

Abstract
Background and Purpose
Recent evidence suggested that urotensin II (UII) and its paralog peptide UII-related peptide (URP) might exert common but also divergent physiological actions. Unfortunately, none of the existing antagonists were designed to discriminate specific UII- or URP-associated actions, and our understanding, on how these two endogenous peptides can trigger different, but also common responses, is limited.
Experimental Approach
Ex vivo rat and monkey aortic ring contraction as well as dissociation kinetics studies using transfected CHO cells expressing the human urotensin (UT) receptors were used in this study.
Key Results
Ex vivo rat and monkey aortic ring contraction studies revealed the propensity of [Pep4]URP to decrease the maximal response of human UII (hUII) without any significant change in potency, whereas no effect was noticeable on the URP-induced vasoconstriction. Dissociation experiments demonstrated the ability of [Pep4]URP to increase the dissociation rate of hUII, but not URP. Surprisingly, URP, an equipotent UII paralog, was also able to accelerate the dissociation rate of membrane-bound 125I-hUII, whereas hUII had no noticeable effect on URP dissociation kinetics. Further experiments suggested that an interaction between the glutamic residue at position 1 of hUII and the UT receptor seems to be critical to induce conformational changes associated with agonistic activation. Finally, we demonstrated that the N-terminal domain of the rat UII isoform was able to act as a specific antagonist of the URP-associated actions.
Conclusion
Such compounds, that is [Pep4]URP and rUII(1–7), should prove to be useful as new pharmacological tools to decipher the specific role of UII and URP in vitro but also in vivo.
Keywords: urotensin II, urotensin II-related peptide, insurmountable antagonism, GPCR, human UT receptors, aortic ring bioassay
Introduction
Despite the currently available drug therapies, cardiovascular diseases remain one of the major causes of ill health in the western world. Therefore, identification of additional targets, such as GPCRs, that may modulate pathological state is of particular interest. Following its desorphanization, the urotensin II (UII) receptor, initially termed sensory epithelium neuropeptide-like receptor or GPR14, has been studied for its implication in the cardiovascular homeostasis in either health or disease state (Ross et al., 2010). Initially isolated more than 20 years ago from the caudal neurosecretory system of teleost fish, UII has been subsequently characterized in several species (Vaudry et al., 2010) including human (Chartrel et al., 2004). This highly conserved cyclic peptide exerts a broad spectrum of biological actions in mammals including the modulation of cardiorenal, pulmonary, central nervous systems and endocrine functions (Vaudry et al., 2010). In particular, UII has been described as the most potent vasoconstrictor to date, being up to two orders of magnitude more potent than endothelin-1 (ET-1) (Douglas and Ohlstein, 2000). Recent works in mammals revealed the existence of a second gene encoding a precursor of a UII paralog, called UII-related peptide (URP) (Sugo and Mori, 2008). This octapeptide, identified in humans, mice and rats (Sugo and Mori, 2008), also comprises the highly conserved cyclic hexapeptide sequence found in UII. UII- and URP-associated actions are mediated by the activation of a specific GPCR, urotensin (UT), which plays a seminal role in the physiological regulation of major mammalian organ systems, including the cardiovascular system (Vaudry et al., 2010). As a matter of fact, UII exerts potent haemodynamic effects (Krum and Kemp, 2007), positive inotropic and chronotropic responses (Watson et al., 2003) and osmoregulatory actions (Song et al., 2006), induces collagen and fibronectin accumulation (Dai et al., 2007; Zhang et al., 2008), modulates the inflammatory response (Shiraishi et al., 2008), plays a role in the induction of cardiac and vascular hypertrophy (Papadopoulos et al., 2008), causes a strong angiogenic effect (Guidolin et al., 2010) and inhibits the glucose-induced insulin release (Silvestre et al., 2004). Thus, the urotensinergic system has been linked to numerous pathophysiological states including atherosclerosis, heart failure, hypertension, pre-eclampsia, diabetes, renal and liver disease, variceal bleeding, ulcers and psychological and neurological disorders (Ross et al., 2010). Therefore, the development of potent compounds that are able to modulate UT activity is mandatory.
Recent evidence suggested that UII and its paralog peptide URP might exert common but also divergent physiological actions (Prosser et al., 2008; Jarry et al., 2010). For instance, studies have reported a differential action for these two peptides on cell proliferation (Jarry et al., 2010), and distinctive myocardial contractile activities (Prosser et al., 2008). In isolated ischaemic heart experiments, both peptides were able to reduce myocardial injury through creatine kinase reduction, but only UII was able to reduce atrial natriuretic peptide (ANP) production (Prosser et al., 2008). More recently, a differential transcriptional modulation upon UII or URP activation was observed in isolated heart nuclei (Doan et al., 2012). Moreover, distinct pathophysiological roles for UII and URP in hypertension have been suggested (Hirose et al., 2009). Indeed, the mRNA expression of both UII and URP were up-regulated in the atrium of spontaneously hypertensive rats (SHR) when compared with age-matched Wistar Kyoto (WKY) rats. However, the specific up-regulation of URP but not UII mRNA in the aorta and kidney of SHR rats supports the idea that these peptides may act individually in biological system (Hirose et al., 2009). Key questions remain regarding the specific role associated with each peptide in this system. Over the past decade, development of non-peptide UT antagonists has allowed investigators to begin to delineate the (patho)physiological roles of the UII/UT system (Maryanoff and Kinney, 2010). None of the existing antagonists (peptidic and non-peptidic) were designed to discriminate specific UII- or URP-associated actions. During the course of structure–activity relationship studies on URP derivatives, we identified a new antagonist [Bip4]URP, that is urocontrin, characterized by a specific behaviour that sets it apart from known UT antagonists (Chatenet et al., 2012). Indeed, this compound was able to specifically reduce the maximal efficacy of human UII (hUII)- but not URP-associated vasoconstriction. Unfortunately, [Bip4]URP is also able to exert a weak but significant agonistic activity both in vitro and in vivo (Chatenet et al., 2012). Based on this atypical behaviour, we thus initiated structure–activity relationship studies through the conception of Pd-catalysed URP derivatives libraries, for example Sonogashira cross-coupling, and identified [Pep4]URP, with no agonistic activity, as a promising lead for the conception of allosteric modulators of the urotensinergic system. Next, we investigated the presence of specific peptide–receptor interactions responsible for hUII- and URP-mediated action and discovered that the N-terminal region of the rat UII (rUII) isoform could serve as a selective URP antagonist. Altogether, these results offer new tools to decipher the specific physiological roles of each peptide and provide future strategies for the specific treatment of UII- or URP-associated pathologies.
Methods
Peptide synthesis
hUII, URP, hUII(4–11) and ET-1 as well as Ala-substituted hUII derivatives were synthesized as previously described (Brkovic et al., 2003; Bourgault et al., 2007). UII derivatives, that is [Pep4]URP (urocontrin A; UCA) and [Pep7]hUII, were synthesized manually by the solid-phase approach, starting with tert-butoxycarbonyl (Boc)-Val-chloromethylated (CM) resin (1 g, 0.54 mmol·g−1). The C-terminally amidated rUII(1–7) was synthesized on a 4-methylbenzhydrylamine resin. Couplings were mediated for 1 h by diisopropylcarbodiimide (DIC) in dichloromethane (DCM) or DIC with N-hydroxybenzotriazole in DCM and dimethylformamide (DMF) (1:1). A three-equivalent excess of the protected amino acids based on the original substitution of the resin was used. Trifluoroacetic acid (TFA), 60% in DCM (2% meta-cresol), was applied for 10–15 min to remove the Boc group. The phenylacetylenyl side chain was introduced on resin via a modified version of the Sonogashira cross-coupling reaction according to a previously reported method (Hoffmanns and Metzler-Nolte, 2006). To do so, the N-terminally Boc protected iodinated-peptide-resin (1 equiv.), phenylacetylene (3 equiv.), PdCl2(PPh3)2 (0.1 equiv.), copper iodide (CuI) (0.1 equiv.) and triethylamine (3 equiv.) in degassed DMF (20 mL) were stirred overnight at 40°C. After successive washings with DMF (×3), H2O (×3), MeOH (×2), DMF (×3) and DCM (×3), peptides were cleaved and deprotected in hydrogen fluoride in the presence of 10% anisole and 10% methyl sulfide for 90 min at 0°C. The diethyl ether-precipitated crude peptides were solubilized in 70% acetic acid (1 mg·mL−1) and then cyclized by the addition of iodine [10% (w/v) solution in methanol] until appearance of a stable orange colour (Erchegyi et al., 2009). Thirty minutes later, ascorbic acid was added to quench the excess of iodine. Crude lyophilized peptides were purified using a preparative reversed-phase (RP) HPLC protocol using a linear gradient from eluant A to B with 1% B per 2 min increments (Eluent A = H2O, 0.1% TFA, Eluent B = 60% CH3CN/40% A, 0.1% TFA). Homogeneity of purified fractions was assessed by analytical RP-HPLC and matrix-assisted desorption ionization time-of-flight mass spectrometry in linear mode using α-cyanohydroxycinnamic acid as matrix. Pure fractions (≥98%) containing the product were pooled and subjected to lyophilization.
Animals
Adult male Sprague-Dawley rats (Charles Rivers, St-Constant, QC, Canada) weighing 250–300 g or 350–400 g were housed in group cages under controlled illumination (12:12 h light-dark cycle), humidity and temperature (21–23°C) and had free access to tap water and Purina rat chow. All experimental procedures were performed in accordance with regulations and ethical guidelines from the Canadian Council for the Care of Laboratory Animals and received approvals by the respective institutional animal care and use committee of the Institut National de la Recherche Scientifique-Institut Armand-Frappier and of the Montreal Heart Institute. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Cell culture
Transfected CHO cells (generous gift from Drs H. Vaudry and C. Dubessy, Rouen, France) expressing the human urotensin II receptor (CHO-UT) were maintained in Ham-F12 medium with 10% FBS, 2 mM L-glutamine, 100 UI·mL−1 each of penicillin and streptomycin and 400 μg·mL−1 G418 (Chatenet et al., 2004).
Peptide iodination
Synthetic hUII or URP (10 μg) was radiolabelled with 0.5 mCi Na125I by the chloramine-T technique as previously described (Doan et al., 2012). Iodinated 125I-hUII and 125I-URP were purified on a C18 cartridge, collected and stored at −20°C until use.
Organ bath experiments
Male rats (Sprague Dawley, 250–300 g) were killed by CO2 asphyxiation. As previously described (Brkovic et al., 2003), the thoracic aorta was then cleared of surrounding tissues and excised from the aortic arch to the diaphragm. From each vessel, conjunctive tissues were removed and the clean cylindrical ring was cut into several pieces. The endothelium was removed by gently rubbing the vessel intimal surface. All preparations were placed in 5 mL organ baths filled with oxygenated normal Krebs-Henselheit solution. Contractile responses to 40 mM KCl were used as control at the beginning and at the end of each experiment except for ET-1 for which 80 mM KCl was used as a reference. Agonistic activity was measured by increasing the concentration of each peptide in the organ chamber (3.10−11–3.10−6 M). For antagonist behaviour, thoracic aortic rings were first exposed to UCA or rUII(1–7) for 15 min, to ensure that the peptide reached equilibrium and that no agonistic effect is observed (Rossowski et al., 2002), and then cumulative concentration-response curves to hUII, rUII, URP or ET-1 (10−10–3.10−6 M) were constructed. The amplitude of the contraction induced by each peptide concentration was expressed as a percentage of the KCl-induced contraction.
Experiments involving cynomolgus monkey thoracic aortic rings (generous gift from Pr. Veronika Von Messling, INRS-IAF, Laval, QC, Canada) were performed as previously described with rat aortic rings except that a 60 mM KCl solution was used as a control at the beginning and at the end of each experiment. These animals, killed via sodium pentobarbital overdose, were non-infected control animals used in a study on measles infections and that was approved by the Institutional Animal Care Committee.
The median effective concentrations (EC50) were expressed as the mean ± SEM, and the n-values, representing the total number of animals from which the vessels were isolated, varied from 4 to 10 animals.
Haemodynamic assessment
Male Sprague-Dawley rats, weighing 350–400 g, were anaesthetized by isoflurane inhalation delivered in 100% oxygen (1 L·min−1) from an Ohmeda Tec 4 anaesthetic vaporizer (Somatechnology, Inc., Bloomfield, CT, USA). Anaesthesia was maintained by mask inhalation of isoflurane vaporized at concentrations of up to 3% in the induction phase, at 1.5% during acute surgical procedures and at 0.8–1.0% during experimental observations. After anaesthesia, a polyethylene catheter (PE 10) was inserted into the right jugular vein to inject saline (0.9% NaCl) or drugs. To measure arterial blood pressure, an incision was made at the common right carotid artery where a microtip pressure transducer catheter (model SPR-407, 2F, Millar Instruments, Houston, TX, USA) was inserted. A period of stabilization of 30 min was observed and the following experimental protocols were then performed and arterial blood pressure was evaluated as previously described (Gendron et al., 2005). During isoflurane inhalation, no breathing complication or change in blood pressure was observed. Mean arterial pressure was 114 ± 3 mmHg at basal condition and was not modified by saline bolus injections.
In the control group, rats received two doses of hUII or URP (10 nmol·kg−1) with a 30 min interval between the two injections. In the treated groups, the same protocol was applied except that for the second injection, hUII was replaced by URP and URP was replaced by hUII, respectively.
In vitro receptor binding studies
The UT-binding potencies of the different compounds used in this study were measured by using in vitro receptor binding assay as reported (Chatenet et al., 2004). For dissociation-binding experiments, performed with CHO cells stably transfected with UT, cells were first equilibrated for 2 h with either 125I-hUII or 125I-URP (0.2 nM) (Castel et al., 2006). Dissociation of receptor-bound radioligand, measured at different intervals, was then initiated by the addition of a supra-maximal concentration of hUII (10−6 M) or URP (10−6 M) alone, or by the simultaneous addition of hUII (10−6 M) and UCA (10−6 M), or URP (10−6 M) and UCA (10−6 M). Cells were then washed, lysed and the radioactivity counted on a γ-counter (1470 Automatic Gamma Counter, Perkin Elmer, Montreal, QC, Canada).
Data analysis
Binding and functional experiments were performed at least in triplicate and data, expressed as mean ± SEM, were analysed with the Prism software (Graph Pad Software, San Diego, CA, USA). In all experiments, n represents the total number of animals studied or individual assays performed. EC50, pEC50, pIC50 as well as Emax values were determined from corresponding concentration–response curves obtained through a sigmoidal dose–response fit with variable slope. Non-competitive antagonist affinities (pKb) were determined as previously described using the method of Gaddum where equiactive concentrations of agonist, in the absence or presence of UCA, were compared in a linear regression (Behm et al., 2010). Statistical comparisons of binding affinities and contractile potencies of URP and its analogues were analysed using unpaired Student's t-test and differences were considered significant when *P < 0.05, **P < 0.01 and ***P < 0.001.
Results
Peptides characterization and in vitro competitive binding assay
The sequences of UCA, hUII and URP are shown in Supporting Information Table S1, whereas the 2D structure of UCA is shown in Figure 1. Included in Supporting Information Table S1 are chromatographic data describing the high level of purity of the analogs and their characterization by mass spectrometry. All peptides were tested in at least three separate experiments for their ability to bind to the hUII receptor stably transfected in CHO cells. Radioligand binding assay also showed that UCA is able to consistently and fully displace both radioligands, that is 125I-hUII and 125I-URP with an apparent IC50 value of 237 nM and 252 nM, respectively (Table 1).
Figure 1.
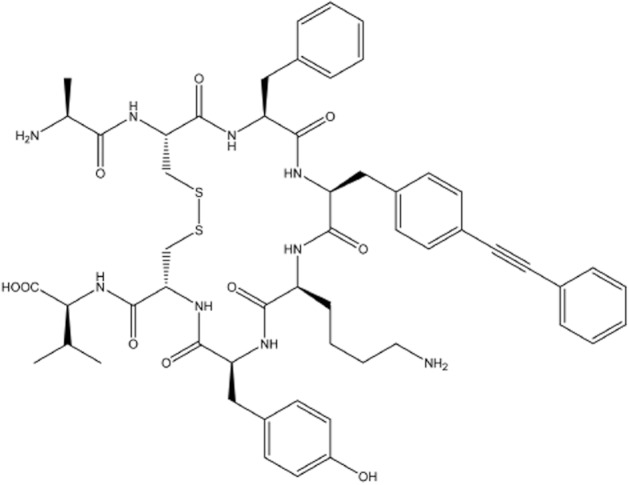
Structure of the novel UT allosteric modulator urocontrin A, that is [Pep4]URP.
Table 1.
IC50 and pIC50 values hUII, URP and UCA binding to recombinant human UT
| Name | Binding (125I-hUII) | Binding (125I-URP) | ||
|---|---|---|---|---|
| IC50 (nM)a | pIC50 | IC50 (nM)a | pIC50 | |
| hUII | 13.2 | 7.88 ± 0.07 | 32.5 | 7.49 ± 0.11 |
| URP | 12.4 | 7.90 ± 0.11 | 31.8 | 7.50 ± 0.11 |
| UCA | 237.2 | 6.62 ± 0.20 | 251.7 | 6.60 ± 0.18 |
IC50 values represent concentration giving 50% of binding inhibition.
Effect of UCA on hUII- or URP-induced rat and monkey aortic ring contraction
Ex vivo, hUII and URP evoked a dose-dependent contraction of rat aortic rings with a pEC50 of 8.89 ± 0.23 (Emax of 120 ± 8%; n = 10) and 8.12 ± 0.12 (Emax = 113 ± 5%; n = 10), respectively (Figure 2A). In opposition to hUII or URP, exposure to increasing concentrations of UCA, up to 10 μM, did not alter basal contractile tone (Figure 2A). Used as an antagonist, UCA suppressed concentration-dependently the maximum contractile response to hUII but not URP (Figure 2B and C; Table 2). For instance, pre-treatment with UCA at 10−8 M or 10−6 M produced a significant suppression of the maximum contractile response to hUII (Emax = 61% and 28%, respectively) with a non-significant shift in the concentration–response curve (Table 2). A further analysis of these results was performed based on a recent report of a non-peptidic UII insurmountable antagonist (Behm et al., 2010). Consistent with non-competitive antagonism, the slope of the double reciprocal plot of agonist equiactive concentrations in the presence and absence of 10 nM UCA was linear with a slope of 2.36 ± 0.06, equating to a pKb of 8.13 (Figure 2D). Since no significant rightward shift or reduction of efficacy associated with URP contractile response was observed following pre-treatment with UCA at either 10−8 M or 10−6 M (Figure 2B), this compound might be of pharmacologic/therapeutic value in order to discriminate hUII- and URP-associated signalling pathway and/or biological activity. Interspecies variability of several peptide and non-peptide ligands have complicated the interpretation of in vivo and in vitro studies. Therefore, functional experiments with non-rodent species, that is cynomolgus monkey, have been carried out with UCA. Consistent with the data generated in rat aorta, UCA significantly suppressed (P < 0.01) the maximal contractile response to hUII but not URP in cynomolgus monkey isolated aorta (Figure 3; Table 3).
Figure 2.

(A) Representative concentration–response curves obtained with rat thoracic aorta rings after adding cumulative concentrations of hUII, URP and UCA. Effects of UCA on (B) URP- and (C) hUII-induced contraction of rat aortic ring. (D) Double reciprocal plot of equiactive concentrations of hUII in the absence (A) and presence (A′) of 10 nM UCA is linear, consistent with non-competitive antagonism. Data represent the mean ± SEM and n = 5 to 10 animals.
Table 2.
Concentration-dependent inhibition of hUII-induced contraction of rat isolated aorta by urocontrin A
| Urocontrin A (nM) | hUII Emax (% KCl) | hUII pEC50 | n |
|---|---|---|---|
| Vehicle | 120 ± 8 | 8.89 ± 0.23 | 10 |
| 1 | 89 ± 5* | 8.63 ± 0.15 | 5 |
| 10 | 61 ± 6* | 8.94 ± 0.35 | 6 |
| 100 | 45 ± 4** | 8.59 ± 0.34 | 5 |
| 1 000 | 28 ± 3** | 8.41 ± 0.47 | 7 |
| 10 000 | – | – | 3 |
All values are expressed as mean ± SEM. Statistical comparisons for both pEC50 and Emax values were performed using anova analysis with a Dunnett's post-test where
P < 0.05 and
P < 0.01 versus vehicle control values.
Figure 3.
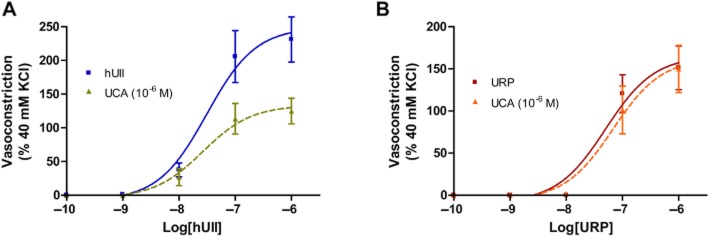
Effects of UCA on (A) hUII- and (B) URP-induced contraction of cynomolgus monkey aortic ring. Data represent the mean ± SEM and n = 7 animals.
Table 3.
Concentration-dependent inhibition of hUII-induced contraction of cynomolgus monkey isolated aorta by urocontrin A
| Urocontrin A (μM) | hUII Emax (% KCl) | hUII pEC50 | n |
|---|---|---|---|
| Vehicle | 249 ± 24 | 7.50 ± 0.22 | 7 |
| 1 | 134 ± 14** | 7.57 ± 0.25 | 7 |
All values are expressed as mean ± SEM. Statistical comparisons for both pEC50 and Emax values were performed using unpaired Student's t-test where
P < 0.01 versus vehicle control values.
Influence of UCA on the dissociation rate of bound 125I-hUII or 125I-URP
To further characterize the insurmountable antagonism produced by UCA on hUII-induced vasoconstriction, dissociation rates of hUII and URP were determined on CHO-UT cells. Cells were incubated with either 125I-hUII or 125I-URP until binding equilibrium was reached. Dissociation of the bound radioligand, either 125I-hUII or 125I-URP, was initiated by high concentration (10−6 M) of the corresponding unlabeled peptide in the absence or presence of an excess of UCA (10−6 M). The addition of unlabeled hUII or URP, to the corresponding radioligand, provoked a gradual dissociation of their corresponding tracer from its receptor site with a dissociation constant (koff) of 0.050 ± 0.006 and 0.052 ± 0.006, respectively (Figure 4A and C). In the presence of UCA, a significant increase (P < 0.01) of 125I-hUII dissociation rate was observed (0.193 ± 0.026), while no change was noticeable for 125I-URP (0.057 ± 0.009) (Figure 4A and C). Since the dissociation experiments were performed using low concentrations, compared to the apparent IC50, an additional experiment with a higher concentration of UCA (10−5 M) versus URP was realized and led to the same observation (data not shown). We next performed competition experiments with UCA using increasing concentrations of either 125I-hUII or 125I-URP from 0.1 to 0.5 nM (Figure 4B and D). Indeed, it has been reported that the relationship between the observed IC50 value and the concentration of radioactive tracer in displacement experiments can inform whether the inhibition of binding is competitive or non-competitive (Watson et al., 2005). The IC50 value is expected to increase linearly with the radioligand concentration for competitive inhibition, whereas an unchanged IC50 value may indicate that non-competitive inhibition occurs and, if so, is an estimate of the equilibrium dissociation constant value of the inhibitor–receptor complex (Watson et al., 2005). We found that the IC50 values do not change with concentration increases of radioactive 125I-hUII or 125I-URP (Figure 4B and D, Supporting Information Table S2), thus indicating that UCA inhibits hUII and URP binding by means of a non-competitive mechanism.
Figure 4.

Influence of UCA on the dissociation rate of 125I-hUII (A) or 125I-URP (B). Displacement of 125I-hUII (B) and 125I-URP (D) binding by UCA. Dissociation time-courses of bound 125I-hUII or 125I-URP and binding experiments were assessed on living CHO cells over-expressing the human urotensin II receptor. Significant differences (**P < 0.01) were determined by unpaired Student's t-test.
Influence of hUII and URP on the dissociation rate of bound 125I-hUII or 125I-URP
The unusual pattern observed with UCA, a URP derivative, prompted us to evaluate the propensity of URP to exert a similar effect on hUII dissociation rate. As shown in Table 1, URP and hUII bind hUT receptors with high affinity (Table 1). Dissociation of the bound radioligand, either 125I-hUII or 125I-URP, was initiated by high concentration (10−6 M) of the corresponding unlabeled peptide in the absence or presence of an excess of URP or hUII (10−6 M). The presence of URP induced a significant (P < 0.01) acceleration of the 125I-hUII dissociation rate (koff = 0.050 ± 0.006 vs. 0.174 ± 0.026; Figure 5A). Surprisingly, no change in the dissociation rate of 125I-URP (koff = 0.052 ± 0.006 vs. 0.055 ± 0.009) was observed following the addition of a supra-maximal concentration of hUII (Figure 5B).
Figure 5.
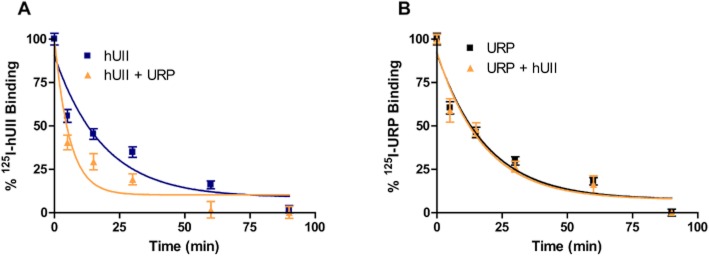
(A) Influence of URP on the dissociation rate of 125I-hUII. (B) Influence of hUII on the dissociation rate of 125I-URP. Dissociation time-courses of bound 125I-hUII or 125I-URP were assessed on living CHO cells over-expressing the human urotensin II receptor. Significant differences (**P < 0.01) were determined by unpaired Student's t-test.
Effect of hUII or URP on hUII and URP haemodynamic action in anaesthetized rats
The hypotensive action of UII and URP in anaesthetized rats was previously investigated (Hassan et al., 2003; Sugo et al., 2003). It was then observed that hUII and URP at a dose of 10 nmol·kg−1 were able to significantly alter the blood pressure. Therefore, this concentration was used in the subsequent studies. As shown in Figure 6A and B, repeated injections of hUII or URP produced equivalent biphasic responses for the respective peptide. Noteworthy, the URP-associated haemodynamic response was significantly lower than that of hUII. Interestingly, following a single injection of hUII, the hypotensive effect of URP, injected 30 min later, was significantly reduced (Figure 6C). However, such observation was not made when hUII was administered 30 min after a bolus injection of URP (Figure 6D).
Figure 6.

Effect of repeated hUII (A) or URP (B) injections on haemodynamism in anaesthetized rats. (C) Effect of hUII on the URP-associated haemodynamic function. (D) Effect of URP on the hUII-associated haemodynamic function. Significant differences (*P < 0.05) were determined by unpaired Student's t-test. Data represent the mean ± SEM and n = 4 to 6 animals.
Insight into putative interaction between hUII and its cognate receptor
As previously reported, replacement of the N-terminal exocyclic residues of hUII by an alanine moiety, that is [Ala1]hUII, [Ala2]hUII, [Ala3]hUII, [Ala4]hUII, generated analogs that appeared as very potent ligands of the hUT receptor (Brkovic et al., 2003). Dissociation of the bound radioligand, either 125I-hUII or 125I-URP, was initiated by high concentration (10−6 M) of the corresponding unlabeled peptide in the absence or presence of an Ala-substituted hUII analog (10−6 M). As shown, [Ala2]hUII, [Ala3]hUII, [Ala4]hUII had no effect on the dissociation rate of either 125I-hUII or 125I-URP (Figure 7A and B). However, [Ala1]hUII was able to produce a significant variation (P < 0.01) of the 125I-hUII dissociation rate (koff = 0.050 ± 0.006 vs. koff = 0.198 ± 0.034). Moreover, hUII(4–11), considered as the minimal equiactive sequence of hUII, was also able to accelerate the dissociation of 125I-hUII from the receptor but had no influence on 125I-URP dissociation kinetic (Figure 7C and D).
Figure 7.

Impact of Ala-substituted hUII analogs on the dissociation rate of 125I-hUII (A) and 125I-URP (B). Influence of hUII(4–11) on the dissociation rate of 125I-hUII (C) or 125I-URP (D). Significant differences (**P < 0.01) were determined by unpaired Student's t-test.
Effect of the N-terminal domain of rUII on rUII- or URP-induced rat aortic ring contraction
Based on the abovementioned putative interaction between UT and the N-terminal segment of rUII, we investigated the impact of this N-terminal peptide segment on rUII- and URP-induced vasoconstriction. Since our functional bioassay used rat aortic ring contraction, we decided to use the homologous N-terminal UII region, that is rUII(1–7), encompassing all the residues adjacent to the cyclic core (Pyr-His-Gly-Thr-Ala-Pro-Glu-amide; Supporting Information Table S1). As an agonist, this compound was unable to induce the contraction of rat aortic rings at a concentration up to 10−6 M (Figure 8A). Pre-treatment with rUII(1–7) (10−6 M, 15 min) did not significantly alter rUII-induced contraction but seems to induce a non-significant increase in efficacy (Figure 8B). However, we observed a significant rightward shift of the URP-associated (P < 0.05) contraction curve, potentially associated with a reduction in efficacy, under the same condition suggesting that the N-terminal segment of rUII might act as a URP-selective antagonist (Figure 8C).
Figure 8.

(A) Concentration–response curves obtained with rat thoracic aorta rings after adding cumulative concentrations of hUII, URP and rUII(1–7). Effects of rUII(1–7) on (B) rUII-, and (C) URP-induced contraction of rat aortic ring. Data represent the mean ± SEM and n = 5 to 8 animals.
Discussion
Recently, our group reported the characterization of a new ligand of the urotensinergic system, termed [Bip4]URP or urocontrin, with a unique pharmacological profile (Chatenet et al., 2012). This derivative, which possess a residual agonistic activity, is able to specifically reduce the maximal efficacy of hUII-, but not URP-associated action both ex vivo and in vivo. Based on this unusual pharmacological profile, we initiated the design of Pd-catalysed URP derivatives through the Sonogashira cross-coupling reaction and identified UCA as a potential candidate for the conception of ligands aimed at discriminating UII- and URP-mediated biological activity.
In the ex vivo rat aorta bioassay, UCA was unable to induce any contractile activity but presented a dual pharmacological profile when used as an antagonist. Indeed, at 10−6 M, UCA was able to selectively and significantly reduce the hUII-induced contraction without altering URP-mediated vasoconstriction. This effect was also observed at low concentrations since pre-treatment of rat aortic rings with UCA at 10−9 M was able to significantly reduce the efficacy (∼31%) of hUII-induced vasoconstriction. This effect appeared to be dose-dependent since increasing concentrations of UCA correlated with a decrease in hUII contractile efficacy, the hUII-induced vasoconstriction being completely abolished with a 10−5 M UCA pre-treatment. Interspecies variability regarding the biological activity of peptidic and non-peptidic ligands has complicated the interpretation of antagonist in vivo studies in preclinical animal disease models. As such, the (patho)physiological significance of the urotensinergic system remains ambiguous. Functional experiments with non-rodent (ideally human) UT receptors are therefore mandatory to further identify UT antagonists with pan-species activity. It is noteworthy that rat and human UT receptors share around 75% of sequence homology, whereas human and cynomolgus monkey receptor isoforms are almost identical (97%) (Elshourbagy et al., 2002). Moreover, human and cynomolgus monkey UII and URP isoforms were found to be strictly identical (Elshourbagy et al., 2002). We therefore investigated the effect of our antagonist on the hUII- or URP-induced vasoconstriction on cynomolgus monkey aortic ring. Confirming the results observed in the rat preparation, UCA was also able to selectively and significantly reduce the hUII-induced contraction without altering URP-mediated vasoconstriction. To the best of our knowledge, only two UT ligands exerted insurmountable activity (Herold et al., 2003; Behm et al., 2010). However, none of them differentially alter hUII and URP biological activity.
Insurmountable blockade may occur if the antagonist binds to an allosteric site and induces a conformational change in the receptor that compromises the ability of the agonist–receptor complex to elicit a response (Kaumann and Frenken, 1985). Strong evidence for allosteric interactions can be observed when the dissociation rate of the radiolabelled agonist (with dissociation initiated by the addition of a receptor-saturating concentration of the endogenous agonist) is modified in the presence of the allosteric modulator (Vauquelin et al., 2002). On the basis of our results showing that IC50 values for 125I-hUII or 125I-URP displacement by UCA does not vary with the radioligand concentration (Figure 4B and D), we provided evidence that this compound acts as a non-competitive antagonist of UT. Moreover, 125I-hUII dissociation kinetics from UT in the presence of UCA is consistent with a radioligand binding inhibition through an allosteric mechanism (Figure 4A). Indeed, in contrast to an orthosteric ligand, that is hUII, an excess concentration of UCA accelerated the 125I-hUII dissociation rate, thus suggesting that UCA can bind to 125I-hUII-occupied UT, and then change the receptor conformation in such a way that the radioligand is released from the receptor. Surprisingly, no difference in 125I-URP dissociation kinetics was observed in similar conditions. The apparent absence of effect on the URP pharmacological profile by UCA might reflect its ability, acting as a functional allosteric modulator, to select a receptor conformation that may impair hUII-associated actions but not URP-mediated biological activities. For a given receptor, a change in the nature and extent of allosteric modulation dependent on the type of orthosteric ligand used is referred to as ‘probe dependence’ (Kenakin, 2005; Keov et al., 2011). This probe dependence phenomenon suggests that the two endogenous ligands, despite depicting a high structure homology and recognizing a similar binding pocket, represent chemically distinct entities that can adopt different orientations within the orthosteric pocket such that they are differentially affected by the receptor conformational change induced by UCA.
As recently reported, UII or URP could act as an endogenous biased agonist of the urotensinergic system, inducing conformational changes within UT that can lead to precisely directed signalling (Doan et al., 2012). As reported, the binding affinity of hUII and URP in UT-transfected cells appeared to be equivalent, while in the rat aortic ring assay, the potency of URP was reduced (Chatenet et al., 2004). Moreover, the in vivo hUII-associated hypotension is significantly different from that observed with URP as depicted in this publication (Figure 5A and B) as well as in another article (Chatenet et al., 2012). The observed differences could reflect the activation of different signalling pathways involved in the UT-associated haemodynamic response as well as differences in coupling efficiency of specific intracellular pathway (Canals et al., 2012). As recently reported, the degree of allosteric modulation depends on the ligand-induced coupling efficiency between the receptor and the recruited signalling proteins (Canals et al., 2012). As such, amplified responses will appear to be modulated to a greater extent than less amplified responses. It is well known that the rat isolated aorta presents a low UT receptor signal transduction/coupling efficiency in which low-efficacy agonists appear to function as antagonists (Behm et al., 2006). As such, the N-terminus of UII isoforms may afford greater binding time to the receptor compared to URP and thus enable the activation of a greater number of intracellular pathways for an extended period of time. As such, by selecting specific receptor conformations, UII is probably able to affect the coupling efficiency of specific signalling pathways in a different manner than URP. It is thus possible that the masked effect of UCA on URP-mediated vasoconstriction is related to different coupling efficiency of the receptor following UII or URP activation as well as specific URP-associated pathway modulation compared to UII.
These intriguing results prompted us to evaluate the effect of hUII or URP on hUII or URP dissociation kinetics. Surprisingly, URP, an equipotent UII paralog, was able to accelerate the dissociation rate of membrane-bound 125I-hUII, while hUII had no noticeable effect on URP dissociation kinetics. Altogether, these results support to some extent the presence of specific pockets/interactions within UT, aimed at selecting a specific UT conformation that can differentiate UII and URP biological activity. Moreover, these observations seem to be in accordance with the concept of hUII or URP being an endogenous biased agonist of the urotensinergic system. The concept of biased agonist has recently emerged from various studies, putting forward the notion that specific ligand-induced conformational changes can lead to precisely directed signalling (Patel et al., 2010). Subsequent experiments investigating the ability of hUII and/or URP to modulate in vivo hUII- or URP-mediated haemodynamism in anaesthetized rats were performed. In anaesthetized rats, hUII and URP exert a rapid and transient pressor phase with the maximal effect occurring almost 1 min after the administration. This first phase is followed by a hypotensive effect reaching its maximum 5 min after injection and with a rather slow offset as previously demonstrated in anaesthetized WKY and SHR rats (Gendron et al., 2005). Interestingly, the hUII-associated hypotension is significantly different from that observed with URP (Figure 5A and B). This difference could reflect the activation of different signalling pathways involved in the UT-associated haemodynamic response. Since no change in their respective haemodynamic profile was observed following repeated injections of hUII or URP, it appears that repeated injections do not induce a tolerance effect, that is the organism does not build up a resistance to the effects of a substance after repeated exposure. However, as shown in Figure 5, hUII pre-treatment induced a marked reduction of the URP-associated hypotensive action. Surprisingly, such behaviour was not observed following pre-treatment with URP. Since desensitization of the urotensinergic system can not be involved in this phenomenon (Du et al., 2010), the observed effect could reflect the inability of URP to properly access its binding pocket due to topological changes induced by hUII. Because hUII and URP differ only by the length and composition of their N-terminal domain (Vaudry et al., 2010), it is conceivable that this region could be responsible for the putative differential binding mode of hUII and URP. Corroborating the role of the N-terminal domain in the recognition and activation processes, it should be noted that the hUII counterpart of the abovementioned UCA antagonist, that is [Pep7]hUII, acted as a weak but full agonist of the UT receptor (Supporting Information Figure S1). This result suggests that upon binding, the [Pep7]hUII N-terminal domain favours a UT conformation associated with agonistic activation reflected by the weak but still consequent [Pep7]hUII-induced contraction. Further experiments revealed that an interaction between the glutamic residue at position 1 of hUII and the UT receptor seems to be critical in order to induce specific conformational changes associated with hUII agonistic activation. Indeed, replacement of this residue by an alanine moiety, that is [Ala1]hUII, provoked an increase of the dissociation rate of hUII but not URP. However, since this compound was reported to exert almost equipotent contractile activity compared to hUII and URP, it is probable that by losing the specific interaction between its first residue and UT, the Ala-substituted analog might behave as a URP derivative acting therefore at its orthosteric binding site. Supporting this hypothesis, another truncated version of hUII, that is hUII(4–11), considered as the minimal fragment to exert full biological activity, was also able to alter hUII but not URP dissociation rate. In accordance with our results, an important electrostatic interaction between Glu1 of hUII and its receptor was observed in docking studies (Lescot et al., 2008).
It thus seems that the N-terminal region plays a crucial role in the recognition and activation process. Besides, as demonstrated in vivo, hUII pre-treatment is able to alter URP-associated biological activity. One explanation could rely on the inability of URP, injected at the same concentration, to displace the N-terminal domain of hUII in order to properly trigger its mediated action. We therefore evaluated the propensity of the N-terminal segment to block/alter URP-mediated vasoconstriction. Since UII isoforms differ only by their N-terminal region, we therefore examine the role of rUII(1–7) on rUII- or URP-induced contraction on rat aortic ring. Striking results demonstrated the propensity of such analog to selectively block URP- but not rUII-associated vasoconstriction. As observed in Figure 8, pre-treatment of aortic rings with rUII(1–7) induces a representative but non-significant increase in rUII contractile efficacy, while it reduces the potency and the efficacy of URP-mediated vasoconstriction. These results clearly suggest a probe-dependent effect of rUII(1–7), thus acting as an allosteric modulator, on rUII- and URP-mediated vasoconstriction. These observations, compared to the possible outcomes of effect for allosteric modulators (Kenakin, 2012), suggest that rUII(1–7) could modulate the contractile efficacy of rUII but not its affinity as reflected by Figure 8B. In contrast, this ligand could reduce both the efficacy and the affinity of the URP as illustrated by Figure 8C. As such, rUII(1–7) represents of new allosteric modulator of the urotensinergic which is able to differentially modulate the biological activity of these endogenous peptides. Although preliminary, these results support the existence of an initial interaction between the UII isoform N-terminal domains and their cognate receptor leading to a specific topological change associated with UT activation. Such initial interaction is missing or at least is different with URP giving rise to a slightly different signalling pathway, this view being in agreement with the concept of URP acting as a biased agonist of the urotensinergic system.
Based on these overall results, we finally propose a schematic representation of a possible mechanism of action for hUII, URP, UCA and rUII(1–7) as depicted in Figure 9. The results presented in this article suggest the existence of an initial interaction between the N-terminal domain of UII and its receptor. Upon this first interaction, UT undergoes a conformational change aimed at welcoming the C-terminal domain of UII, characterized by an intracyclic β-turn (Carotenuto et al., 2004), in a specific binding pocket. However, URP, lacking this N-terminal portion and characterized by the presence of an intracyclic γ-turn (Chatenet et al., 2004), is also able to bind UT but in a slightly different manner, probably characterized by the activation of a different subset of signalling pathways. Supporting this view, isolated ischaemic heart experiments revealed that UII and URP were both able to reduce myocardial damages through creatine kinase reduction, but only UII was able to reduce ANP production (Prosser et al., 2008). Based on the results presented herein, it appears that UCA probably binds to an allosteric binding site. As such, and probably through specific interactions that need to be determined, UCA is able to modify the receptor topography preventing the proper interaction with the linear UII N-terminal region and ultimately leading to an inefficient activation characterized by a reduced efficacy. On the opposite, such receptor conformational change has no effect on URP-mediated action. Conversely, binding of the rUII(1–7) N-terminal segment initiates a topographical change that antagonizes the effect of URP, but not UII. Since all UII isoforms present different N-terminal domains, it might be hypothesized that these regions could act in a species-selective manner as specific URP antagonist.
Figure 9.

Schematic representation of a proposed binding mode for hUII, URP, UCA and rUII(1–7). Following the initial interaction between the N-terminal domain of UII and its receptor, UT undergoes a conformational change aimed at welcoming the C-terminal domain of UII, characterized by an intracyclic β-turn, in a specific binding pocket. However, URP, lacking this N-terminal portion and characterized by the presence of an intracyclic γ-turn, is also able to bind UT but in a slightly different manner, probably characterized by the activation of a different subset of signalling pathways. By acting to an allosteric binding site, UCA is able to modify the receptor topography preventing the proper interaction of UT with the linear UII N-terminal region ultimately leading to an inefficient activation characterized by a reduced efficacy. On the opposite, such receptor conformational change has no effect on URP-mediated action. Conversely, binding of the rUII(1–7) N-terminal segment initiates a topographical change that antagonizes the effect of URP, but not UII.
Conclusions
In this study, we have presented data demonstrating the ability of UCA, a functional allosteric modulator, to reduce the efficacy of hUII- but not URP-induced vasoconstriction in rat and monkey aorta. Commonly proposed advantages of targeting receptor allosteric sites include the possibility of greater selectivity due to lower sequence conservation within allosteric pockets across subtypes of a given GPCR, as well as the potential to fine-tune physiological signalling in a more spatial and temporally selective manner (Christopoulos and Kenakin, 2002; May et al., 2007). Therefore, even with the high structural homology between UII, URP and somatostatin ligands, it is unlikely that UCA acts as a somatostatin receptor ligand. Besides, since this new ligand is unable to potentiate ET-1-induced effects (Supporting Information Figure S2) in the rat aorta (a phenomenon closely connected to somatostatin receptor affinity) (Behm et al., 2002), it is unlikely that UCA binds to the orthosteric site of the somatostatin receptor. Moreover, pharmacological characterization suggested the existence of different binding patterns associated with UT activation by either UII- or URP. Finally, we also demonstrated that the N-terminal domain of UII might act as a key determinant for binding specificity and that this fragment is able to act as a specific antagonist of URP-associated actions. Such compounds, that is UCA and rUII(1–7), should prove to be useful as new pharmacological tools aimed at deciphering the specific role of UII and URP in vitro but also in vivo.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research. N.D.D. is the recipient of a studentship from the Heart and Stroke Foundation of Canada and the Fondation Armand-Frappier. The authors declare no competing financial interests.
Glossary
- Boc
tert-butoxycarbonyl
- CM
resin, chloromethylated resin
- hUII
human urotensin II; hUT, human urotensin II receptor;
- Pep
(phenylethynyl)phenylalanine;
- RP
reversed-phase
- SHR
spontaneously hypertensive rat
- URP
urotensin II-related peptide
- WKY
Wistar Kyoto rat
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Concentration-response curves obtained with rat thoracic aorta rings after adding cumulative concentrations of hUII, URP and [Pep7]hUII. Data represent the mean ± SEM and n = 4 to 8 animals.
Figure S2 Effects of UCA on ET-1-induced contraction of rat aortic ring. In the rat isolated aorta bioassay, UCA was unable to produce a leftward shift of the endothelin-1-induced vasoconstriction. Indeed, mean pEC50 values with and without UCA (10−6 M), respectively, were as follows: ET-1 pEC50 = 8.05 ± 0.12 (Emax = 171 ± 7%; n = 8) and 8.19 ± 0.22 (Emax = 150 ± 11%; n = 4).
Table S1 Physicochemical properties of synthetic peptides.
Table S2 IC50 and pIC50 values of UCA binding to recombinant human UT.
References
- Behm DJ, Herold CL, Ohlstein EH, Knight SD, Dhanak D, Douglas SA. Pharmacological characterization of SB-710411 (Cpa-c[D-Cys-Pal-D-Trp-Lys-Val-Cys]-Cpa-amide), a novel peptidic urotensin-II receptor antagonist. Br J Pharmacol. 2002;137:449–458. doi: 10.1038/sj.bjp.0704887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behm DJ, Stankus G, Doe CP, Willette RN, Sarau HM, Foley JJ, et al. The peptidic urotensin-II receptor ligand GSK248451 possesses less intrinsic activity than the low-efficacy partial agonists SB-710411 and urantide in native mammalian tissues and recombinant cell systems. Br J Pharmacol. 2006;148:173–190. doi: 10.1038/sj.bjp.0706716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behm DJ, Aiyar NV, Olzinski AR, McAtee JJ, Hilfiker MA, Dodson JW, et al. GSK1562590, a slowly dissociating urotensin-II receptor antagonist, exhibits prolonged pharmacodynamic activity ex vivo. Br J Pharmacol. 2010;161:207–228. doi: 10.1111/j.1476-5381.2010.00889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgault S, Letourneau M, Fournier A. Development of photolabile caged analogs of endothelin-1. Peptides. 2007;28:1074–1082. doi: 10.1016/j.peptides.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Brkovic A, Hattenberger A, Kostenis E, Klabunde T, Flohr S, Kurz M, et al. Functional and binding characterizations of urotensin II-related peptides in human and rat urotensin II-receptor assay. J Pharmacol Exp Ther. 2003;306:1200–1209. doi: 10.1124/jpet.103.052415. [DOI] [PubMed] [Google Scholar]
- Canals M, Lane JR, Wen A, Scammells PJ, Sexton PM, Christopoulos A. A Monod-Wyman-Changeux mechanism can explain G protein-coupled receptor (GPCR) allosteric modulation. J Biol Chem. 2012;287:650–659. doi: 10.1074/jbc.M111.314278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carotenuto A, Grieco P, Campiglia P, Novellino E, Rovero P. Unraveling the active conformation of urotensin II. J Med Chem. 2004;47:1652–1661. doi: 10.1021/jm0309912. [DOI] [PubMed] [Google Scholar]
- Castel H, Diallo M, Chatenet D, Leprince J, Desrues L, Schouft MT, et al. Biochemical and functional characterization of high-affinity urotensin II receptors in rat cortical astrocytes. J Neurochem. 2006;99:582–595. doi: 10.1111/j.1471-4159.2006.04130.x. [DOI] [PubMed] [Google Scholar]
- Chartrel N, Leprince J, Dujardin C, Chatenet D, Tollemer H, Baroncini M, et al. Biochemical characterization and immunohistochemical localization of urotensin II in the human brainstem and spinal cord. J Neurochem. 2004;91:110–118. doi: 10.1111/j.1471-4159.2004.02698.x. [DOI] [PubMed] [Google Scholar]
- Chatenet D, Dubessy C, Leprince J, Boularan C, Carlier L, Segalas-Milazzo I, et al. Structure-activity relationships and structural conformation of a novel urotensin II-related peptide. Peptides. 2004;25:1819–1830. doi: 10.1016/j.peptides.2004.04.019. [DOI] [PubMed] [Google Scholar]
- Chatenet D, Nguyen QT, Letourneau M, Dupuis J, Fournier A. Urocontrin, a novel UT receptor ligand with a unique pharmacological profile. Biochem Pharmacol. 2012;83:608–615. doi: 10.1016/j.bcp.2011.12.009. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- Dai HY, Kang WQ, Wang X, Yu XJ, Li ZH, Tang MX, et al. The involvement of transforming growth factor-beta1 secretion in urotensin II-induced collagen synthesis in neonatal cardiac fibroblasts. Regul Pept. 2007;140:88–93. doi: 10.1016/j.regpep.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Doan NG, Nguyen TTM, Létourneau M, Turcotte K, Fournier A, Chatenet D. Biochemical and pharmacological characterization of nuclear urotensin II binding sites in rat heart. Br J Pharmacol. 2012;166:243–257. doi: 10.1111/j.1476-5381.2011.01710.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas SA, Ohlstein EH. Human urotensin-II, the most potent mammalian vasoconstrictor identified to date, as a therapeutic target for the management of cardiovascular disease. Trends Cardiovasc Med. 2000;10:229–237. doi: 10.1016/s1050-1738(00)00069-4. [DOI] [PubMed] [Google Scholar]
- Du AT, Onan D, Dinh DT, Lew MJ, Ziogas J, Aguilar MI, et al. Ligand-supported purification of the urotensin-II receptor. Mol Pharmacol. 2010;78:639–647. doi: 10.1124/mol.110.065151. [DOI] [PubMed] [Google Scholar]
- Elshourbagy NA, Douglas SA, Shabon U, Harrison S, Duddy G, Sechler JL, et al. Molecular and pharmacological characterization of genes encoding urotensin-II peptides and their cognate G-protein-coupled receptors from the mouse and monkey. Br J Pharmacol. 2002;136:9–22. doi: 10.1038/sj.bjp.0704671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erchegyi J, Cescato R, Grace CR, Waser B, Piccand V, Hoyer D, et al. Novel, potent, and radio-iodinatable somatostatin receptor 1 (sst1) selective analogues. J Med Chem. 2009;52:2733–2746. doi: 10.1021/jm801314f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron G, Gobeil F, Jr, Belanger S, Gagnon S, Regoli D, D'Orleans-Juste P. Urotensin II-induced hypotensive responses in Wistar-Kyoto (Wky) and spontaneously hypertensive (Shr) rats. Peptides. 2005;26:1468–1474. doi: 10.1016/j.peptides.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Guidolin D, Albertin G, Ribatti D. Urotensin-II as an angiogenic factor. Peptides. 2010;31:1219–1224. doi: 10.1016/j.peptides.2010.03.022. [DOI] [PubMed] [Google Scholar]
- Hassan GS, Chouiali F, Saito T, Hu F, Douglas SA, Ao Z, et al. Effect of human urotensin-II infusion on hemodynamics and cardiac function. Can J Physiol Pharmacol. 2003;81:125–128. doi: 10.1139/y03-004. [DOI] [PubMed] [Google Scholar]
- Herold CL, Behm DJ, Buckley PT, Foley JJ, Wixted WE, Sarau HM, et al. The neuromedin B receptor antagonist, BIM-23127, is a potent antagonist at human and rat urotensin-II receptors. Br J Pharmacol. 2003;139:203–207. doi: 10.1038/sj.bjp.0705251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose T, Takahashi K, Mori N, Nakayama T, Kikuya M, Ohkubo T, et al. Increased expression of urotensin II, urotensin II-related peptide and urotensin II receptor mRNAs in the cardiovascular organs of hypertensive rats: comparison with endothelin-1. Peptides. 2009;30:1124–1129. doi: 10.1016/j.peptides.2009.02.009. [DOI] [PubMed] [Google Scholar]
- Hoffmanns U, Metzler-Nolte N. Use of the Sonogashira coupling reaction for the ‘two-step’ labeling of phenylalanine peptide side chains with organometallic compounds. Bioconjug Chem. 2006;17:204–213. doi: 10.1021/bc050259c. [DOI] [PubMed] [Google Scholar]
- Jarry M, Diallo M, Lecointre C, Desrues L, Tokay T, Chatenet D, et al. The vasoactive peptides urotensin II and urotensin II-related peptide regulate astrocyte activity through common and distinct mechanisms: involvement in cell proliferation. Biochem J. 2010;428:113–124. doi: 10.1042/BJ20090867. [DOI] [PubMed] [Google Scholar]
- Kaumann AJ, Frenken M. A paradox: the 5-HT2-receptor antagonist ketanserin restores the 5-HT-induced contraction depressed by methysergide in large coronary arteries of calf. Allosteric regulation of 5-HT2-receptors. Naunyn Schmiedebergs Arch Pharmacol. 1985;328:295–300. doi: 10.1007/BF00515556. [DOI] [PubMed] [Google Scholar]
- Kenakin T. New concepts in drug discovery: collateral efficacy and permissive antagonism. Nat Rev Drug Discov. 2005;4:919–927. doi: 10.1038/nrd1875. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br J Pharmacol. 2012;165:1659–1669. doi: 10.1111/j.1476-5381.2011.01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keov P, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology. 2011;60:24–35. doi: 10.1016/j.neuropharm.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krum H, Kemp W. Therapeutic potential of blockade of the urotensin II system in systemic hypertension. Curr Hypertens Rep. 2007;9:53–58. doi: 10.1007/s11906-007-0010-x. [DOI] [PubMed] [Google Scholar]
- Lescot E, Sopkova-de Oliveira Santos J, Colloc'h N, Rodrigo J, Milazzo-Segalas I, Bureau R, et al. Three-dimensional model of the human urotensin-II receptor: docking of human urotensin-II and nonpeptide antagonists in the binding site and comparison with an antagonist pharmacophore model. Proteins. 2008;73:173–184. doi: 10.1002/prot.22050. [DOI] [PubMed] [Google Scholar]
- Maryanoff BE, Kinney WA. Urotensin-II receptor modulators as potential drugs. J Med Chem. 2010;53:2695–2708. doi: 10.1021/jm901294u. [DOI] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos P, Bousette N, Giaid A. Urotensin-II and cardiovascular remodeling. Peptides. 2008;29:764–769. doi: 10.1016/j.peptides.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Patel CB, Noor N, Rockman HA. Functional selectivity in adrenergic and angiotensin signaling systems. Mol Pharmacol. 2010;78:983–992. doi: 10.1124/mol.110.067066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser HC, Forster ME, Richards AM, Pemberton CJ. Urotensin II and urotensin II-related peptide (URP) in cardiac ischemia-reperfusion injury. Peptides. 2008;29:770–777. doi: 10.1016/j.peptides.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Ross B, McKendy K, Giaid A. Role of urotensin II in health and disease. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1156–R1172. doi: 10.1152/ajpregu.00706.2009. [DOI] [PubMed] [Google Scholar]
- Rossowski WJ, Cheng BL, Taylor JE, Datta R, Coy DH. Human urotensin II-induced aorta ring contractions are mediated by protein kinase C, tyrosine kinases and Rho-kinase: inhibition by somatostatin receptor antagonists. Eur J Pharmacol. 2002;438:159–170. doi: 10.1016/s0014-2999(02)01341-9. [DOI] [PubMed] [Google Scholar]
- Shiraishi Y, Watanabe T, Suguro T, Nagashima M, Kato R, Hongo S, et al. Chronic urotensin II infusion enhances macrophage foam cell formation and atherosclerosis in apolipoprotein E-knockout mice. J Hypertens. 2008;26:1955–1965. doi: 10.1097/HJH.0b013e32830b61d8. [DOI] [PubMed] [Google Scholar]
- Silvestre RA, Egido EM, Hernandez R, Leprince J, Chatenet D, Tollemer H, et al. Urotensin-II is present in pancreatic extracts and inhibits insulin release in the perfused rat pancreas. Eur J Endocrinol. 2004;151:803–809. doi: 10.1530/eje.0.1510803. [DOI] [PubMed] [Google Scholar]
- Song W, Abdel-Razik AE, Lu W, Ao Z, Johns DG, Douglas SA, et al. Urotensin II and renal function in the rat. Kidney Int. 2006;69:1360–1368. doi: 10.1038/sj.ki.5000290. [DOI] [PubMed] [Google Scholar]
- Sugo T, Mori M. Another ligand fishing for G protein-coupled receptor 14. Discovery of urotensin II-related peptide in the rat brain. Peptides. 2008;29:809–812. doi: 10.1016/j.peptides.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Sugo T, Murakami Y, Shimomura Y, Harada M, Abe M, Ishibashi Y, et al. Identification of urotensin II-related peptide as the urotensin II-immunoreactive molecule in the rat brain. Biochem Biophys Res Commun. 2003;310:860–868. doi: 10.1016/j.bbrc.2003.09.102. [DOI] [PubMed] [Google Scholar]
- Vaudry H, Do Rego JC, Le Mevel JC, Chatenet D, Tostivint H, Fournier A, et al. Urotensin II, from fish to human. Ann N Y Acad Sci. 2010;1200:53–66. doi: 10.1111/j.1749-6632.2010.05514.x. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Van Liefde I, Birzbier BB, Vanderheyden PM. New insights in insurmountable antagonism. Fundam Clin Pharmacol. 2002;16:263–272. doi: 10.1046/j.1472-8206.2002.00095.x. [DOI] [PubMed] [Google Scholar]
- Watson AM, Lambert GW, Smith KJ, May CN. Urotensin II acts centrally to increase epinephrine and ACTH release and cause potent inotropic and chronotropic actions. Hypertension. 2003;42:373–379. doi: 10.1161/01.HYP.0000084633.85427.E6. [DOI] [PubMed] [Google Scholar]
- Watson C, Jenkinson S, Kazmierski W, Kenakin T. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol Pharmacol. 2005;67:1268–1282. doi: 10.1124/mol.104.008565. [DOI] [PubMed] [Google Scholar]
- Zhang YG, Li J, Li YG, Wei RH. Urotensin II induces phenotypic differentiation, migration, and collagen synthesis of adventitial fibroblasts from rat aorta. J Hypertens. 2008;26:1119–1126. doi: 10.1097/HJH.0b013e3282fa1412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.