

Abstract
Cardiac troponin C (cTnC) is the calcium binding subunit of the troponin complex that triggers the thin filament response to calcium influx into the sarcomere. cTnC consists of two globular EF-hand domains (termed the N- and C-domains) connected by a flexible linker. While the conformation of each domain of cTnC has been thoroughly characterized through NMR studies involving either the isolated N-domain (N-cTnC) or C-domain (C-cTnC), little attention has been paid to the range of interdomain orientations possible in full-length cTnC that arises as a consequence of the flexibility of the domain linker. Flexibility in the domain linker of cTnC is essential for effective regulatory function of troponin. We have therefore utilized paramagnetic relaxation enhancement (PRE) NMR to assess the interdomain orientation of cTnC. Ensemble fitting of our interdomain PRE measurements reveals that isolated cTnC has considerable interdomain flexibility and preferentially adopts a bent conformation in solution, with a defined range of relative domain orientations.
Keywords: cardiac troponin C, solution NMR, paramagnetic relaxation enhancement, site-directed spin labeling, ensemble states
Introduction
Cardiac troponin C (cTnC) is the 18 kDa calcium (Ca2+)-binding subunit of the troponin complex that is responsible for initiating muscle contraction in response to Ca2+ influx into the sarcomere. Both the cardiac (cTnC) and skeletal (skTnC) isoforms are highly α-helical and comprise two EF-hand domains (the N- and C-domains) connected by a highly conserved nine-residue linker [Fig. 1(D)].1-3 The structural C-domain contains two metal binding sites (site III and site IV), which bind Ca2+ or Mg2+ with high affinity. The regulatory N-domain also contains two metal binding sites (site I and site II) which are Ca2+ specific. However, in the cardiac isoform, site I is inactive. Thus, in contrast to skTnC where two Ca2+ ions bind the regulatory N-domain, it is binding of only one Ca2+ ion to site II which triggers the series of conformational changes that then propagate through troponin to the thin filament to allow for actomyosin interaction and muscle contraction to occur.5
Figure 1.
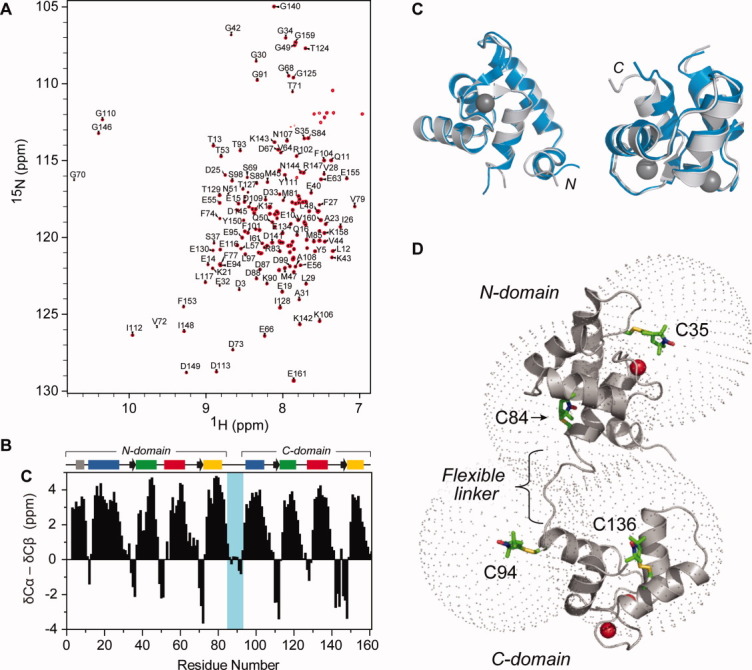
A backbone assignment of “cysless” cTnC (cTnC*) was completed, and the assigned 15N-HSQC spectrum of cTnC* is shown in (A). (B) A combined 13Cα, 13Cβ secondary chemical shift plot, where the Cβ secondary shift is subtracted from the Cα secondary shift and a three-point smoothing function applied.4 Positive stretches are indicative of α-helical structure and negative stretches indicate β-strand structure. Secondary structure is shown where arrows indicate β-strands and bars on the left represent helices from the N-domain (helix N, grey; helix A, blue; helix B, green; helix C, red and helix D, yellow). Likewise, helices E to H are shown on the top right. (C) Chemical shifts were used to obtain a model of cTnC* using CS23D (blue). Both the N-domain (left) and the C-domain (right) of our CS23D generated structure align well with the NMR solution structure 1AJ4 (grey). (D) TnC consists of two Ca2+-binding EF hand domains connected by a flexible linker. To examine the conformation of cTnC in its entirety, four single cysteine cTnC constructs were created and covalently labeled with MTSL, as indicated on the solution structure of the Ca2+-saturated cTnC (PDB 1AJ418). C35 and C84 are located in the N-domain; C94 and C136 are located in the C-domain. A sphere with a 20 Å radius is shown for each spin label site to indicate the range within which PRE distances can most accurately be measured.
The linker region [residues 86–94, Fig. 1(D)] connecting the N- and C-domains of isolated TnC is reported to be highly flexible in solution.6, 7 Previous NMR relaxation studies have demonstrated the existence of significant interdomain flexibility within TnC with motions in the nanosecond timescale,8 on the same timescale as interdomain motions described for calmodulin (∼ 3 ns).9, 10 While flexibility of this region in TnC is observed to decrease in the presence of binding partners TnI and TnT in the troponin complex, a high degree of mobility is still retained for the linker region in the complex allowing movement between the N and C domains.11, 12 The flexibility of the linker is also functionally significant as TnC mutants with altered central linker lengths, or reduced linker flexibility, are ineffective in the regulation of the actomyosin ATPase.13-15 The expected consequence of the intrinsic flexibility of the central linker is that the two globular domains can adopt a wide range of orientations relative to one other. However, despite the wealth of structural data on the isolated TnC from NMR, a clear description of the orientational dynamics of the two domains of TnC in solution, critical for understanding the muscle regulatory model, is still lacking.
Conventional NMR structure determination relies heavily on semiquantitative distance measurements derived from interproton nuclear Overhauser effects (NOEs) and problems arise when such NOEs are not observed between structural elements. As noted by others, this can result in difficulties in defining the relative orientations between helical elements,16 as well as determining the relative packing or orientation between domains.17 In TnC, no NOEs are observed between the N and C globular domains,18, 19 and we therefore have little understanding of the conformational relationship between these two domains in solution.
We therefore sought to use paramagnetic relaxation enhancement (PRE) NMR, which allows the measurement of long-range distances (up to 25 Å),20 to assess the interdomain orientation of cardiac TnC. Visualization of the conformational states of TnC is possible by PRE-NMR as the interdomain motion within TnC occurs on the nanosecond timescale. The PRE approach is a highly sensitive technique for examining conformational changes and dynamics occurring within the fast exchange regime, including the detection of sparsely populated species as low as 0.5% in solution.9, 21 We generated four spin-labeled variants of full-length cTnC and used PRE measurements to define the interdomain orientation of the protein. Our data show that isolated cTnC preferentially adopts a relatively compact conformation in solution, with a defined range of relative domain orientations that are comparable to the conformation observed for TnC in the crystal structure of the cardiac troponin core complex.22
Results and Discussion
Initial NMR characterization of C35S/C84S cardiac TnC
The site-specific spin labeling of cTnC with the nitroxide spin label requires mutagenesis of the two native cysteine residues to serine (C35S, C84S) to create a cysteine-less cTnC construct, referred to hereafter as cTnC*, before the introduction of a cysteine residue at the desired location. Comparison of 1H and 15N chemical shifts from the 15N-HSQC spectra between native cTnC and cTnC* revealed mostly small chemical shifts changes (Δδav)23 of less than 0.1 ppm for each amide peak (Supporting Information Fig. S1). However, 16 large chemical shift changes (<1 standard deviation from the mean chemical shift change) resulting from the double mutation to replace the native cysteine residues were noted to occur within the regulatory N-domain. The large chemical shift perturbations were clustered around the two mutation sites; the defunct site I in the vicinity of the C35S mutation and the D-helix of site II for the C84S mutation. As the chemical shift perturbations were confined to residues surrounding the mutation sites, the global fold of cTnC* was not expected to differ significantly from that of the native protein. cTnC* has been produced previously and the functional activity shown to be preserved, with at most, a modest reduction for the ATPase activity as measured in reconstituted fibres.24 Further, functional impairment of Ca2+ binding to site II of cTnC* was reported to be minimal.25, 26
The assignment of our 15N-HSQC cTnC* spectrum is shown in Figure 1(A). Almost complete backbone assignments were obtained for our cTnC* construct, with the exception of Met1, Asp2, and the two prolines (Pro52 and Pro54). The deviation of 13Cα and 13Cβ chemical shifts of cTnC* from random coil values were used to identify the secondary structural elements by utilizing the well-established correlations of these 13C shifts to secondary structure4 [Fig. 1(B)]. The assigned positions of the secondary structural elements of cTnC* are in good agreement with those demonstrated previously for cTnC*,18 with the exception of the β1 strand (residues 35–37). While chemical shift deviations just fell short of the criteria employed for assignment of secondary structural elements, this region did display propensity for a β-strand.
Chemical shifts were further used to generate a model of cTnC* using the prediction program CS23D.27 The model was then compared with an NOE-derived solution structure of cTnC* [Fig. 1(C).]18 The alignment of both the N- and C-domains of our CS23D generated structure with each respective domain from PDB 1AJ4 showed excellent agreement, supporting the conclusion that the two cysteine-to-serine mutations did not significantly alter the conformation of cTnC. This result also highlights the value in using a program such as CS23D to assess structural changes, rather than simply comparing chemical shifts. The structure prediction algorithm is able to “flatten out” the high sensitivity of the chemical shift to small changes in chemical environment that can be observed even in the absence of significant conformational changes; in this case, the replacement of a cysteine with a serine.
In order to explore the interdomain dynamics of TnC, nitroxide spin-labeled single cysteine mutants of TnC were engineered at four carefully chosen sites and PRE-NMR performed to obtain long-range distances in the Ca2+ saturated state [Fig. 1(D)]. The four sites were selected to gain overlapping structural information spanning the entire TnC protein. Two of these sites were the two native cysteine residues, Cys35 and Cys84, located near the nonfunctional metal binding loop of site I in the N-domain between the A-helix and the B-helix, and at the C-terminal end of the D-helix linking the two EF-hand domains, respectively. The two additional introduced sites, Cys94 and Cys136, lie at the N-terminal end of the E-helix and at the centre of the G helix, respectively, and thereby encompass the entire C-terminal structural domain and the linker region.
Site-specific spin labeling of cTnC
Accurate interpretation of the PRE distances from a single label site requires that the labeling efficiency with the nitroxide spin label to be as close to unity as possible. Complete spin labeling (<95%) was achieved for the four 15N-labeled single cysteine TnC constructs (C35, C84, C94, and C136), as determined via double-integrating the EPR resonances from each labeled sample relative to MTSL standards. Additionally, complete derivatization of free sulfhydryl groups by the nitroxide label is further supported by the absence of any dimerization of the spin-labeled samples in the absence of DTT, as judged by nonreducing SDS-PAGE and analytical SEC.
Our assignments of the cTnC* sample were transferred to the 15N-HSQC spectra of each of the four MTSL labeled TnC constructs. Superimposing the 15N-HSQC spectrum for the two N-domain spin labeled constructs (for which the nitroxide has been reduced to its diamagnetic equivalent), C35 and C84, onto the cTnC* spectrum showed that ∼ 85% of the amide peaks could be overlaid with the cTnC* with a reasonable level of confidence. For the C-domain constructs, C94 and C136, ∼ 95% of backbone amides overlaid with confidence. While some chemical shift perturbations were observed from the cumulative process of introducing the cysteine residue and spin labeling each TnC construct, we do not consider that the structure of any construct used in this study was significantly perturbed, as the chemical shift changes mapped mostly to the local vicinity of the labeling site (Supporting Information Figs. S2 and S3).
Detection of interdomain PRE effects in cTnC
15N-HSQC spectra were recorded for each paramagnetic spin-labeled construct and for the corresponding diamagnetic molecule following the complete reduction of the nitroxide label with an excess of ascorbic acid. A region of the paramagnetic and diamagnetic spectra is shown for each mutant in Figure 2. The observed PRE effects can broadly be grouped into three categories, each of which is mapped onto the TnC structures in Figure 2. The first class includes resonance peaks that are broadened beyond detection in the paramagnetic spectrum, such as residue K90 for C84-MTSL. All assignable residues in this class (red in Fig. 2) are clustered in close proximity (∼ 10 Å) to the corresponding spin label location, as expected. The second, most useful class of PRE effects (yellow in Fig. 2) includes detectable yet significantly broadened peaks (observed as reduced intensity), such as E19. Measurable PREs are derived from this class using the peak intensity ratio (Ipara/Idia) to calculate the magnitude of the relaxation enhancement (R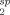 ) to obtain the time-averaged distance between the residue and the paramagnetic centre (rmeas). Distance-dependent broadening due to the interaction with the spin label is detectable for this group above the 10 Å “blind zone” limit and up to ∼ 25 Å.20 Beyond 25 Å, peak intensity changes are generally too small to reliably quantify (Ipara/Idia > 0.95) (Supporting Information Fig. S4).
) to obtain the time-averaged distance between the residue and the paramagnetic centre (rmeas). Distance-dependent broadening due to the interaction with the spin label is detectable for this group above the 10 Å “blind zone” limit and up to ∼ 25 Å.20 Beyond 25 Å, peak intensity changes are generally too small to reliably quantify (Ipara/Idia > 0.95) (Supporting Information Fig. S4).
Figure 2.
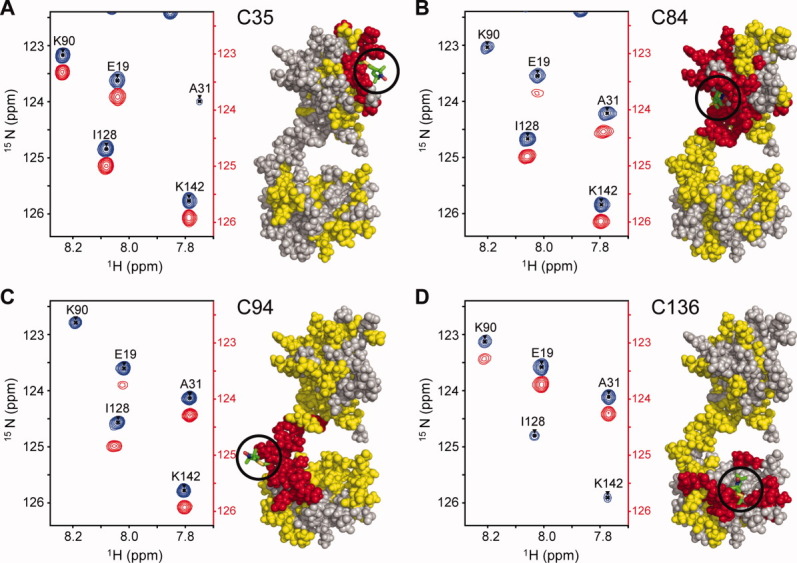
Strong peak broadening due to the PRE effect was observed in the vicinity of each spin label. A representative section of the paramagnetic (red) and diamagnetic (blue) 15N-HSQC spectrum of each spin-labeled cTnC construct; (A) TnC C35, (B) TnC C84, (C) TnC C94, (D) TnC C136. The superimposed paramagnetic spectrum is offset by 0.30 ppm in the 15N dimension for clarity. The spin label location is shown on the structures to the right of each spectrum (stick representation, green) and residues affected by the spin label are coloured onto cTnC accordingly, where residues broadened beyond detection are in red (<10 Å), residues with significant detectable PREs in yellow (<25 Å) and residues to which no significant PRE was assigned are coloured grey. An interactive view is available in the electronic version of the article.
While most of the detectable PREs can be seen to be located within the same domain as the spin label (intradomain), there are also a significant number of PREs detected for residues that are not in the same domain as the spin label (interdomain PREs) (Fig. 2). The distance between the centre of mass of the N and C-domains is 36 Å in the single conformer deposited in the NMR cTnC structure 1AJ4; interdomain PRE effects would therefore not be expected if TnC were a single extended static entity. The observation of significant interdomain PRE effects immediately suggests that the two domains of TnC spend at least some time in reasonably close proximity. The question then arises as to whether the interdomain effects arise from linker flexibility producing an ensemble of various conformational states or whether a previously uncharacterized compact conformational state exists.
The experimental distances derived from our PRE data for each of the four spin-labeled samples were initially compared with the distances calculated from the cTnC* NMR structure PDB 1AJ418 after positioning of the spin label. The agreement between the PRE-measured distances and distances calculated within the cTnC cysless model (1AJ4) were then quantified using the Q-factor statistic. Comparison of all measured distances to 1AJ4 gave rise to a poor linear correlation overall, and high overall Q-factor values (Qall) (Fig. 3). The distance plots (left panels in Fig. 3) show that the distances measured to residues located in the same domain as the spin label (red) are comparable to those calculated from 1AJ4. The greatest differences between the measured and calculated distances, and those that most strongly contribute to the somewhat high Qall values, are those distances measured to residues outside the spin-labeled domain (blue). The separation of Q-factor values into two subpopulations within each correlation plot, that is intradomain and interdomain, is thus necessary for interpretation of the interdomain orientation using available dynamic structural models of TnC.
Figure 3.
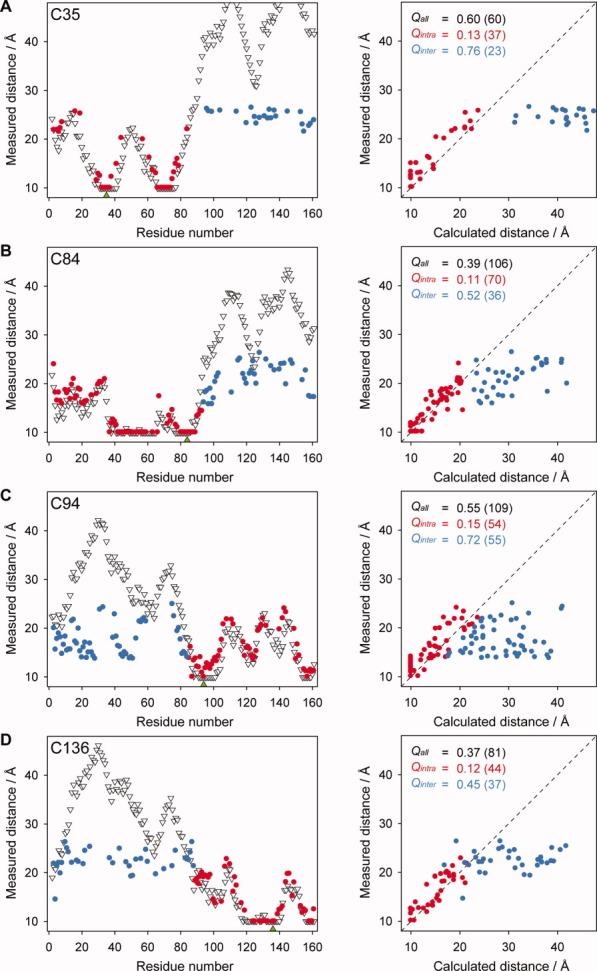
Measured PRE distances were compared with distances calculated from the coordinates of the cTnC structure 1AJ4.18 Left: The distance plots show the distance measured between the spin label and the amide group of each residue in cTnC, where intra- and interdomain measured distances are colored red and blue, respectively. The distances calculated from the coordinates of the energy minimized solution structure 1AJ4 are also shown (white triangles). The location of the spin label is indicated by a green triangle on the x-axis. For the N-domain spin label sites, (A) TnC C35 and (B) TnC C84, the intradomain distances are those measured within the N-domain. For the C-domain label sites, (C) TnC C94 and (D) TnC C136, the intradomain distances are those measured within the C-domain. Right:Q-factors were used to report the correlation between our measured distances and those calculated from 1AJ4. The Q-factors calculated for each mutant are also indicated where Qall represents all measured distances (both red and blue points) and Qintra and Qinter, represent the intradomain and interdomain distances, respectively. The number of distances contributing to each set is given in brackets. Residues broadened beyond detection in the paramagnetic spectrum and all calculated distances <10 Å are plotted as 10 Å.
Upon separation of the data, there is significant improvement for the comparison of the intradomain PRE distances for all four constructs with distances from the TnC structure 1AJ4, with Qintra values ranging from 0.11 to 0.15 (Fig. 3). In contrast, treatment of interdomain distances as a separate subset led to a general worsening of Q-factor values for all four probe sites, as expected, with Qinter values increasing in comparison with Qall values to values ranging from 0.45 to 0.76 (Fig. 3). The low values observed for all four intradomain Q-factors clearly indicates that that NMR structure 1AJ4 is an accurate representation of the individual conformation of both the N- and C-domains in solution and that the mutation and the spin label did not give rise to significant conformational perturbation. On the other hand, the data also show that the interdomain orientation in 1AJ4 is a poor representation of the orientation between the two globular domains.
Interdomain dynamics visualized by an ensemble state of TnC
In order to model the interdomain orientation, we used 10 structural models (c1, c2, …, c10) for the 3Ca2+-loaded cTnC structure (PDB 2JT319) as a basis set and asked whether a linear combination of any subset of these conformers can reproduce the observed interdomain PRE data. These 10 available structural models, calculated primarily using intradomain NOE data, essentially represent a random set of structures with respect to their interdomain orientation and result in a broad spatial region through which cTnC is likely to sample.
Interdomain dynamics for TnC is on the nanosecond timescale. As such, in this fast exchanging system, the observed PRE is the population-weighted average of all possible conformational states. Therefore, ensemble Qe-factors comparing the R2-weighted distances (ravg) to our measured interdomain distances were calculated for each unique set of conformers, which ranged in size from N = 1 (a single structure) to N = 10 (all 10 structures). In general, the mean Qe-values are observed to rapidly decrease, that is improve, with an increasing ensemble size for all four spin labeled constructs, with the least improvement observed for the C35 construct [Fig. 4(A)]. Despite the location of the C35 spin label at the “top” of the N-domain, such that it is predicted to be >30 Å away from the majority of C-domain residues in the cTnC conformations examined, the small data set of interdomain PREs measured for this construct [n = 23, Fig. 3(A)] was still adequate to allow selection of conformers of best fit.
Figure 4.
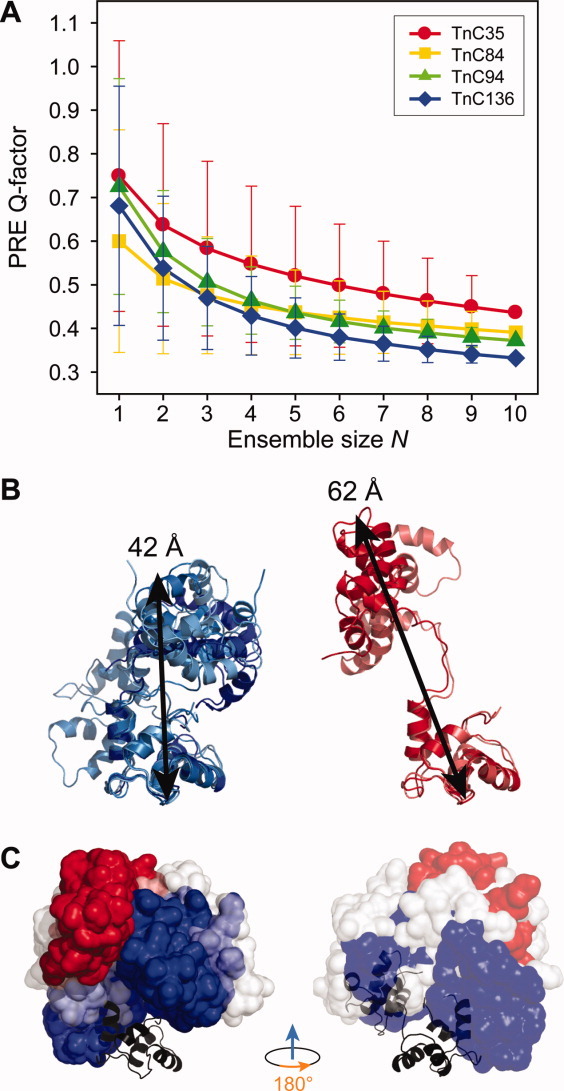
Qe values were used to compare interdomain distances to ensemble sets derived from PDB 2JT3.19 (A) Interdomain Qe-factors as a function of number of conformers (N) of cTnC (PDB 2JT319) for each of the four MTSL-labeled cTnC constructs. The plotted Qe values represent the mean Qe for all combinations of N conformers and error bars indicate one standard deviation. The Qe-factors for TnC C35, C84, C94, C136 are colored red, yellow, green, and blue, respectively. The 10 conformers (from 2JT3) were ranked based on their frequency of occurrence in the combinations that produced the (B) lowest interdomain Qe values (i.e. c1, c2, c5, and c7, left, colored blue), and highest Qe values (i.e. c3 and c8, right, colored red). The highest ranking conformers (ie lowest Q-factor) demonstrate a more compact collapsed conformation than the lowest ranking set of conformers. (C) The N-domains of the most (blue) and least (red) favorable orientation of the N-domain with respect to the C-domain for the TnC conformers depicted in B are represented as surfaces structures aligned at their C-domains. For completeness, conformers c4, c6, c9, and c10 are shown in white. The conformation of cTnC within the crystal structure of the core complex (TnC + TnI + TnT2, PDB 1J1D22) is also shown (black), and can be seen to lie within the favorable region. Figures on left in panel (C) are in the same orientation as figures in panel (B). An interactive view is available in the electronic version of the article.
While the mean interdomain Qe-values shown in Figure 4 were typically found to monotonically decrease with increasing ensemble size N and not level off, the standard deviation of the Qe-values (indicated with error bars) shows that, for each ensemble size, not every combination of structures leads to an improved fit for each increment in N. That is, the addition of certain structures to an ensemble set leads to deterioration in the agreement with the measured interdomain PREs rather than an improvement. This is also observed by viewing the Qe-values for the best scoring individual combinations of conformers which are generally observed to increase in value for ensemble sets representing three or more conformers (Supporting Information Fig. S5). Therefore, inspections of the best outlier combinations of conformers with the lowest Qe-values, independent of ensemble size, were used to determine the conformers of best fit. The Qe-values were used to rank the 20 best ensemble combinations and a weighted scoring system based on position revealed that conformers appearing with the highest frequency of occurrence were c1, c2, c5, and c7. Likewise, the conformers that most frequently led to the poorest fit to the interdomain PREs were c3 and c8. Consistent with this finding, the interdomain Qinter values for each individual conformer were lowest for c1, c2, c5, and c7 and highest for c3 and c8 (Supporting Information Table 1).
Structural inspection of the set of “favorable structures,” that is c1, c2, c5, and c7, show that these four conformers of cTnC represent, in general, a compact interdomain arrangement [Fig. 4(B), left panel]. In contrast, conformers c3 and c8, which contributed negatively to the interdomain Q-factors, are more notably extended in nature [Fig. 4(B), right panel]. Additionally, conformers c4, c6, c9, and c10, despite their compact interdomain arrangement, were not selected for by the PRE data. Alignment of the C-domains of the favorable and unfavorable structures at their C-domains visibly demonstrates distinct differences in the interdomain spacing and orientations between each set [Fig. 4(C)]. The N-domains of the favorable conformers (blue) can be seen to sample a relatively broad spatial volume in comparison with the unfavorable conformers (red) which are more tightly clustered in the extended conformation. Although the flexible domain linker of isolated TnC allows for a large range of interdomain motion, these data suggest that there is a degree of restriction of the domain motion within isolated cTnC favoring a more compact as opposed to an extended conformation. Also overlayed in Figure 4(C) (right) is the conformation of cTnC obtained from the crystallisation of the Ca2+ saturated core of the cardiac troponin complex (Protein Data Bank code 1J1D22). This crystallized conformation of cTnC lies almost entirely within the allowed conformational space selected by our PRE analysis.
Our PRE measurements presented in this study clearly demonstrate that TnC does not exist as a single species but rather identifies a defined region, predominately compact in nature, through which TnC can transit. Our present results are also consistent with an earlier spin label study of [methyl-13C] methionine-labeled TnC, where paramagnetic effects from the spin-labeled residue Cys84, as used in our study, detected significant interdomain broadening events, suggesting a close interaction between the two lobes of TnC.28 While we can identify the dominant conformational states for isolated TnC, our approach is not overly sensitive to low populations of the extended conformational state, if present.29 That is, the presence of TnC within these extended states may go undetected due to the small reduction in the observed PRE effect, and complementary approaches, such as residual dipolar coupling measurements,30 should be sought to obtain a more precise quantitative description of the population of each individual structure within the conformational space.
While our approach with PRE NMR has allowed us to obtain an experimentally based representation of the conformations for TnC in solution, we believe that a similar range of collapsed TnC conformations are also likely to be dominant in the troponin complex. Fluorescence resonance energy transfer (FRET) measurements of the interdomain distance within cTnC are similar in isolated cTnC and cTnC within the ternary complex,31 indicating that complex formation is unlikely to further refine selection of the collapsed conformations we have identified. Previous small angle neutron scattering data have also described a partially collapsed conformation for cTnC in the ternary complex with the two lobes of cTnC close together but not in contact.32 On the other hand, the distribution between the multiple possible conformational states within the stabilizing environment of the ternary complex is more likely regulated by calcium binding to the N-domain of TnC, where the TnC may alternate between weakly and tightly constrained environments. This has been observed for the ternary troponin complex in the skeletal isoform, where calcium binding to TnC triggers a large rotation (∼ 38°) of the regulatory N-domain relative to its position in the Ca2+ free state.33
Taken together with previous NMR studies of cTnC, our PRE strategy has allowed us to delineate the possible range of interdomain orientations of cTnC to provide us with a reliable description of the conformational space sampled by cTnC arising from the flexibility of its interdomain linker region. The preference for TnC to adopt more compact conformations may well have implications for cardiac Ca2+-signaling. A PRE analysis of cTnC in the troponin complex may ultimately be able to provide further understanding of the role and molecular basis of cTnC in muscle regulation.
Materials and Methods
Preparation of cTnC constructs
Rat cardiac TnC was expressed in the pET-3d expression vector (Novagen) with cardiac nomenclature, as previously described.34 cTnC mutants were constructed with the Quick-Change II site-directed mutagenesis kit (Stratagene) using complementary oligonucleotides containing the desired mutations into the cysteine-less background (C35S, C84S). The cTnC cysless clone (cTnC*) was used to generate four single cysteine mutants at positions 35, 84, 94, and 136. Thus, the four single cysteine mutant constructs are denoted as C35, C84, C94, and C136. Sequences of all mutant clones were verified by DNA sequencing and DNA transformed into Escherichia coli BL21 (DE3) cells.
cTnC mutants were expressed in M9 minimal media as described by Sambrook et al.,35 containing 2.5 g/L glucose and 1 g/L 15NH4Cl, using the isopropyl β-d-1-thiogalactopyranoside (IPTG) induction protocol of Studier et al.36 After two passes through a chilled French Press (10,000 psi) and removal of insoluble cellular debris by centrifugation, the cell lysate was dialyzed against Buffer A (0.2M NaCl, 1 mM DTT, 5 mM CaCl2 and 50 mM Tris, pH 7.5). After dialysis, the sample was loaded onto a 1.5 cm × 15 cm Phenyl Sepharose 6 Fast Flow column (GE Healthcare) equilibrated in Buffer A. cTnC was eluted upon application of Buffer B to the column (0.2M NaCl, 1 mM DTT, 10 mM EDTA, and 50 mM Tris-HCl pH 7.5). cTnC fractions identified by SDS-PAGE were pooled and dialyzed against Buffer C (0.35M KCl, 1 mM DTT, 0.2 mM CaCl2, 1 mM EDTA, and 50 mM Tris pH 7.5) before loading onto a 2.5 cm × 7.5 cm DEAE-Sephadex A25 column (Sigma-Aldrich) equilibrated with Buffer C. TnC was eluted with a linear KCl gradient from 0.35M to 0.6M KCl. Pure TnC fractions (eluted at ∼ 0.5M KCl) were concentrated using an Amicon Ultra-15 centrifugal filtration device and protein concentrations determined using a BCA Assay Kit (Pierce).
Paramagnetic labeling of cTnC
The paramagnetic nitroxide moiety was introduced onto the sulfhydryl group of the cysteine for each single cysteine TnC mutant by the covalent attachment of the spin label 1-oxyl-2,2,5,5-tetra-methylpyrroline-3-methyl-16 methanethiosulfonate (MTSL) (Toronto Research Chemicals). cTnC was dialyzed against labeling buffer (100 mM KCl, 20 mM KPO4 buffer pH 8) and immediately before labeling, the protein was incubated with 10 mM DTT for 2 h at room temperature to ensure complete reduction of the cysteine sulfhydryl groups, confirmed by nonreducing SDS-PAGE (data not shown). DTT was then removed from the sample using a 5 mL HiTrap Desalting column (GE Healthcare) and reduced cTnC was incubated with a 10-fold molar excess of MTSL overnight with gentle stirring at room temperature. Excess MTSL was then removed by exhaustive dialysis against NMR buffer.
EPR spectroscopy
EPR spectroscopy was used to determine the spin-labeling efficiency of each spin-labeled cTnC sample. All EPR experiments were performed on a Bruker EMX X-Band (9.5 GHz) spectrometer using a standard rectangular TE cavity at room temperature. First derivative EPR absorption spectra were collected with a microwave power of 5.0 mW, modulation amplitude of 1.0 G and sweep width of 140 G. EPR spectra were analyzed with a LabView package (National Instruments) of EPR spectral analysis programs. Spin labeling yields for each sample were determined from the integrated area under the EPR absorbance spectrum by interpolation from a calibration curve constructed from MTSL standards. In all cases, complete modification of each single cysteine mutant was achieved (<95%), also evident from the 15N-HSQC spectra which showed complete broadening of peaks for residues within the immediate vicinity of the spin labeled site.
cTnC sample preparation for NMR spectroscopy
All NMR samples were 500 μL in volume in 50 mM KCl, 1 mM CaCl2, 20 mM KPO4 pH 6.7, 10% D2O, and 0.02 mM 2,2-dimethyl-2-silapentane-5-sulfonate (DSS). For resonance assignment of the cTnC* “cysless” sample, a 500 μL sample of uniformly 15N,13C-labeled cTnC* at a concentration of 420 μM was prepared in NMR buffer supplemented with 0.03% sodium azide. NMR samples were checked for homogeneity by size exclusion chromatography on an analytical Superdex 75 10/300 column (GE Lifesciences) and nonreducing SDS-PAGE.26 Aggregation was not observed in any instance. TnC concentrations ranged from 130 μM to 230 μM for PRE-NMR samples. These low TnC concentrations were required to prevent intermolecular PRE effects.37
NMR spectroscopy
NMR data were collected on a Bruker Avance 600 MHz spectrometer equipped with a cryoprobe. All experiments were performed at 303 K and spectra were processed using Topspin 1.3 (Bruker, Inc). Spectral assignment and analysis was performed with the program Sparky.38 Backbone assignments of cTnC cysless were made using HNCA, HNCACB, CBCA(CO)NH, HNCO, and HN(CA)CO experiments.39–4115N-HSQC spectra were acquired for native cTnC, cTnC* (Supporting Information Fig. S1) and each single cysteine TnC mutant in the absence of the spin label to ensure that the removal of native cysteines and subsequent introduction of cysteines did not perturb tertiary structure. For NMR of samples in the absence of the spin label, all NMR spectra were recorded in the presence of 10 mM DTT to prevent possible dimerization of the unlabeled constructs via cysteine residues. Comparison of 1H and 15N chemical shifts from the 15N-HSQC spectra showed no significant perturbations to the cTnC structural fold were induced by the introduction of cysteine residues at any of the four sites (Supporting Information Fig. S2), or from the spin labeling with MTSL (Supporting Information Fig. S3).
For the paramagnetic labeled samples, 15N-HSQC spectra were collected for each of the four spin labeled TnC constructs. After acquiring each paramagnetic spectrum, the nitroxyl radical of the MTSL label was reduced to its hydroxylamine equivalent by the addition of ∼ 1/250 volume of a fivefold molar excess of ascorbic acid (from a 0.5M stock) directly to the NMR sample. One hour at room temperature was sufficient time to allow for the reduction reaction to go to completion as confirmed by EPR. 15N-HSQC spectra were then collected for the reduced diamagnetic sample. The small volume of ascorbic acid enabled the direct comparison of peak intensities between paramagnetic and diamagnetic spectra used in the calculation of the relaxation enhancement. Peak assignments in the diamagnetic and paramagnetic spectra of each TnC construct were made using the sequentially assigned 15N-HSQC spectrum of TnC*.
PRE data analysis and distance determinations
For each amide peak assigned in the 15N-HSQC spectra of the cTnC constructs, the PRE caused by the MTSL spin label (R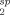 ) was calculated from peak intensities in the paramagnetic state (Ipara), and in the diamagnetic state after reduction of the spin label (Idia), using Eq. (1). Peak heights were used as measures of peak intensities, and the intrinsic transverse relaxation rate (R2) was estimated from the peak width at half-height (R2 = πΔv1/2).20, 42 All spectra were recorded with the single evolution time point (t) of 10 ms.
) was calculated from peak intensities in the paramagnetic state (Ipara), and in the diamagnetic state after reduction of the spin label (Idia), using Eq. (1). Peak heights were used as measures of peak intensities, and the intrinsic transverse relaxation rate (R2) was estimated from the peak width at half-height (R2 = πΔv1/2).20, 42 All spectra were recorded with the single evolution time point (t) of 10 ms.
| (1) |
The calculated PRE rates (R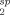 ) were converted to distances (r) using a modified form of the Solomon-Bloembergen equation:20, 42, 43
) were converted to distances (r) using a modified form of the Solomon-Bloembergen equation:20, 42, 43
| (2) |
where ωH is the Larmor frequency of the proton, K is a constant (1.23 × 10−32 cm6 s−2) which describes the spin properties of the nitroxide spin label,20 and τc is the correlation time of the electron-proton interaction [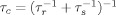 ]. Due to the long electronic relaxation time of the nitroxide spin label (τs) compared with the protein rotational correlation time (τr), the value of τc is effectively equal to the overall correlation time of the protein44 and was taken as 7 ns for intact TnC.45, 46 This approximation is close to the NMR derived value of 5 ns for an individual domain of TnC.47 Due to the steep distance dependence of the PRE effect, the τc value used in Eq. (2) has a relatively minor impact on distance determinations; with a 2 ns uncertainty in τc causing, at most, <6% change in the PRE calculated distance.20
]. Due to the long electronic relaxation time of the nitroxide spin label (τs) compared with the protein rotational correlation time (τr), the value of τc is effectively equal to the overall correlation time of the protein44 and was taken as 7 ns for intact TnC.45, 46 This approximation is close to the NMR derived value of 5 ns for an individual domain of TnC.47 Due to the steep distance dependence of the PRE effect, the τc value used in Eq. (2) has a relatively minor impact on distance determinations; with a 2 ns uncertainty in τc causing, at most, <6% change in the PRE calculated distance.20
PRE back-calculations and assessment of PRE distances
All PRE measured distances were compared with available Ca2+ saturated solution structures, namely the NMR structure of 3Ca2+-cTnC Cysless (PDB 1AJ418), and the ensemble of 10 cTnC structures (c1, c2, …, c10) in PDB 2JT3,19 following the optimization of the position of the nitroxide group onto each structure at the desired residue. The paramagnetic group of the spin label is tethered to the protein backbone at the Cα atom via a flexible linker. As the structures of both the N- and C-domains of cTnC are relatively well defined, a multiple conformer representation of the dynamic spin label group can be described using the intradomain PRE distances.9 In brief, the conformational space occupied by the nitroxide group was optimized via comparative analysis of the calculated and observed PRE measured distances, using the Q-factor statistic [Eq. (3)]. An evenly distributed generic set of points (1 Å spacing intervals) was initially generated between two concentric spheres of 5 Å and 10 Å centered at the Cα atom of the labeled site, reflecting the region for which the nitroxide could occupy, as described by X-ray crystallography.48 Points were then screened for steric clashes and distances calculated from each remaining sterically feasible point to the NH atoms in the relevant PDB structure. Comparison between our PRE-measured (rmeas) and calculated spin label distances (rcalc) was performed using the Q-factor statistic [Eq. (3)],49 where the conformational space of the nitroxide group was defined by all points with a Q-factor <0.2. The number of points (N) defining this region was unrestricted to accommodate the varying mobility of each spin label with the resulting angular spread obtained comparing well with the experimental EPR spectra for each label site.
| (3) |
Since the correlation time of the spin label motion (measured by EPR for all four sites to be ∼ 1 to 2 ns) is much faster than the PRE relaxation rate, the observed PRE rate represents the population weighted average (R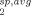 ) of the PRE rates derived from each of the individual points (R
) of the PRE rates derived from each of the individual points (R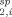 ) within the area sampled by the spin label.21 This can be expressed as:
) within the area sampled by the spin label.21 This can be expressed as:
| (4) |
where the contribution from each of the N points is equally weighted (1/N).
Using Eq. (2), distances between the optimized spin label position and each NH were then calculated (rcalc) from the average PRE rates obtained in Eq. (4). An overall Q-factor (Qall) was then individually calculated for each of the four spin-labeled constructs for all assignable peaks [Eq. (3)]. The dataset was further divided into intradomain for distances within the same domain as the spin label, and interdomain for distances measured between domains (where the N-domain domain is defined as residues 1 to 85, and the C-domain residues 93–161) and the Q-factors Qintra and Qinter calculated for each, respectively. For TnC84 and TnC94, the entire central linker (residues 85–93) was also considered as intradomain. Calculation of Qall, Qintra, and Qinter was performed for all structures, namely PDB 1AJ4 and the 10 cTnC structures in PDB 2JT3. In general, small Q-factor values of <0.2 indicate good correlation between experimental data (rmeas) and the reference structure (rcalc).50
Ensemble analysis of cTnC models
Due to the complete lack of NOEs between the two globular domains of TnC, each of the various orientations of the N-domain of cTnC relative to the C-domain used in this analysis are essentially arbitrary, but satisfy the condition that they are sterically possible. The observed paramagnetic effects in the fast exchange regime represent a population-weighted average of the contributions from the ensemble of orientations of the N-domain relative to the C-domain.
Ensemble conformer sets were generated using the 10 conformers within PDB 2JT3 with ensemble sizes (N) ranging from 1 to 10, resulting in 1023 unique sets [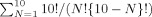 ]. For each of the 1023 sets, the average PRE rates (R
]. For each of the 1023 sets, the average PRE rates (R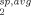 ) of each conformer comprising the set (R
) of each conformer comprising the set (R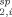 ) were calculated using Eq. (4) where all conformers within an ensemble set represented a predefined number of equally weighted structures (1/N). Equation (2) was used to convert the average PRE rates (R
) were calculated using Eq. (4) where all conformers within an ensemble set represented a predefined number of equally weighted structures (1/N). Equation (2) was used to convert the average PRE rates (R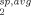 ) to distances (ravg) and a second ensemble Q-factor (Qe) was then used to compare each ensemble conformer set against the measured interdomain PRE distances for each residue i according to:
) to distances (ravg) and a second ensemble Q-factor (Qe) was then used to compare each ensemble conformer set against the measured interdomain PRE distances for each residue i according to:
| (5) |
Acknowledgments
The authors are grateful to Professor Paul Curmi, Professor Timothy Logan, and Dr James Cooke for many helpful discussions and thank Mr. Michael Howell for technical assistance.
Glossary
Abbreviations
- c1, c2, …, c10
structures within PDB 2JT3
- cTnC
cardiac troponin C
- cTnC*
cysteine-less cTnC construct
- PRE
paramagnetic relaxation enhancement
- Qall
overall Q-factor
- Qe
interdomain Q-factor statistic for an ensemble of structures
- Qinter
interdomain Q-factor
- Qintra
intradomain Q-factor
- skTnC
skeletal troponin C
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Herzberg O, James MN. Structure of the calcium regulatory muscle protein troponin-C at 2.8 Å resolution. Nature. 1985;313:653–659. doi: 10.1038/313653a0. [DOI] [PubMed] [Google Scholar]
- 2.Li MX, Robertson IM, Sykes BD. Interaction of cardiac troponin with cardiotonic drugs: a structural perspective. Biochem Biophys Res Commun. 2008;369:88–99. doi: 10.1016/j.bbrc.2007.12.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sundaralingam M, Bergstrom R, Strasburg G, Rao ST, Roychowdhury P, Greaser M, Wang BC. Molecular structure of troponin C from chicken skeletal muscle at 3-angstrom resolution. Science. 1985;227:945–948. doi: 10.1126/science.3969570. [DOI] [PubMed] [Google Scholar]
- 4.Metzler WJ, Constantine KL, Friedrichs MS, Bell AJ, Ernst EG, Lavoie TB, Mueller L. Characterization of the three-dimensional solution structure of human profilin: proton, carbon-13, and nitrogen-15 NMR assignments and global folding pattern. Biochemistry. 1993;32:13818–13829. doi: 10.1021/bi00213a010. [DOI] [PubMed] [Google Scholar]
- 5.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 6.Heidorn DB, Trewhella J. Comparison of the crystal and solution structures of calmodulin and troponin C. Biochemistry. 1988;27:909–915. doi: 10.1021/bi00403a011. [DOI] [PubMed] [Google Scholar]
- 7.Slupsky CM, Sykes BD. NMR solution structure of calcium-saturated skeletal muscle troponin C. Biochemistry. 1995;34:15953–15964. doi: 10.1021/bi00049a010. [DOI] [PubMed] [Google Scholar]
- 8.Blumenschein TMA, Stone DB, Fletterick RJ, Mendelson RA, Sykes BD. Calcium-dependent changes in the flexibility of the regulatory domain of troponin C in the troponin complex. J Biol Chem. 2005;280:21924–21932. doi: 10.1074/jbc.M500574200. [DOI] [PubMed] [Google Scholar]
- 9.Anthis NJ, Doucleff M, Clore GM. Transient, sparsely populated compact states of apo and calcium-loaded calmodulin probed by paramagnetic relaxation enhancement: interplay of conformational selection and induced fit. J Am Chem Soc. 2011;133:18966–18974. doi: 10.1021/ja2082813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baber JL, Szabo A, Tjandra N. Analysis of slow interdomain motion of macromolecules using NMR relaxation data. J Am Chem Soc. 2001;123:3953–3959. doi: 10.1021/ja0041876. [DOI] [PubMed] [Google Scholar]
- 11.Abbott MB, Gaponenko V, Abusamhadneh E, Finley N, Li G, Dvoretsky A, Rance M, Solaro RJ, Rosevear PR. Regulatory domain conformational exchange and linker region flexibility in cardiac troponin C bound to cardiac troponin I. J Biol Chem. 2000;275:20610–20617. doi: 10.1074/jbc.M909252199. [DOI] [PubMed] [Google Scholar]
- 12.Gaponenko V, Abusamhadneh E, Abbott MB, Finley N, Gasmi-Seabrook G, Solaro RJ, Rance M, Rosevear PR. Effects of troponin I phosphorylation on conformational exchange in the regulatory domain of cardiac troponin C. J Biol Chem. 1999;274:16681–16684. doi: 10.1074/jbc.274.24.16681. [DOI] [PubMed] [Google Scholar]
- 13.Babu A, Rao VG, Su H, Gulati J. Critical minimum length of the central helix in troponin C for the Ca2+ switch in muscular contraction. J Biol Chem. 1993;268:19232–19238. [PubMed] [Google Scholar]
- 14.Dobrowolski Z, Xu GQ, Hitchcock-DeGregori SE. Modified calcium-dependent regulatory function of troponin C central helix mutants. J Biol Chem. 1991;266:5703–5710. [PubMed] [Google Scholar]
- 15.Ramakrishnan S, Hitchcock-DeGregori SE. Investigation of the structural requirements of the troponin C central helix for function. Biochemistry. 1995;34:16789–16796. doi: 10.1021/bi00051a029. [DOI] [PubMed] [Google Scholar]
- 16.Zheng D, Aramini JM, Montelione GT. Validation of helical tilt angles in the solution NMR structure of the Z domain of Staphylococcal protein A by combined analysis of residual dipolar coupling and NOE data. Protein Sci. 2004;13:549–554. doi: 10.1110/ps.03351704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deshmukh MV, John M, Coles M, Peters J, Baumeister W, Kessler H. Inter-domain orientation and motions in VAT-N explored by residual dipolar couplings and 15N backbone relaxation. Magn Reson Chem. 2006;44:S89–S100. doi: 10.1002/mrc.1837. [DOI] [PubMed] [Google Scholar]
- 18.Sia SK, Li MX, Spyracopoulos L, Gagne SM, Liu W, Putkey JA, Sykes BD. Structure of cardiac muscle troponin C unexpectedly reveals a closed regulatory domain. J Biol Chem. 1997;272:18216–18221. doi: 10.1074/jbc.272.29.18216. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Mercier P, Letourneau PJ, Sykes BD. Effects of Phe-to-Trp mutation and fluorotryptophan incorporation on the solution structure of cardiac troponin C, and analysis of its suitability as a potential probe for in situ NMR studies. Protein Sci. 2005;14:2447–2460. doi: 10.1110/ps.051595805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 21.Clore GM, Iwahara J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem Rev. 2009;109:4108–4139. doi: 10.1021/cr900033p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 23.Farmer BT, Constantine KL, Goldfarb V, Friedrichs MS, Wittekind M, Yanchunas J, Robertson JG, Mueller L. Localizing the NADP+ binding site on the MurB enzyme by NMR. Nat Struct Mol Biol. 1996;3:995–997. doi: 10.1038/nsb1296-995. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura M, Ueki S, Hara H, Arata T. Calcium structural transition of human cardiac troponin C in reconstituted muscle fibres as studied by site-directed spin labelling. J Mol Biol. 2005;348:127–137. doi: 10.1016/j.jmb.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 25.Dong W-j, Wang C-K, Gordon AM, Cheung HC. Disparate fluorescence properties of 2-[4′-(iodoacetamido)anilino]-naphthalene-6-sulfonic acid attached to Cys-84 and Cys-35 of troponin C in cardiac muscle troponin. Biophys J. 1997;72:850–857. doi: 10.1016/s0006-3495(97)78719-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Putkey JA, Dotson DG, Mouawad P. Formation of inter- and intramolecular disulfide bonds can activate cardiac troponin C. J Biol Chem. 1993;268:6827–6830. [PubMed] [Google Scholar]
- 27.Wishart DS, Arndt D, Berjanskii M, Tang P, Zhou J, Lin G. CS23D: a web server for rapid protein structure generation using NMR chemical shifts and sequence data. Nucleic Acids Res. 2008;36:W496–W502. doi: 10.1093/nar/gkn305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleerekoper Q, Howarth JW, Guo X, Solaro RJ, Rosevear PR. Cardiac Troponin I induced conformational changes in cardiac troponin C as monitored by NMR using site-directed spin and isotope labeling. Biochemistry. 1995;34:13343–13352. doi: 10.1021/bi00041a010. [DOI] [PubMed] [Google Scholar]
- 29.Tang C, Schwieters CD, Clore GM. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature. 2007;449:1078–1082. doi: 10.1038/nature06232. [DOI] [PubMed] [Google Scholar]
- 30.Bertini I, Luchinat C, Nagulapalli M, Parigi G, Ravera E. Paramagnetic relaxation enhancement for the characterization of the conformational heterogeneity in two-domain proteins. Phys Chem Chem Phys. 2012;14:9149–9156. doi: 10.1039/c2cp40139h. [DOI] [PubMed] [Google Scholar]
- 31.Dong W-J, Robinson JM, Xing JUN, Umeda PK, Cheung HC. An interdomain distance in cardiac troponin C determined by fluorescence spectroscopy. Protein Sci. 2000;9:280–289. doi: 10.1110/ps.9.2.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heller WT, Abusamhadneh E, Finley N, Rosevear PR, Trewhella J. The solution structure of a cardiac troponin C-troponin I-troponin T complex shows a somewhat compact troponin C interacting with an extended troponin I-troponin T component. Biochemistry. 2002;41:15654–15663. doi: 10.1021/bi026687c. [DOI] [PubMed] [Google Scholar]
- 33.Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, Fletterick RJ. Ca(2+)-regulated structural changes in troponin. Proc Natl Acad Sci USA. 2005;102:5038–5043. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown LJ, Sale KL, Hills R, Rouviere C, Song L, Zhang X, Fajer PG. Structure of the inhibitory region of troponin by site directed spin labeling electron paramagnetic resonance. Proc Natl Acad Sci USA. 2002;99:12765–12770. doi: 10.1073/pnas.202477399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 36.Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 37.Donaldson LW, Skrynnikov NR, Choy WY, Muhandiram DR, Sarkar B, Forman-Kay JD, Kay LE. Structural characterization of proteins with an attached ATCUN motif by paramagnetic relaxation enhancement NMR spectroscopy. J Am Chem Soc. 2001;123:9843–9847. doi: 10.1021/ja011241p. [DOI] [PubMed] [Google Scholar]
- 38.Goddard TD, Kneller DG. SPARKY 3. San Francisco: University of California; 2007. [Google Scholar]
- 39.Grzesiek S, Bax A. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J Am Chem Soc. 1992;114:6291–6293. [Google Scholar]
- 40.Ikura M, Kay LE, Bax A. A novel approach for sequential assignment of proton, carbon-13, and nitrogen-15 spectra of larger proteins: heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry. 1990;29:4659–4667. doi: 10.1021/bi00471a022. [DOI] [PubMed] [Google Scholar]
- 41.Wittekind M, Mueller L. HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha- and beta-carbon resonances in proteins. J Magn Reson B. 1993;101:201–205. [Google Scholar]
- 42.Volkov AN, Worrall JAR, Holtzmann E, Ubbink M. Solution structure and dynamics of the complex between cytochrome c and cytochrome c peroxidase determined by paramagnetic NMR. Proc Natl Acad Sci USA. 2006;103:18945–18950. doi: 10.1073/pnas.0603551103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang B, Bushweller JH, Tamm LK. Site-directed parallel spin-labeling and paramagnetic relaxation enhancement in structure determination of membrane proteins by solution NMR spectroscopy. J Am Chem Soc. 2006;128:4389–4397. doi: 10.1021/ja0574825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iwahara J, Schwieters CD, Clore GM. Ensemble approach for NMR structure refinement against (1)H paramagnetic relaxation enhancement data arising from a flexible paramagnetic group attached to a macromolecule. J Am Chem Soc. 2004;126:5879–5896. doi: 10.1021/ja031580d. [DOI] [PubMed] [Google Scholar]
- 45.Cantor CR, Schimmel PR. Biophysical chemistry. New York: W. H. Freeman & Co; 1980. p. 461. [Google Scholar]
- 46.Li H-C, Fajer PG. Orientational changes of troponin C associated with thin filament activation. Biochemistry. 1994;33:14324–14332. doi: 10.1021/bi00251a046. [DOI] [PubMed] [Google Scholar]
- 47.Gagné SM, Tsuda S, Spyracopoulos L, Kay LE, Sykes BD. Backbone and methyl dynamics of the regulatory domain of troponin C: anisotropic rotational diffusion and contribution of conformational entropy to calcium affinity. J Mol Biol. 1998;278:667–686. doi: 10.1006/jmbi.1998.1723. [DOI] [PubMed] [Google Scholar]
- 48.Guo Z, Cascio D, Hideg K, Hubbell WL. Structural determinants of nitroxide motion in spin-labeled proteins: solvent-exposed sites in helix B of T4 lysozyme. Protein Sci. 2008;17:228–239. doi: 10.1110/ps.073174008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of Protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc. 1998;120:6836–6837. [Google Scholar]
- 50.Bermejo GA, Strub M-P, Ho C, Tjandra N. Determination of the solution-bound conformation of an amino acid binding protein by NMR paramagnetic relaxation enhancement: use of a single flexible paramagnetic probe with improved estimation of its sampling space. J Am Chem Soc. 2009;131:9532–9537. doi: 10.1021/ja902436g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.