

Abstract
Background and Purpose
Infections with a strain of Escherichia coli producing Shiga toxins could be one of the causes of fetal morbidity and mortality in pregnant women. We have previously reported that Shiga toxin type 2 (Stx2) induces preterm delivery in pregnant rats. In this study, we evaluate the role of TNF-α, PGs and NO in the Stx2-induced preterm delivery.
Experimental Approach
Pregnant rats were treated with Stx2 (0.7 ng g−1) and killed at different times after treatment. Placenta and decidua were used to analyse NOS activity by the conversion of L-[14C]arginine into L-[14C]citrulline, levels of PGE2 and PGF2α assessed by radioimmunoassay, and cyclooxygenase (COX) proteins by Western blot. TNF-α level was analysed in serum by ELISA and by cytotoxicity in L929 cells. The inhibitor of inducible NOS, aminoguanidine, the COX-2 inhibitor, meloxicam, and the competitive inhibitor of TNF-α, etanercept, were used alone or combined to inhibit NO, PGs and TNF-α production respectively, to prevent Stx2-induced preterm delivery.
Key Results
Stx2 increased placental PGE2 and decidual PGF2α levels as well as COX-2 expression in both tissues. Aminoguanidine and meloxicam delayed the preterm delivery time but did not prevent it. Etanercept blocked the TNF-α increase after Stx2 treatment and reduced the preterm delivery by approximately 30%. The combined action of aminoguanidine and etanercept prevented Stx2-induced preterm delivery by roughly 70%.
Conclusion and Implications
Our results demonstrate that the increased TNF-α and NO induced by Stx2 were the predominant factors responsible for preterm delivery in rats.
Keywords: preterm delivery, Stx2 treatment, nitric oxide, prostaglandins, tumour necrosis factor-α, pregnant rats, cyclooxygenases, nitric oxide synthase
Introduction
Gastrointestinal infection with Shiga toxin (Stx)-producing Escherichia coli (STEC) strains causes diarrhoea and haemorrhagic colitis and, in addition, it is the leading cause of the haemolytic uremic syndrome (HUS; Richardson et al., 1988). HUS is characterized by a triad of haemolytic anaemia, thrombocytopenia and acute renal failure (Gianantonio et al., 1964; Repetto, 1997). HUS is most commonly seen in young children, but it is occasionally present in adults (Scully et al., 1995; Repetto, 1997; Frank et al., 2011), including post partum women. Most of the post partum cases had preceding upper respiratory or gastrointestinal symptoms (Strauss and Alexander, 1976; Steele et al., 1984) and at least in one case, HUS was detected after a preterm delivery at 32 weeks (Steele et al., 1984). However, to our knowledge, an increased risk of preterm delivery linked to STEC infection has not yet been evaluated.
HUS is a systemic complication attributed to the expression of Stx (Karmali et al., 1983). Two types of Stx may be produced by STEC strains: Stx1 and/or Stx2 with their variants (Paton and Paton, 1998). In Argentina, STEC strain O157 : H7 producing only Stx2 is highly prevalent (Rivas et al., 2006). Stx is an AB5 holotoxin possessing a single A subunit in non-covalent association with five B subunits (Fraser et al., 1994). The B subunits form a pentameric ring and mediate toxin binding to the glycolipid receptor globotriaosylceramide (Gb3) expressed in vascular endothelial cells, podocytes, mesangial cells and epithelial cells of renal proximal tubules (Lingwood et al., 1987; Lingwood, 1996). The A subunit is an N-glycosidase, which removes an adenine of the 28S ribosomal RNA resulting in cell death (Endo et al., 1988).
We have previously reported that i.p. administration of Stx2 in the late stage of pregnancy induces preterm delivery of dead fetuses (Burdet et al., 2009). Overproduction of NO and damage in placenta which was prevented by aminoguanidine, an inhibitor of inducible NOS (iNOS), demonstrated that NO plays an important role in placental toxicity and fetal mortality induced by Stx2 (Burdet et al., 2010).
During inflammation, PGs are released simultaneously with NO and their overproduction could be detrimental. PGs promote uterine contractions contributing to embryonic expulsion (Aisemberg et al., 2007). Stx may act in concert with bacterial lipopolysaccharide (LPS) and induce the production of TNF-α, IL-1 and IL-6 by macrophages rendering the vascular endothelial cells are more sensitive to the toxin (Louise and Obrig, 1991; Tesh et al., 1994). Furthermore, high concentrations of many cytokines in the vaginal or cervical secretions, including TNF-α and IL-6 in women with symptoms of preterm labour, are associated with early preterm delivery (Inglis et al., 1994).
These observations led us to investigate whether PGs and TNF-α could be involved in Stx2-induced preterm delivery in rats.
Methods
Animals
All animal care and experimental procedures in this study were in strict accordance with the recommendations detailed in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Protocols were approved by the Committee for the Care and Use of Laboratory Animals of the University of Buenos Aires (CICUAL, Permit Number 1209–10). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of N animals were used in the experiments described here. The animals received food and water ad libitum and were housed under controlled conditions of light (12 h light, 1 h dark) and temperature (23–25°C). To obtain timed pregnant females, male and virgin female Sprague-Dawley rats between 250 and 300 g of body weight (bwt) were acquired from the animal facility of the School of Veterinary, University of Buenos Aires. Mating was performed placing the female rats in the cages of the male rats from the same strain for several days. Day 1 of gestation was determined when sperm was observed in the vaginal smear.
Experimental protocols
Pregnant rats on day 15 of gestation (gd) were randomly divided into groups of at least three rats each. The Stx2 injury was induced as previously described (Burdet et al., 2009). Briefly, Stx2-treated rats were injected (i.p.) with 0.5 mL of culture supernatant from recombinant E. coli, equal to a dose of 0.7 ng Stx2 and 50 pg LPS per gram. Control rats were inoculated with 0.5 mL of culture supernatant containing only LPS (50 pg LPS g−1). In separate experiments, control and Stx2-treated rats were injected (i.p.) with aminoguanidine (100 μg g−1) 24 h before and 4 h after toxin injection; meloxicam (2 μg g−1) simultaneously with the toxins and 12, 24 and 36 h later; etanercept (100 μg g−1) 6 h before toxins; a combination of aminoguanidine and meloxicam, or aminoguanidine and etanercept according the protocol previously described for each one. Rats from the different experimental groups were observed daily to evaluate delivery time and fetal status. Some of them were anaesthetized and killed by cervical dislocation at different times after treatment. Placenta and decidua tissues were removed to evaluate PGs synthesis, NOS activity, COX-1 and COX-2 protein expression. Serum and amniotic fluid were obtained to measure TNF-α production.
Determination of PGs
Placenta and decidua were used to measure PGE2 and PGF2α levels as previously detailed (Ribeiro et al., 2005). Briefly, tissues were incubated for 1 h in Krebs-Ringer bicarbonate solution under a 5% CO2 atmosphere at 37°C. Afterwards, medium was acidified and PGs were extracted twice with ethyl acetate. PG concentration was determined by radioimmunoassay. Sensitivity was 5–10 pg per tube and values are expressed as pg PG (mg protein)−1.
Determination of NOS activity
NOS enzyme activity in placenta was quantified by measuring the conversion of L-[14C]arginine into L[14C]citrulline as described before (Burdet et al., 2010). Samples were weighed, homogenized and incubated with 10 μM L-[14C]arginine (0.3 μCi). After 15 min, samples were centrifuged for 10 min at 10 000 × g and applied to a Dowex AG50-X8 column. L-[14C]citrulline was eluted and measured by liquid scintillation counting. Enzyme activity is reported as fmol L-[14C]citrulline formed (mg protein)−1 over 15 min.
TNF-α bioassay and immunoassay
TNF bioactivity in serum and amniotic fluid samples was assayed employing the TNF-sensitive cell line L929 (Shiau et al., 2001). In brief, 1 × 104 L929 cells per well were seeded into a 96-well tissue culture plate and cultured in RPMI 1640 medium supplemented with 5 μg mL−1 streptomycin, 5 U mL−1 penicillin and 10% fetal calf serum. The medium was replaced by 0.1 mL of medium alone, or with samples from control or Stx2-treated rats. The L929 cells were grown in arrested-growth conditions (actinomycin D, 10 μg mL−1). After 24 h incubation, the cells were washed with PBS and viability was assayed by using the neutral red uptake technique (Goldstein et al., 2007). Bioactive TNF-α was expressed as percentage of L929 cell viability. Concentrations of TNF-α were measured with the Biotrak rat TNF-α elisa system according to the manufacturer's instructions.
Western blot analysis
Tissues were processed according to the method described by Burdet et al., (2010). In brief, placental and decidual tissues were fragmented and homogenized on ice in an appropriate buffer with protease inhibitors. Homogenates were pre-centrifuged at 2500 × g for 10 min at 4°C and the collected supernatants were additionally centrifuged at 7800 × g for 10 min at 8°C. The supernatants were collected and stored at −70°C until Western blotting was performed. Protein concentrations were determined with the BCA Protein Assay Kit. Equal amounts of protein (100 μg ) were loaded in each line. Samples were separated on 10% (w/v) sodium dodecyl sulphate-polyacrylamide gel by electrophoresis and transferred to a PVDF membrane. The blots were incubated 48 h at 4°C with anti-COX-1 or anti-COX-2 rabbit polyclonal antibody diluted 1:250 in PBS. The membranes were then incubated for 1 h at room temperature with HRP-conjugated goat anti-rabbit IgG antibody (1:3000). Proteins were detected through ECL detection system. To determine the uniformity of loading, protein blots were probed with a monoclonal anti-β-actin antibody (1:4000). Band intensities were measured using the Quantity One densitometry software package (Bio-Rad Lab, USA). Protein bands were normalized to their respective β-actin bands.
Data analysis
Statistical analysis was performed using the Graph Pad Prism Software (San Diego, CA, USA). Comparison between values of different groups was performed using one-way anova. Significance was determined using Tukey's multiple comparison test for unequal replications, or Student's t-test. Fisher exact test was used to compare the term and preterm delivery under different treatments. Statistical significance was set at P < 0.05.
Materials
L-[14C]arginine (specific activity 360 mCi mmol−1), [5,6,8,11,12,14,15-3H(N)]- PGE2 (specific activity 150 Ci mmol−1) and [5,6,8,9,11,12,14,15-3H(N)]-PGF2α (specific activity 200 Ci mmol−1) were from Perkin Elmer Life and Analytical Sciences, Walthman, MA, USA. ECL detection system, Biotrak rat TNF-α elisa and actinomycin D were from GE Healthcare Life Sciences, Piscataway, NJ, USA. Meloxicam was purchased from Boehringer Ingelheim, Mainz Bingen, Germany. Aminoguanidine, PGE2 and PGF2α standards, monoclonal β-actin antibody, secondary antibodies, Luria-Bertani broth and ampicillin were from Sigma Chemical Corp, St Louis, MO, USA. Etanercept was from Wyeth Lab, Madison, NJ, USA. The Dowex AG50-X8 column (Na+ form), nitrocellulose membranes and all other Western blot reagents were from Bio-Rad Lab, Hercules, CA, USA. Filters were from Millipore Corp, Billerica, MA, USA. Primary COX antibodies were from Cayman Chemical, Ellsworth Road, MI, USA. The HEK-Blue LPS Detection Kit was from InvivoGen, San Diego, CA, USA. The BCA Protein Assay Kit was from Pierce Biotechnology Inc, Rockford, IL, USA. All other chemicals were analytical grade.
Results
Stx2 increases PGs levels in placenta and decidua
Taking into account that during inflammation, PGs are released simultaneously with NO, we examined PGE2 and PGF2α formation in placenta and decidua from rats treated with Stx2 alone or in the presence of aminoguanidine. In placenta, PGE2 showed an increase 12 h after Stx2 injection (P < 0.01, n = 18) while PGF2α remained constant. Treatment with aminoguanidine plus Stx2 caused a greater increase in PGE2 (P < 0.001, n = 18) than the increase detected only with Stx2 and also caused an increase in PGF2α (P < 0.01, n = 18; Figure 1A). In decidua, PGE2 decreased and PGF2α increased (P < 0.05, n = 18) in Stx2-treated rats while both PGs were similar to the controls after aminoguanidine plus Stx2 treatment (Figure 1B).
Figure 1.
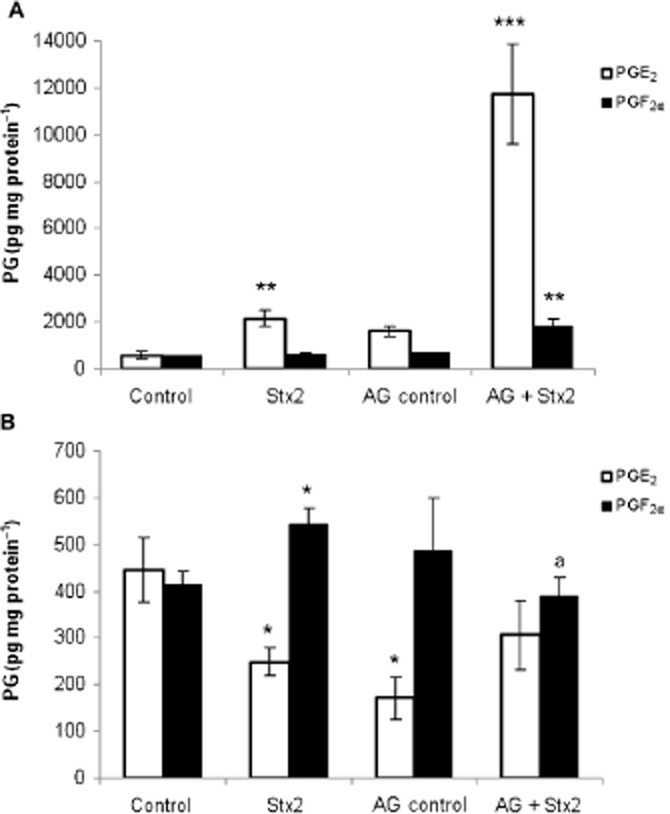
Effects of Stx2 and aminoguanidine (AG) on PG production. Pregnant rats (gd 15) were treated with LPS alone (50 pg g−1) (shown as control) or with 0.7 ng Stx2 and 50 pg LPS g−1 (shown as Stx2) and killed 12 h post-treatment. Some animals were injected with aminoguanidine (i.p., 100 μg g−1) 24 h before and 4 h after toxin injection. PGE2 and PGF2α levels were evaluated in placenta (A) and decidua (B) by RIA. Data corresponding to at least two separate experiments are shown. Values are expressed as means ± SEM (n = 18). *P < 0.05, **P < 0.01, ***P < 0.001 versus control; aP < 0.05 versus Stx2.
Stx2 stimulates the expression of COX-2 in placenta and decidua
An increase in COX-2 protein expression was detected in placenta from rats treated with Stx2 alone or in the presence of aminoguanidine (Figure 2A and C, P < 0.05, n = 6). An increase in COX-2 expression was also detected in decidua from Stx2-treated rats (P < 0.05, n = 6) although the expression was similar to the controls after aminoguanidine treatment (Figure 2B and D). None of the applied treatments changed the expression level of COX-1 in placental and decidual tissues of Stx2-treated rats (data not shown). These results suggest that changes in the levels of PGs caused by Stx2 were mediated by COX-2 and modulated by NO production.
Figure 2.

Effect of Stx2 and aminoguanidine (AG) on COX-2 protein expression. Pregnant rats were injected with LPS alone (50 pg g−1) (shown as control) or with 0.7 ng Stx2 and 50 pg LPS g−1 (shown as Stx2) and killed 12 h post-treatment. Some animals were injected with aminoguanidine (i.p., 100 μg g−1) 24 h before and 4 h after toxin treatment. Placenta and deciduas were removed for Western blot analysis. To determine the uniformity of loading, blots were probed with a monoclonal anti-β-actin antibody. Representative gel for COX-2 protein expression in placenta (A) and decidua (B) and the corresponding densitometry of the bands (C and D, respectively) are shown. Values correspond to the mean of two different pools of six animals ± SEM. *P < 0.05 versus control; #P < 0.05 versus Stx2 + aminoguanidine.
Aminoguanidine and meloxicam delay the preterm delivery induced by Stx2
In order to determine whether the alterations in the PG levels detected in placenta and decidua from Stx2-treated rats may be in part responsible for preterm delivery, meloxicam, a selective inhibitor of COX-2, was used. Table 1 shows that meloxicam did not prevent preterm delivery in Stx2-treated rats. All the rats treated with meloxicam and Stx2 had premature delivery of dead fetuses while control rats treated with meloxicam delivered normal live pups at term (Table 1, row 4 vs. 3). Furthermore, we studied the ability of aminoguanidine combined with meloxicam to prevent Stx2-induced preterm delivery. Previously, we have showed that aminoguanidine administration did not prevent the premature delivery of dead fetuses (Table 1, row 6; Burdet et al., 2010). Instead, co-administration of aminoguanidine and meloxicam delayed preterm delivery by around 2 days in 50% of the rats, 3 days in 17% of the rats and 4 days in 33% of the rats but did not prevent the preterm delivery of dead fetuses (Table 1, row 8). These results suggest that both NO and PGs are involved in the action of Stx2 on pregnant rats, although their blockade was not enough to reverse the preterm delivery induced by Stx2.
Table 1.
Effect of meloxicam (Melo), aminoguanidine (AG) and etanercept (ETA) on delivery time and pups status
| Preterm delivery (%) | Term delivery (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Treatment | *n | gd 16–17 | gd 18 | gd 19 | gd 20 | gd 21 | gd 22 | gd 23 | Live/dead pups (%) |
| Control | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 100/0 |
| Stx2 | 8 | 100 | 0 | 0 | 0 | 0 | 0 | 0a | 0/100 |
| †Melo control | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 100/0 |
| †Melo + Stx2 | 8 | 100 | 0 | 0 | 0 | 0 | 0 | 0b | 0/100 |
| #AG control | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 100/0 |
| #AG + Stx2 | 6 | 100 | 0 | 0 | 0 | 0 | 0 | 0c | 0/100 |
| #AG+ †Melo control | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 100/0 |
| #AG+ †Melo + Stx2 | 6 | 0 | 0 | 50 | 17 | 33 | 0 | 0d | 0/100 |
| ¥ETA control | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 100/0 |
| ¥ETA + Stx2 | 10 | 30 | 30 | 0 | 0 | 10 | 0 | 30 | 30/70 |
| #AG + ¥ETA control | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 100/0 |
| #AG + ¥ETA + Stx2 | 10 | 0 | 0 | 0 | 0 | 30 | 0 | 70e | 70/30 |
Meloxicam: 2 μg g−1; #aminoguanidine 100 μg g−1; ¥etanercept 100 μg g−1. *n = number of rats.
aP < 0.005 versus control. bP < 0.005 versus meloxicam control. cP < 0.005 versus aminoguanidine control. dP < 0.005 versus aminoguanidine + meloxicam control. eP < 0.005 versus Stx2. a,b,c,d,eCalculated by Fisher exact test.
Stx2 induces deregulation of serum TNF-α production
TNF-α is one of the main cytokines mediating the inflammatory response to Stx2 and its deregulation, either systemic or locally, could contribute to the changes observed during the progress of pregnancy. To test this hypothesis, we determined the TNF-α levels in serum and amniotic fluid of Stx2-treated rats as well as its toxicity on the TNF-sensitive cell line L929.
An increase in the level of TNF-α protein in serum of Stx2-treated rats was maximal at 2 h after Stx2 injection, after which levels returned to normal at 6 h and 12 h (Figure 3A). Consequently, the toxic activity of TNF-α showed a reduction in the viability of L929 cells to approximately 47% at 2 h after Stx2 injection (Figure 3B). However, the TNF-α level did not change in the amniotic fluid of pregnant rats treated with Stx2 (data no shown).
Figure 3.
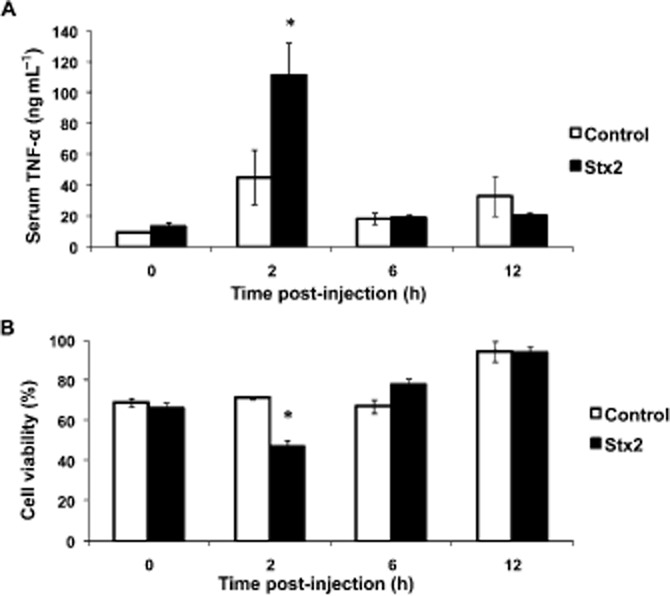
TNF-α production in pregnant rats. Pregnant rats were injected with LPS alone (50 pg g−1) (shown as control) or with 0.7 ng Stx2 and 50 pg LPS g−1 (shown as Stx2) and killed 0, 2, 6 and 12 h after the injection. Serum samples were used to determine amount of TNF-α by elisa assay (A) and TNF-α cytotoxic activity by viability assay in L929 cells (B). Data corresponding to at least two separate experiments are shown. Values are expressed as the mean ± SEM (n = 6). *P < 0.05 versus control.
Pretreatment with etanercept prevented the increase of TNF-α production in serum samples induced by Stx2 (Figure 4). Etanercept also prevented the increase in placental PGE2 (P < 0.001, n = 18) and decidual PGF2α (P < 0.001, n = 18) levels caused by Stx2 (Figure 5 and B). In contrast, no significant difference in placental NOS activity was found in rats treated with etanercept + Stx2 compared with Stx2 alone. In both cases, a significant increase in NOS activity in placental tissues (P < 0.01, n = 6) was observed, compared with the control (Figure 6).
Figure 4.
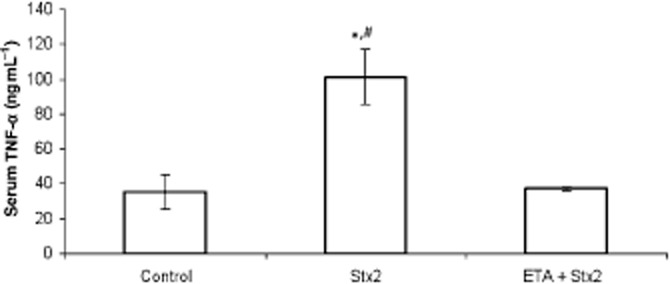
Effects of etanercept (ETA) on TNF-α production. Pregnant rats were injected with LPS alone (50 pg g−1) (shown as control) or with 0.7 ng Stx2 and 50 pg LPS g−1 (shown as Stx2) and killed 2 h after injection. Some animals were injected with etanercept (i.p., 100 μg g−1) 6 h before toxin treatment. Serum samples were used to determine amount of TNF-α by elisa assay. Data corresponding to at least two separate experiments are shown. Values are expressed as the mean ± SEM (n = 4). *P < 0.05 versus control, #P < 0.05 versus etanercept + Stx2.
Figure 5.
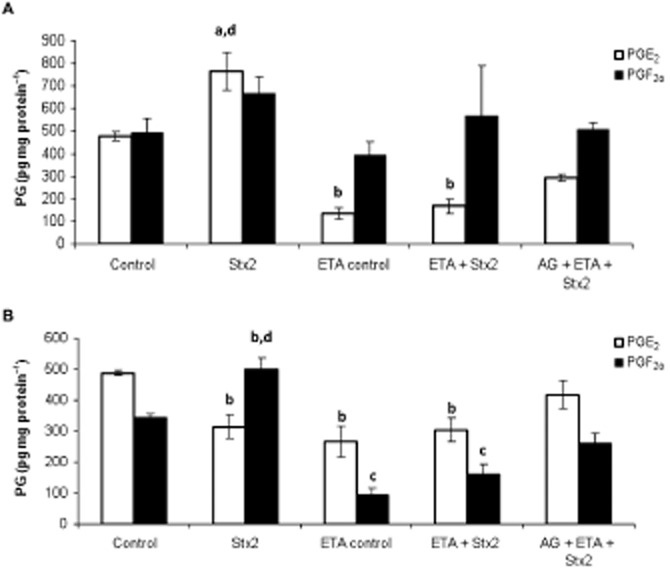
Effects of etanercept (ETA) and aminoguanidine (AG) on PG production. Pregnant rats (gd 15) were injected with LPS alone (50 pg g−1) (shown as control) or with 0.7 ng Stx2 and 50 pg LPS g−1 (shown as Stx2) and killed 12 h post-injection. Some animals were injected with etanercept (i.p., 100 μg g−1) 6 h before toxin treatment. Others were treated with a combination of aminoguanidine and etanercept according the protocol described before for each one. PGE2 and PGF2α levels were evaluated in placenta (A) and decidua (B) by RIA. Data corresponding to at least two separate experiments are shown. Values are expressed as the mean ± SEM (n = 18). aP < 0.05 versus control, bP < 0.01 versus control, cP < 0.001 versus control, dP <0.001 versus etanercept control, etanercept + Stx2 and aminoguanidine + etanercept + Stx2.
Figure 6.
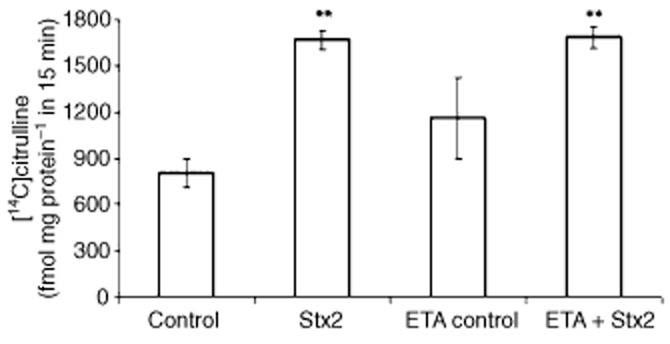
Effect of etanercept (ETA) on NOS activity. Pregnant rats (gd 15) were injected with LPS alone (50 pg g−1) (shown as control) or with 0.7 ng Stx2 and 50 pg LPS g−1 (shown as Stx2) and killed 12 h post-injection. Some animals were treated with etanercept (i.p., 100 μg g−1) 6 h before toxin treatment. NOS activity in placenta was quantified by measuring the conversion of L-[14C]arginine to L-[14C] citrulline. Data corresponding to at least two separate experiments are shown. Values are expressed as the mean ± SEM (n = 6). **P < 0.01 versus control.
Etanercept and aminoguanidine prevent the preterm delivery induced by Stx2
To determine whether the increase in TNF-α production and NOS activity triggers the Stx2-induced preterm delivery, we evaluated the action of etanercept on the delivery time and fetal status of Stx2-treated rats. Administration of etanercept 6 h before Stx2 injection prevented the preterm delivery by roughly 30% compared with their controls (Table 1 row 10 vs. 9). Furthermore, we evaluated the combined action of aminoguanidine and etanercept in Stx2-treated rats compared with control rats. For the first time, we observed that the treatment with both inhibitors, aminoguanidine plus etanercept, significantly (P < 0.005, n = 10) prevented Stx2-induced preterm delivery in 70% of the cases (Table 1, row 12). The pups of these rats were born alive and had normal development compared with their controls (Table 1, row 11). These results suggest that overproduction of TNF-α and NO are the major factors responsible for the preterm delivery of dead fetuses induced by Stx2.
Discussion
In this study, a significant increase in placental PGE2 and decidual PGF2α was found in Stx-treated rats. Additionally, in both tissues, COX-2 protein expression was stimulated by Stx2, although meloxicam, a specific inhibitor of COX-2, was unable to prevent placental toxicity and fetal mortality. Here, we found that production of PGs and expression of COX-2 protein were modulated by NOS activity in Stx2-treated rats. Thus, it is reasonable to infer that NO could elicit different effects on the progression of pregnancy, some directly related with its overproduction and others related with the regulation of PG production (Aisemberg et al., 2007). We reported before that aminoguanidine prevented placental damage in Stx2-treated rats but did not prevent preterm delivery (Burdet et al., 2010). Here, the administration of aminoguanidine, together with meloxicam, delayed preterm delivery but did not prevent it, which indicates that other factors are involved in the Stx2-induced preterm delivery. It is well known that Stx may act in concert with LPS to elicit cellular dysfunction (Louise and Obrig, 1992). Stx is able to bind to Gb3 receptors expressed on the membrane of monocytes/macrophages and leads to cellular activation and secretion of cytokines such as TNF-α, IL-1β and IL-8 that increase endothelial susceptibility to Stx (Ibarra and Palermo, 2010). Many studies conducted in human and experimental animals have established that a correct balance of cytokines at the maternal-fetal interface is an essential requirement for proper placental development and therefore reproductive success (Peltier, 2003; Gravett et al., 2007). In pregnant rats, administration of TNF-α produced placental injury and fetal death similar to the effects observed after LPS exposure (Silen et al., 1989). In this study, we showed that a significant increase in serum TNF-α observed 2 h after Stx2 treatment may be responsible for preterm delivery. Pregnancy loss was triggered by abnormal inflammation as reported by other researchers (Grigsby et al., 2003; Renaud et al., 2011). TNF-α in amniotic fluid was not detected suggesting that maternal, more than fetal, TNF-α exerted its detrimental effects on pregnancy. Administration of etanercept completely prevented the increase in serum TNF-α level caused by Stx2 as previously demonstrated in astrocytes treated with LPS and Stx1 (Landoni et al., 2010). Moreover, etanercept prevented by 30% of the preterm delivery caused by Stx2 and, although this was not significant, it showed a clear trend towards prevention of the effects of Stx2. Our results indicate that TNF-α may play a causal role in Stx2 pregnancy loss, consistent with previous studies reported in human and animal models (Silver et al., 1994; Raghupathy et al., 2000). Etanercept also inhibited the induction of placental PGE2 and decidual PGF2α synthesis in Stx2-treated rats, even in the presence of aminoguanidine. These findings indicate that TNF-α stimulates PG production independent of NO production as already described (Romero et al., 1988; Sato et al., 2003). Etanercept did not inhibit the increase of placental NOS activity caused by Stx2. Thus, it is reasonable to infer that Stx2-induced increase in NO production is not mediated by TNF-α. We assayed the combined action of aminoguanidine and etanercept on fetal status and delivery time in Stx2-treated rats. Co-administration of aminoguanidine and etanercept significantly prevented preterm delivery caused by Stx2 in approximately 70% of the cases. The pups of these rats were similar in size and weight to those observed in the controls. These results indicate that the preterm delivery of dead fetuses induced by Stx2 is triggered by TNF-α and mediated by an increase in NOS production. Both placental PGE2 and decidual PGF2α increase secondarily to the increase in TNF-α and output of iNOS and contribute to the mechanisms that lead to the preterm delivery.
In conclusion, the model presented in this study is relevant because it shows that Stx2 increases TNF-α production, which renders the feto-maternal unit more susceptible to Stx2 through the stimulation of local PG synthesis, which in turn may activate uterine contractions and cervical dilation. This proinflammatory environment could promote the contraction of the uterus and the expulsion of the fetuses. Additionally, Stx2 induces the overproduction of NO as a consequence of an increase in the levels of iNOS protein in placental tissues, which plays an important role in placental toxicity and fetal mortality.
It is important to note that the administration of aminoguanidine and etanercept partially prevented preterm delivery of dead fetuses induced by Stx2. This suggests that they could be used in the future as therapeutic agents in this pathological consequence of HUS.
Acknowledgments
This paper was presented at the 8th International Symposium on Shiga toxin (Verocytotoxin) producing Escherichia coli infection (VTEC2012), Amsterdam, The Netherlands, 6–9 May 2012.
This work was supported by grants to Cristina Ibarra from the University of Buenos Aires: (UBACYT-M095), the National Council of Research of Argentina (CONICET-PIP 344) and ANPCYT-PICT 642.
Glossary
- Gb3
globotriaosylceramide
- gd
gestation day
- HUS
haemolytic uremic syndrome
- iNOS
inducible nitric oxide synthase
- STEC
Shiga toxin-producing Escherichia coli
- Stx
Shiga toxin
- Stx2
Shiga toxin type 2
Conflict of interest
None.
References
- Aisemberg J, Vercelli C, Billi S, Ribeiro ML, Ogando D, Meiss R, et al. Nitric oxide mediates prostaglandins' deleterious effect on lipopolysaccharide-triggered murine fetal resorption. Proc Natl Acad Sci USA. 2007;104:7534–7539. doi: 10.1073/pnas.0702279104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdet J, Zotta E, Franchi AM, Ibarra C. Intraperitoneal administration of Shiga toxin type 2 in rats in the late stage of pregnancy produces premature delivery of dead fetuses. Placenta. 2009;30:491–496. doi: 10.1016/j.placenta.2009.03.012. [DOI] [PubMed] [Google Scholar]
- Burdet J, Zotta E, Cella M, Franchi AM, Ibarra C. Role of nitric oxide in Shiga toxin-2-induced premature delivery of dead fetuses in rats. Plos ONE. 2010;5:e15127. doi: 10.1371/journal.pone.0015127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo Y, Tsurugi K, Yutsudo T, Takeda Y, Ogasawara T, Igarashi K. Site of action of a Vero toxin (VT2) from Escherichia coli O157 : H7 and Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur J Biochem. 1988;171:45–50. doi: 10.1111/j.1432-1033.1988.tb13756.x. [DOI] [PubMed] [Google Scholar]
- Frank C, Werber D, Cramer JP, Askar M, Faber M, an der Heiden M, et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104 : H4 outbreak in Germany. N Engl J Med. 2011;365:1771–1780. doi: 10.1056/NEJMoa1106483. [DOI] [PubMed] [Google Scholar]
- Fraser ME, Chernaia MM, Kozlov YV, James MNG. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 Å resolution. Nat Struct Biol. 1994;1:59–64. doi: 10.1038/nsb0194-59. [DOI] [PubMed] [Google Scholar]
- Gianantonio C, Vitacco M, Mendizharzu F, Rutty A. The hemlytic-uremic syndrome. J Pediatr. 1964;64:478–491. doi: 10.1016/s0022-3476(64)80337-1. [DOI] [PubMed] [Google Scholar]
- Goldstein J, Loidl CF, Pistone Creydt V, Boccoli J, Ibarra C. Intracerebroventricular administration of Shiga toxin type 2 induces striatal neuronal death and glial alterations: an ultrastructural study. Brain Res. 2007;1161:106–115. doi: 10.1016/j.brainres.2007.05.067. [DOI] [PubMed] [Google Scholar]
- Gravett MG, Adams KM, Sadowsky DW, Grosvenor AR, Witkin SS, Axthelm MK, et al. Immunomodulators plus antibiotics delay preterm delivery after experimental intraamniotic infection in a nonhuman primate model. Am J Obstet Gynecol. 2007;197:518.e1–518.e8. doi: 10.1016/j.ajog.2007.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigsby PL, Hirst JJ, Scheerlinck JP, Phillips DJ, Jenkin G. Fetal responses to maternal and intra-amniotic lipopolysaccharide administration in sheep. Biol Reprod. 2003;68:1695–1702. doi: 10.1095/biolreprod.102.009688. [DOI] [PubMed] [Google Scholar]
- Ibarra C, Palermo M. Host responses to pathogenic Escherichia coli. Cap 9. In: Torres A, editor. Pathogenic Escherichia Coli in Latin America. 1st edn. University of Texas, TX: Betham Publ; 2010. pp. 122–141. Available at: http://www.bentham.org/ebooks/9781608051922/index.htm (accessed 28 November 2012) [Google Scholar]
- Inglis SR, Jeremias J, Kuno K, Lescale K, Peeper Q, Chervenak FA, et al. Detection of tumor necrosis factor-alpha, interleukin-6, and fetal fibronectin in the lower genital tract during pregnancy: relation to outcome. Am J Obstet Gynecol. 1994;171:5–10. doi: 10.1016/s0002-9378(94)70069-9. [DOI] [PubMed] [Google Scholar]
- Karmali MA, Steele BT, Petric M, Lim C. Sporadic cases of haemolytic-uraemic syndrome associated with faecal cytotoxin and cytotoxin-producing Escherichia coli in stools. Lancet. 1983;1:619–620. doi: 10.1016/s0140-6736(83)91795-6. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landoni VI, de Campos-Nebel M, Schierloh P, Calatayud C, Fernandez GC, Lapponi ML, et al. Shiga toxin 1-induced inflammatory response in lipopolysaccharide-sensitized astrocytes is mediated by endogenous tumor necrosis factor alpha. Infect Immun. 2010;78:1193–1201. doi: 10.1128/IAI.00932-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood CA. Role of verotoxin receptors in pathogenesis. Trends Microbiol. 1996;4:147–153. doi: 10.1016/0966-842x(96)10017-2. [DOI] [PubMed] [Google Scholar]
- Lingwood CA, Law H, Richardson S, Petric M, Brunton JL, De Grandis S, et al. Glycolipid binding of purified and recombinant Escherichia coli-produced verotoxin in vitro. J Biol Chem. 1987;262:8834–8839. [PubMed] [Google Scholar]
- Louise CB, Obrig TG. Shiga toxin-associated hemolytic-uremic syndrome: combined cytotoxic effects of Shiga toxin, interleukin-1 beta, and tumor necrosis factor alpha on human vascular endothelial cells in vitro. Infect Immun. 1991;59:4173–4179. doi: 10.1128/iai.59.11.4173-4179.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louise CB, Obrig TG. Shiga toxin-associated hemolytic uremic syndrome: combined cytotoxic effects of Shiga toxin and lipopolysaccharide (endotoxin) on human vascular endothelial cells in vitro. Infect Immun. 1992;60:1536–1543. doi: 10.1128/iai.60.4.1536-1543.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton JC, Paton AW. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin Microbiol Rev. 1998;11:450–479. doi: 10.1128/cmr.11.3.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltier MR. Immunology of term and preterm labor. Reprod Biol Endocrinol. 2003;1:122. doi: 10.1186/1477-7827-1-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghupathy R, Makhseed M, Azizieh F, Omu A, Gupta M, Farhat R. Cytokine production by maternal lymphocytes during normal human pregnancy and in unexplained recurrent spontaneous abortion. Hum Reprod. 2000;15:713–718. doi: 10.1093/humrep/15.3.713. [DOI] [PubMed] [Google Scholar]
- Renaud SJ, Cotechini T, Quirt JS, Macdonald-Goodfellow M, Othman M, Graham CH. Spontaneous pregnancy loss mediated by abnormal maternal inflammation in rats is linked to deficient uteroplacental perfusion. J Immunol. 2011;186:1799–1808. doi: 10.4049/jimmunol.1002679. [DOI] [PubMed] [Google Scholar]
- Repetto HA. Epidemic hemolytic-uremic syndrome in children. Nephrology Forum. Kidney Int. 1997;52:1708–1719. doi: 10.1038/ki.1997.508. [DOI] [PubMed] [Google Scholar]
- Ribeiro ML, Aisemberg J, Billi S, Farina MG, Meiss R, McCann S, et al. Epidermal growth factor prevents prepartum luteolysis in the rat. Proc Natl Acad Sci USA. 2005;102:8048–8053. doi: 10.1073/pnas.0502899102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SE, Karmali MA, Becker LE, Smith CR. The histopathology of the hemolytic uremic syndrome associated with verocytotoxin-producing Escherichia coli. Hum Pathol. 1988;19:1102–1110. doi: 10.1016/s0046-8177(88)80093-5. [DOI] [PubMed] [Google Scholar]
- Rivas M, Miliwebsky E, Chinen I, Deza N, Leotta G. The epidemiology of hemolytic uremic syndrome in Argentina. Diagnosis of the etiologic agent, reservoirs and routes of transmission. Medicina (B Aires) 2006;66:27–32. [PubMed] [Google Scholar]
- Romero R, Mazor M, Wu YK, Sirtori M, Oyarzum E, Mitchell MD, et al. Infection in the pathogenesis of preterm labor. Semin Perinatol. 1988;12:262–279. [PubMed] [Google Scholar]
- Sato TA, Keelan JA, Mitchell MD. Critical paracrine interactions between TNF-α and IL-10 regulate pipopolysaccharide-stimulated human choriodecidual cytokine and prostaglandin E2 production. J Immunol. 2003;170:158–166. doi: 10.4049/jimmunol.170.1.158. [DOI] [PubMed] [Google Scholar]
- Scully RE, Mark EJ, McNeely WF, Ebeling SH, Phillips LD. Case records of the Massachusetts General Hospital. N Engl J Med. 1995;336:1587–1594. doi: 10.1056/NEJM199706263362608. [DOI] [PubMed] [Google Scholar]
- Shiau MY, Chiou HL, Lee YL, Kuo TM, Chang YH. Establishment of a consistent L929 bioassay system for TNF-alpha quantitation to evaluate the effect of lipopolysaccharide, phytomitogens and cytodifferentiation agents on cytotoxicity of TNF-alpha secreted by adherent human mononuclear cells. Mediators Inflamm. 2001;10:199–208. doi: 10.1080/09629350123139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silen ML, Firpo A, Morgello S, Lowry SF, Francus T. Interleukin-1 alpha and tumor necrosis factor alpha cause placental injury in the rat. Am J Pathol. 1989;135:239–244. [PMC free article] [PubMed] [Google Scholar]
- Silver RM, Lohner WS, Daynes RA, Mitchell MD, Branch DW. Lipopolysaccharide-induced fetal death: the role of tumor-necrosis factor alpha. Biol Reprod. 1994;50:1108–1112. doi: 10.1095/biolreprod50.5.1108. [DOI] [PubMed] [Google Scholar]
- Steele BT, Goldie J, Alexopoluou I, Shimizu A. Post-partum haemolytic uraemic syndrome and verotoxin-producing Escherichia coli. Lancet. 1984;1:511. doi: 10.1016/s0140-6736(84)92877-0. [DOI] [PubMed] [Google Scholar]
- Strauss RC, Alexander RW. Post-partum hemolytic uremic syndrome. Obstet Gynecol. 1976;47:169–173. [PubMed] [Google Scholar]
- Tesh VL, Ramegowda B, Samuel JE. Purified Shiga-like toxins induce expression of proinflammatory cytokines from murine peritoneal macrophages. Infect Immun. 1994;62:5085–5094. doi: 10.1128/iai.62.11.5085-5094.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]