Requisite volumes of stock solution of substrate PNPB and the inhibitor in acetonitrile were injected into reaction buffer via a pipet. CEase was dissolved in sodium phosphate buffer (0.1 M, pH 7.0). The reaction was followed until 85% of substrate consumption was completed. Inhibitors 1–17 were all characterized as the pseudo or alternate substrate inhibitors of CEase (Fig. 1).12–21
In the presence of a carbamate inhibitor, time courses for hydrolysis of PNPB are biphasic, and kapp values can be calculated as Eq. (1).12–14
| (1) |
In Eq. (1), A0, kapp, vo, and vss are the absorbance at t = 0, the observed first-order inhibition rate constant, the initial velocity, and the steady-state velocity, respectively.
The carbamylation stage was rapid compared to subsequent decarbamylation (k2 >> k3); thus, the two steps are easily resolved kinetically. The apparent inhibition constant (1 + [S]/Km) Ki and carbamylation constant (k2) were obtained from the nonlinear least-square curve fitting of the kapp versus [I] plot against Eq. (2) (Fig. 6). The inhibition constant Ki was then calculated from the apparent inhibition constant when both [S] and Km values for the CEase-catalyzed hydrolysis of PNPB were known (Tables I and II). The Km value for the CEase catalyzed hydrolysis of PNPB was 100 ± 20 μM obtained from Michaelis–Menten equation. The bimolecular rate constant, ki = k2/Ki, was related to overall inhibitory potency.
| (2) |
Duplicate sets of data were collected for each inhibitor concentration.
Molecular modeling
Molecular structures of tridentate inhibitor 1, TG, cholesterol ester shown in Figures 1 and 4 were depicted from the molecular structures after MM-2 energy minimization (minimum root mean square gradient was set to be 0.01) by CS Chem 3D (version 6.0).
Conformational analysis of inhibitor 1
Conformational analysis of inhibitor 1 was performed by energy minimization of each conformer from the semiempirical method of GAUSSIAN 03 (Fig. 10).42 Conformational analysis of TG was made by the MM-2 energy minimization of CS Chem 3D (version 6.0; Fig. 4).
Automated docking inhibitors into CEase
Molecular structures of (1,3,5)-(cis, trans, trans)- and (trans, trans, trans)- tricarbamate rotamers of inhibitor 1 were depicted from the molecular structures after MM-2 energy minimization (minimum root mean square gradient was set to be 0.01) by CS Chem 3D (version 6.0). Of all the entries for lipase from RCSB protein data bank, CEase (1AQL) [6] was taken for docking analysis. Protein-inhibitor docking studies were performed to evaluate the algorithm and scoring function efficiency between a standalone AutoDock 4.241, 44–46 and experimental activities. Automated docking was used to locate the appropriate binding orientations and conformations of various inhibitors in the 1AQL-binding pocket. All water molecules were removed from the original Protein Data Bank file. Polar hydrogen atoms were added, and Kollman charge, atomic solvation parameters, and fragmental volumes were assigned to the protein using Auto Dock Tools. For docking calculations, Gasteiger partial charges were assigned to the tested derivatives, and nonpolar hydrogen atoms were merged. The torsions of C–O and carbamate CN partial double bonds were allowed to rotate during docking. The program AutoGrid used to generate the grid maps. Each grid was centered at the crystal structure of the corresponding 1AQL. The grid dimensions were 40 × 40 × 40 Å3 with points separated by 0.375 Å. For all inhibitors, random starting positions, random orientations, and torsions were used. The translation, quaternion, and torsion steps were taken from default values in Auto Dock. The Lamarckian genetic algorithm and the pseudo-Solids and Wets methods were applied for minimization using default parameters. The standard docking protocol for rigid and flexible inhibitor docking consisted of 50 independent runs per inhibitor, using an initial population of 150 randomly placed individuals, with 2.5 × 106 energy evaluations, a maximum number of 27,000 iterations, a mutation rate of 0.02, a crossover rate of 0.80, and an elitism value of 1. The probability of performing a local search on an individual in the population was 0.06, using a maximum of 300 iterations per local search. After docking, the 10 solutions were clustered into groups with RMS deviations lower than 1.0 Å. The clusters were ranked by the lowest energy representative of each cluster. The interactive visualization and analysis of molecular structures and hydrogen bonds between protein and inhibitor were performed by UCSF Chimera.47
Glossary
Abbreviations
- a
anti
- (a, +g)
sn-1 and sn-2 oxygen atoms are in anticonformation; sn-2 and sn-3 oxygen atoms are in gauche conformation
- ACS
acyl chain-binding site
- CEase
cholesterol esterase
- +g
gauche conformation with a positive dihedral angle
- −g
gauche conformation with a negative dihedral angle
- LDL
low-density lipoprotein
- LGS
leaving group-binding site
- PC
phosphatidylcholine
- PNPB
p-nitrophenyl butyrate
- QSAR
quantitative structure activity relationship
- SACS
second acyl chain-binding site
- TACS
third acyl chain-binding site
- TG
triacylglycerol
- TX
triton-X 100
References
- 1.Hui DY. Molecular biology of enzymes involved with cholesterol esterase hydrolysis in mammalian tissues. Biochim Biophys Acta. 1996;1303:1–14. doi: 10.1016/0005-2760(96)00085-9. [DOI] [PubMed] [Google Scholar]
- 2.Lopewz-Candales A, Bosner MS, Spilburg CA, Lange LG. Cholesterol transport function of pancreatic cholesterol esterase: directed sterol uptake and esterification in entercytes. Biochemistry. 1993;32:12085–12089. doi: 10.1021/bi00096a019. [DOI] [PubMed] [Google Scholar]
- 3.Brockerhoff H, Jensen RG. Lipolytic enzymes. New York: Academic Press; 1974. Cholesterol esterase; pp. 176–193. [Google Scholar]
- 4.Brodt-Eppley J, White P, Jenkins S, Hui D. Plasma cholesterol esterase level is a determinant for an atherogenic lipoprotein profile in normolipidemic human subjects. Biochim Biophys Acta. 1995;1272:69–72. doi: 10.1016/0925-4439(95)00083-g. [DOI] [PubMed] [Google Scholar]
- 5.Chen JCH, Miercke LJW, Krucinski J, Starr JR, Saenz G, Wang X, Spilburg CA, Lange LG, Ellsworth JL, Stroud RM. Structure of bovine pancreatic cholesterol esterase at 1.6 Å: novel structural features involved in lipase activation. Biochemistry. 1998;37:5107–5117. doi: 10.1021/bi972989g. [DOI] [PubMed] [Google Scholar]
- 6.Wang X, Wang CS, Tang J, Dyda F, Zhang XC. The crystal structure of bovine bile salt activated lipase: insights into the bile salt activation mechanism. Structure. 1997;5:1209–1218. doi: 10.1016/s0969-2126(97)00271-2. [DOI] [PubMed] [Google Scholar]
- 7.Cygler M, Schrag JD, Sussman JL, Harel M, Silman I, Gentry MK, Doctor BP. Relationship between sequence conservation and three-dimensional structure in a large family of esterases, lipases, and related proteins. Protein Sci. 1993;2:366–382. doi: 10.1002/pro.5560020309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow FSM, Harel M, Remington SJ, Silman I, Schrag JD, Sussman JL, Vershueren KHG, Goldman A. The α/β hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 9.Svendsen A, Lipases . In: Their structure biochemistry and application. Woolley P, Petersen SB, editors. Cambridge, UK: Cambridge University Pres; 1994. pp. 1–21. [Google Scholar]
- 10.Chiou SY, Lai GW, Lin LY, Lin G. Kinetics and mechanisms of cholesterol esterase inhibition by cardiovascular drugs in vitro. Ind J Biochem Biophys. 2006;43:52–55. [PubMed] [Google Scholar]
- 11.Pioruńska-Stolzmann M, Piroruńska-Mikołajczak A. The influence of simvastatin on lipase and cholesterol esterase activity in the serum of men with coronary heart disease. Pharm Res. 2002;43:359–362. doi: 10.1006/phrs.2000.0787. [DOI] [PubMed] [Google Scholar]
- 12.Hosie L, Sutton LD, Quinn DM. p-Nitrophenyl and cholesteryl-N-alkyl carbamates as inhibitors of cholesterol esterase. J Biol Chem. 1987;262:260–264. [PubMed] [Google Scholar]
- 13.Feaster SR, Lee K, Baker N, Hui DY, Quinn DM. Molecular recognition by cholesterol esterase of active site ligands: structure-reactivity effects for inhibition by aryl carbamates and subsequent carbamylenzyme turnover. Biochemistry. 1996;35:16723–16734. doi: 10.1021/bi961677v. [DOI] [PubMed] [Google Scholar]
- 14.Feaster SR, Quinn DM. Mechanism-based inhibitors of mammalian cholesterol esterase. Methods Enzymol. 1997;286:231–252. doi: 10.1016/s0076-6879(97)86013-2. [DOI] [PubMed] [Google Scholar]
- 15.Lin G, Shieh CT, Ho HC, Chouhwang JY, Lin WY, Lu CP. Structure-reactivity relationships for the inhibition mechanism at the second alkyl chain binding site of cholesterol esterase and lipase. Biochemistry. 1999;38:9971–9981. doi: 10.1021/bi982775e. [DOI] [PubMed] [Google Scholar]
- 16.Lin G, Shieh CT, Tsai YC, Hwang CI, Lu CP. Structure-reactivity probes for active site shapes of cholesterol esterase by carbamate inhibitors. Biochim Biophys Acta. 1999;1431:500–511. doi: 10.1016/s0167-4838(99)00073-4. [DOI] [PubMed] [Google Scholar]
- 17.Lin G, Lee YR, Liu YC, Wu YG. Ortho effects for inhibition mechanisms of butyrylcholinesterase by o-substituted phenyl N-butylcarbamates and comparison with acetylcholinesterase, cholesterol esterase, and lipase. Chem Res Toxicol. 2005;18:1124–1131. doi: 10.1021/tx050014o. [DOI] [PubMed] [Google Scholar]
- 18.Lin G, Lai CY. Hammett analysis of the inhibition of pancreatic cholesterol esterase by substituted phenyl-N-butylcarbamate. Tetrahedron Lett. 1995;36:6117–6120. [Google Scholar]
- 19.Lin G, Lai CY. Linear free energy relationships of the inhibition of pancreatic cholesterol esterase by 4-nitrophenyl-N-alkylcarbamate. Tetrahedron Lett. 1996;37:193–196. [Google Scholar]
- 20.Pietsch M, Gütschow M. Alternate substrate inhibition of cholesterol esterase by thieno[2,3-d][1,3]oxazin-4-ones. J Biol Chem. 2002;277:24006–24013. doi: 10.1074/jbc.M112252200. [DOI] [PubMed] [Google Scholar]
- 21.Pietsch M, Gütschow M. Synthesis of tricyclic 1,3-oxazin-4-ones and kinetic analysis of cholesterol esterase and acetylcholinesterase inhibition. J Med Chem. 2005;48:8270–8288. doi: 10.1021/jm0508639. [DOI] [PubMed] [Google Scholar]
- 22.Quinn DM, Sutton LD, Stout JS, Calogeropoulou T, Wiemer DF. Phospholipase A2 activity and catalytic mechanism of pancreatic cholesterol esterase. Phosphorus Sulfur Silicon. 1990;51/52:43–46. [Google Scholar]
- 23.Lin G, Noel J, Loffredo W, Sable HZ, Tsai MD. Use of short-chain cyclopentano phosphatidylcholines to probe the mode of activation of phospholipase A2 from bovine pancreas and bee venom. J Biol Chem. 1988;263:13208–13214. [PubMed] [Google Scholar]
- 24.Lin G, Chen GH, Ho HC. Conformationally restricted carbamate inhibitors of horse serum butyrylcholinesterase. Bioorg Med Chem Lett. 1998;8:2747–2750. doi: 10.1016/s0960-894x(98)00484-3. [DOI] [PubMed] [Google Scholar]
- 25.Lin G, Yu GY. QSAR for phospholipase A2 inhibitions by 1-acyloxy-3-Nn-octylcarbamylbenzenes. Bioorg Med Chem Lett. 2005;15:2405–2408. doi: 10.1016/j.bmcl.2005.02.092. [DOI] [PubMed] [Google Scholar]
- 26.Lin G, Lai FH, Tsai BI, Hsieh CW, Tsai HJ. Probing conformations of the glycerol backbones of triacyglycerols in the active site of lipase by 1,2-cyclopentanecarbamates: the meso effect for the enzyme inhibition. J Mol Catal B Enzym. 2006;40:86–92. [Google Scholar]
- 27.Lin MC, Hwang MT, Chang HG, Lin CS, Lin G. Benzene-1,2-, 1,3-, and 1,4-di-N-substituted carbamates as conformationally constrained of inhibitors of acetylcholinesterase. J Biochem Mol Toxicol. 2007;21:348–353. doi: 10.1002/jbt.20202. [DOI] [PubMed] [Google Scholar]
- 28.Lin MC, Hwang MT, Chang HG, Lin CS, Lin G. Benzene-di-N-substituted-carbamates as conformationally constrained analogs of lipase substrate. Eur J Lipid Sci Technol. 2007;109:1104–1110. [Google Scholar]
- 29.Chiou SY, Chen YR, Lu CP, Lin LY, Lin G. The conformational freeze for the ethylene glycol backbone of 1,2-ethylene-di-Nn-propylcarbamate in triton X-100 mixed micelle. J Phys Chem C. 2008;112:2325–2329. [Google Scholar]
- 30.Chiou SY, Lin MC, Hwang MT, Chang HG, Lin G. Benzene-di-N-substituted carbamates as conformationally constrained substrate analogs of cholesterol esterase. Protein J. 2008;27:274–282. doi: 10.1007/s10930-008-9135-2. [DOI] [PubMed] [Google Scholar]
- 31.Chiou SY, Huang CF, Hwang MT, Lin G. Comparison of active sites of butyrylcholinesterase and acetylcholinesterase based on inhibition by geometric isomers of benzene-di-N-substituted carbamates. J Biochem Mol Toxicol. 2009;23:303–308. doi: 10.1002/jbt.20286. [DOI] [PubMed] [Google Scholar]
- 32.Chiou SY, Yu GY, Lin G. QSAR for inhibition of Pseudomonas species lipase by 1-acyloxy-3-Nn-octylcarbamyl-benzenes. QSAR Comb Sci. 2009;28:267–273. [Google Scholar]
- 33.Jencks WP. Economics of enzyme catalysis. Cold Spring Harbor Symb Quant Biol. 1987;52:65–73. doi: 10.1101/sqb.1987.052.01.010. [DOI] [PubMed] [Google Scholar]
- 34.Abeles RH, Frey PA, Jencks WP. Biochemistry. Boston, USA: Jones and Barlett; 1992. [Google Scholar]
- 35.Isaacs N. Physical organic chemistry. 2nd ed. Essex, UK: Longman; 1995. [Google Scholar]
- 36.Lowry TH, Richardson KS. Mechanism and theory in organic chemistry. 3rd ed. New York: Harper & Row; 1992. [Google Scholar]
- 37.Graham LP. An introduction to medicinal chemistry. 4th ed. Oxford, UK: Oxford University Press; 2009. [Google Scholar]
- 38.Järv J, Kesvatera T, Aavikasar A. Structure-activity relationships in acetylcholinesterase reactions. Hydrolysis of non-ionic acetic esters. Eur J Biochem. 1976;67:315–322. doi: 10.1111/j.1432-1033.1976.tb10694.x. [DOI] [PubMed] [Google Scholar]
- 39.Fujita T, Iwasa J, Hansch C. A new substituent constant, π, derived from partition coefficients. J Am Chem Soc. 1964;86:5175–5180. [Google Scholar]
- 40.Lin G, Lai CY, Liao WC, Liao PS, Chan CH. Structure-activity relationships as probes to the inhibition mechanisms of acetylcholinesterase by aryl carbamates. I. The steady-state kinetics. J Chin Chem Soc. 2003;50:1259–1265. [Google Scholar]
- 41.Goodsell DS, Lauble H, Stout CD, Olson AJ. Automated docking in crystallography: analysis of the substrates of aconitase. Proteins. 1993;17:1–10. doi: 10.1002/prot.340170104. [DOI] [PubMed] [Google Scholar]
- 42.Frisch MJ, Trucks GW, Schlerel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayaara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. GAUSSIAN 03 (Revision B.01) Pittsburgh PA: Gaussian, Inc; 2003. [Google Scholar]
- 43.Moraczewski A, Banaszynski LA, From AM, White CE, Smith BD. Using hydrogen bonding to control carbamate C-N rotamer equilibria. J Org Chem. 1998;63:7258–7262. doi: 10.1021/jo980644d. [DOI] [PubMed] [Google Scholar]
- 44.Goodsell DS, Olson AJ. Automated docking of substrates to proteins by simulated annealing. Proteins. 1990;8:195–202. doi: 10.1002/prot.340080302. [DOI] [PubMed] [Google Scholar]
- 45.Goodsell DS, Morris GM, Olson AJ. Docking of flexible ligands. Applications of AutoDock. J Mol Recogn. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 46.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 47.Leo A, Hansch C, Elkins D. Partition coefficients and their uses. Chem Rev. 1971;71:525–616. [Google Scholar]