

Abstract
Background And Purpose
Recent studies suggest a primary role for aldehyde dehydrogenase 2 (ALDH2) in mediating the biotransformation of organic nitrates, such as glyceryl trinitrate (GTN), to the proximal activator of soluble guanylyl cyclase (sGC), resulting in increased cGMP accumulation and vasodilation. Our objective was to assess the role of ALDH2 in organic nitrate action using a cell culture model.
Experimental Approach
Porcine renal epithelial (LLC-PK1) cells possess an intact NO-sGC-cGMP signaling system, and can be used as a biochemical model of organic nitrate action. We used a pcDNA3.1-human ALDH2 expression vector to establish a stably transfected cell line (PK1ALDH2) that overexpressed ALDH2, or small interfering RNA (siRNA) to deplete endogenous ALDH2, and assessed GTN biotransformation and GTN-induced cGMP formation.
Key Results
ALDH2 activity in the stably transfected cells was approximately sevenfold higher than wild-type cells or cells stably transfected with empty vector (PK1vector); and protein expression, as assessed by immunoblot analysis, was markedly increased. In PK1ALDH2, GTN biotransformation was significantly increased as a result of increased glyceryl-1,2-dinitrate formation compared to wild-type or PK1vector. However, the incubation of PK1ALDH2 with 1 or 10 μM GTN did not alter GTN-induced cGMP accumulation compared with wild-type or PK1vector cells. Furthermore, siRNA-mediated depletion of ALDH2 had no effect on GTN-induced cGMP formation.
Conclusions And Implications
In an intact cell system, neither overexpression nor depletion of ALDH2 affects GTN-induced cGMP formation, indicating that ALDH2 does not mediate the mechanism-based biotransformation of GTN to an activator of sGC.
Keywords: glyceryl trinitrate, NO, aldehyde dehydrogenase, nitrate tolerance, cGMP
Introduction
A major limitation with the chronic use of organic nitrates is the development of tolerance. Details of the mechanism of tolerance are lacking; however, decreased biotransformation is likely involved. It is generally accepted that GTN and other nitrates require bioactivation to an active metabolite prior to initiating their vasodilator effect. Two pathways have been proposed for the biotransformation of nitrates; mechanism-based biotransformation results in the formation of an activator (presumably NO or a related species) of soluble guanylyl cyclase (sGC), resulting in cGMP accumulation and subsequent vascular smooth muscle relaxation. Alternatively, the clearance-based pathway results in the formation of the inorganic nitrite anion (NO2–) and lacks sGC activation (Bennett et al., 1994).
The enzymatic bioactivation of GTN has been widely studied, involving enzymes such as cytochrome P450 (McDonald and Bennett, 1990), NADPH cytochrome-P450 reductase (McGuire et al., 1998), cytosolic (Tsuchida et al., 1990; Nigam et al., 1996) and microsomal (Ji et al., 2009) glutathione transferases and xanthine oxidoreductase (Doel et al., 2001); but their specific role in GTN bioactivation and tolerance remains equivocal. The enzyme aldehyde dehydrogenase 2 (ALDH2) has been shown to mediate GTN biotransformation and has been implicated as a GTN bioactivating enzyme (reviewed in Daiber et al., 2008; Mayer and Beretta, 2008). During the ALDH2-mediated biotransformation of GTN, which yields glyceryl-1,2-dinitrate (1,2-GDN) and inorganic nitrite anion (NO2−), the active site of ALDH2 is inactivated through the oxidation of sulfhydryl groups (Beretta et al., 2008). This is further supported by Wenzl et al. (2009), who showed that the mutation of an active site Cys (C302S) resulted in a 90% reduction of ALDH2 esterase and dehydrogenase activity. This inactivation is considered to be a major mechanism of GTN tolerance, and regeneration of ALDH2 has been suggested as a viable therapeutic strategy for the reversal of nitrate tolerance. A physiological reductant has yet to be identified, although dihydrolipoic acid has been proposed as a leading candidate (Wenzel et al., 2007; Beretta et al., 2008).
Although numerous studies have suggested a role for ALDH2 in GTN bioactivation and tolerance development, two recent studies have questioned the role of ALDH2 in mechanism-based GTN biotransformation. We have shown that ALDH2 expression and activity are dissociated from the development and reversal of GTN tolerance in an in vivo rat model. Furthermore, the ALDH inhibitor daidzin did not significantly affect GTN biotransformation or GTN-induced relaxation of rat aorta (D'Souza et al., 2011). Second, in a clinical study of 117 normal Japanese volunteers, 48 of whom were heterozygous and 22 of whom were homozygous for the loss-of-function ALDH2 point mutation Glu504Lys, the maximal vasodilator response to sublingual GTN was the same, regardless of ALDH2 genotype (Sakata et al., 2011).
The human ALDH superfamily consists of at least 19 isozymes that participate in the detoxification of endogenous and exogenous compounds (Vasiliou and Nebert, 2005). Different isozymes of ALDH have different physiochemical and enzymatic properties, subcellular localization and tissue distribution. The ALDH2 isozyme has a very low Km value (<1.0 μM) for short-chain aliphatic aldehydes such as acetaldehyde and propionaldehyde. Thus, ALDH2 activity and tissue distribution may be relevant to the toxicology of ethanol and to a number of endogenous substances. Several studies have shown ALDH2 deficiency to increase oxidative stress, suggesting ALDH2 may play a role in reducing oxidative damage (Ohsawa et al., 2003; 2008; Wenzel et al., 2008; Endo et al., 2009), and Fadel et al. (2012) found continuous GTN exposure results in increased oxidative stress, which selectively alters blood vessel responsiveness to sympatholytics. Furthermore, other studies have shown an association between ALDH2 deficiency and a higher incidence of certain cancers (Seitz and Meier, 2007; Gao et al., 2008). Thus, the inactivation of ALDH2 in nitrate tolerance could be of pathological significance. Interestingly, Choi et al. (2011) showed that intracellular delivery of ALDH2 into mouse aorta reduces the augmented vascular contraction observed in angiotensin II hypertensive mice and suggested this occurs through ALDH2-mediated decreases in reactive oxygen species.
Porcine renal proximal tubular epithelial cells (LLC-PK1) have been used in the study of membrane transport mechanisms, xenobiotic cytotoxicity and drug metabolism. LLC-PK1 cells are one of the few cell lines that maintain an intact NO-sGC-cGMP signaling system, and continuous exposure of these cells to GTN results in attenuated GTN-induced cGMP accumulation (Bennett et al., 1989; Hinz and Schröder, 1998; Ji et al., 2009). Thus, this cell line is a useful biochemical model of nitrate tolerance. In the current study, we developed a stable LLC-PK1 cell line that overexpressed the human ALDH2 protein and utilized small interfering RNA (siRNA) constructs to deplete endogenous ALDH2, to examine whether increased or decreased expression of ALDH2 altered GTN biotransformation and GTN-induced cGMP accumulation.
Methods
Test system
LLC-PK1 cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD). The cells were inoculated at a density of 3.0 × 105 cells per well in six-well plates in 2.0 mL of DMEM/Ham's F12 medium (1:1) supplemented with 10% FBS, 5 μg·mL−1 insulin, 2 mM glutamine, 10 mM HEPES (pH 6.9), 50U mL−1 penicillin and 50 μg·mL−1 streptomycin, and grown at 37°C in an atmosphere of 5% CO2 in air until confluent. The cells were transfected with pcDNA-3.1(–) containing human ALDH2 cDNA (a gift from Dr Vasilis Vasiliou, Department of Pharmaceutical Sciences, University of Colorado Health Sciences Center, Denver, CO) or empty vector using FuGene 6 according to the manufacturer's instructions (Roche Molecular Biochemicals, Basel, Switzerland). Twenty-four hours later, G-418 (1.5 mg·mL−1) was added to the culture medium to select for transfected cells, and selection was continued for 2 weeks with regular changes of media. Stably transfected cells were cloned by limiting dilution; and two subclonal cell lines, designated PK1ALDH2 and PK1vector, were expanded and characterized using immunoblot analysis, confocal microscopy and enzyme activity.
Immunoblot analysis of ALDH2
Cells grown to confluence in T75 flasks were washed twice with ice-cold PBS (pH 7.4), harvested with a disposable cell scraper and homogenized in lysis buffer (50 mM Tris–HCl pH 7.4, 1 mM EDTA, 0.1 mM PMSF, 1 mM DTT, 1% Triton X-100 and protease inhibitors) (Roche Diagnostics, Mannheim, Germany), and differentially centrifuged to obtain mitochondrial and cytosolic fractions as previously described (DiFabio et al., 2003); 20 μg mitochondrial or cytosolic protein from wild-type and stably transfected cells were separated on 10% gels by SDS-PAGE and transferred electrophoretically to PVDF membranes. Blots were probed with a polyclonal antibody to human ALDH2, and immunoreactive bands were visualized by enhanced chemiluminescence. Membranes were then stripped and re-probed with antibodies to both β-actin and cytochrome c. Purified recombinant human ALDH2 was used as a positive control (gift from Dr Vasilis Vasiliou). In siRNA knock-down experiments, whole cell lysates were used.
Confocal fluorescence microscopy
PK1ALDH2, PK1vector and wild-type cells were grown on coverslips, fixed with 4% formalin and permeabilized with 1% Triton-X 100. Cells were incubated with 10% normal goat serum followed by incubation with affinity-purified rabbit anti-human ALDH2 IgG (1:1000) (a gift from Dr Vasilis Vasiliou) for 30 min at room temperature. Cells were then incubated with Alexa-Fluor-546-conjugated goat anti-rabbit IgG. Nuclei were stained using the DNA binding dye, DAPI (5 μM). Prior to fixation with formalin, the cells were pre-incubated with MitoTracker Orange CM-H2TMRos to stain mitochondria, according to the manufacturer's instructions. Confocal microscopy was performed using a Leica TCS SP2 Multi Photon instrument.
ALDH activity measurement
Cells grown in T75 flasks were washed twice with ice-cold PBS (pH 7.4) and harvested with a disposable cell scraper. The cell suspension was sonicated, and mitochondrial and cytosolic fractions were prepared by differential centrifugation as described (DiFabio et al., 2003). Cell preparations were solubilized with deoxycholate (2.5 mg·mg−1 protein), and ALDH activity measured in either mitochondrial or cytosolic fractions as the change in A340 during incubation with 1 mM NAD+ in 50 mM sodium pyrophosphate, pH 8.8 containing 2 μM rotenone, 1 mM 4-methylpyrazone, 200 μg protein and substrate (0.05 or 5 mM propionaldehyde) (Tottmar et al., 1973; Loomis and Brien, 1983). Specific activity was calculated using the molar extinction coefficient for NADH of 6306 M·cm−1. ALDH2 activity is reflected by changes in A340 at low substrate concentration.
GTN biotransformation and GTN-induced cGMP accumulation
Cells grown to confluence in six-well plates were washed twice with PBS and incubated in serum-free medium for 2 h. For assessment of GTN biotransformation, cells were incubated with 1 μM GTN for 20 min, and 1 mL aliquots of the incubation medium were extracted with diethyl ether and the GTN metabolites, 1,2-GDN and 1,3-GDN, quantitated by gas chromatography with electron capture detection as described (McDonald and Bennett, 1990). For cGMP determinations, cells were incubated for 3 min at 37°C in serum-free medium containing 0.5 mM isobutylmethylxanthine and GTN (1 or 10 μM) or DEA/NO (10 μM). After incubation, the medium was aspirated, and the cells were immediately treated with 6% ice-cold trichloroacetic acid, and cGMP was determined by radioimmunoassay (Steiner et al., 1972). In some experiments, cells were pre-incubated with 10 μM GTN for 2 h prior to assessment of GTN biotransformation or cGMP determinations, in order to simulate GTN tolerance.
siRNA transfection
siRNA were designed to target sites specific for ALDH2 mRNA, based on the porcine ALDH2 sequence (accession no. DQ266356). Two siRNAs, designated siRNA-1 (sense, 5′-CAUCUCUUACCUGGUAGAUtt-3′; antisense, 5′-AUCUACCAGGUAAGAGAUGtt-3′) and siRNA-2 (sense, 5′-AGAUUCUUGGCUACAUCAAtt-3′; antisense, 5′-UUGAUGUAGCCAAGAAUCUtt-3′) were used. Allstars Negative Control siRNA was used as a negative control (Qiagen, Mississauga, ON, Canada). This control siRNA has no homology to any known mammalian gene. siRNA transfections were performed using Lullaby® transfection reagent according to the manufacturer's instructions (OZ Biosciences, Marseille, France). Briefly, cells were plated in six-well plates 1 day before transfection; 50 nM of siRNA was prepared in 100 μL of serum-free media and then added to 100 μL of serum-free media containing 14 μL of Lullaby reagent, and the mixture was incubated for 20 min at room temperature. The mixture was then added dropwise directly onto the cells, and the cells were incubated for 72 h prior to experimentation.
Data analysis
All data are expressed as the mean ± SD of n independent experiments. Data were analysed by two-way anova with a Bonferroni post hoc test and Student's t-test for unpaired data, as indicated. A P-value of 0.05 or less was considered statistically significant.
Drugs and solutions
β-Nicotinamide adenine dinucleotide (NAD+), DMEM, nutrient mixture F-12 HAM, Triton X-100 and siRNA constructs were purchased from Sigma-Aldrich Canada Ltd (Oakville ON, Canada). MitoTracker Orange CM-H2TMRos, DAPI, Alexa Fluor 546 and goat anti-rabbit IgG were obtained from Cedarlane Laboratories Ltd (Hornby, ON, Canada); GTN was obtained as a solution (TRIDIL®, 5 mg·mL−1) in ethanol, propylene glycol and water (1:1:1.33) from Sabex Inc. (Boucherville, QC, Canada). 1,2- and 1,3-GDN were prepared and quantitated as described (Brien et al., 1986; Bennett et al., 1988). Geneticin (G-418 sulfate) was from Gibco Life Technologies (Burlington, ON, Canada), and 1,1-diethyl-2-hydroxy-2-nitrosohydrazine (DEA/NO) was from Calbiochem (La Jolla, CA). Chemiluminescence reagents were from Kirkegaard and Perry Laboratories (Gaithersburg, MA). The rabbit anti-human ALDH2 antiserum and purified recombinant human ALDH2 were gifts from Dr V. Vasiliou (University of Colorado Health Science Center, Denver, CO). Mouse monoclonal anti-β-actin antibody was obtained from Sigma (St. Louis, MO), and mouse monoclonal anti-cytochrome c antibody was obtained from BD Biosciences (Mississauga, ON). All other chemicals were of reagent grade and were obtained from a variety of commercial sources.
Results
Expression and activity of ALDH2 in PK1 cells
To determine the role of ALDH2 in the bioactivation of GTN, we developed a stably transfected cell line that overexpressed ALDH2. Immunoblot analysis indicated that in wild-type LLC-PK1 cells as well as in cells transfected with empty vector, ALDH2 was predominantly localized in mitochondrial fraction (Figure 1). In PK1ALDH2, ALDH2 expression was markedly increased compared with wild-type or PK1vector. Furthermore, the increased expression of ALDH2 in PK1ALDH2 occurred in both the mitochondrial and cytosolic fractions. Data from confocal microscopy experiments (Figure 2) were in agreement with the immunoblot data. ALDH activity was measured in the cytosolic and mitochondrial fractions at a low and high substrate concentration to assess ALDH2 and total ALDH activity. In the cytosolic fraction, ALDH2 activity was increased approximately sevenfold in PK1ALDH2 compared with wild-type or PK1vector cells (P < 0.001) (Figure 3A). Total ALDH activity in PK1ALDH2 also increased in a similar manner (Figure 3B). In the mitochondrial fraction, ALDH2 and total ALDH activity was greater than that in the cytosolic fraction, and a similar increase ALDH2 activity was observed in PK1ALDH2 cells (Figure 3 and D). Pre-incubation of PK1ALDH2 with 10 μM GTN, in order to simulate tolerance, resulted in an 80% and 60% reduction in ALDH2 activity in the cytosolic and mitochondrial fractions respectively (Figure 3 and C). However, ALDH2 activity was 1.2- and 2.2-fold higher in tolerant PK1ALDH2 compared with untreated wild-type activity in the cytosolic and mitochondrial fractions respectively.
Figure 1.
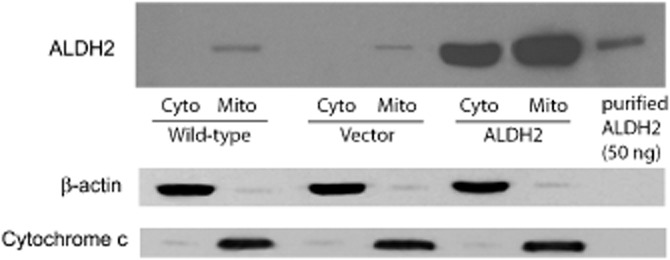
Expression of ALDH2 in PK1 cells. Immunoblot analysis of cytosolic (Cyto) or mitochondrial (Mito) fractions from wild-type, PK1vector (Vector) and PK1ALDH2 cells (ALDH2); 20 μg of cytosolic or mitochondrial proteins was resolved on a 10% SDS-PAGE gel under reducing conditions, transferred to a PVDF membrane and probed with a rabbit polyclonal antibody to human ALDH2, and then to antibodies to the cytosolic and mitochondrial markers, β-actin and cytochrome c. Note that ALDH2 was detected in both the cytosol and mitochondria in PK1ALDH2 cells, whereas in wild-type or PK1vector cells, ALDH2 was only detected in the mitochondrial fraction.
Figure 2.
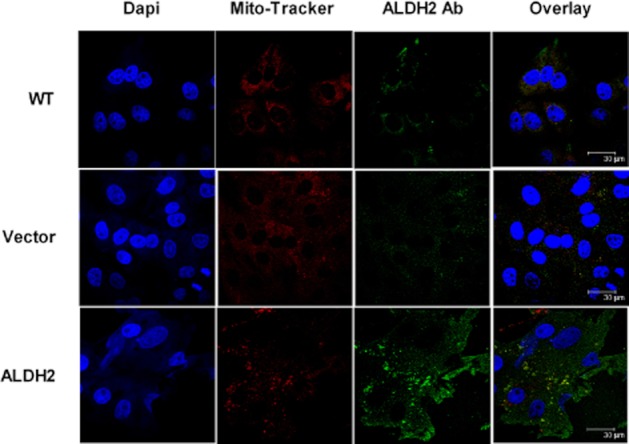
Subcellular location of ALDH2 in PK1 cells. Cells grown on coverslips were fixed with 4% formalin. Nuclei were visualized by staining with DAPI, mitochondria by staining with MitoTracker and ALDH2 by labelling with goat anti-rabbit antibody conjugated with Alexa Fluor 546, after incubation with an antibody directed against ALDH2. Images were obtained by confocal microscopy. Note the increased expression of ALDH2 in PK1ALDH2 cells, both in the cytosol, and co-localized with MitoTracker.
Figure 3.
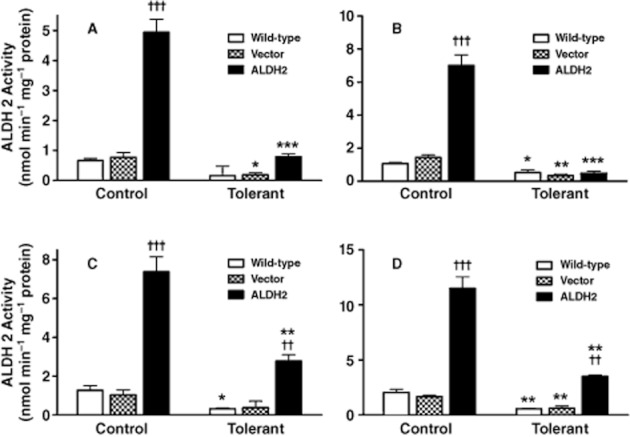
ALDH activity in the cytosolic and mitochondrial fractions from PK1 cells. Cytosolic ALDH2-specific activity (A) and total ALDH activity (B) and mitochondrial ALDH2-specific activity (C) and total ALDH activity (D) was assessed from Wild-type, PK1vector (Vector) and PK1ALDH2 (ALDH2) cells. Cells were either untreated (Control) or incubated with 10 μM GTN for 2 h prior to harvest (Tolerant). Data are presented as the mean ± SD (n = 4) and were analysed by two-way anova and Student's t-test for unpaired data. * indicates significant difference from Control (***P < 0.001, **P < 0.01, *P < 0.05), and † indicates significant difference between cell types (†††P < 0.001 and ††P < 0.01 vs. Wild-type and Vector).
GTN biotransformation in PK1ALDH2 cells
GTN biotransformation by wild-type, PK1vector and PK1ALDH2 cells resulted in the formation of 3.5- to 4.5-fold more 1,2-GDN compared with 1,3-GDN (Figure 4). Pre-incubation of wild-type or PK1vector cells with 10 μM GTN for 2 h resulted in a significant decrease in 1,2-GDN formation, whereas 1,3-GDN formation was unchanged. In PK1ALDH2 cells, there was a significant increase in GTN biotransformation relative to the other two cell types, primarily attributable to an increase in 1,2-GDN formation. Desensitization of PK1ALDH2 cells by pre-incubation with 10 μM GTN for 2 h significantly decreased 1,2-GDN formation. However, 1,2-GDN formation in desensitized PK1ALDH2 cells was significantly greater than that in desensitized wild-type or PK1vector cells (P < 0.01).
Figure 4.
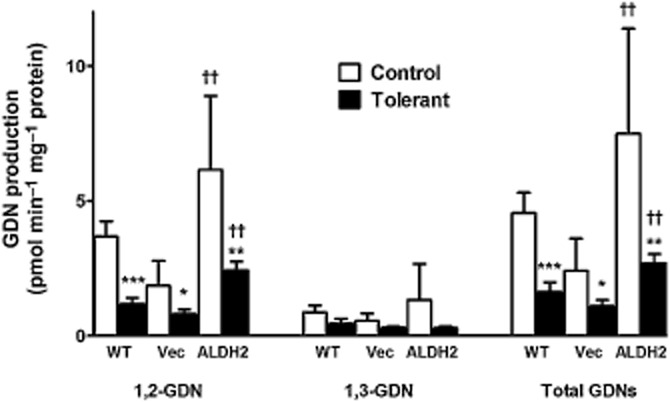
Effect of overexpression of ALDH2 on GTN biotransformation in PK1 cells. Wild-type (WT), PK1vector (Vec) or PK1ALDH2 (ALDH2) cells, were exposed to 1.0 μM GTN for 20 min at 37°C (Control). Aliquots of the incubation medium were extracted with diethyl ether and the GTN metabolites, 1,2-and 1,3-GDN, quantitated by gas chromatography with electron capture detection. GTN-tolerant cells (Tolerant) were pre-incubated with 10 μM GTN for 2 h prior to assessment of GTN biotransformation. Data are presented as the mean ± SD (n = 6–8) and were analysed by two-way anova and Student's t-test for unpaired data. * indicates significant difference from control after pre-incubation with GTN (***P < 0.001, **P < 0.01, *P < 0.05), and † indicates significant differences between cell types (††P < 0.01 vs. WT and Vec).
GTN-induced cGMP accumulation in PK1ALDH2 cells
Cellular cGMP levels were measured in each cell line in response to different concentrations of GTN (Figure 5 and B). Incubation with 1 μM GTN resulted in a sixfold increase in cGMP accumulation in all cell types, whereas a 10 μM incubation of GTN resulted in a 15-fold increase in cGMP levels. Pre-incubation of each cell type with 10 μM GTN for 2 h resulted in desensitization of cells to GTN-induced cGMP accumulation, and cGMP levels increased only two- and sixfold after cells were exposed to 1 and 10 μM GTN respectively (Figure 5 and B). There were no differences in GTN-induced cGMP accumulation in cells overexpressing ALDH2 compared with wild-type or PK1vector cells. We also assessed increases in cGMP accumulation after exposure to the NO donor, DEA/NO (Figure 5C). Similar to the findings with GTN, there were no differences in cGMP accumulation in the different cell types (P > 0.05). However, the cGMP responses to DEA/NO were unaltered in cells pretreated with GTN for 2 h, indicating that the ability of sGC to respond to NO was not affected by the GTN tolerance protocol.
Figure 5.
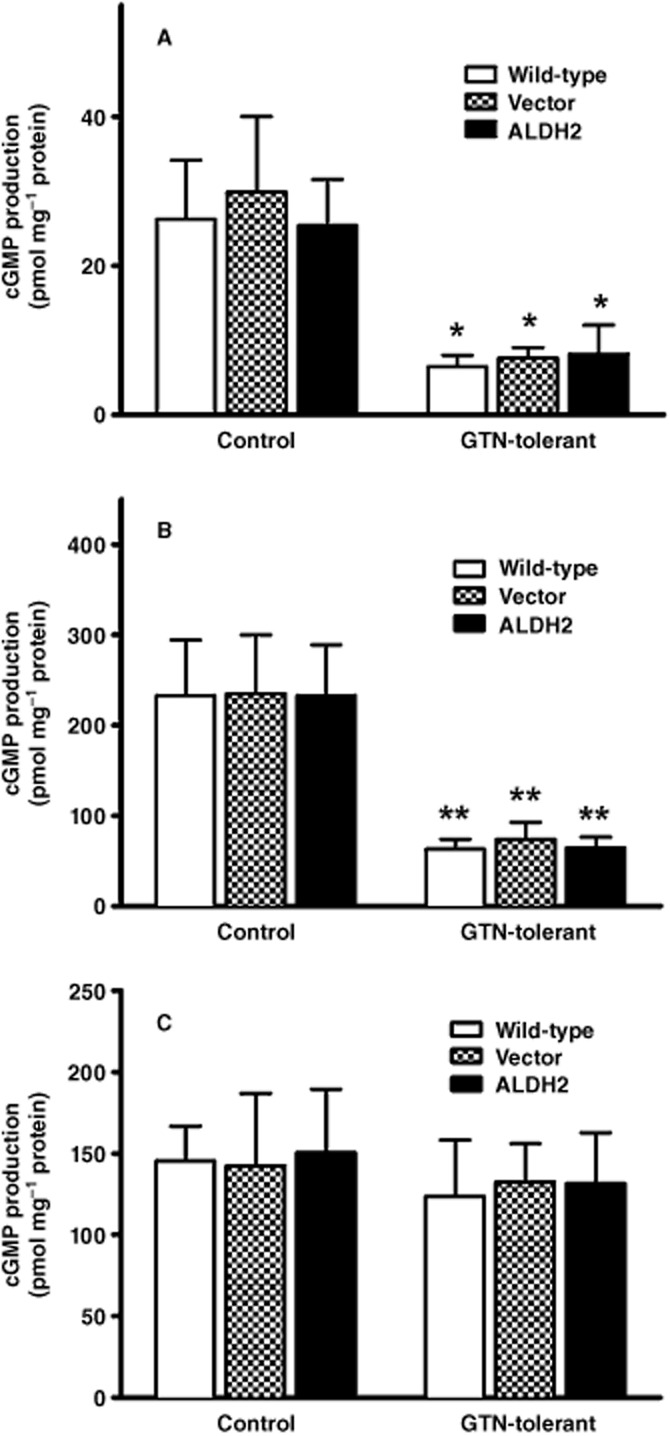
Effect of overexpression of ALDH2 on GTN-induced or DEA/NO-induced cGMP accumulation in PK1 cells. Wild-type cells (Wild-type), PK1vector cells (Vector) or PK1ALDH2 cells (ALDH2) were exposed to 1.0 μM GTN (A), 10 μM GTN (B) or 10 μM DEA/NO (C) for 3 min at 37°C in the presence of 0.5 mM isobutylmethylxanthine, after which cGMP accumulation was measured by radioimmunoassay. GTN tolerance was induced by pre-incubation of cells with 10 μM GTN for 2 h prior to the assessment of GTN-induced cGMP accumulation. Data are presented as the mean ± SD (n = 3) and were analysed by two-way anova and Student's t-test for unpaired data. * indicates significant difference from control after pre-incubation with GTN (***P < 0.001, **P < 0.01).
siRNA-mediated knockdown of ALDH2
Immunoblot analysis indicated that the transfection of siRNA-1 and siRNA-2 resulted in a dose-dependent decrease in ALDH2 protein (data not shown). Treatment of cells with 50 nM siRNA-1 or siRNA-2 for 72 h resulted in an approximate 95–100% decrease in ALDH2 protein expression compared with control (Figure 6, middle panel). Treatment with Allstars Negative Control resulted in no change in ALDH2 expression, whereas a slight decrease was observed with treatment with Lullaby® transfection reagent.
Figure 6.
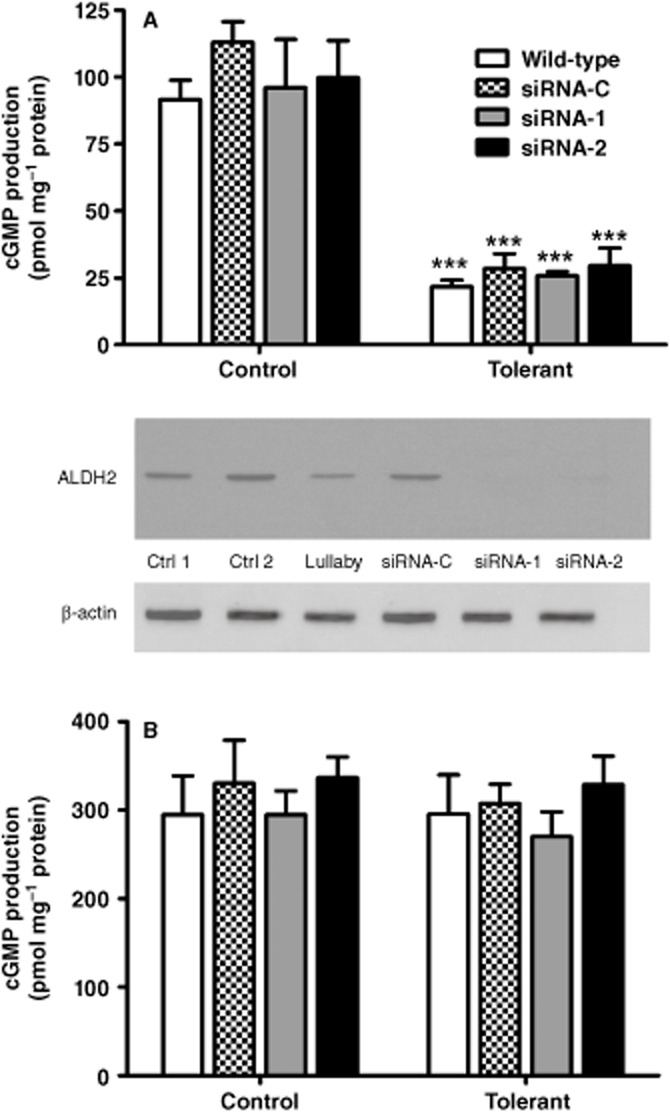
Effect of ALDH2 knockdown on GTN-induced and DEA/NO-induced cGMP formation. Wild-type cells and cells treated with 50 nM Allstars Negative Control (siRNA-C); 50 nM siRNA-1 (siRNA-1) or 50 nM siRNA-2 (siRNA-2) for 72 h were exposed to 1.0 μM GTN (A) or 10 μM DEA/NO (B) for 3 min at 37°C in the presence of 0.5 mM isobutylmethylxanthine, after which cGMP accumulation was measured by radioimmunoassay. GTN tolerance was induced by pre-incubation of cells with 10 μM GTN for 2 h prior to the assessment of GTN-induced cGMP accumulation. Data are presented as the mean ± SD (n = 4–5) and were analysed by two-way anova and Student's t-test for unpaired data. * indicates significant difference from control after pre-incubation with GTN (***P < 0.001). Middle panel: siRNA induced knockdown of ALDH2 in LLC-PK1 cells. Immunoblot analysis of whole cell fractions from wild-type PK1 cells treated with 14 μL of lullaby transfection reagent (Lullaby), 50 nM Allstars Negative Control (siRNA-C), 50 nM siRNA-1 (siRNA-1), 50 nM siRNA-2 (siRNA-2) for 72 h and no treatment (Ctrl 1, Ctrl 2). 20 μg of proteins were resolved on a 10% SDS-PAGE gel under reducing conditions, transferred to a PVDF membrane, and probed with antibodies as indicated. Note that ALDH2 was barely detectable in PK1 cells treated with siRNA-1 and siRNA-2, whereas in the other treatment groups ALDH2 was readily visible.
GTN-induced cGMP accumulation after ALDH2 knockdown
Incubation of cells with 1 μM GTN resulted in a five- to sixfold increase in cGMP in all treatment groups (Figure 6A). No differences were observed in the GTN-induced cGMP response in PK1 cells treated with Allstars Negative Control, siRNA-1 or siRNA-2 compared with non-transfected wild-type cells. Pre-incubation of cells from each treatment group with 10 μM GTN for 2 h resulted in desensitization of the cGMP response, and incubation of GTN tolerant cells with 1 μM GTN resulted in only a twofold increase in cGMP. No differences were observed with respect to the GTN-induced cGMP responses in each treatment group during GTN tolerance. Furthermore, DEA/NO-induced cGMP formation did not differ between treatment groups (Figure 6B).
Discussion
A number of enzymes or proteins catalysing GTN biotransformation have been identified, including haemoglobin and myoglobin, cytochromes P450, glutathione transferases, NADPH cytochrome P450 reductase, xanthine oxidoreductase and ALDH2 (Bennett et al., 1986; McDonald and Bennett, 1990; Tsuchida et al., 1990; Nigam et al., 1996; McGuire et al., 1998; Doel et al., 2001; Chen et al., 2002). It still remains unclear whether a specific enzyme bioactivates GTN and is inactivated during GTN tolerance, or whether GTN tolerance is a multi-factorial phenomenon involving a number of enzymes. In 2002, a prominent role for ALDH2 in the bioactivation of GTN was proposed by Stamler's group and has been supported by some studies, but not all (Chen et al., 2002; 2005; DiFabio et al., 2003; Kollau et al., 2005; Hink et al., 2007; Beretta et al., 2008). Furthermore, decreased bioactivation of GTN as a mechanism of tolerance due to the inactivation of ALDH2 also has been proposed.
The development of a stably transfected cell line that overexpressed ALDH2 (PK1ALDH2) enabled us to examine the role of ALDH2 in GTN bioactivation and tolerance. LLC-PK1 cells were chosen because they are one of the few cell lines that maintain an intact NO-sGC-cGMP signalling system. The overexpression of ALDH2 resulted in a marked increase in ALDH2 protein (Figure 1). Examination by confocal microscopy confirmed this observation, as well as the location of ALDH2 in both the mitochondria and cytosol. This is quite different than the location of ALDH2 in wild-type cells, where the majority of ALDH2 is localized to mitochondria. Previous data from our laboratory indicated that in rat or rabbit aorta, ALDH2 is expressed primarily in the cytosol, whereas in rat liver, it is only present in mitochondria (DiFabio et al., 2003). Furthermore, a recent study by Beretta et al. (2012) concluded that cytosolic ALDH2 mediates the bioactivation of GTN. Given the suggested therapeutic relevance of the subcellular distribution of ALDH2, it was important to characterize this in PK1ALDH2 cells. However, our data show that neither the intracellular location nor abundance of ALDH2 has any impact on GTN bioactivation or on GTN-induced sGC activation (Figures 5 and 6).
Examination of ALDH2 activity in PK1ALDH2 cells indicated that ALDH2 activity was approximately sevenfold higher in these cells compared with wild-type or empty vector cells. Since the ALDH2 isozyme has a low Km value (<1.0 μM) relative to other ALDH isozymes, it allowed us to distinguish between ALDH2-specific activity (low Km) and total ALDH activity (high Km). In PK1ALDH2 cells, ALDH2 and total ALDH activities were increased, the latter being most likely attributable to the increase in ALDH2 activity. Thus, the ALDH2-overexpressing cell line exhibited a marked increase in functional ALDH2. A 2 h incubation with 10 μM GTN resulted in a significant decrease in ALDH2 activity in all cell lines tested and is consistent with other findings (Chen et al., 2002; DiFabio et al., 2003; Sydow et al., 2004; Hink et al., 2007). Although pre-incubation with GTN decreased ALDH2 activity, ALDH2 activity in PK1ALDH2 cells was significantly higher than wild-type or PK1vector cells pretreated with GTN (Figure 3 and C). Furthermore, ALDH2 activity after pretreatment with GTN in PK1ALDH2 cells was higher than ALDH2 activity in untreated wild-type or PK1vector cells. Thus, some ALDH2 activity was preserved in PK1ALDH2 cells after simulating GTN tolerance.
In cells overexpressing ALDH2, incubation with 1 μM GTN resulted in a significant increase in GTN biotransformation as well as a significant increase in 1,2-GDN formation, compared with wild-type or PK1vector cells (Figure 4). The formation of 1,3-GDN was not affected in the PK1ALDH2 cell line. The selective formation of 1,2-GDN by ALDH2 has been previously shown by others using purified ALDH2 (Chen et al., 2002; Kollau et al., 2005; Tsou et al., 2011). However, despite the sevenfold increase in ALDH2 activity in the PK1ALDH2 cells, GTN biotransformation activity was only increased 1.5- to 3-fold compared with wild-type or PK1vector cells, suggesting a relatively minor contribution of ALDH2 to the overall biotransformation of GTN. This is consistent with biotransformation data obtained in intact blood vessels, in which pretreatment with the ALDH2 inhibitor, daidzin, had very little effect on GTN biotransformation (D'Souza et al., 2011). Chronic in vivo exposure to GTN results in tolerance, which is associated with reduced GTN biotransformation and reduced GTN-induced cGMP accumulation. The pre-incubation of PK1 cells with GTN resulted in decreased GTN biotransformation, consistent with responses observed during in vivo GTN tolerance. Although PK1ALDH2 had an attenuated GTN biotransformation response after pre-incubation with 10 μM GTN, 1,2-GDN formation and total biotransformation were still increased compared to GTN-tolerant wild-type and PK1vector cells. This was not unexpected since our tolerance protocol did not completely inhibit ALDH2 activity in PK1ALDH2 cells (Figure 3).
We next assessed the effect of ALDH2 overexpression on the cGMP response to GTN. No significant differences were observed with respect to GTN-induced cGMP accumulation in response to 1 or 10 μM GTN in PK1ALDH2 cells compared with wild-type or PK1vector cells. Thus, despite the marked increase in ALDH2 activity and GTN biotransformation, and the significant increase in 1,2-GDN formation observed in PK1ALDH2 cells, GTN-induced cGMP responses remained unchanged relative to wild-type or PK1vector cells (Figure 5). Thus, it appears that ALDH2-specific biotransformation does not significantly contribute to the mechanism-based biotransformation of GTN to an activator of sGC. A 2 h incubation of each cell line with 10 μM GTN resulted in a significant decrease in the cGMP responses to GTN, indicating that desensitization had occurred. As mentioned previously, GTN-tolerant PK1ALDH2 cells exhibit ALDH2 activity and 1,2-GDN formation that is similar to that in untreated wild-type or PK1vector cells. However, despite the increase in ALDH2-specific GTN biotransformation, GTN-induced cGMP formation did not significantly differ in tolerant PK1ALDH2 cells compared with tolerant wild-type or PK1vector cells.
Since overexpression of ALDH2 did not alter GTN-induced cGMP formation, we hypothesized that GTN bioactivation may occur at a maximal rate in wild-type cells, and thus overexpressing ALDH2 would have no further impact on GTN bioactivation. To investigate this, we used siRNA to deplete endogenous ALDH2, followed by assessment of GTN-induced cGMP formation. The siRNA-induced knockdown of ALDH2 had no effect on GTN-induced cGMP formation; furthermore, a 2 h incubation with 10 μM GTN led to desensitization of the cGMP response to GTN, but not to DEA/NO (Figure 6). These results indicate that ALDH2 is not necessary for GTN bioactivation and that GTN tolerance can occur in the absence of ALDH2.
A suggested biomarker for GTN bioactivation is the ratio of 1,2/1,3-GDN formation, since it has been suggested that 1,2-GDN formation is associated with NO bioactivity, and 1,3-GDN formation is associated with GTN clearance. The vascular biotransformation of GTN is selective for 1,2-GDN formation as seen in the isolated rabbit aorta (Brien et al., 1986), rat aorta (Fung and Poliszczuk, 1986; McGuire et al., 1994) and human saphenous vein (Sage et al., 2000). Furthermore, 1,2-GDN formation occurs almost exclusively during initial GTN exposure to vascular tissues, and 1,2-GDN-selective formation is decreased in GTN-tolerant tissues (Brien et al., 1986). Whereas these data may be taken to suggest 1,2-GDN is associated with mechanism-based biotransformation, we have suggested previously that regioselective denitration itself is not sufficient for mechanism-based biotransformation (DiFabio et al., 2003). The results from the current study indicate that although increased 1,2-GDN formation is associated with overexpression of ALDH2, it is not associated with bioactive ‘NO’, since the increases in 1,2-GDN formation had no effect on GTN-induced cGMP accumulation. A previous study from our laboratory demonstrated that the overexpression of microsomal glutathione transferase 1 in LLC-PK1 cells resulted in a marked increase in GTN biotransformation via selective 1,3-GDN formation. However, GTN-induced cGMP accumulation did not significantly differ from that observed in wild-type or cells transfected with empty vector (Ji et al., 2009), indicating that selective 1,3-GDN formation had no impact on the formation of bioactive ‘NO’. Together, these data indicate that regioselective 1,3-GDN or 1,2-GDN formation does not reflect clearance or mechanism-based GTN biotransformation per se, but rather, reflects the selectivity for denitration characteristic of the particular enzyme being studied, or in the case of intact tissues, the composite of multiple enzymes that mediate GTN biotransformation, some portion of which is mechanism-based. Thus, selective 1,2-GDN formation cannot be used as a surrogate marker for the formation of NO bioactivity from GTN, but rather, that assessment of GTN-induced cGMP formation is required to demonstrate that the formation of NO bioactivity from GTN has occurred.
In summary, we have developed a stable cell line that overexpresses ALDH2 and have used this cell line to evaluate the role of ALDH2 in GTN bioactivation. Although the overexpression of ALDH2 resulted in significant increases in 1,2-GDN formation and total GTN biotransformation, increased expression of ALDH2 in either the cytosolic or mitochondrial fractions had no effect on GTN-induced cGMP accumulation. Furthermore, GTN-induced cGMP accumulation occurred in cells in which endogenous ALDH2 had been depleted, indicating that GTN bioactivation occurs in the absence of ALDH2. Together, these data indicate that ALDH2-mediated biotransformation is not associated with the mechanism-based biotransformation of GTN to an activator of sGC.
Acknowledgments
The authors wish to thank Mrs Diane Anderson for technical assistance. This work was supported by a grant from the Canadian Institutes of Health Research (MOP 81175).
Glossary
- 1,2-GDN
glyceryl-1,2-dinitrate
- 1,3-GDN
glyceryl-1,3-dinitrate
- ALDH
aldehyde dehydrogenase
- DEA/NO
1,1-diethyl-2-hydroxy-2-nitrosohydrazine
- GTN
glyceryl trinitrate
- LLC-PK1
porcine renal proximal tubular epithelial cell
- sGC
soluble guanylyl cyclase
Conflict of interest
None.
References
- Bennett BM, Kobus SM, Brien JF, Nakatsu K, Marks GS. Requirement for reduced, unliganded hemoprotein for the hemoglobin- and myoglobin-mediated biotransformation of glyceryl trinitrate. J Pharmacol Exp Ther. 1986;237:629–635. [PubMed] [Google Scholar]
- Bennett BM, Schroder H, Hayward LD, Waldman SA, Murad F. Effect of in vitro organic nitrate tolerance on relaxation, cyclic GMP accumulation and guanylate cyclase activation by glyceryl trinitrate and the enantiomers of isoidide dinitrite. Circ Res. 1988;63:693–701. doi: 10.1161/01.res.63.4.693. [DOI] [PubMed] [Google Scholar]
- Bennett BM, Leitman DC, Schroder H, Kawamoto JH, Nakatsu K, Murad F. Relationship between biotransformation of glyceryl trinitrate and cyclic GMP accumulation in various cultured cell lines. J Pharmacol Exp Ther. 1989;250:316–323. [PubMed] [Google Scholar]
- Bennett BM, McDonald BJ, Nigam R, Simon WC. Biotransformation of organic nitrates and vascular smooth muscle cell function. Trends Pharmacol Sci. 1994;15:245–249. doi: 10.1016/0165-6147(94)90319-0. [DOI] [PubMed] [Google Scholar]
- Beretta M, Sottler A, Schmidt K, Mayer B, Gorren ACF. Partially irreversible inactivation of mitochondrial aldehyde dehydrogenase by nitroglycerin. J Biol Chem. 2008;45:30735–30744. doi: 10.1074/jbc.M804001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beretta M, Wölkart G, Schernthaner M, Griesberger M, Neubauer R, Schmidt K, et al. Vascular bioactivation of nitroglycerin is catalyzed by cytosolic aldehyde dehydrogenase-2. Circ Res. 2012;110:385–393. doi: 10.1161/CIRCRESAHA.111.245837. [DOI] [PubMed] [Google Scholar]
- Brien JF, McLaughlin BE, Breedon TH, Bennett BM, Nakatsu K, Marks GS. Biotransformation of glyceryl trinitrate occurs concurrently with relaxation of rabbit aorta. J Pharmacol Exp Ther. 1986;237:608–614. [PubMed] [Google Scholar]
- Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhang J, Foster MW, Stamler JS. An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2005;102:12159–12164. doi: 10.1073/pnas.0503723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Tostes RC, Webb RC. Mitochondrial aldehyde dehydrogenase prevents ROS-induced vascular contraction in angiotensin-II hypertensive mice. J Am Soc Hypertens. 2011;5:154–160. doi: 10.1016/j.jash.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza Y, Dowlatshahi S, Bennett BM. Changes in aldehyde dehydrogenase 2 expression in rat blood vessels during glyceryl trinitrate tolerance development and reversal. Br J Pharmacol. 2011;164:632–643. doi: 10.1111/j.1476-5381.2011.01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiber A, Wenzel P, Oelze M, Münzel T. New insights into bioactivation of organic nitrates, nitrate tolerance and cross-tolerance. Clin Res Cardiol. 2008;97:12–20. doi: 10.1007/s00392-007-0588-7. [DOI] [PubMed] [Google Scholar]
- DiFabio J, Ji Y, Vasiliou V, Thatcher GR, Bennett BM. Role of mitochondrial aldehyde dehydrogenase in nitrate tolerance. Mol Pharmacol. 2003;64:1109–1116. doi: 10.1124/mol.64.5.1109. [DOI] [PubMed] [Google Scholar]
- Doel JJ, Godber BL, Eisenthal R, Harrison R. Reduction of organic nitrates catalysed by xanthine oxidoreductase under anaerobic conditions. Biochem Biophys Acta. 2001;1527:81–87. doi: 10.1016/s0304-4165(01)00148-9. [DOI] [PubMed] [Google Scholar]
- Endo J, Sano M, Katayama T, Hishiki T, Shinmura K, Morizane S, et al. Metabolic remodeling induced by mitochondrial aldehyde stress stimulates tolerance to oxidative stress in the heart. Circ Res. 2009;105:1118–1127. doi: 10.1161/CIRCRESAHA.109.206607. [DOI] [PubMed] [Google Scholar]
- Fadel PJ, Farias M, Gallagher KM, Wang Z, Thomas GD. Oxidative stress and enhanced sympathetic vasoconstriction in contracting muscles of nitrate-tolerant rats and humans. J Physiol. 2012;590:395–407. doi: 10.1113/jphysiol.2011.218917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung H-L, Poliszczuk R. Nitrosothiol and nitrate tolerance. Z Kardiol. 1986;75:25–27. [PubMed] [Google Scholar]
- Gao CM, Takezaki T, Wu JZ, Zhang XM, Cao HX, Ding JH, et al. Polymorphisms of alcohol dehydrogenase 2 and aldehyde dehydrogenase 2 and colorectal cancer risk in Chinese males. World J Gastroenterol. 2008;14:5078–5083. doi: 10.3748/wjg.14.5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hink U, Daiber A, Kayhan N, Trischler J, Kraatz C, Oelze M, et al. Oxidative inhibition of the mitochondrial aldehyde dehydrogenase promotes nitroglycerin tolerance in human blood vessels. J Am Coll Cardiol. 2007;50:2226–2232. doi: 10.1016/j.jacc.2007.08.031. [DOI] [PubMed] [Google Scholar]
- Hinz B, Schröder H. Vitamin C attenuates nitrate tolerance independently of its antioxidant effect. FEBS Lett. 1998;428:97–99. doi: 10.1016/s0014-5793(98)00506-7. [DOI] [PubMed] [Google Scholar]
- Ji Y, Anderson DJ, Bennett BM. Role of microsomal glutathione transferase 1 in the mechanism-based biotransformation of glyceryl trinitrate in LLC-PK1 cells. Biochem Pharmacol. 2009;77:1702–1708. doi: 10.1016/j.bcp.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Kollau A, Hofer A, Russwurm M, Koesling D, Keung WM, Schmidt K, et al. Contribution of aldehyde dehydrogenase to mitochondrial bioactivation of nitroglycerin: evidence for the activation of purified soluble guanylate cyclase through direct formation of nitric oxide. Biochem J. 2005;385:769–777. doi: 10.1042/BJ20041354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis CW, Brien JF. Inhibition of hepatic aldehyde dehydrogenases in the rat by calcium carbimide (calcium cyanamide) Can J Physiol Pharmacol. 1983;61:1025–1034. doi: 10.1139/y83-153. [DOI] [PubMed] [Google Scholar]
- McDonald BJ, Bennett BM. Cytochrome P-450 mediated biotransformation of organic nitrates. Can J Physiol Pharmacol. 1990;68:1552–1557. doi: 10.1139/y90-236. [DOI] [PubMed] [Google Scholar]
- McGuire JJ, Anderson DJ, Bennett BM. Inhibition of the biotransformation and pharmacological actions of glyceryl trinitrate by the flavoprotein inhibitor, diphenyleneiodonium sulfate. J Pharmacol Exp Ther. 1994;271:708–714. [PubMed] [Google Scholar]
- McGuire JJ, Anderson DJ, McDonald BJ, Narayanasami R, Bennett BM. Inhibition of NADPH-cytochrome P450 reductase and glyceryl trinitrate biotransformation by diphenyleneiodonium sulfate. Biochem Pharmacol. 1998;56:881–893. doi: 10.1016/s0006-2952(98)00216-0. [DOI] [PubMed] [Google Scholar]
- Mayer B, Beretta M. The enigma of nitroglycerin bioactivation and nitrate tolerance: news, views, and troubles. Br J Pharmacol. 2008;155:170–184. doi: 10.1038/bjp.2008.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigam R, Anderson DJ, Bennett BM. Isoform-specific biotransformation of glyceryl trinitrate by rat aortic glutathione S-transferases. J Pharmacol Exp Ther. 1996;279:1527–1534. [PubMed] [Google Scholar]
- Ohsawa I, Nishimaki K, Yasuda C, Kamino K, Ohta S. Deficiency in a mitochondrial aldehyde dehydrogenase increases vulnerability to oxidative stress in PC12 cells. J Neurochem. 2003;84:1110–1117. doi: 10.1046/j.1471-4159.2003.01619.x. [DOI] [PubMed] [Google Scholar]
- Ohsawa I, Nishimaki K, Murakami Y, Suzuki Y, Ishikawa M, Ohta S. Age-dependent neurodegeneration accompanying memory loss in transgenic mice defective in mitochondrial aldehyde dehydrogenase 2 activity. J Neurosci. 2008;28:6239–6249. doi: 10.1523/JNEUROSCI.4956-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage PR, de la Lande IS, Stafford I, Bennett CL, Phillipov G, Stubberdfield J, et al. Nitroglycerin tolerance in human vessels. Evidence for impaired nitroglycerin bioconversion. Circulation. 2000;102:2810–2815. doi: 10.1161/01.cir.102.23.2810. [DOI] [PubMed] [Google Scholar]
- Sakata S, Yoshihara T, Arima H, Shiraishi F, Oniki H, Takahashi-Yanaga F, et al. Differential effects of organic nitrates on arterial diameter among healthy Japanese participants with different mitochondrial aldehyde dehydrogenase 2 genotypes: randomised crossover trial. BMJ Open. 2011;1:e000133. doi: 10.1136/bmjopen-2011-000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz HK, Meier P. The role of acetaldehyde in upper digestive tract cancer in alcoholics. Transl Res. 2007;149:293–297. doi: 10.1016/j.trsl.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Steiner AL, Parker CW, Kipnis DM. Radioimmunoassay for cyclic nucleotides. J Biol Chem. 1972;247:1106–1113. [PubMed] [Google Scholar]
- Sydow K, Daiber A, Oezle M, Chen Z, August M, Wendt M, et al. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J Clin Invest. 2004;113:482–489. doi: 10.1172/JCI19267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottmar SOC, Pettersson H, Kiessling K-H. The subcellular distribution and properties of aldehyde dehydrogenases in rat liver. Biochem J. 1973;135:577–586. doi: 10.1042/bj1350577a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou PS, Page NA, Lee SG, Fung SM, Keung WM, Fung HL. Differential metabolism of organic nitrates by aldehyde dehydrogenase 1a1 and 2: substrate selectivity, enzyme inactivation, and active cysteine sites. AAPS J. 2011;13:548–555. doi: 10.1208/s12248-011-9295-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida S, Maki J, Sato K. Purification and characterization of glutathione transferases with an activity towards nitroglycerin from human aorta and heart: multiplicity of the human class mu forms. J Biol Chem. 1990;265:7150–7157. [PubMed] [Google Scholar]
- Vasiliou V, Nebert DW. Analysis and update of the human aldehyde dehydrogenase(ALDH) gene family. Hum Genomics. 2005;2:138–143. doi: 10.1186/1479-7364-2-2-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel P, Hink U, Oelze M, Schuppan S, Schaeuble K, Schildknecht S, et al. Role of reduced lipoic acid in the redox regulation of mitochondrial aldehyde dehydrogenase (ALDH-2) activity. Implications for mitochondrial oxidative stress and nitrate tolerance. J Biol Chem. 2007;282:792–799. doi: 10.1074/jbc.M606477200. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Muller J, Zurmeyer S, Schuhmacher S, Schulz E, Oelze M, et al. ALDH-2 deficiency increases cardiovascular oxidative stress-Evidence for indirect antioxidative properties. Biochem Biophys Res Commun. 2008;367:137–143. doi: 10.1016/j.bbrc.2007.12.089. [DOI] [PubMed] [Google Scholar]
- Wenzl MV, Beretta M, Gorren AC, Zeller A, Baral PK, Gruber K, et al. Role of the general base Glu-268 in nitroglycerin bioactivation and superoxide formation by aldehyde dehydrogenase-2. J Biol Chem. 2009;284:19878–19886. doi: 10.1074/jbc.M109.005652. [DOI] [PMC free article] [PubMed] [Google Scholar]