

Abstract
Background and Purpose
Evidence suggests that phosphorylation of TRPV1 is an important component underlying its aberrant activation in pathological pain states. To date, the detailed pharmacology of diverse TRPV1 receptor agonists and antagonists has yet to be reported for native TRPV1 under phosphorylating conditions. Our goal was to optimize a relatively high-throughput methodology to allow pharmacological characterization of the native TRPV1 receptor using a spinal cord neuropeptide release assay under naive and phosphorylating states.
Experimental Approach
Herein, we describe characterization of rodent TRPV1 by measurement of CGRP release from acutely isolated lumbar (L1-L6) spinal cord using a 96-well technique that combines use of native, adult tissue with quantitation of CGRP release by elisa.
Key Results
We have studied a diverse panel of TRPV1 agonists and antagonists under basal and phosphorylating conditions. We show that TRPV1-mediated CGRP release is evoked, in a temperature-dependent manner, by a PKC activator, phorbol 12,13-dibutyrate (PDBu); and that treatment with PDBu increases the potency and efficacy of known TRPV1 chemical agonists, in an agonist-specific manner. We also show that the pharmacological profile of diverse TRPV1 antagonists is dependent on whether the stimulus is PDBu or capsaicin. Of note, HPPB was identified as an antagonist of capsaicin-evoked, but a potentiator of PDBu-evoked, CGRP release.
Conclusions and Implications
Our findings indicate that both TRPV1 agonist and antagonist profiles can be differentially altered by PKC activation. These findings may offer new insights for targeting TRPV1 in pain states.
Keywords: TRPV1, CGRP release, phosphorylation, spinal cord
Introduction
Transient receptor potential vanilloid subtype 1 (TRPV1) is a Ca2+-permeable cation channel that acts as a polymodal receptor responding to noxious chemical and physical stimuli, including capsaicin, heat (>43°C) and acidic pH ≤5.9 (Caterina et al., 1997; Tominaga et al., 1998). Additionally, TRPV1 is activated by endogenous lipid mediators such as anandamide and N-arachidonyldopamine (NADA) and lipoxegenase products including 12-HPETE and leukotriene B4 (Zygmunt et al., 1999; Hwang et al., 2000; Huang et al., 2002; Huang and Walker, 2006). TRPV1 is also sensitized by protein kinases, including PKC and PKA (Premkumar and Ahern, 2000; De Petrocellis et al., 2001; Vellani et al., 2001; Bhave et al., 2002; 2003; Crandall et al., 2002; Premkumar et al., 2004).
Consistent with a role in nociception, TRPV1 is dominantly expressed in a subset of primary afferent fibres, with activation of TRPV1 eliciting release of neuropeptides (e.g. CGRP, Substance P) and neurotransmitters (e.g. glutamate) from the peripheral and central terminals (Wardle et al., 1997; Kanai et al., 2005; Schicho et al., 2005; Lappin et al., 2006; Puttfarcken et al., 2010).
Following tissue injury, there is local inflammation caused by the release of endogenous growth factors, protons and neuropeptides. Inflammatory mediators such as ATP, trypsin, bradykinin, PGE2 and nerve growth factor act at their cognate G-protein-coupled receptors/tyrosine kinase receptors to activate signalling cascades leading to generation of lipid metabolites and activation of protein kinases. Integration of these diverse signals leads to a dynamic reduction in the threshold of TRPV1 activation (Ma and Quirion, 2007; Szallasi et al., 2007). This molecular integration likely underlies the aberrant activation of TRPV1 seen under in vivo pathological injury or inflammatory states (Kanai et al., 2005; Schicho et al., 2005; Lappin et al., 2006; Puttfarcken et al., 2010). In line with this, thermal hyperalgesia under inflammatory conditions is strongly attenuated in TRPV1 knock-out mice (Caterina et al., 2000; Davis et al., 2000) and a number of highly selective TRPV1 antagonists are efficacious across multiple pre-clinical models of pain (Pomonis et al., 2003; Honore et al., 2005; Kanai et al., 2007; Lehto et al., 2008; Puttfarcken et al., 2010).
In addition to TRPV1 antagonists, TRPV1 agonists are also of significant interest for alleviating pain in humans. TRPV1 agonists, including capsaicin and resiniferatoxin are utilized clinically, or are being clinically assessed for efficacy in treating some conditions of chronic pain; however, the route of administration may limit efficacy and broad applicability (Iadarola and Mannes, 2011). For both TRPV1 agonist and antagonist approaches, adverse effects have significantly hindered clinical progression. Hyperthermia and impaired noxious heat sensation are key concerns for TRPV1 antagonist therapies, whilst systemic toxicity is a limiting issue for agonist approaches (Wong and Gavva, 2009; Trevisani and Szallasi, 2011). Recent work has focused on identifying safer ways to target TRPV1, such as stimulus-mode specific antagonists devoid of hyperthermia (Garami et al., 2010; Watabiki et al., 2010), allosteric modulators (Kaszas et al., 2012) or ligands that selective target pain state TRPV1. For the latter, evidence is accumulating that phosphorylated TRPV1 is highly relevant to pain states.
In preclinical animal models, increased phosphorylation of TRPV1 appears to be a feature common to diverse pain states. In particular, a growing body of data implicates phosphorylation by PKC/PKCε as a key step in TRPV1 sensitization under pathological conditions. PKCε is activated downstream of numerous inflammatory mediators (Cesare et al., 1999; Amadesi et al., 2006; Plant et al., 2006; Zhang et al., 2007; Pan et al., 2010) and directly phosphorylates and sensitizes TRPV1 (Numazaki et al., 2002; Mandadi et al., 2006). PKC-induced phosphorylation underlies the reduction in heat threshold required for TRPV1 activation in the presence of inflammatory mediators such as bradykinin and ATP and is also reported to reverse TRPV1 desensitization and up-regulate TRPV1 channel expression (Premkumar and Ahern, 2000; Chuang et al., 2001; Mandadi et al., 2004; Morenilla-Palao et al., 2004; Sculptoreanu et al., 2008). PKC-induced TRPV1 sensitization is associated with pain states, including diabetic neuropathy (Hong and Wiley, 2005), bladder cystitis (Sculptoreanu et al., 2005) and bone cancer (Pan et al., 2010), with altered phosphorylation and/or function reported in dorsal root ganglion from these animals. In vivo, pain behaviours induced by injection of a PKC activator are absent in TRPV1 knock-out mice (Bolcskei et al., 2005); conversely, PKCε has been shown to be a requirement for capsaicin and acid induced pain behaviours (Khasar et al., 1999; Jung et al., 2004; Srinivasan et al., 2008).
If, as these findings indicate, TRPV1 in pathological pain conditions exists in a hyper-phosphorylated state, it would be important to assess TRPV1 agonist and antagonist profiles under these conditions. Whilst there are several studies in recombinant systems on phosphorylated TRPV1 (Wang et al., 2003; El-Kouhen et al., 2005; Swanson et al., 2005; Pearce et al., 2008), the pharmacological characterization performed to date in native systems is limited (Fischer and Reeh, 2007; Sikand and Premkumar, 2007).
We have studied TRPV1 evoked CGRP release from acutely isolated rat lumbar spinal cord in response to a structurally diverse set of TRPV1 agonists and antagonists under basal and phosphorylating conditions. Phosphorylation by PKC increased the potency of known TRPV1 chemical agonists, in an agonist specific manner, in addition to significantly altering the inhibitory profile of structurally diverse TRPV1 antagonists. The data provide new insights into the pharmacology of native TRPV1 under phosphorylating conditions, which may be highly relevant to pain states.
Methods
Materials
Standard buffer components were of analytical grade and were purchased from Fisher Scientific (Loughborough, UK). Commercially available compounds were purchased from Sigma Aldrich (Gillingham, UK) or Tocris-Cookson (Bristol, UK). TRPV1 antagonists, with the exception of capsazepine, HPPB, SB366791, and AMG9810, were obtained from the chemical synthesis laboratory (Eli Lilly & Company, Indianapolis, IN). HPPB was from Asinex (Moscow, Russia). Ninety-six-well multi-screen filter plates were from Millipore Inc. (Abingdon, UK). CGRP release elisa kits were obtained from SPI BIO (Montigny le Bretonneux, France). TRPV1 knock-out mice were from Jackson Laboratories (Sacramento, CA, USA). C57BL/6 wild-type mice and Lister-Hooded rats were obtained from Charles River (Margate, UK). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). All animal procedures and experiments were conducted in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986 and conformed to institutional regulations at Eli Lilly & Company Ltd.
Methodology for 96-well CGRP release assay
Experiments were conducted in Krebs buffer composed of (in mM) 118 NaCl, 2.4 KCl, 2.4 CaCl2.2H2O, 1.2 MgSO4.7H2O, 1.2 KH2PO4, 25 NaHCO3, 10 Glucose, 0.1% DMSO (v/v) ±10 μM thiorphan (to prevent CGRP breakdown by endogenous enkephalinases). Buffer was gassed with 5% CO2/95% O2 for 1 h to pH 7.4 and was kept at 37°C using a heated water bath. For Ca2+-free experiments, CaCl2.2H2O was omitted from the buffer and 1 mM EGTA added. MgSO4·7H2O concentration was increased to a concentration of 3.6 mM in order to maintain osmolarity. Compound stocks were prepared in DMSO and then diluted to the required concentrations in Krebs buffer containing thiorphan.
Male, adult (250–350 g), Lister-Hooded rats or C57BL/6 mice (wild type or TRPV1 knock-out) were killed by exposure to a rising concentration of CO2, followed by cervical dislocation. The lumbar portion (L1–L6) of the spinal cord and vertebrae was rapidly dissected out and the whole cord (including lumbar enlargement) obtained by hydraulic extrusion. This was immediately placed into ice-cold buffer in the absence of thiorphan and weighed (180–220 mg or 20–25 mg wet weight/cord for rat or mouse, respectively) and homogenized using 20 strokes of a glass-Teflon hand homogenizer. The crude homogenate was diluted to an optimal 4 mg mL−1 (rat or mouse) wet weight with Krebs buffer in the absence of thiorphan and spun for 1 min at 1500 rpm (363× g) in a bench-top centrifuge. It was determined that tissue from one rat or eight mice was sufficient for four 96-well plates and could be diluted further with only slight loss of signal. Supernatant containing the synaptosomal preparation was carefully decanted and 100 μL (0.2–0.3 mg mL−1 protein) placed in each well of a 96-well filter plate. Use of these filter plates allowed stimulating ligands to be applied and resulting CGRP release removed without disturbance to the synaptosomes on the filters of the plate. Plates were incubated at 37°C for 30 min prior to compound addition. After the incubation period, filter plates were removed, and the buffer filtered to waste using a vacuum manifold. The contents of the basal plate were added (100 μL per well), and the filter plate was incubated for 10 min at 37°C. The contents of the filter plate were then vacuumed to waste, and the contents of the stimulation plate were added (100 μL per well) and incubated for a further 10 min at 37°C.
At the end of the 10 min stimulation period, the contents of the filter plate were transferred to the elisa immunoplate by centrifugation at 200× g. Tracer was then added, and the immunoplate was covered with plastic film and incubated at 4°C for 16–20 h. The following day the immunoplate was washed and Ellman's reagent added, and the plate was left at room temperature to develop in the dark for 45 min. Limit of detection for the elisa was approximately 3 pg mL−1 of CGRP. After the development time, the absorbance of each well was measured at 405 nm using a 96-well spectrophotometer (Molecular Devices Corporation, Sunnyvale, CA).
Data analysis
Raw absorbance values were imported into SigmaPlot v10, and the amount of CGRP released reported as a percentage of the capsaicin (0.3 μM) or 40 mM KCl control minus the basal release (the amount of CGRP released upon addition of buffer or buffer plus 10 nM PDBu). Graphs of percentage release were drawn using SigmaPlot, and curves were fitted using the four-parameter Hill equation. Where stated, n refers to the number of independent experiments performed using spinal cord tissue from different animals. Significant differences in curve-fitting parameters (EC50/IC50/% efficacy/% inhibition) were calculated in GraphPad Prism 5 (La Jolla, CA, USA) using the extra sum of squares F-test.
Assay characteristics
Statistical assessment of assay performance was performed using capsaicin as the stimulus. Plate uniformity and signal variability assessment were performed over 3 days with Z′ values (a measure of statistical effect size) ranging between 0.53 to 0.84 for replicates of four wells, meaning robust data generation was possible with a minimum number of experimental repeats. There was no evidence of significant drift or edge effects. The 96-well format enabled construction of full concentration response curves and thus comparison of pEC50/IC50 and efficacy/inhibition values for the compounds tested.
Measurement of intracellular Ca2+
Recombinant CHO stably expressing rat TRPV1 were cultured in DMEM : F-12 Ham (1:3) plus 10% (v/v) FBS, 0.02 M HEPES, penicillin/streptomycin/glutamate (10 000 units, 10 000 μg and 29.2 mg mL−1, respectively) and 1.25 μg mL−1 Geneticin G418 sulphate, and incubated in a 37°C humidified incubator with 5% CO2 in air. They were plated into black clear-bottomed 96-well plates 24 h prior to experiments, at a concentration of 40 K per well.
Receptor-mediated changes in intracellular Ca2+ concentration were determined using a Ca2+-sensitive fluorescent dye, Fluo3-AM (Invitrogen, Paisley, UK) and a fluorometric imaging plate reader with heated stage (FLIPR, Molecular Devices Corporation). All assays were performed using HBSS assay buffer, supplied by Invitrogen (Gibco 14025-050) supplemented with 10 mM HEPES and adjusted to pH 7.2. At the start of the assay, growth media was removed from cell plates by inversion and gentle tapping and replaced with assay buffer containing 10 μM Fluo-3AM/0.05% pluronic F-127. Cell plates were stored in the dark at room temperature for 60 min, to allow dye loading into cells. Subsequently, the dye solution was removed and replaced with assay buffer (at 21°C or 37°C), and cell plates were transferred to the FLIPR for assay. Intracellular Ca2+ levels were monitored before and after the addition of compounds (at 21°C or 37°C). Responses were measured as the maximal peak height in relative fluorescent units (RFUs), and the assay window was defined as the maximal response obtained by capsaicin (10 μM)-stimulated wells. All RFU values were corrected for basal fluorescence in the absence of agonist. Graphs of percentage stimulation or inhibition were drawn using SigmaPlot, and curves were fitted using the four-parameter Hill equation. Where stated, n refers to the number of independent experiments performed using cells plated on different days. Significant differences in curve-fitting parameters (EC50/IC50/% efficacy/% inhibition) were calculated in GraphPad Prism 5 using the extra sum of squares F-test.
Results
Capsaicin and PDBu-induced CGRP release are mediated by TRPV1 activation
The prototypic TRPV1 agonist capsaicin (0.001–1 μM) evoked a concentration-dependent increase in release of CGRP from adult rat spinal cord homogenate. The dose–response curve obtained for capsaicin appeared monophasic with an pEC50 of 6.99 ± 0.19 M and a maximal response at 300 nM (Figure 1A). In the absence of extracellular Ca2+, or in the presence of a TRPV1 antagonist, N-(4-t-butylphenyl)-4-(3-chloropyridin-2-yl) tetrahydropryazine-1(2H)-carboxamide (BCTC), capsaicin failed to evoke CGRP release (Figure 1A).
Figure 1.

Initial characterization of capsaicin and PDBu evoked CGRP release from rat spinal cord homogenate. (A and B) Concentration–response curves to capsaicin or PDBu were inhibited by the TRPV1 antagonist BCTC and by removal of extracellular Ca2+. (C) Responses to PDBu were inhibited by the PKC inhibitor Ro-31,3220. (D) The structural analogue of PDBu, 4α-PDD did not evoke significant CGRP release above basal. (E) Responses to PDBu were enhanced at 30°C and 37°C as compared with 21°C. Responses were calculated as a percentage of 0.3 μM capsaicin in buffer containing Ca2+. Each data point represents the mean release ± SEM from at least three separate experiments.
To assess if PKC activation had any effects on basal, or TRPV1 agonist evoked CGRP release, homogenate was exposed to an activator of PKC, phorbol 12,13-dibutyrate (PDBu), for 10 min, followed by TRPV1 agonist addition. Application of PDBu alone was able to elicit CGRP release in a concentration-dependent manner, with an pEC50 of 6.77 ± 0.07 M and a maximal efficacy near to that of a maximally effective concentration of capsaicin (94 ± 8%). PDBu-stimulated CGRP release was inhibited by the TRPV1 antagonist BCTC and by removal of extracellular Ca2+ (Figure 1B). PDBu-stimulated release was also fully blocked by a PKC inhibitor, Ro-31-8220, whilst a close analogue of PDBu, 4α-PDD, which does not activate PKC, failed to evoke CGRP release (Figure 1C,D). There was a clear temperature-dependent increase in efficacy of PDBu-evoked CGRP release, which was significantly greater at 30°C and 37°C compared with 21°C [Figure 1E; 49 ± 2% efficacy at 21°C vs. 67 ± 4% at 30°C (P < 0.01) vs. 89 ± 5% at 37°C (P < 0.05)]. Additional experiments conducted in a recombinant system gave results mirroring those obtained in the native release experiments. In CHO cells stably expressing rat TRPV1, PDBu caused a concentration dependent increase in [Ca2+]i, as measured using fluo-3 and a FLIPR, which was significantly greater at 37°C versus room temperature [65 ± 3% and 24 ± 1%, respectively (P < 0.01)], and which was fully blocked by incubation with the TRPV1 antagonist BCTC. The potencies of PDBu to evoke functional responses in the recombinant system (pEC50 = 7.03 ± 0.1 M) and of BCTC to block the response (pIC50 = 8.81 ± 0.1 M) corresponded closely to values observed in the native preparation (Figure S1).
Release of CGRP was also assessed in spinal cord tissue from wild-type and TRPV1 null-mutant mice (Figure 2). In tissue from wild-type mice, capsaicin and PDBu both evoked CGRP release in a concentration-dependent manner with pEC50 values of 7.64 ± 0.23 M and 7.66 ± 0.06 M respectively (Figure 2A). In contrast, in spinal cord tissue from TRPV1 null-mutant mice, neither PDBu nor capsaicin elicited CGRP release above the basal control (Figure 2B).
Figure 2.
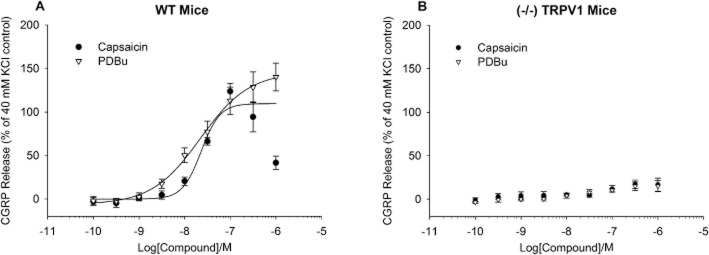
Release of CGRP from mouse spinal cord homogenate. Responses to capsaicin or PDBu were assessed using tissue from either wild-type (A) or TRPV1 knockout mice (B). Responses were calculated as a percent of the response to 40 mM KCl. Each data point represents the mean release ± SEM from three separate experiments.
Sub-maximal PKC activation enhances TRPV1 sensitivity to agonists in an agonist specific manner
To assess the effect of sub-maximal PKC activation on the pharmacological profile of a panel of TRPV1 agonists, responses to agonists were investigated under basal or phosphorylating conditions (Figure 3). In preliminary experiments, 10 nM PDBu was selected as whilst it produced only a small increase in basal CGRP release when applied alone, it significantly sensitized responses to capsaicin (pEC50 = 7.57 ± 0.14 M, P < 0.001, Figure 3A). Addition of the PKC inhibitor Ro-31-8220 prevented this PDBu-induced increase in TRPV1 agonist potency (pEC50 = 7.05 ± 0.05 M; Figure 3A).
Figure 3.

TRPV1 agonists evoke CGRP release with higher potency and efficacy after pre-incubation with PDBu. (A–H) Effect of PDBu pre-treatment (10 nM for 10 min) on agonist-evoked CGRP release from rat spinal cord homogenate. All agonists were incubated for 10 min. Pre-treatment with PDBu caused a leftward shift in the curve for most agonists, with degree of shift being agonist dependent and shown in Table 1. Agonist effects on tissue alone shown for reference. Responses to agonists in the presence of 1 μM of the TRPV1 antagonist BCTC are also shown. Each data point represents the mean release ± SEM from at least three separate experiments.
The additional TRPV1 agonists tested all evoked CGRP release in a concentration-dependent manner (Figure 3B–H) with a rank order of potency of resiniferatoxin > capsaicin > arvanil > n-arachidonoyl dopamine (NADA) > anandamide > n-oleoyldopamine (OLDA) > olvanil > 9-HODE. OLDA, anandamide, olvanil, NADA and 9-HODE appeared to be partial agonists with a rank order of efficacy of OLDA > anandamide > olvanil > NADA > 9-HODE (Table 1). Addition of the TRPV1 antagonist BCTC (1 μM) largely inhibited TRPV1 agonist evoked CGRP release to the level of the basal control (Figure 3B–H). In the presence of 10 nM PDBu, increases in potency and efficacy were seen for most TRPV1 agonists (Table 1). However, the PKC-mediated potency shift appeared to be agonist dependent, with potency shifts ranging from 1.1-fold (olvanil) to 8.8-fold (arvinal) and increases in efficacy ranging from 1.1-fold (RTX) to 2.3-fold (anandamide) (Table 1).
Table 1.
Summary of the effects of TRPV1 agonists on CGRP release from rat spinal cord homogenate in the absence and presence of the PKC activator PDBu
| Compound | Agonist alone | Agonist + 10 nM PDBu | Fold shift in EC50 | Fold increase in efficacy | ||||
|---|---|---|---|---|---|---|---|---|
| pEC50 ± SEM (M) | Efficacy (%) | n | pEC50 ± SEM (M) | Efficacy (%) | n | |||
| Capsaicin | 6.99 ± 0.19 | 98 ± 6 | 14 | 7.46 ± 0.14 | 97 ± 14 | 7 | 2.9** | 1.3 |
| RTX | 8.72 ± 0.06 | 84 ± 6 | 9 | 8.95 ± 0.13 | 100 ± 14 | 3 | 1.7 | 1.2 |
| Arvanil | 5.48 ± 0.13 | 89 ± 9 | 6–8 | 6.42 ± 0.21 | 135 ± 25 | 4 | 8.8** | 1.5* |
| OLDA | 4.29 ± 0.08 | 81 ± 14 | 5 | 5.03 ± 0.03 | 72 ± 3 | 3 | 5.5** | 0.9 |
| NADA | 5.29 ± 0.18 | 30 ± 6 | 7 | 5.52 ± 0.16 | 68 ± 12 | 4 | 1.7 | 2.3** |
| Anandamide | 4.46 ± 0.01 | 59 ± 1 | 12 | 4.74 ± 0.03 | 137 ± 5 | 3 | 1.9** | 2.3** |
| Olvanil | 4.98 ± 0.1 | 39 ± 7 | 8 | 5.00 ± 0.28 | 64 ± 24 | 4 | 1.1 | 1.6&* |
| 9-HODE | >4.5 | 37 ± 13 @ 33μM | 4 | >4.5 | 51 ± 7 @ 33μM | 3 | N/A | N/A |
| HPPB | >5 | 14 ± 1 @ 10μM | 2 | ∼5.44 | ∼115 | 2 | N/A | N/A |
P < 0.01,
P < 0.05.
Effect of PKA activation on TRPV1 function in rat spinal cord tissue
The effect of PKA activation on basal and stimulated CGRP release was examined using forskolin to stimulate cAMP production and activate PKA. Forskolin, in the presence of 100 μM IBMX to inhibit breakdown of cAMP, evoked a small increase in basal CGRP release that was highest at 100 μM (19 ± 3% of 300 nM capsaicin response) and inhibited by 1 μM BCTC (Figure 4A). Pre-incubation (10 min) of rat spinal cord homogenate with 30 μM forskolin/100 μM IBMX, followed by application of capsaicin induced a small leftward shift in the potency for capsaicin (Figure 4B; pEC50 = 7.11 ± 0.04 M vs. 7.36 ± 0.08 M (P < 0.05), in the absence and presence of forskolin respectively). Interestingly, a small increase in potency of PDBu was also seen in the presence of forskolin (Figure 4C; pEC50 = 6.71 ± 0.16 M vs. 7.06 ± 0.13 M, P = 0.051, in the absence and presence of forskolin respectively).
Figure 4.
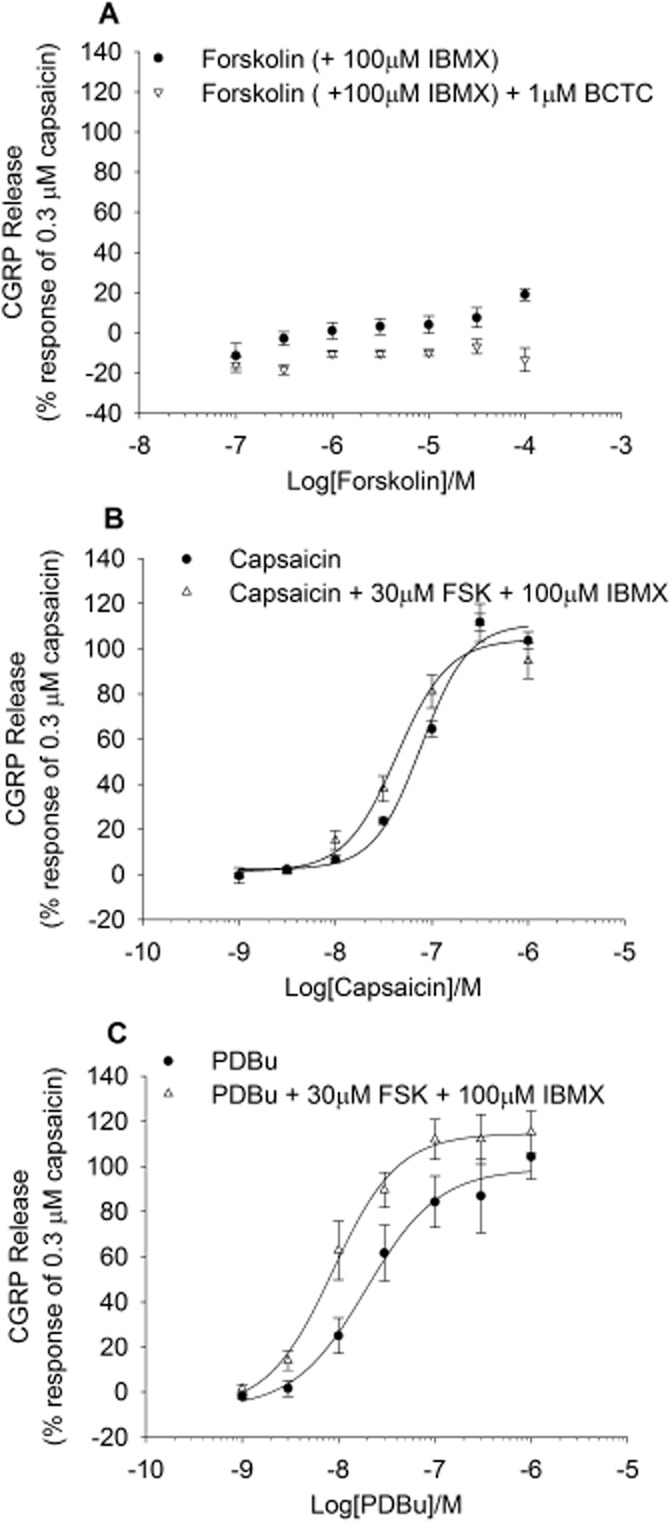
Effects of PKA activation on CGRP release. (A) Effects of the PKA activator forskolin in the presence of the PDE inhibitor IBMX in the spinal cord homogenate. IBMX alone did not evoke CGRP release (data not shown). Forskolin evoked CGRP release was inhibited by BCTC (1 μM). (B) Responses to capsaicin in the absence and in the presence of 30 μM forskolin + IBMX (100 μM). In the presence of forskolin + IBMX there is a 1.8-fold shift in the curve to capsaicin. 30 μM FSK and IBMX evoked a basal CGRP release of 12.7 ± 2.5%. Each data point represents the mean release ± SEM from at least three separate experiments. (C) Forskolin potentiation of PDBu responses – PDBu tested alone and in the presence of 30 μM forskolin and 100 μM IBMX. Data are displayed as a percentage of 0.3 μM capsaicin control minus relevant basal. Each data point represents the mean release ± SEM from at least three separate experiments.
TRPV1 antagonists can differentially block capsaicin versus PKC stimulated TRPV1
The pharmacology of rat spinal TRPV1 under basal and phosphorylating conditions was further explored using a panel of TRPV1 antagonists (Figure 7). TRPV1 antagonists were assessed for their ability to inhibit capsaicin and PDBu-evoked CGRP release (Figure 6). All antagonists tested fully inhibited capsaicin-evoked CGRP release (Table 2); however, effects on PDBu-mediated release were more divergent (Tables 3).
Figure 7.
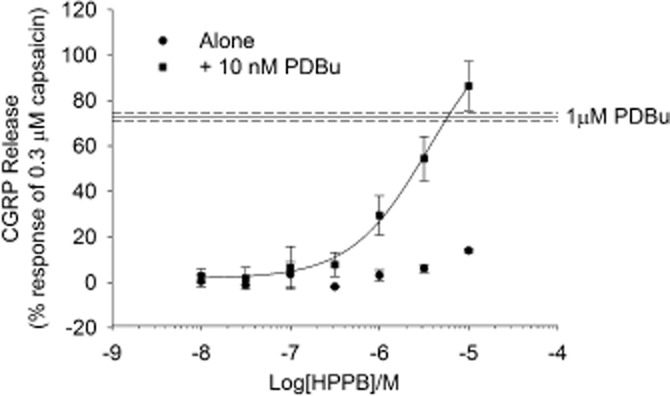
TRPV1 antagonist exhibiting an agonistic-type profile on CGRP release. Antagonist (HPPB) was applied alone or after 10 min pre-incubation with 10 nM PDBu. Data are displayed as a percentage of 0.3 μM capsaicin control minus relevant basal. Each data point represents the mean release ± SEM from two separate experiments.
Figure 6.
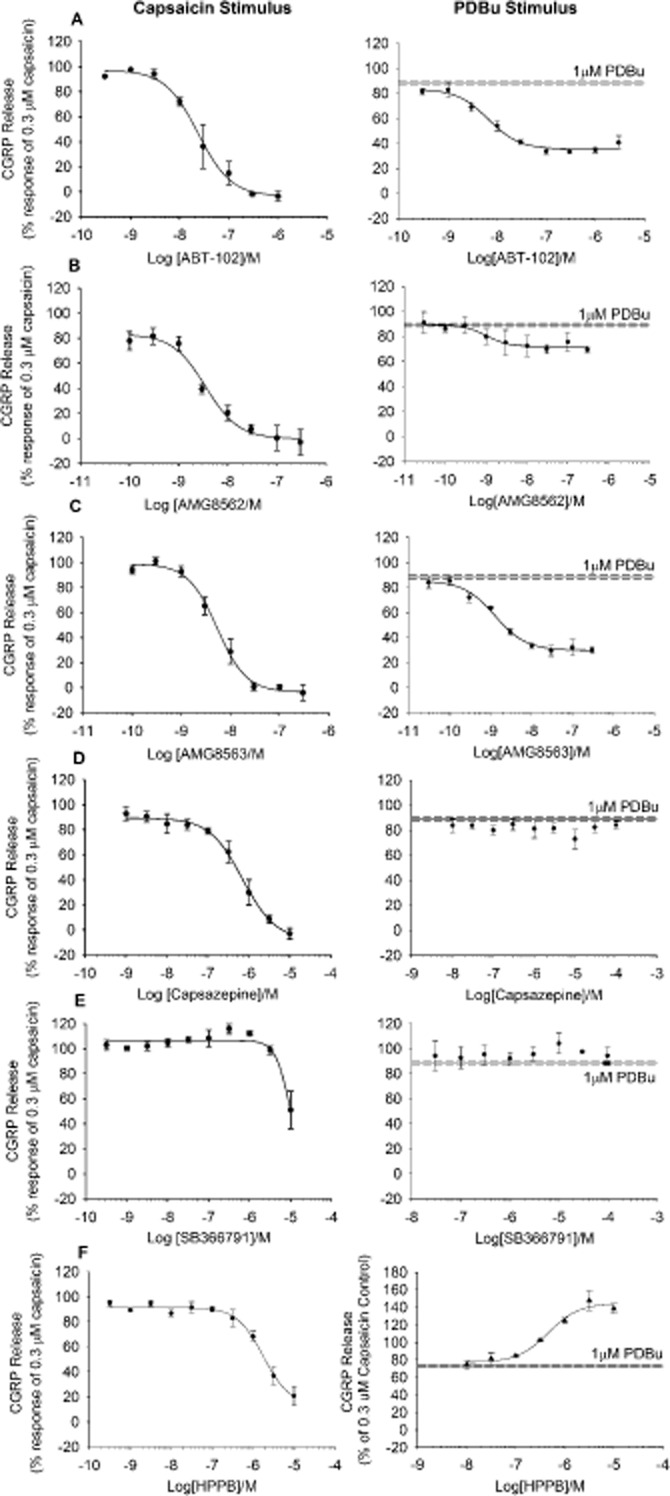
TRPV1 antagonists showing partial inhibition of the response to PDBu. Antagonists were pre-incubated for 10 min and subsequently applied together with 0.3 μM (A) Capsaicin or 1 μM PDBu (B). Data are displayed as a percentage of 0.3 μM capsaicin control minus relevant basal. Each data point represents the mean release ± SEM from at least three separate experiments.
Table 2.
Summary of the effects of TRPV1 antagonists on CGRP release elicited by maximally effective concentrations of capsaicin or PDBu
| Compound | Inhibition of 300nM capsaicin response | n | Inhibition of 1 μM PDBu response | n | Reference | ||
|---|---|---|---|---|---|---|---|
| pIC50 ± SEM (M) | Inhibition (%) | pIC50 ± SEM (M) | Inhibition (%) | ||||
| BCTC | 8.46 ± 0.09 | 102 ± 6 | 3 | 8.93 ± 0.05** | 98 ± 4 | 3 | (Valenzano et al., 2003) |
| AMG-517 | 7.33 ± 0.02 | 106 ± 1 | 3 | 7.92 ± 0.05** | 94 ± 2 | 3–4 | (Gavva et al., 2007) |
| Neurogen | 8.02 ± 0.07 | 100 ± 4 | 2 | 8.52 ± 0.1**4 | 101 ± 14 | 3–6 | (Zheng et al., 2006) |
| A425619 | 7.12 + 0.08 | 92 ± 4 | 3 | 6.63 ± 0.08* | 90 ± 4 | 3–7 | (El-Kouhen et al., 2005) |
| A784168 | 6.98 ± 0.04 | 104 ± 3 | 3 | 6.80 ± 0.08** | 95 ± 3 | 3–7 | (Cui et al., 2006) |
| AMG 49a | 6.78 ± 0.08 | 102 ± 4 | 3 | 6.55 ± 0.08* | 97 ± 3 | 3–6 | (Doherty et al., 2005) |
| SB705498 | 5.97 + 0.12 | 102 ± 9 | 3 | 5.46 ± 0.16 | 116 ± 12 | 3–6 | (Gunthorpe et al., 2007) |
| JNJ17203212 | 6.59 ± 0.09 | 98 ± 6 | 3 | 6.75 ± 0.08 | 98 ± 18 | 3 | (Swanson et al., 2005) |
| AMG-9810 | 5.89 ± 0.09 | 88 ± 5 | 3 | 6.81 ± 0.04* | 84 ± 1 | 3 | (Gavva et al., 2010) |
P < 0.01,
P < 0.05.
Table 3.
Summary of the effects of TRPV1 antagonists displaying differential effects on CGRP release elicited by maximally effective concentrations of capsaicin or PDBu
| Compound | Inhibition of 300 nM capsaicin response | Inhibition of 1 μM PDBu response | Reference | ||||
|---|---|---|---|---|---|---|---|
| pIC50 ± SEM | Inhibition (%) | n | pIC50 ± SEM | Inhibition (%) | n | ||
| ABT-102 | 7.62 ± 0.05 | 104 ± 4 | 3 | 8.18 ± 0.1** | 60 ± 2** | 2 | (Surowy et al., 2008) |
| AMG-8562 | 8.45 ± 0.08 | 100 ± 4 | 3 | 9.00 ± 0.18 | 19 ± 15** | 3 | (Lehto et al., 2008) |
| AMG-8563 | 8.27 ± 0.05 | 104 ± 3 | 3 | 8.90 ± 0.08** | 67 ± 2** | 3 | (Lehto et al., 2008) |
| Capsazepine | 6.15 ± 0.06 | 108 ± 5 | 3–6 | >4 | 0 | 3–6 | (Bevan et al., 2005) |
| SB366791 | ∼5 | ∼50% @ 10μM | 4 | >4 | 0 | 3 | (Gunthorpe et al., 2004) |
| HPPB | 5.76 ± 0.08 | 86 ± 9 | 3 | 6.34 ± 0.17 (EC50) | 162 ± 9 (Efficacy) | 2–3 | See methods |
P < 0.01, *P < 0.05.
The majority of antagonists tested (including BCTC, AMG517, A425619, A784168, AMG 49a, AMG9810, JNJ 17203212, SB705498, Neurogen) fully inhibited CGRP release evoked by 300 nM capsaicin or 1 μM PDBu with similar potency values (Figure 5; Table 2; Supporting Information, Figure S2). A correlation plot comparing the potency of all antagonists tested versus capsaicin or PDBu showed a similar rank order of potency and a strong positive correlation (R = 0.91; Supporting Information, Figure S3).
Figure 5.
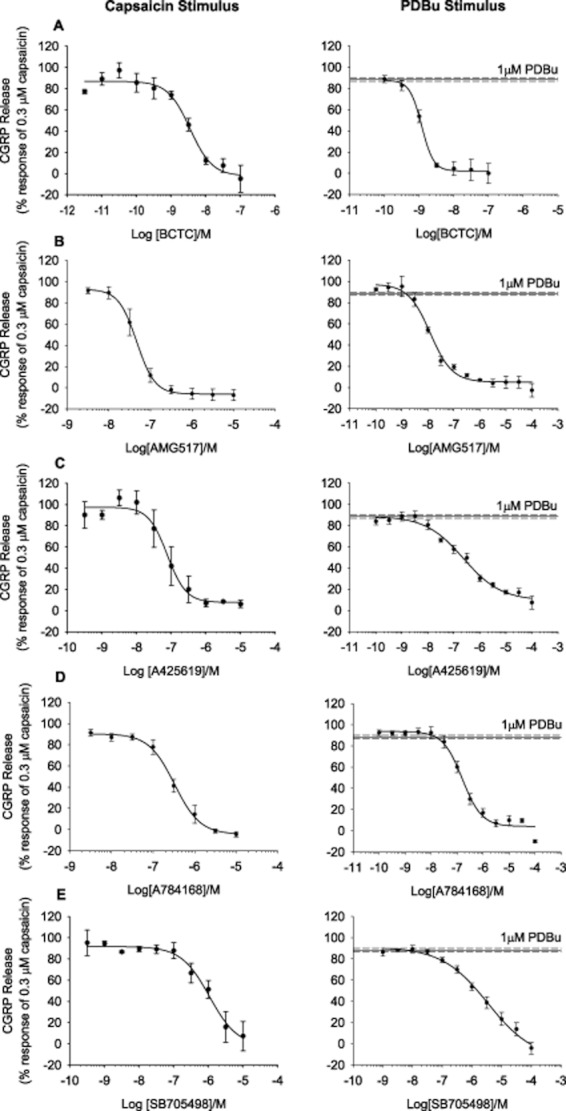
TRPV1 antagonists eliciting full inhibition of capsaicin or PDBu-evoked CGRP release. (A–E) Antagonists were pre-incubated for 10 min and subsequently applied together with 0.3 μM Capsaicin or 1 μM PDBu. Data are displayed as a percentage of 0.3 μM capsaicin control minus relevant basal with the response to 1 μM PDBu shown for comparison. Each data point represents the mean release ± SEM from at least three separate experiments.
An additional set of TRPV1 antagonists, including ABT-102, AMG8562 and AMG8563, (Figure 6A–C) partially inhibited PDBu evoked CGRP release or failed to antagonize PDBu activation of TRPV1 (Figure 6D–E; capsazepine and SB366791), despite fully inhibiting capsaicin-evoked release (Figure 6; Table 3).
Finally, one TRPV1 antagonist, N-(3-hydroxyphenyl0-4-phenyl-benzamide (HPPB), which blocked capsaicin induced CGRP release (pIC50 = 5.76 ± 0.08 M), actually increased 1 μM PDBu-evoked release (pEC50 = 6.34 ± 0.17 M) to 162% (Figure 6; Table 3). This compound was subsequently tested as an agonist of TRPV1 in the absence of PDBu (pEC50 > 5 M, 14 ± 0.8% stimulation at 10 μM) or in the presence of sub-maximal PKC stimulation (pEC50 ∼5.44 M, ∼115% stimulation at 10 μM; Figure 7).
Discussion
In the current study, we have utilized a relatively high-throughput in vitro CGRP release assay to pharmacologically characterize native rat TRPV1 receptor function. The assay is statistically robust, straightforward to perform and enables the construction of full concentration–response curves for both TRPV1 agonist and antagonists. We show that functional TRPV1 receptors are present in rat and mouse spinal cord homogenate and are directly activated by a number of endogenous and synthetic TRPV1 ligands. In addition, spinal cord TRPV1 receptors are activated and/or sensitized by conditions promoting phosphorylation by PKA and PKC. Phosphorylation by PKC increased the potency of known TRPV1 chemical agonists in an agonist-specific manner, in addition to significantly altering the inhibitory profile of structurally diverse TRPV1 antagonists. A comparison of such a wide range of TRPV1 ligands in a preparation containing native receptor–effector mechanisms coupled to release in the absence, or presence of phosphorylation has not previously been performed.
The prototypic TRPV1 agonist, capsaicin, evoked CGRP release in a Ca2+-dependent manner with a similar pEC50 to that found in CGRP release studies from spinal cord slices (Wardle et al., 1997). Responses elicited by capsaicin were fully inhibited by a TRPV1 antagonist, BCTC, and were absent in tissue from mice lacking the TRPV1 receptor, confirming that capsaicin-induced CGRP release was indeed mediated by the TRPV1 receptor. The 96-well format of the assay made it possible to directly compare the potencies and efficacies of a number of other TRPV1 agonists including olvanil, anandamide, OLDA, arvanil, NADA, 9-HODE and RTX. Application of a sub-maximal concentration of PDBu resulted in increases in potency and efficacy of these agonists at TRPV1. The degree of shift in pEC50 was agonist dependent, with arvanil showing the largest shift and RTX, NADA and 9-HODE shifting the least. RTX is structurally related to phorbol esters and has been shown to directly activate PKC in dorsal root ganglion neurones (Harvey et al., 1995); however, we do not believe that this is the case here, as pre-incubation with a PKC inhibitor, Ro 31-8220, did not affect RTX induced CGRP release (data not shown). In line with our findings, alteration of TRPV1 agonist pharmacology by phosphorylation has been reported previously in studies utilizing recombinant TRPV1. A PKC activator, phorbol myristate acid (PMA), was shown to increase the efficacy of two partial agonists, JYL827 and JYL1511 (Wang et al., 2003). Whilst a protein phosphatase inhibitor, cyclosporine A, employed to inhibit TRPV1 de-phosphorylation, led to increased potencies (1.1- to 7.8-fold) of a panel of TRPV1 agonists and increased efficacy of JYL1511 (Pearce et al., 2008).
The PKC activator PDBu, applied alone, evoked a concentration-dependent increase in CGRP release of comparable magnitude with capsaicin and was inhibited by BCTC or a PKC inhibitor, Ro 31-8220. PDBu did not elicit release above basal levels in experiments performed using tissue from mice deficient in the TRPV1 receptor. In addition, the inactive analogue 4α-PDD did not elicit significant release. PKC activation may act to sensitize CGRP synaptic release in a number of ways, including direct phosphorylation of the TRPV1 channel. As the findings in the present study replicate in a simpler recombinant system and are consistent with the findings of Premkumar et al. (2004), who reported that the sensitizing/activating effects of PDBu on recombinant TRPV1 were significantly reduced or lost in a TRPV1 double PKC phosphorylation site mutant, we hypothesize that direct phosphorylation of TRPV1 likely underlies the effects observed in the native system as well. Of note in the present study, the maximal PDBu-evoked response is lower in the recombinant (65 ± 3%) versus the native (94 ± 8 or 89 ± 5%) preparation, relative to a maximal concentration of capsaicin, perhaps suggesting a contribution from additional elements in the latter preparation.
Whilst the effects of a PKA activator, forskolin, were not as pronounced as the effects of PDBu, at high concentrations, forskolin induced BCTC-sensitive CGRP release. Moreover, when PDBu was assayed in the presence of forskolin and IBMX, there was a twofold increase in potency, suggesting a synergistic relationship between PKA and PKC. Taken together, these results imply that in CGRP-releasing terminals in the spinal cord, there is a tight relationship between PKC/PKA and TRPV1; and that phosphorylation of the receptor is sufficient to induce activation at temperatures at or below body temperature, as shown previously (Sikand and Premkumar, 2007). In line with this, aberrant TRPV1 activity has been shown to underlie the increased basal neurotransmitter release observed in spinal cord from pain state animals (Tohda et al., 2001; Kanai et al., 2005; Schicho et al., 2005; Puttfarcken et al., 2010).
By comparing the profile of a panel of literature TRPV1 antagonists to inhibit capsaicin or PDBu-induced responses, distinct categories of TRPV1 antagonist were revealed. Pharmacological characterizations of TRPV1 antagonists routinely employ chemical agonists (e.g. capsaicin), pH and heat to activate TRPV1. Utilizing these activators has led to the identification of diverse TRPV1 antagonist profiles, from TRPV1 antagonists that block all three modes of activation to antagonists that block capsaicin, but vary in their profiles against pH or heat responses. In the current study, antagonists previously reported to inhibit TRPV1 activation by capsaicin, pH and heat (Gavva et al., 2005; 2007; Gunthorpe et al., 2007; Broad et al., 2008) generally also fully inhibited phosphorylated TRPV1, with IC50 values comparable with those for inhibition of capsaicin responses (Figure 5). SB366791 appears to be an exception as it was reported to block capsaicin, heat and pH activated TRPV1 (Gunthorpe et al., 2004) but failed to inhibit phosphorylated TRPV1 in the current study, despite inhibition of capsaicin mediated responses. However, in our hands, SB366791 failed to block pH activated TRPV1 and only partially inhibited heat activated TRPV1 (Broad et al., 2008). Likewise, whilst capsazepine failed to inhibit rat TRPV1 in some reports (Bevan et al., 1992; Vyklicky et al., 1998), it fully blocked all three activation modes at rat TRPV1 in others (Broad et al., 2008). In the present study, capsazepine failed to reduce phosphorylated TRPV1. Another anomaly was seen with A425619, which gave comparable potency and extent of block of capsaicin and phosphorylation activated TRPV1, but is reported as a more potent and/or efficacious blocker of capsaicin versus pH (Broad et al., 2008; Wong and Gavva, 2009), but see also El-Kouhen et al., 2005. Finally, AMG 8562, AMG 8563 and ABT-102, partially inhibited PDBu-evoked CGRP release, despite full inhibition of capsaicin responses. In line with this, previous data indicate that these ligands all block capsaicin-induced responses in recombinant systems; however, their profile versus heat and pH varies from full block through to potentiation (Lehto et al., 2008; Surowy et al., 2008). The incomplete overlap of antagonist pharmacology for phosphorylation activated TRPV1 compared with other activation modes suggests phosphorylation may promote a distinct state of activation, although further work would be required to confirm this.
The most striking finding was the contrasting profile of HPPB, which inhibited capsaicin-evoked CGRP release, but potentiated phosphorylation-evoked CGRP release. When assessed as an agonist of TRPV1 in its naïve state, there was little discernable agonism, but in the presence of sub-maximal PKC stimulation, the efficacy of release was increased 8-fold. In contrast, other full and partial agonists studied showed only a 0.9- to 2.3-fold increase in efficacy, respectively, in the presence of sub-maximal PKC stimulation. We have previously reported a similar finding with the CB2 agonist GW405833 under naive and phosphorylating conditions (Schuelert et al., 2010). Underlying mechanisms may include an increase in the sensitivity and maximal response of the receptor, activation of silent channels or recruitment of new channels to the plasma membrane as well as changes in channel desensitization (Premkumar et al., 2004; Sculptoreanu et al., 2008; Studer and McNaughton, 2010).
In summary, the results presented herein demonstrate that this assay is a valuable tool in the analysis of both agonist and antagonist responses at native TRPV1 under naive or phosphorylating conditions, employed to simulate a pain state. The data suggest that in addition to capsaicin, heat and pH, a distinct mode of TRPV1 activation, phosphorylation, may exist. Profiling of phosphorylated TRPV1 may be important to include when designing TRPV1 ligands, as it may provide further insights into differential in vivo efficacy and adverse profiles. Additionally, by identification of ligands with agonist properties differentially modulated by, or apparent only, in a context-dependent manner, the possibility of selectively targeting pathological state TRPV1 moves closer.
Acknowledgments
We thank Roger Moore for statistical analysis of CGRP release data and Dr Emanuele Sher for helpful discussions and comment.
Glossary
- BCTC
N-(4-t-butylphenyl)-4-(3-chloropyridin-2-yl) tetrahydropryazine-1(2H)-carboxamide
- CGRP
calcitonin-gene-related peptide
- HPPB
N-(3-hydroxyphenyl0-4-phenyl-benzamide
- NADA
N-arachidonoyl dopamine
- OLDA
N-oleoyldopamine
- PDBu
phorbol 12,13-dibutyrate
- RTX
resiniferatoxin
- TRPV1
transient receptor potential vanilloid subtype 1
Conflict of interest
The authors declare no conflicts of interest. Eli Lilly does not sell any of the drugs or devices mentioned in this article.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 (A) Comparison of PDBu-evoked Ca2+ influx responses at 21°C or 37°C in CHO cells transfected with the rTRPV1 receptor, recorded at either 21°C (•) or 37°C (r) (B) Inhibition of responses to 1μM PDBu with BCTC.
Data are displayed as a percentage of the 10μM capsaicin control response minus the response to buffer alone. Each data point represents the mean response ± SEM from 2–5 separate experiments.
Figure S2 TRPV1 antagonists eliciting full inhibition of capsaicin or PDBu-evoked CGRP release. Antagonists were pre-incubated for 10 minutes and subsequently applied together with 0.3 μM Capsaicin or 1 μM PDBu. Data are displayed as a percentage of 0.3 μM capsaicin control minus relevant basal with the response to 1 μM PDBu shown for comparison. Each data point represents the mean release ± SEM from at least 3 separate experiments.
Figure S3 Correlation plot comparing potency of antagonists eliciting full inhibition of capsaicin and PDBu-evoked CGRP release.
References
- Amadesi S, Cottrell GS, Divino L, Chapman K, Grady EF, Bautista F, et al. Protease-activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent mechanisms in rats and mice. J Physiol. 2006;575:555–571. doi: 10.1113/jphysiol.2006.111534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan S, Hothi S, Hughes G, James IF, Rang HP, Shah K, et al. Capsazepine: a competitive antagonist of the sensory neurone excitant capsaicin. Br J Pharmacol. 1992;107:544–552. doi: 10.1111/j.1476-5381.1992.tb12781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW. cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721–731. doi: 10.1016/s0896-6273(02)00802-4. [DOI] [PubMed] [Google Scholar]
- Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, et al. Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1) Proc Natl Acad Sci U S A. 2003;100:12480–12485. doi: 10.1073/pnas.2032100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolcskei K, Helyes Z, Szabo A, Sandor K, Elekes K, Nemeth J, et al. Investigation of the role of TRPV1 receptors in acute and chronic nociceptive processes using gene-deficient mice. Pain. 2005;117:368–376. doi: 10.1016/j.pain.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Broad LM, Keding SJ, Blanco MJ. Recent progress in the development of selective TRPV1 antagonists for pain. Curr Top Med Chem. 2008;8:1431–1441. doi: 10.2174/156802608786264254. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- Crandall M, Kwash J, Yu W, White G. Activation of protein kinase C sensitizes human VR1 to capsaicin and to moderate decreases in pH at physiological temperatures in Xenopus oocytes. Pain. 2002;98:109–117. doi: 10.1016/s0304-3959(02)00034-9. [DOI] [PubMed] [Google Scholar]
- Cui M, Honore P, Zhong C, Gauvin D, Mikusa J, Hernandez G, et al. TRPV1 receptors in the CNS play a key role in broad-spectrum analgesia of TRPV1 antagonists. J Neurosci. 2006;26:9385–9393. doi: 10.1523/JNEUROSCI.1246-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Harrison S, Bisogno T, Tognetto M, Brandi I, Smith GD, et al. The vanilloid receptor (VR1)-mediated effects of anandamide are potently enhanced by the cAMP-dependent protein kinase. J Neurochem. 2001;77:1660–1663. doi: 10.1046/j.1471-4159.2001.00406.x. [DOI] [PubMed] [Google Scholar]
- Doherty EM, Fotsch C, Bo Y, Chakrabarti PP, Chen N, Gavva N, et al. Discovery of potent, orally available vanilloid receptor-1 antagonists. Structure-activity relationship of N-aryl cinnamides. J Med Chem. 2005;48:71–90. doi: 10.1021/jm049485i. [DOI] [PubMed] [Google Scholar]
- El-Kouhen R, Surowy CS, Bianchi BR, Neelands TR, McDonald HA, Niforatos W, et al. A-425619 [1-isoquinolin-5-yl-3-(4-trifluoromethyl-benzyl)-urea], a novel and selective transient receptor potential type V1 receptor antagonist, blocks channel activation by vanilloids, heat, and acid. J Pharmacol Exp Ther. 2005;314:400–409. doi: 10.1124/jpet.105.084103. [DOI] [PubMed] [Google Scholar]
- Fischer MJ, Reeh PW. Sensitization to heat through G-protein-coupled receptor pathways in the isolated sciatic mouse nerve. Eur J Neurosci. 2007;25:3570–3575. doi: 10.1111/j.1460-9568.2007.05582.x. [DOI] [PubMed] [Google Scholar]
- Garami A, Shimansky YP, Pakai E, Oliveira DL, Gavva NR, Romanovsky AA. Contributions of different modes of TRPV1 activation to TRPV1 antagonist-induced hyperthermia. J Neurosci. 2010;30:1435–1440. doi: 10.1523/JNEUROSCI.5150-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang TJ, Immke D, et al. AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther. 2005;313:474–484. doi: 10.1124/jpet.104.079855. [DOI] [PubMed] [Google Scholar]
- Gavva NR, Bannon AW, Hovland DN, Jr, Lehto SG, Klionsky L, Surapaneni S, et al. Repeated administration of vanilloid receptor TRPV1 antagonists attenuates hyperthermia elicited by TRPV1 blockade. J Pharmacol Exp Ther. 2007;323:128–137. doi: 10.1124/jpet.107.125674. [DOI] [PubMed] [Google Scholar]
- Gunthorpe MJ, Hannan SL, Smart D, Jerman JC, Arpino S, Smith GD, et al. Characterization of SB-705498, a potent and selective vanilloid receptor-1 (VR1/TRPV1) antagonist that inhibits the capsaicin-, acid-, and heat-mediated activation of the receptor. J Pharmacol Exp Ther. 2007;321:1183–1192. doi: 10.1124/jpet.106.116657. [DOI] [PubMed] [Google Scholar]
- Gunthorpe MJ, Rami HK, Jerman JC, Smart D, Gill CH, Soffin EM, et al. Identification and characterisation of SB-366791, a potent and selective vanilloid receptor (VR1/TRPV1) antagonist. Neuropharmacol. 2004;46:133–149. doi: 10.1016/s0028-3908(03)00305-8. [DOI] [PubMed] [Google Scholar]
- Harvey JS, Davis C, James IF, Burgess GM. Activation of protein kinase C by the capsaicin analogue resiniferatoxin in sensory neurones. J Neurochem. 1995;65:1309–1317. doi: 10.1046/j.1471-4159.1995.65031309.x. [DOI] [PubMed] [Google Scholar]
- Hong S, Wiley JW. Early painful diabetic neuropathy is associated with differential changes in the expression and function of vanilloid receptor 1. J Biol Chem. 2005;280:618–627. doi: 10.1074/jbc.M408500200. [DOI] [PubMed] [Google Scholar]
- Honore P, Wismer CT, Mikusa J, Zhu CZ, Zhong C, Gauvin DM, et al. A-425619 [1-isoquinolin-5-yl-3-(4-trifluoromethyl-benzyl)-urea], a novel transient receptor potential type V1 receptor antagonist, relieves pathophysiological pain associated with inflammation and tissue injury in rats. J Pharmacol Exp Ther. 2005;314:410–421. doi: 10.1124/jpet.105.083915. [DOI] [PubMed] [Google Scholar]
- Huang SM, Walker JM. Enhancement of spontaneous and heat-evoked activity in spinal nociceptive neurons by the endovanilloid/endocannabinoid N-arachidonoyldopamine (NADA) J Neurophysiol. 2006;95:1207–1212. doi: 10.1152/jn.00395.2005. [DOI] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De PL, Fezza F, et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci U S A. 2002;99:8400–8405. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, et al. Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci U S A. 2000;97:6155–6160. doi: 10.1073/pnas.97.11.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadarola MJ, Mannes AJ. The Vanilloid Agonist Resiniferatoxin for Interventional-Based Pain Control. Curr Top Med Chem. 2011;11:2171–2179. doi: 10.2174/156802611796904942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Shin JS, Lee SY, Hwang SW, Koo J, Cho H, et al. Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin-dependent kinase II regulates its vanilloid binding. J Biol Chem. 2004;279:7048–7054. doi: 10.1074/jbc.M311448200. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Nakazato E, Fujiuchi A, Hara T, Imai A. Involvement of an increased spinal TRPV1 sensitization through its up-regulation in mechanical allodynia of CCI rats. Neuropharmacology. 2005;49:977–984. doi: 10.1016/j.neuropharm.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Hara T, Imai A, Sakakibara A. Differential involvement of TRPV1 receptors at the central and peripheral nerves in CFA-induced mechanical and thermal hyperalgesia. J Pharm Pharmacol. 2007;59:733–738. doi: 10.1211/jpp.59.5.0015. [DOI] [PubMed] [Google Scholar]
- Kaszas K, Keller JM, Coddou C, Mishra SK, Hoon MA, Stojilkovic S, et al. Small Molecule Positive Allosteric Modulation of TRPV1 Activation by Vanilloids and Acidic pH. J Pharmacol Exp Ther. 2012;340:152–160. doi: 10.1124/jpet.111.183053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, et al. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PubMed] [Google Scholar]
- Lappin SC, Randall AD, Gunthorpe MJ, Morisset V. TRPV1 antagonist, SB-366791, inhibits glutamatergic synaptic transmission in rat spinal dorsal horn following peripheral inflammation. Eur J Pharmacol. 2006;540:73–81. doi: 10.1016/j.ejphar.2006.04.046. [DOI] [PubMed] [Google Scholar]
- Lehto SG, Tamir R, Deng H, Klionsky L, Kuang R, Le A, et al. Antihyperalgesic effects of (R,E)-N-(2-hydroxy-2,3-dihydro-1H-inden-4-yl)-3-(2-(piperidin-1-yl)-4-(tri fluoromethyl)phenyl)-acrylamide (AMG8562), a novel transient receptor potential vanilloid type 1 modulator that does not cause hyperthermia in rats. J Pharmacol Exp Ther. 2008;326:218–229. doi: 10.1124/jpet.107.132233. [DOI] [PubMed] [Google Scholar]
- Ma W, Quirion R. Inflammatory mediators modulating the transient receptor potential vanilloid 1 receptor: therapeutic targets to treat inflammatory and neuropathic pain. Expert Opin Ther Targets. 2007;11:307–320. doi: 10.1517/14728222.11.3.307. [DOI] [PubMed] [Google Scholar]
- Mandadi S, Numazaki M, Tominaga M, Bhat MB, Armati PJ, Roufogalis BD. Activation of protein kinase C reverses capsaicin-induced calcium-dependent desensitization of TRPV1 ion channels. Cell Calcium. 2004;35:471–478. doi: 10.1016/j.ceca.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Mandadi S, Tominaga T, Numazaki M, Murayama N, Saito N, Armati PJ, et al. Increased sensitivity of desensitized TRPV1 by PMA occurs through PKCepsilon-mediated phosphorylation at S800. Pain. 2006;123:106–116. doi: 10.1016/j.pain.2006.02.016. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reportingexperiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morenilla-Palao C, Planells-Cases R, Garcia-Sanz N, Ferrer-Montiel A. Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J Biol Chem. 2004;279:25665–25672. doi: 10.1074/jbc.M311515200. [DOI] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Toyooka H, Tominaga M. Direct phosphorylation of capsaicin receptor VR1 by protein kinase Cepsilon and identification of two target serine residues. J Biol Chem. 2002;277:13375–13378. doi: 10.1074/jbc.C200104200. [DOI] [PubMed] [Google Scholar]
- Pan HL, Zhang YQ, Zhao ZQ. Involvement of lysophosphatidic acid in bone cancer pain by potentiation of TRPV1 via PKCepsilon pathway in dorsal root ganglion neurons. Mol Pain. 2010;6:85. doi: 10.1186/1744-8069-6-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LV, Toth A, Ryu H, Kang DW, Choi HK, Jin MK, et al. Differential modulation of agonist and antagonist structure activity relations for rat TRPV1 by cyclosporin A and other protein phosphatase inhibitors. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:149–157. doi: 10.1007/s00210-007-0258-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant TD, Zollner C, Mousa SA, Oksche A. Endothelin-1 potentiates capsaicin-induced TRPV1 currents via the endothelin A receptor. Exp Biol Med (Maywood) 2006;231:1161–1164. [PubMed] [Google Scholar]
- Pomonis JD, Harrison JE, Mark L, Bristol DR, Valenzano KJ, Walker K. N-(4-Tertiarybutylphenyl)-4-(3-cholorphyridin-2-yl)tetrahydropyrazine -1(2H)-carbox-amide (BCTC), a novel, orally effective vanilloid receptor 1 antagonist with analgesic properties: II. in vivo characterization in rat models of inflammatory and neuropathic pain. J Pharmacol Exp Ther. 2003;306:387–393. doi: 10.1124/jpet.102.046268. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C. Nature. 2000;408:985–990. doi: 10.1038/35050121. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Qi ZH, Van Buren J, Raisinghani M. Enhancement of potency and efficacy of NADA by PKC-mediated phosphorylation of vanilloid receptor. J Neurophysiol. 2004;91:1442–1449. doi: 10.1152/jn.00745.2003. [DOI] [PubMed] [Google Scholar]
- Puttfarcken PS, Han P, Joshi SK, Neelands TR, Gauvin DM, Baker SJ, et al. A-995662 [(R)-8-(4-methyl-5-(4-(trifluoromethyl)phenyl)oxazol-2-ylamino)-1,2,3,4-te trahydronaphthalen-2-ol], a novel, selective TRPV1 receptor antagonist, reduces spinal release of glutamate and CGRP in a rat knee joint pain model. Pain. 2010;150:319–326. doi: 10.1016/j.pain.2010.05.015. [DOI] [PubMed] [Google Scholar]
- Schicho R, Donnerer J, Liebmann I, Lippe IT. Nociceptive transmitter release in the dorsal spinal cord by capsaicin-sensitive fibers after noxious gastric stimulation. Brain Res. 2005;1039:108–115. doi: 10.1016/j.brainres.2005.01.050. [DOI] [PubMed] [Google Scholar]
- Schuelert N, Zhang C, Mogg AJ, Broad LM, Hepburn DL, Nisenbaum ES, et al. Paradoxical effects of the cannabinoid CB2 receptor agonist GW405833 on rat osteoarthritic knee joint pain. Osteoarthritis Cartilage. 2010;18:1536–1543. doi: 10.1016/j.joca.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Sculptoreanu A, de Groat WC, Buffington CA, Birder LA. Protein kinase C contributes to abnormal capsaicin responses in DRG neurons from cats with feline interstitial cystitis. Neurosci Lett. 2005;381:42–46. doi: 10.1016/j.neulet.2005.01.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sculptoreanu A, Aura KF, de Groat WC. Neurokinin 2 receptor-mediated activation of protein kinase C modulates capsaicin responses in DRG neurons from adult rats. Eur J Neurosci. 2008;27:3171–3181. doi: 10.1111/j.1460-9568.2008.06267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikand P, Premkumar LS. Potentiation of glutamatergic synaptic transmission by protein kinase C-mediated sensitization of TRPV1 at the first sensory synapse. J Physiol. 2007;581:631–647. doi: 10.1113/jphysiol.2006.118620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Wolfe D, Goss J, Watkins S, de Groat WC, Sculptoreanu A, et al. Protein kinase C epsilon contributes to basal and sensitizing responses of TRPV1 to capsaicin in rat dorsal root ganglion neurons. Eur J Neurosci. 2008;28:1241–1254. doi: 10.1111/j.1460-9568.2008.06438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer M, McNaughton PA. Modulation of single-channel properties of TRPV1 by phosphorylation. J Physiol. 2010;588:3743–3756. doi: 10.1113/jphysiol.2010.190611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surowy CS, Neelands TR, Bianchi BR, McGaraughty S, El KR, Han P, et al. R)-(5-tert-butyl-2,3-dihydro-1H-inden-1-yl)-3-(1H-indazol-4-yl)-urea (ABT-102) blocks polymodal activation of transient receptor potential vanilloid 1 receptors in vitro and heat-evoked firing of spinal dorsal horn neurons in vivo. J Pharmacol Exp Ther. 2008;326:879–888. doi: 10.1124/jpet.108.138511. [DOI] [PubMed] [Google Scholar]
- Swanson DM, Dubin AE, Shah C, Nasser N, Chang L, Dax SL, et al. Identification and biological evaluation of 4-(3-trifluoromethylpyridin-2-yl)piperazine-1-carboxylic acid (5-trifluoromethylpyridin-2-yl)amide, a high affinity TRPV1 (VR1) vanilloid receptor antagonist. J Med Chem. 2005;48:1857–1872. doi: 10.1021/jm0495071. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Cortright DN, Blum CA, Eid SR. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat Rev Drug Discov. 2007;6:357–372. doi: 10.1038/nrd2280. [DOI] [PubMed] [Google Scholar]
- Tohda C, Sasaki M, Konemura T, Sasamura T, Itoh M, Kuraishi Y. Axonal transport of VR1 capsaicin receptor mRNA in primary afferents and its participation in inflammation-induced increase in capsaicin sensitivity. J Neurochem. 2001;76:1628–1635. doi: 10.1046/j.1471-4159.2001.00193.x. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, et al. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Trevisani M, Szallasi A. Targeting TRPV1 for pain relief: should we quench or reignite the fire? J Pain Man. 2011;4:229–247. [Google Scholar]
- Valenzano KJ, Grant ER, Wu G, Hachicha M, Schmid L, Tafesse L, et al. N-(4-tertiarybutylphenyl)-4-(3-chloropyridin-2-yl) tetrahydropyrazine-1(2H)-carbox-amide (BCTC), a novel, orally effective vanilloid receptor 1 antagonist with analgesic properties: I. in vitro characterization and pharmacokinetic properties. J Pharm Exp Ther. 2003;306:377–386. doi: 10.1124/jpet.102.045674. [DOI] [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol. 2001;534:813–825. doi: 10.1111/j.1469-7793.2001.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyklicky L, Knotkova-Urbancova H, Vitaskova Z, Vlachova V, Kress M, Reeh PW. Inflammatory mediators at acidic pH activate capsaicin receptors in cultured sensory neurons from newborn rats. J Neurophysiol. 1998;79:670–676. doi: 10.1152/jn.1998.79.2.670. [DOI] [PubMed] [Google Scholar]
- Wang Y, Toth A, Tran R, Szabo T, Welter JD, Blumberg PM, et al. High-affinity partial agonists of the vanilloid receptor. Mol Pharmacol. 2003;64:325–333. doi: 10.1124/mol.64.2.325. [DOI] [PubMed] [Google Scholar]
- Wardle KA, Ranson J, Sanger GJ. Pharmacological characterization of the vanilloid receptor in the rat dorsal spinal cord. Br J Pharmacol. 1997;121:1012–1016. doi: 10.1038/sj.bjp.0701199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabiki T, Kiso T, Kuramochi T, Yonezawa K, Tsuji N, Kohara A, et al. Amelioration of neuropathic pain by novel TRPV1 antagonist (R)-N-(1-methyl-2-oxo-1,2,3,4-tetrahydro-7-quinolyl)-2-[(2-methylpyrrolidin-1-yl)methyl]biphenyl-4-carboxamide (AS1928370) in rats without hyperthermic effect. J Pharmacol Exp Ther. 2010;336:743–750. doi: 10.1124/jpet.110.175570. [DOI] [PubMed] [Google Scholar]
- Wong GY, Gavva NR. Therapeutic potential of vanilloid receptor TRPV1 agonists and antagonists as analgesics: recent advances and setbacks. Brain Res Rev. 2009;60:267–277. doi: 10.1016/j.brainresrev.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cang CL, Kawasaki Y, Liang LL, Zhang YQ, Ji RR, et al. Neurokinin-1 receptor enhances TRPV1 activity in primary sensory neurons via PKCepsilon: a novel pathway for heat hyperalgesia. J Neurosci. 2007;27:12067–12077. doi: 10.1523/JNEUROSCI.0496-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Hodgetts KJ, Brielmann H, Hutchison A, Burkamp F, Brian JA, et al. From arylureas to biarylamides to aminoquinazolines: discovery of a novel, potent TRPV1 antagonist. Bioorg Med Chem Lett. 2006;16:5217–5221. doi: 10.1016/j.bmcl.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di MV, et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.