

Abstract
Background and Purpose
Hydrogen sulphide (H2S) is a gas that has recently been shown to have biological activity. In the majority of blood vessels studied so far, H2S has been shown to cause vasorelaxation, although contractile responses have been reported. In the present study, we have made a pharmacological assessment of the effects of H2S in mesenteric small arteries isolated from rats.
Experimental Approach
Rat mesenteric small arteries were studied using pressure myography. In pressurised arteries, responses were obtained to the H2S donor, sodium hydrogen sulphide (NaHS), in the absence and presence of the NOS inhibitor L-NAME, raised extracellular potassium, the KATP channel inhibitor glibenclamide, the Cl– channel blockers DIDS, NPPB and A9C, the TRPV1 receptor desensitizing agent, capsaicin, the CGRP antagonist, olcegepant, the TRPV1 channel blocker capsazepine and the TRPA1 channel blocker HC-030031.
Key Results
NaHS produced a vasodilator response in rat mesenteric small arteries held at 90 mmHg. Responses to NaHS were not reproducible. Neither, glibenclamide nor, L-NAME inhibited responses to NaHS. DIDS abolished vasodilator responses to NaHS, but these were unaffected by the chloride channel blockers, NPPB and A9C. Responses to NaHS were attenuated after capsaicin pre-treatment, by a CGRP receptor antagonist and an inhibitor of TRPA1 channels.
Conclusions and Implications
In small arteries isolated from the rat mesentery, NaHS caused a vasodilatation. This response was not reproducible in vitro, since it was mediated by the release of sensory neurotransmitters in a capsaicin-like action. This release was mediated by a H2S-induced activation of TRPA1 channels.
Keywords: hydrogen sulphide, sensory nerves, CGRP, vascular
Introduction
Hydrogen sulphide (H2S) is a toxic gas that has become the subject of intense investigation over the past few years since it was discovered to be produced in mammals by specific synthetic enzymes (for review see: Li and Moore, 2008). In the vasculature, H2S has a complex profile. Initial studies showed that H2S caused a vasorelaxation in both the rat aorta and portal vein (Hosoki et al., 1997) in which the presence of the synthetic enzyme cystathionine-γ-lyase (CSE) was also shown. Subsequent studies suggested that the mechanism of vasorelaxation in the rat aorta and the rat perfused mesenteric arterial bed was due to activation of KATP channels (Zhao et al., 2001; Cheng et al., 2004). Additional evidence from single cells isolated from mesenteric arteries showed that H2S directly increased current flow through KATP channels (Zhao et al., 2001; Tang et al., 2005). However, it is now apparent that the mechanism of vasorelaxation varies considerably dependent upon the vessel studied and/or the experimental conditions employed. In the rat aorta, the H2S-induced vasorelaxation has been reported to involve the release of NO (Zhao et al., 2001), intracellular acidification (after activation of the Cl–/HCO3– exchanger) (Lee et al., 2007; Kiss et al., 2008), inhibition of phosphodiesterase activity (Bucci et al., 2010), metabolic inhibition (Kiss et al., 2008) and to be influenced by the prevailing level of oxygen use to gas the tissue (Koenitzer et al., 2007; Kiss et al., 2008). Interestingly, H2S has been reported to be an adipocyte-derived relaxing factor (ADRF) in the rat aorta via activation of voltage-dependent potassium (KCNQ) channels (Schliefenbaum et al., 2010). In the mouse aorta, H2S produces an endothelium-independent relaxation that involves activation of a variety of potassium and chloride channels (Kubo et al., 2007; Al-Magableh and Hart, 2011). In the human internal mammary artery, H2S produced a vasorelaxation that was modestly attenuated by the KATP channel blocker, glibenclamide (Webb et al., 2008). In rat coronary arteries, H2S causes vasorelaxation by activating Kv channels (Cheang et al., 2010). In some instances, the effects of H2S can be mimicked using the precursor, L-cysteine, an effect blocked by inhibitors of CSE, showing that H2S may also be involved in the physiological control of vascular tone (Cheng et al., 2004; Bucci et al., 2010; Leffler et al., 2011).
The vascular effects of H2S are complicated by the fact that it can also cause a contraction in some blood vessels (Dombkowski et al., 2004; Ali et al., 2006; Kubo et al., 2007). In the rat aorta and human internal mammary artery, it was proposed that the contraction resulted from H2S interacting with NO to form an inactive nitrosothiol compound, with the contraction following the removal of the vasorelaxatory influence of NO (Ali et al., 2006; Webb et al., 2008). These differential effects have resulted in H2S producing both a pressor and depressor response in vivo (Zhao et al., 2001; Ali et al., 2006). The predominant mechanism for endogenous H2S, however, is likely to be a vasodilatation since mice, which lack the most abundant vascular H2S synthetic enzyme, CSE, are hypertensive (Yang et al., 2008).
There are fewer studies that have examined responses to H2S in smaller arteries that are more important for determining vascular resistance. H2S has been shown to produce a vasorelaxation in mice mesenteric arteries, and it has been suggested that it may act as an endothelium-derived hyperpolarizing factor (EDHF) in these vessels by modifying the activity of several potassium channels (Yang et al., 2008; Mustafa et al., 2011). In rat mesenteric arterioles exposed to intermittent hypoxia, endogenous H2S has been shown to modulate myogenic tone by activating large-conductance Ca2+-activated potassium channels (BKCa) channels (Jackson-Weaver et al., 2011). In cerebral arterioles in new born pigs, H2S induced a vasodilatation mediated by activation of KATP channels and was proposed to mediate the dilator response to hypercapnia (Leffler et al., 2011).
Given that relatively few studies have examined the effect of H2S in small arteries, and given the complex profile of responses to this agent, we hypothesized that H2S would regulate vascular tone in rat mesenteric small arteries held under pressurized conditions, in vitro. We report a novel mechanism for H2S-induced vasodilatation involving activation of sensory nerves and the release of vasodilator neuropeptides, in a capsaicin-like action.
Methods
Male Wistar rats (150–200 g) were killed by stunning and bleeding. The gastrointestinal tract with the mesenteric arcade attached was excised and placed in physiological salt solution (PSS). Second-order arteries [mean diameter at 90 mmHg (± SD) 357 ± 48 μm, n = 192] were dissected and cleaned of connective tissue before being secured between two glass cannulas of a pressure myograph (Living Systems Instrumentation, Burlington, VT) (Rummery et al., 2007). One cannula was attached to a pressure-servo system containing PSS, allowing the control of intraluminal pressure and measurement of diameter changes under isobaric conditions. After residual blood was flushed from the lumen, the other cannula was sealed, and the vessel checked for leaks. Arteries were superfused, at a rate of approximately 50 mL·min−1 to ensure rapid delivery of drugs, with PSS gassed with 5% CO2 : 95% O2 and the temperature of the organ bath was maintained at 36–37°C. Vessels were imaged using a video camera, and the internal diameter was measured using a dimension analyser (Living Systems Instrumentation), linked to a MacLab data acquisition system.
Experimental procedure
Intraluminal pressure was set at 90 mmHg and vessels were allowed to equilibrate for 60 min. After this vessels were pre-constricted by 40–60% of the initial diameter using either, the thromboxane mimetic U46619 (9,11-dideoxy-9α,11α-methanoepoxy prostaglandin F2α), the α1-adrenoceptor agonist methoxamine or raised extracellular KCl. Once a stable pre-constriction was obtained, vessels were exposed to increasing concentrations of the H2S donor, NaHS (1–300 μM) in threefold increments. Each concentration was applied for 5 min before the addition of the next concentration. This time table was adhered to strictly, to limit the possibility of loss of H2S gas from the tissue bath during the construction of the concentration–response curve.
The reproducibility of responses was assessed by washing the tissue with fresh PSS, before pre-constricting vessels again (with U46619) and repeating the concentration–response curve to NaHS. Using this protocol, responses to NaHS were shown not to be reproducible. Therefore, these experiments were repeated, but the highest concentration of NaHS used was limited to 30 μM. Even under these conditions, responses to NaHS were attenuated upon second exposure. All experiments thereafter only involved obtaining a single concentration–response curve to NaHS in a single artery. The effects of antagonists were assessed in vessels obtained from the same animal and compared with a control experiment in an artery obtained from the same rat. The order of control and antagonist experiments was randomized.
The effects of the following drugs were assessed on NaHS responses: the NOS inhibitor L-NAME (100 μM); the non-specific K+ channel inhibitor, tetraethylammonium (TEA) (1 mM); the KATP channel inhibitor, glibenclamide (10 μM); the Cl– channel inhibitors; 4,4′-Diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS) (100 μM); 5-nitro-2-(3-phenylpropylamino)-benzoate (NPPB) (10 μM); anthracere-9-carboxylic acid (A9C) (100 μM); the CGRP antagonist, olcegepant (BIBN 4096 BS) (1 μM); the TRPV1 channel blocker, capsazepine (10 μM) and the TRPA1 channel blocker, 2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)-N-(4-isopropylphenyl)acetamide (HC-030031) (10 μM). These drugs were added before pre-constriction of the vessels with U46619, a minimum of 30 min before the generation of the concentration–response curve to NaHS.
Additionally, responses to NaHS were assessed after pre-constricting the arteries with raised extracellular potassium in the PSS (equimolar exchange with sodium). Once a stable vasoconstriction was obtained, amounting to a 40–60% reduction in arterial diameter, a concentration–response curve to NaHS was generated.
Some experiments were designed to desensitize the sensory nerves. In these experiments, vessels were pre-constricted with U46619 by 40–60%. Vessels were then exposed to either capsaicin (1 μM) or methanol, the vehicle control. Vessels were left for 10–20 min during which time capsaicin caused a transient vasodilator response. Vessels were then exposed to capsaicin (1 μM) or methanol for a second time. Thereafter, capsaicin or methanol was washed out of the system with fresh PSS. The vessel was left at least 30 min to re-equilibrate before pre-constriction with U46619 and the generation of a NaHS concentration–response curve. In some experiments, responses to capsaicin (1 μM) were determined after arteries had been exposed to NaHS (30 μM). Similar experiments were conducted using the KATP channel opener, pinacidil (Alexander et al., 2011) as the vasodilator agent.
Drugs and solutions
The composition of PSS was as follows (mM): NaCl 118, NaHCO3 25, glucose 11.1, KCl 4.8, MgSO4 2.5, KH2PO4 1.2 and CaCl2 1.25. Ca2+-free PSS composition was as above with the addition of EGTA (0.5 mM) and the omission of CaCl2. PSS with raised extracellular potassium was prepared by equimolar replacement of NaCl with KCl. All drugs and chemicals were purchased from Sigma-Aldrich (Gillingham, UK) unless otherwise stated. U46619 and pinacidil were obtained from Tocris (Abingdon, UK); HC-030031 and XE 991 were obtained from Abcam Biochemicals (Cambridge, UK).
Drugs were dissolved in double distilled water except as indicated below. NaHS was freshly prepared in PSS on the day of use. Stock solutions of capsaicin were prepared in methanol, while glibenclamide; NPPB, A9C, HC-030031 and DIDS were prepared in dimethyl sulphoxide (DMSO), with further dilutions made using double distilled water.
Data analysis
Vasoconstriction was determined as a % reduction in the initial diameter of the vessel once pressurized to 90 mmHg. Responses to NaHS were measured as the peak change during the 5 min time period of exposure and subsequently expressed as a percentage of the maximum possible vasodilator range, i.e. the difference in vessel diameter in Ca2+-free PSS and after the induction of tone with U46619, methoxamine or raised extracellular potassium. Results are presented as mean ± SEM with n = to the number of rats. The difference between means was considered statistically significant at a value of P < 0.05. Differences between concentration response curves were assessed using two-way anova with a Bonferroni post hoc test to assess difference between individual concentrations.
Results
Generating a cumulative concentration–response curve to NaHS, with additions at 5 min intervals, caused a vasodilator response. Several different patterns of response were observed. In all arteries, NaHS produced a vasodilatation with a threshold concentration of between 3 and 10 μM. In some vessels, NaHS fully dilated arteries, and this effect was maintained through the duration of the concentration–response curve (Figure 1A). In others, the vasodilator response was seen to wane during construction of the concentration–response curve (Figure 1B), before a second phase of vasodilatation completely relaxed the blood vessel at concentrations of NaHS above 100 μM.
Figure 1.
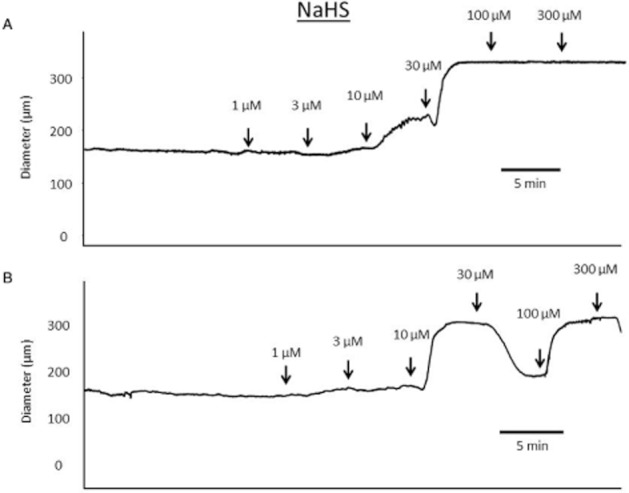
Sample traces showing cumulative concentration–response curves to NaHS (added at 5 min intervals) in rat mesenteric small arteries pressurized to 90 mmHg. U46619 was used to pre-constrict vessels by 40–60% of original vessel diameter and then a concentration–response curve was generated. (A) An example of NaHS producing a concentration-dependent vasodilatation that was maintained over the time course of constructing the concentration–response curve. (B) An example of a complex response to NaHS, with an initial vasodilatation that was not maintained returning towards baseline, before a further vasodilatation was produced by high concentrations of NaHS.
A vasodilator response was produced by NaHS regardless of whether U46619 or methoxamine was used as the agent to induce vasoconstriction (Figure 2A). Attempts to generate consecutive concentration–response curves to NaHS, after inducing tone with U46619, were not successful. Upon second exposure, arteries did not dilate until concentrations of NaHS reached at least 100 μM, resulting in a rightward displacement of the concentration–response curve (Figure 2B). Fearing that the inability to reproduce responses to NaHS was due to desensitization, we reduced the highest concentration used to 30 μM. Even with this change, however, the vasodilator response to NaHS was attenuated after first exposure (Figure 2C). The effects of drugs on responses to NaHS were therefore assessed in separate preparations.
Figure 2.
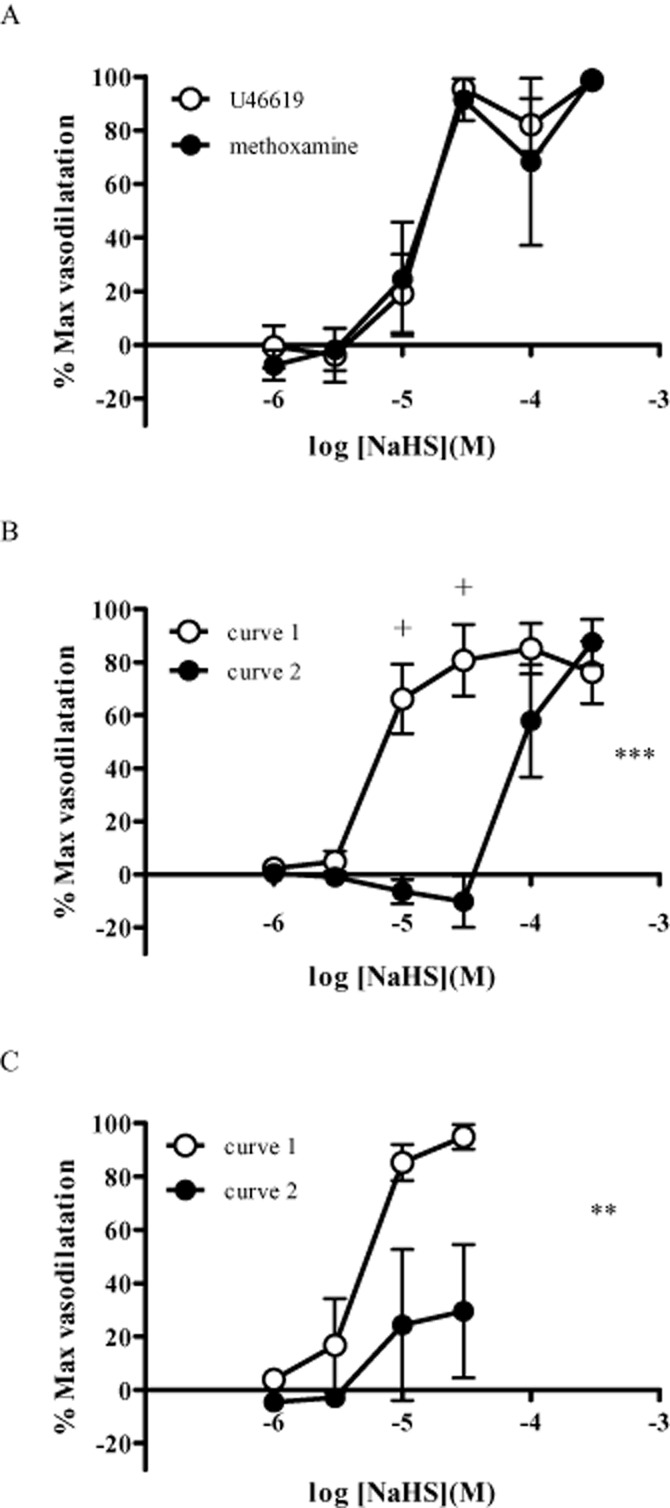
(A) Responses to NaHS after inducing vasoconstriction with either U46619 or methoxamine (n = 4). (B) Reproducibility of responses to NaHS (exposed to a maximum concentration of 100 μM) after pre-constriction with U46619 (n = 6). (C) Reproducibility of responses to NaHS (exposed to a maximum concentration of 10 μM) after pre-constriction with U46619 (n = 6). Each point represents the mean ± SEM. A significant difference between the curves is indicated by **P < 0.01 or ***P < 0.001 (two-way anova), with + representing a significant difference between individual concentrations (Bonferroni's post hoc test).
Responses to NaHS were similar in the absence and presence of L-NAME (100 μM) (Figure 3A). Raising the concentration of extracellular potassium to 31 ± 4 mM caused a vasoconstriction equivalent to a reduction in diameter of 45 ± 4% (n = 7) of the initial value. In the presence of raised extracellular K+, the vasodilator response to NaHS was abolished at concentrations below 100 μM uncovering a vasoconstrictor response. At a concentration of 300 μM, NaHS caused vasodilatation (Figure 3B). In the control experiments for this series, U46619 caused a constriction of 52 ± 4% (n = 7), and a NaHS-induced vasodilator response was evident at low concentrations (<30 μM) (Figure 3B).
Figure 3.
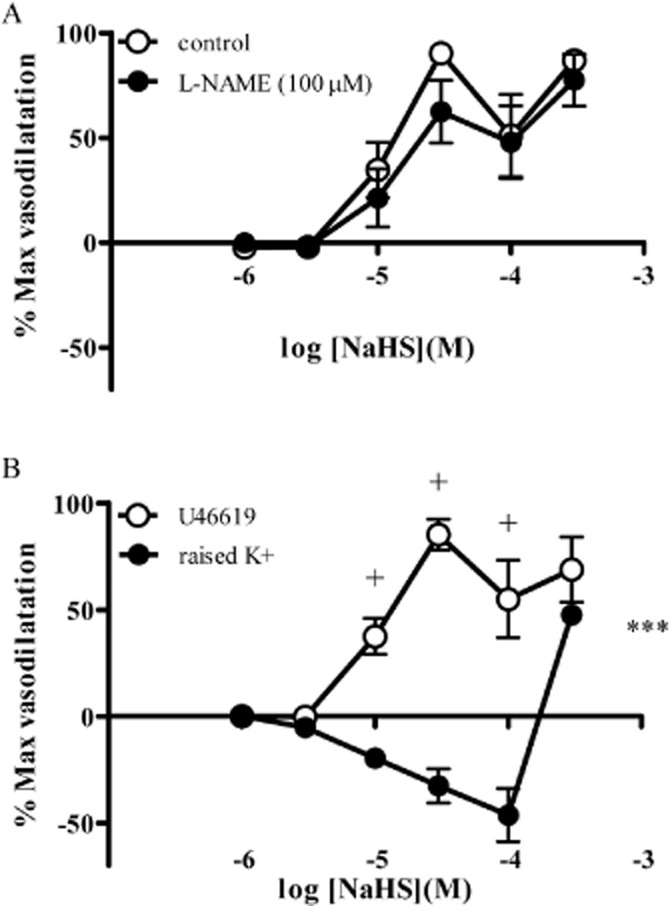
Responses to NaHS in the absence and presence of L-NAME (100 μM) (A) and after inducing tone using raised extracellular potassium (B). Each point represents the mean ± SEM (n = 7). A significant difference between the curves is indicated by ***P < 0.001 (two-way anova), with + representing a significant difference between individual concentrations (Bonferroni's post hoc test).
TEA (1 mM) (Figure 4A), glibenclamide (10 μM) (Figure 4B) and XE991 (10 and 30 μM) (Figure 4C) did not significantly alter responses to NaHS. XE991 caused a transient vasoconstriction in mesenteric arteries. By contrast, DIDS (1 mM) completely abolished responses to NaHS in mesenteric arteries at all concentrations of NaHS (Figure 5A). Despite this, other chloride channel blockers NPPB (10 μM) and A9C (100 μM) were without effect of responses to NaHS (Figure 5B, C).
Figure 4.
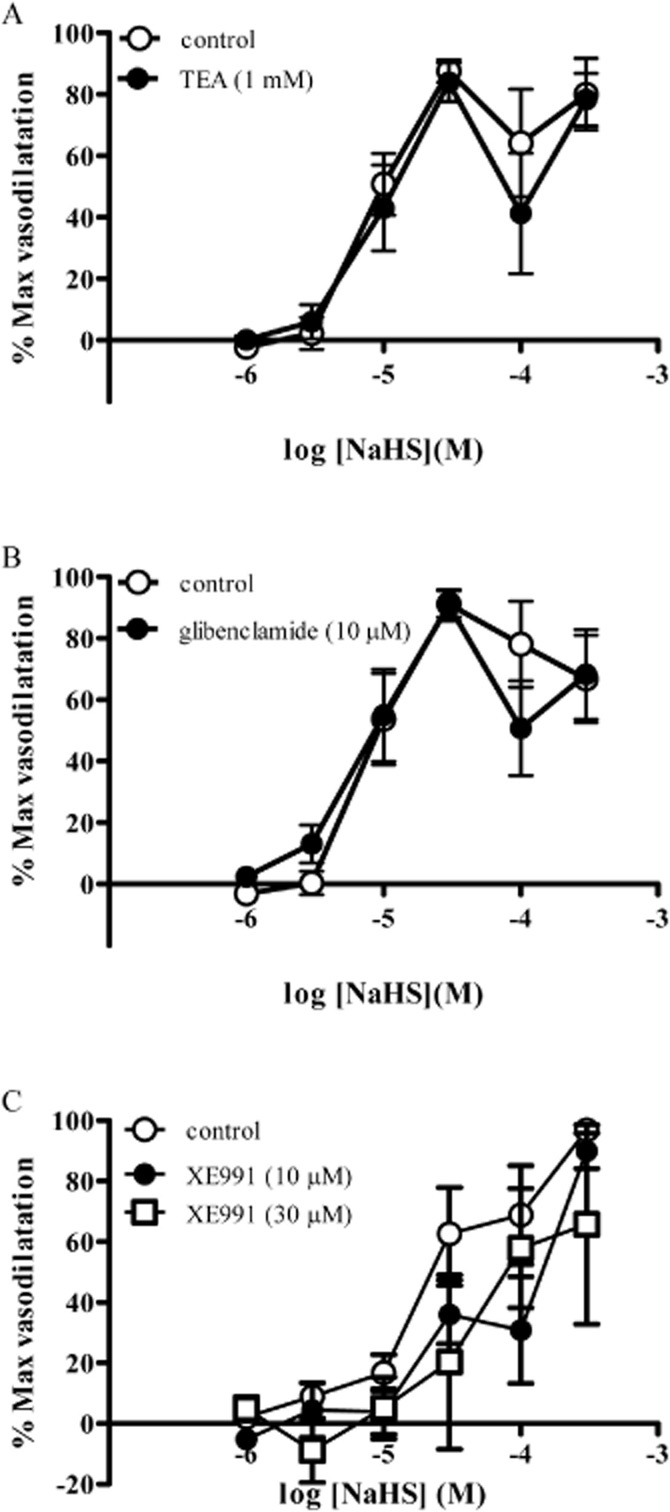
Responses to NaHS in the absence and presence of (A) TEA (1 mM), (B) glibenclamide (10 μM) and (C) XE991 (10 and 30 μM) in rat mesenteric small arteries. Each point represents the mean ± SEM (n = 4–8).
Figure 5.
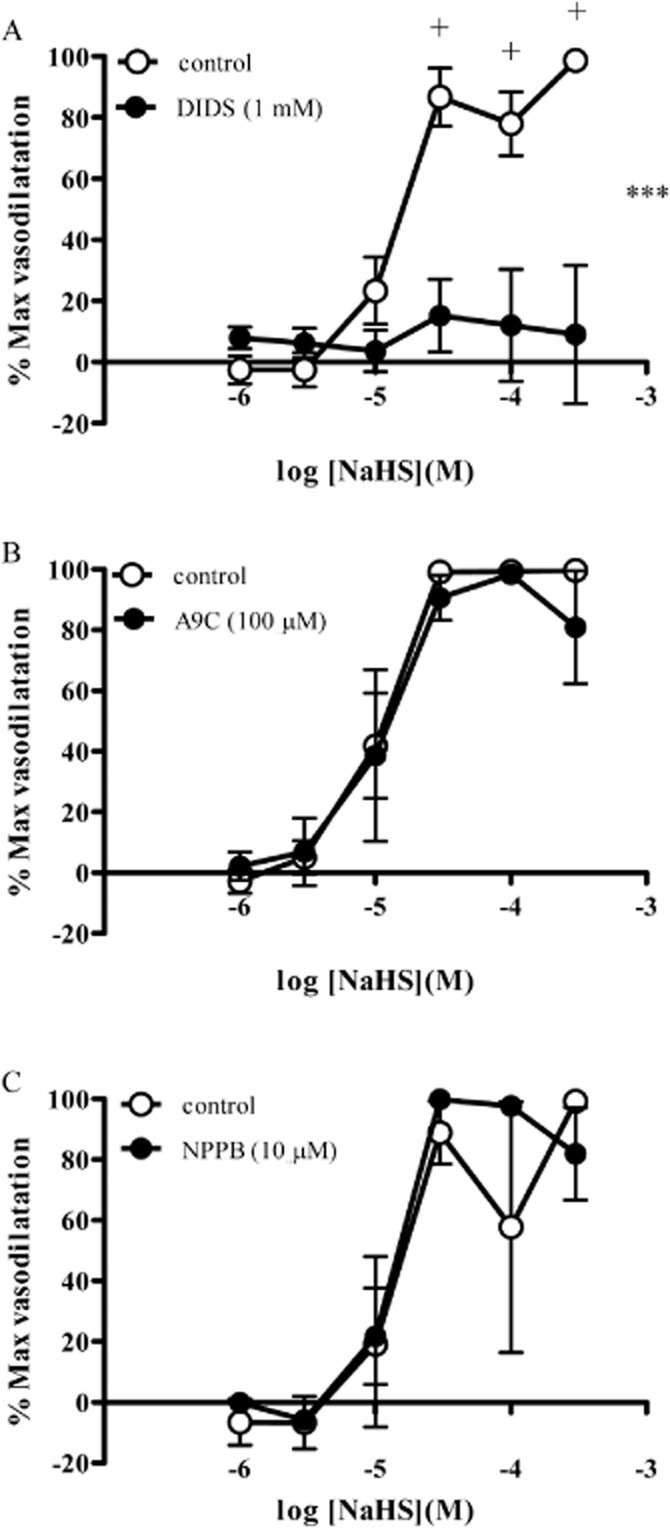
Responses to NaHS in the absence and presence of (A) DIDS (1 mM), (B) A9C (100 μM) and (C) NPPB (10 μM) in rat mesenteric small arteries. Each point represents the mean ± SEM (n = 6). A significant difference between the curves is indicated by ***P < 0.001 (two-way anova), with + representing a significant difference between individual concentrations (Bonferroni's post hoc test).
To investigate the possible role of sensory nerves in mediating NaHS-induced vasodilatation, vessels were exposed to capsaicin. Upon first exposure to capsaicin (1 μM), a transient vasodilator response equivalent to a 74 ± 25% (n = 8) reversal of U46619-induced tone was observed, returning to baseline over a 10–20 min timescale. Subsequent exposure to capsaicin (1 μM) produced no response. After exposure to capsaicin, the NaHS-induced vasodilatation was significantly attenuated, uncovering a small vasoconstrictor response at concentrations up to 100 μM (Figure 6A). In these arteries, vasodilator responses were produced by 300 μM NaHS, mirroring the response observed after repeat exposure to NaHS (Figure 2A). In a separate series of experiments, arteries were first exposed to NaHS (30 μM) which caused a vasodilatation equivalent to a 48 ± 16% (n = 5) reversal of U46619-induced tone. This was a transient response that returned to baseline over a period of between 12 and 29 min. Subsequent exposure to capsaicin did not alter vascular tone [reversal of U46619-induced tone was 1 ± 1% (n = 5)]. The CGRP antagonist olcegepant (1 μM) produced a similar effect to capsaicin pre-treatment since it abolished NaHS-induced vasodilatation at lower concentrations (up to and including 100 μM), uncovering a small vasoconstrictor response, with vasodilator response only apparent at 300 μM NaHS (Figure 6B). Responses to pinacidil (Figure 7) (log EC50 −6.1 ± 0.2) were unaffected by capsaicin pre-treatment (log EC50 −6.2 ± 0.1) and olcegepant (1 μM) (log EC50 −6.4 ± 0.2) (n = 4).
Figure 6.
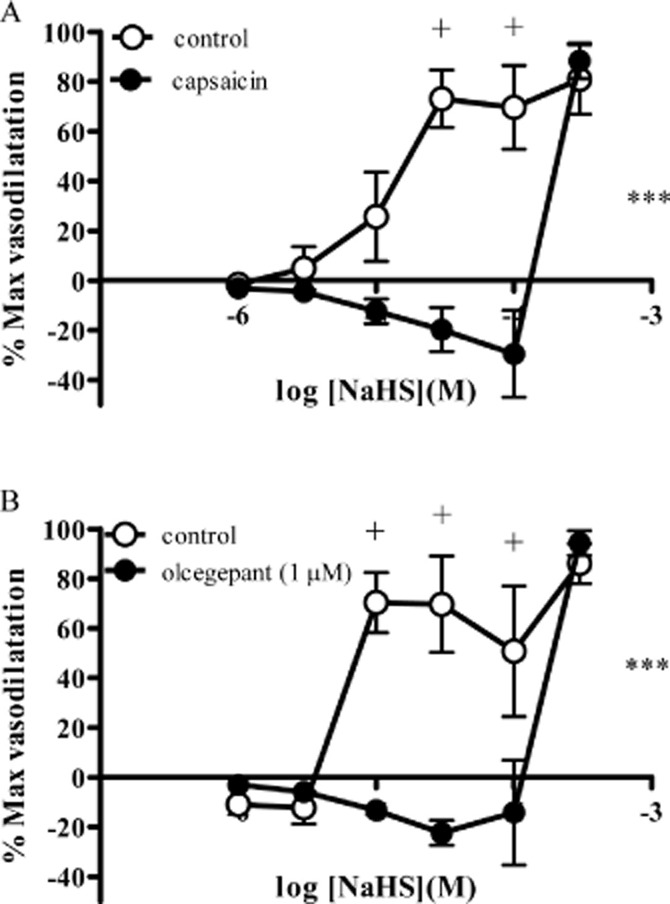
Responses to NaHS (A) after pre-treatment with capsaicin or the vehicle control, methanol (n = 8) or (B) Olcegepant (1 μM) (n = 6) in rat mesenteric small arteries. Each point represents the mean ± SEM. A significant difference between the curves is indicated by ***P < 0.001 (two-way anova), with + representing a significant difference between individual concentrations (Bonferroni's post hoc test).
Figure 7.
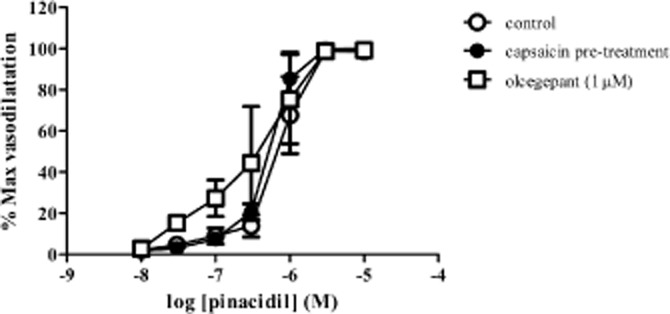
Responses to pinacidil after pre-treatment with capsaicin or in the presence of Olcegepant (1 μM) (n = 4) in rat mesenteric small arteries. Each point represents the mean ± SEM.
The vasodilator response to NaHS was unaffected by the TRPV1 channel blocker capsazepine (10 μM) (Figure 8A) but was antagonized by the TRPA1 channel blocker HC-030031 (10 μM) (Figure 8B).
Figure 8.
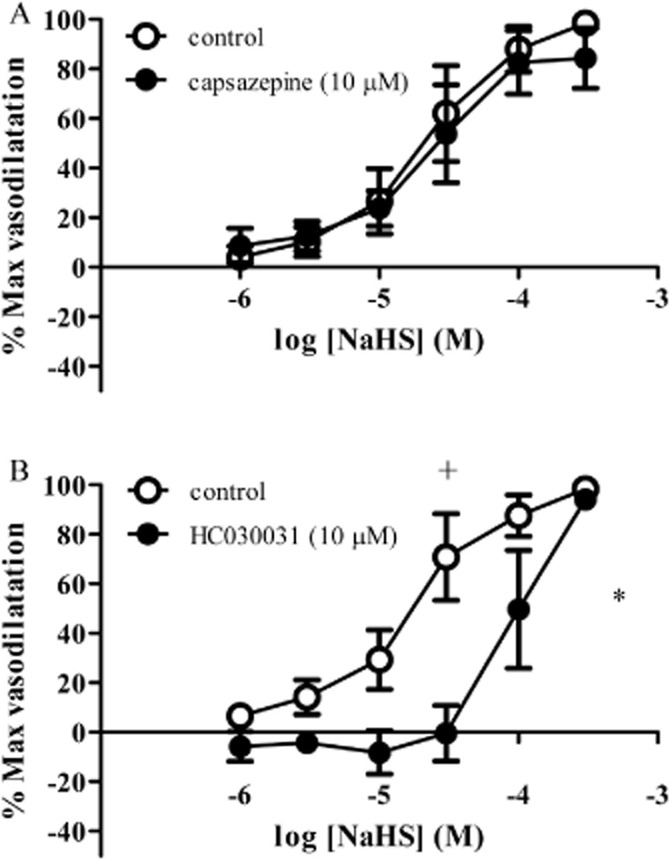
Effect of (A) capsazepine (10 μM) or (B) HC-030031 (10 μM) (n = 6) on responses to NaHS in rat mesenteric small arteries. Each point represents the mean ± SEM. A significant difference between the curves is indicated by *P < 0.05 (two-way anova), with + representing a significant difference between individual concentrations (Bonferroni's post hoc test).
Discussion
The main findings of the current study are that the H2S donor, NaHS, caused a vasodilator response in mesenteric small arteries isolated from rats. These responses were not reproducible, suggesting they involve some mechanism of desensitization. Responses to NaHS were sensitive to capsaicin, which desensitizes TRPV1 channels on sensory nerves causing the release of vasodilator sensory neuropeptides. NaHS-induced vasodilatation was insensitive to the TRPV1 channel blocker but antagonized by the TRPA1 channel blocker HC-030031. Since responses to NaHS were also sensitive to the CGRP antagonist, olcegepant, these data suggest that H2S-induced vasodilatation causes the release of CGRP from sensory nerves after activation of TRPA1 channels.
NaHS caused a concentration-dependent vasodilatation in rat mesenteric small arteries, consistent with the vasorelaxatory response observed in larger blood vessels (Hosoki et al., 1997; Kubo et al., 2007; Cheang et al., 2010), the vasodilator response in the perfused rat mesenteric arterial bed (Cheng et al., 2004) and small arteries (Yang et al., 2008; Jackson-Weaver et al., 2011; Mustafa et al., 2011) and the depressor response observed in vivo (Zhao et al., 2001; Ali et al., 2006). There has been some suggestion that the response can be influenced by the agent used to pre-constrict arteries (Dombkowski et al., 2004), although this was not the case in rat mesenteric small arteries since responses to NaHS were identical whether vasoconstriction was induced with either methoxamine or U46619. Responses to NaHS were not reproducible in rat mesenteric small arteries. Few studies have reported upon the reproducibility of responses to NaHS in isolated blood vessels. Indeed, the majority of the studies seem to report examination of the effects of inhibitors on an unpaired basis (see references from the introduction). In one of the few studies to examine reproducibility, Dombkowski et al. (2004) reported that responses to NaHS were not reproducible in several elasmobranch species. It is possible that the desensitization occurred as a consequence of a non-specific metabolic inhibition via suppression of oxidative phosphorylation produced by high concentrations (>100 μM) of NaHS (Kiss et al., 2008). However, limiting the concentration of NaHS in the first concentration–response curve to 30 μM did not prevent the desensitization. Subsequent experiments were carried out by obtaining a single concentration–response curve to NaHS in the absence and presence of putative inhibitors.
A variety of studies have proposed a role for potassium channels in mediating the vasorelaxation produced by H2S. These include activation of BKCa (Yang et al., 2008; Jackson-Weaver et al., 2011; Mustafa et al., 2011) and, more predominantly, KATP channels (Zhao et al., 2001; Cheng et al., 2004; Leffler et al., 2011). In rat mesenteric small arteries, raising the extracellular potassium concentration attenuated responses to NaHS, indicating the potential involvement of a hyperpolarizing mechanism. However, this was not due to activation of KATP channels, since glibenclamide was without effect, nor was it due to activation of BKCa channels, since responses were also insensitive to TEA (1 mM) at a concentration that inhibits these channels (Fallet et al., 2001). Similarly, it is unlikely that NaHS is activating Kv channels, since the Kv channel blocker XE991 (Yeung and Greenwood, 2005) was largely without effect on responses to NaHS. An alternative explanation for the effect of raising extracellular potassium on responses to NaHS is that the associated depolarization activates sensory nerves, causing the release (and subsequent depletion) of sensory neuropeptides (see below).
Lee et al. (2007) and Kiss et al. (2008) reported that H2S-induced vasodilatation in the rat aorta was sensitive to DIDS, which blocks a variety of chloride channels and the Cl–/HCO3– exchange protein (Alexander et al., 2011). Similarly, DIDS abolished responses to NaHS in rat mesenteric arteries in the present study. This is not due to modulation of the gating of chloride channels, since neither of the non-selective chloride channel blockers, NPPB or A9C affected responses to NaHS. Thus it seems likely that H2S, during some part of the transduction process, causes intracellular acidification by activating the Cl–/HCO3– exchange protein, as has been reported for both the rat aorta (Lee et al., 2007) and also in cultured glial cells (Lu et al., 2010). In mesenteric arteries, it is possible that one of the sites of action is on perivascular sensory nerves (see below), in which intracellular acidification has been shown to be a potent stimulus for the release of neuropeptides such as CGRP (Vause et al., 2007).
In the rat urinary bladder, NaHS has been shown to activate the transient receptor potential vanilloid receptor 1 (TRPV1) on sensory nerves causing the release of neuropeptides and a contractile response in a similar action to that produced by the TRPV1 agonist capsaicin (Trevisani et al., 2005). Furthermore, in the guinea pig and human colon, NaHS has been shown to activate TRPV1 receptors located on afferent nerves to cause the release of neuropeptides, subsequently evoking mucosal secretions (Krueger et al., 2010). We showed that exposure to capsaicin caused a transient vasodilator response in rat mesenteric arteries that was not reproducible since it involves the release of a finite pool of neurotransmitters from sensory nerves (Kawasaki et al., 1988). Since responses to NaHS were frequently transient in nature and not reproducible, we hypothesized that NaHS may be acting in a similar manner. In support of this hypothesis, capsaicin pre-treatment abolished vasodilator responses to NaHS, at concentrations below 100 μM. However, the TRPV1 channel blocker capsazepine was without effect on responses to NaHS in rat mesenteric arteries. H2S has also been shown to activate TRPA1 channels on sensory neurones in the rat bladder (Streng et al., 2008) and dorsal root ganglia (Miyamoto et al., 2011). Since the TRPA1 channel blocker HC-030031 antagonized NaHS-induced vasodilatation, we propose that H2S activates TRPA1 channels to cause the release of vasodilator sensory neuropeptides. Since, the CGRP antagonist olcegepant markedly attenuated responses to low concentrations of NaHS, this indicates that CGRP is the neurotransmitter released from sensory nerves in mesenteric arteries to mediate the vasodilator effect of NaHS.
Under the majority of experimental conditions, a vasodilatation was observed with very high (>100 μM) concentrations of NaHS. This was insensitive to raising extracellular potassium, capsaicin pre-treatment and the CGRP antagonist, olcegepant. It was, however, abolished by DIDS, suggesting that it involved intracellular acidification of vascular smooth muscle cells (Lee et al., 2007; Kiss et al., 2008). Indeed, DIDS was the only agent used that completely abolished responses to NaHS, which could indicate that intracellular acidification may act as the final common pathway downstream of activation of sensory nerves and for direct effects of H2S on vascular smooth muscle.
In summary, NaHS caused a vasodilator response in mesenteric small arteries isolated from rats. Vasodilator responses were sensitive to capsaicin pre-treatment, a TRPA1 channel blocker and a CGRP receptor antagonist suggesting H2S causes the release of CGRP from sensory nerves to cause vasodilatation after activation of TRPA1 channels.
Note added in proof
In support of the data presented here, Pozsgai et al. (2012) have recently shown that hydrogen-sulphide-mediated vasodilatation in the murine ear involves the release of CGRP by activation of TRPA1 receptors.
Acknowledgments
BJOW held a BBSRC studentship during this project.
Glossary
- ADRF
adipocyte-derived hyperpolarizing factor
- BKCA
large conductance Ca2+-activated potassium channels
- CGRP
calcitonin gene related polypeptide
- CSE
cystathionine-γ-lyase
- DMSO
dimethyl sulphoxide
- EDHF
endothelium-derived hyperpolarizing factor
- KCNQ
voltage-gated potassium channels
- TRPA1
transient receptor potential ankyrin 1
- TRPV1
transient receptor potential vanilloid 1
Conflict of interest
There are no conflicts of interest for any of the authors with the material presented in this manuscript.
References
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels. Br J Pharmacol. 2011;164(Suppl. 1):1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali MY, Ping CY, Mok Y-YP, Ling L, Whiteman M, Bhatia M, et al. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br J Pharmacol. 2006;149:625–634. doi: 10.1038/sj.bjp.0706906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Magableh MR, Hart JL. Mechanism of vasorelaxation and role of endogenous hydrogen sulphide production in mouse aorta. Naunyn Schmiedebergs Arch Pharmacol. 2011;383:403–413. doi: 10.1007/s00210-011-0608-z. [DOI] [PubMed] [Google Scholar]
- Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Pyriochou A, Roussos C, et al. Hydrogen sulphide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler Thromb Vasc Biol. 2010;30:1998–2004. doi: 10.1161/ATVBAHA.110.209783. [DOI] [PubMed] [Google Scholar]
- Cheang WS, Wong WT, Shen B, Lau CW, Tian XY, Tsang SY, et al. 4-aminopyridine-sensitive K+ channels contributes to NaHS-induced membrane hyperpolarization and relaxation in the rat coronary artery. Vasc Pharmacol. 2010;53:94–98. doi: 10.1016/j.vph.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. Hydrogen sulphide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol. 2004;287:H2316–H2323. doi: 10.1152/ajpheart.00331.2004. [DOI] [PubMed] [Google Scholar]
- Dombkowski RA, Russell MJ, Schulman AA, Doellman MM, Olson KR. Vertebrate phylogeny of hydrogen sulphide vasoactivity. Am J Physiol. 2004;288:R243–R252. doi: 10.1152/ajpregu.00324.2004. [DOI] [PubMed] [Google Scholar]
- Fallet RW, Bast JP, Fujiwara K, Ishii N, Sansom SC, Carmines PK. Influence of Ca2+-activated K+ channels on rat renal arteriolar responses to depolarizing agents. Am J Physiol. 2001;280:F583–F591. doi: 10.1152/ajprenal.2001.280.4.F583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulphide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Comm. 1997;237:527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- Jackson-Weaver O, Paredes DA, Gonzalez Bosc LV, Walker BR, Kanagy NL. Intermittent hypoxia in rats increases myogenic tone through loss of hydrogen sulphide activation of large-conductance Ca2+-activated potassium channels. Circ Res. 2011;108:1439–1447. doi: 10.1161/CIRCRESAHA.110.228999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Takasaki K, Saito A, Goto K. Calcitonin-gene-related polypeptide acts as a novel vasodilator neurotransmitter in mesenteric resistance vessels of the rat. Nature. 1988;335:164–167. doi: 10.1038/335164a0. [DOI] [PubMed] [Google Scholar]
- Kiss L, Deitch EA, Szabo C. Hydrogen sulphide decreases adenosine triphosphate levels in aortic rings and leads to vasorelaxation via metabolic inhibition. Life Sci. 2008;83:589–594. doi: 10.1016/j.lfs.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenitzer JR, Isbell TS, Patel HD, Benavides GA, Dickinson DA, Patel RP, et al. Hydrogen sulphide mediates vasoactivity in an O2-dependent manner. Am J Physiol. 2007;292:H1953–H1960. doi: 10.1152/ajpheart.01193.2006. [DOI] [PubMed] [Google Scholar]
- Krueger D, Foerster M, Mueller K, Zeller F, Slotta-Huspenina J, Donovan J, et al. Signalling mechanisms involved in the intestinal pro-secretory actions of hydrogen sulphide. Neurogastroenterol Motil. 2010;22:1224–1231. doi: 10.1111/j.1365-2982.2010.01571.x. [DOI] [PubMed] [Google Scholar]
- Kubo S, Doe I, Kurokawa Y, Nishikawa H, Kawabata A. Direct inhibition of endothelial nitric oxide synthase by hydrogen sulphide: contribution to dual modulation of vascular tension. Toxicology. 2007;232:138–146. doi: 10.1016/j.tox.2006.12.023. [DOI] [PubMed] [Google Scholar]
- Lee SW, Cheng Y, Moore PJ, Bian J-S. Hydrogen sulphide regulates intracellular pH in vascular smooth muscle cells. Biochem Biophys Res Commun. 2007;358:1142–1147. doi: 10.1016/j.bbrc.2007.05.063. [DOI] [PubMed] [Google Scholar]
- Leffler CW, Parfenova H, Basuroy S, Jaggar JH, Umstot ES, Fedinec AL. Hydrogen sulphide and cerebral microvascular tone in newborn pigs. Am J Physiol. 2011;300:H440–H447. doi: 10.1152/ajpheart.00722.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Moore PK. Putative biological roles of hydrogen sulphide in health and disease: a breath of not so fresh air? Trends Pharmacol Sci. 2008;29:84–90. doi: 10.1016/j.tips.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Lu M, Choo CH, Hu LF, Tan BH, Hu G, Bian JS. Hydrogen sulphide regulates intracellular pH in rat primary cultured glia cells. Neurosci Res. 2010;66:92–98. doi: 10.1016/j.neures.2009.09.1713. [DOI] [PubMed] [Google Scholar]
- Miyamoto R, Otsuguro K-I, Ito S. Time- and concentration-dependent activation of TRPA1 by hydrogen sulphide in rat DRG neurons. Neurosci Lett. 2011;499:137–142. doi: 10.1016/j.neulet.2011.05.057. [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, et al. Hydrogen sulphide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res. 2011;109:1259–1268. doi: 10.1161/CIRCRESAHA.111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozsgai G, Hajna Z, Bagoly T, Boros M, Kemény Á, Materazzi S, et al. The role of transient receptor potential ankyrin 1 (TRPA1) receptor activation in hydrogen-sulphide-induced CGRP-release and vasodilation. Eur J Pharmacol. 2012;689:56–64. doi: 10.1016/j.ejphar.2012.05.053. [DOI] [PubMed] [Google Scholar]
- Rummery NM, Brock JA, Pakdeechote P, Ralevic V, Dunn WR. ATP is the predominant sympathetic neurotransmitter in rat mesenteric arteries at high pressure. J Physiol. 2007;582:745–754. doi: 10.1113/jphysiol.2007.134825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliefenbaum J, Kohn C, Voblova N, Dubrovska G, Zavarirskaya O, Gloe T, et al. Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulphide. J Hypertens. 2010;28:1875–1882. doi: 10.1097/HJH.0b013e32833c20d5. [DOI] [PubMed] [Google Scholar]
- Streng T, Axelsson HE, Hedlund P, Andersson DA, Jordt S-E BS, et al. Distribution and function of the hydrogen sulphide-sensitive TRPA1 ion channel in rat urinary bladder. J Urol. 2008;53:391–400. doi: 10.1016/j.eururo.2007.10.024. [DOI] [PubMed] [Google Scholar]
- Tang G, Wu L, Liang W, Wang R. Direct stimulation of KATP channels by exogenous and endogenous hydrogen sulphide in vascular smooth muscle cells. Mol Pharmacol. 2005;68:1757–1764. doi: 10.1124/mol.105.017467. [DOI] [PubMed] [Google Scholar]
- Trevisani M, Patacchini NP, Gatti R, Gazzieri D, Lissi N, et al. Hydrogen sulphide causes vanilloid receptor 1-mediated neurogenic inflammation in the airways. Br J Pharmacol. 2005;145:1123–1131. doi: 10.1038/sj.bjp.0706277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vause C, Bowen E, Spierings E, Durham P. Effect of carbon dioxide on calcitonin gene-related polypeptide secretion from trigeminal neurons. Headache. 2007;47:1385–1397. doi: 10.1111/j.1526-4610.2007.00850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb GD, Lim LH, Oh VMS, Yeo SB, Cheong YP, Ali MY, et al. Contractile and vasorelaxant effects of hydrogen sulphide and its biosynthesis in the human internal mammary artery. J Pharmacol Exp Ther. 2008;324:876–882. doi: 10.1124/jpet.107.133538. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, et al. H2S as a physiological vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SYM, Greenwood IA. Electrophysiological and functional effects of the KCNQ channel blocker XE991 on murine portal vein smooth muscle cells. Br J Pharmacol. 2005;146:585–595. doi: 10.1038/sj.bjp.0706342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effects of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]