

Abstract
Background & Aims
Dysregulated glucose homeostasis and lipid accumulation characterize non-alcoholic fatty liver disease (NAFLD), but underlying mechanisms are obscure. We report here that Krüppel-like factor 6 (KLF6), a ubiquitous transcription factor that promotes adipocyte differentiation, also provokes the metabolic abnormalities of NAFLD by post-transcriptionally activating PPARα-signaling.
Methods
Mice with either hepatocyte-specific depletion of KLF6 (‘DeltaHepKlf6’) or global KLF6 heterozygosity (Klf6 +/−) were fed a high fat diet (HFD) or chow for 8 or 16 weeks. Glucose and insulin tolerance tests were performed to assess insulin sensitivity. Overexpression and knockdown of KLF6 in cultured cells enabled the elucidation of underlying mechanisms. In liver samples from a cohort of 28 NAFLD patients, the expression of KLF6-related target genes was quantified.
Results
Mice with global- or hepatocyte-depletion of KLF6 have reduced body fat content and improved glucose and insulin tolerance, and are protected from HFD-induced steatosis. In hepatocytes, KLF6 deficiency reduces PPARα-regulated genes (Trb3, Pepck) with diminished PPARα-protein but no change in Pparα-mRNA which is explained by the discovery that KLF6 represses miRNA 10b, which leads to induction of PPARα. In NAFLD-patients with advanced disease and inflammation, the expression of miRNA 10b is significantly down-regulated, while PEPCK mRNA is up-regulated; KLF6 mRNA expression also correlates with TRB3 as well as PEPCK gene expression.
Conclusions
KLF6 increases PPAR -activity, whereas KLF6 loss leads to PPARα repression and attenuation of lipid and glucose abnormalities associated with a high fat diet. The findings establish KLF6 as a novel regulator of hepatic glucose and lipid metabolism in fatty liver.
Keywords: KLF6, PPARα, TRB3, PEPCK, CD36, miRNA-10b, NAFLD
Introduction
Non-alcoholic fatty liver disease (NAFLD) prevalence in the US and Europe is rapidly rising. NAFLD is characterized by hepatic steatosis, which can progress to non-alcoholic steatohepatitis (NASH). Subsequent progression from NASH to cirrhosis may precipitate liver failure and hepatocellular carcinoma (HCC) [1]. The mechanisms underlying the pathogenesis of NAFLD and its progression to NASH are poorly understood.
Krüppel-like factor 6 (KLF6) is a ubiquitously expressed, multifunctional transcription factor and tumor suppressor gene with emerging roles in glucose and lipid homeostasis [2–4]. KLF6 promotes adipogenesis in preadipocytes and fibroblasts, and interferes with peroxisome proliferator activated receptor γ (PPARγ) signaling [5, 6]. A functional polymorphism in the KLF6 gene that increases the generation of antagonistic KLF6 alternative splice forms protects patients from advanced NAFLD [7]. KLF6 directly transactivates hepatic glucokinase, and modulates hepatic insulin sensitivity in NAFLD [8].
Insulin resistance (IR) is a sentinel feature of the metabolic syndrome and contributes to NASH progression. IR is regulated in part by the actions of peroxisome proliferator activated receptor alpha (PPARα), a xenobiotic and lipid sensor that regulates hepatic steatosis, lipoprotein synthesis, and hepatic gluconeogenesis. PPARα is activated by excess fatty acids (FA), thereby inducing expression of downstream genes that metabolize FA [9], including fatty acid transport proteins [10].
Effects of PPARα activation on IR are multifaceted and incompletely understood. While fibrates, which act as PPARα ligands, improve IR, the PPARα co-activator peroxisome proliferator-activated receptor gamma co-activator 1-α (PGC1α) has a paradoxical effect on IR, since PGC1α knockout mice are protected from high fat diet-induced steatosis and IR [11]. A similar observation was made for farnesoid X receptor (FXR), which also induces PPARα signaling, but FXR agonists increase hepatic gluconeogenesis via PPARα-mediated phosphoenolpyruvate carboxykinase (PEPCK) upregulation and therefore worsen hepatic IR [12]. In contrast, micro-RNA 10b (miRNA 10b) opposes the actions of PPARα signaling by repressing PPARα protein expression [13, 14]. Down-regulation of the PPARα target tribbles-homologue 3 (TRB3) improves IR via increased AKT phosphorylation and PPARγ activation [15, 16].
Here, we describe a functional link between KLF6 expression and hepatic PPARα signaling, in part via miRNA 10b repression. We have uncovered this link by characterizing two mouse models, one with global KLF6 heterozygosity, the second with hepatocyte KLF6 deficiency, as well as by quantifying KLF6 mRNA in a cohort of 28 NAFLD patients. Together these data reveal a regulatory role of KLF6 in insulin signaling and lipid metabolism.
Experimental procedures
Mouse models
Mice with a floxed KLF6 targeting vector (C57BL/6;129Sv, Genentech, San Francisco, CA [17]) were crossed with mice expressing Cre under control of the albumin promoter (B6.Cg-Tg(Alb-cre)21Mgn/J; Jackson Labs, Bar Harbor, Maine). After further backcrossing, male offspring, expressing Cre with two floxed KLF6 alleles was used as the experimental group, while mice with two floxed alleles and no Cre expression were used as controls as previously described [8, 18]. Male Klf6+/− mice (C57BL6) were compared to wildtype littermates as previously described [19]. All mice were maintained in temperature- and humidity-controlled conditions with a 12:12 h light-dark cycle and were allowed food and water ad libitum. Mice were either fed with lab standard chow diet (PicoLab® rodent diet 20, LabDiet, Richmond, Indiana) or an adjusted-calorie high fat diet containing 45% of calories from fat (Teklad TD06415, Harlan, South Easton, MA) for eight or sixteen weeks starting at three weeks of age. Mice were euthanized at the end of the feeding period after an overnight fast. All experiments were approved by the Institutional Animal Care and Use Committee (IACUC).
NAFLD liver tissues
Liver biopsy tissues were collected from 28 patients with NAFLD. Collection was performed in accordance with ethical requirements of the local institution. Histology was evaluated according to modified Brunt criteria [20]. Ethanol consumption, viral or autoimmune liver diseases were exclusion criteria. Messenger-RNA was isolated from human liver tissue as previously described [8].
Glucose tolerance and insulin tolerance tests
For glucose tolerance tests (GTT), mice were fasted overnight and blood glucose levels were measured before and at the indicated time points following an intraperitoneal injection of glucose (2 mg/g body weight). For insulin tolerance tests (ITT), mice were injected intraperitoneally with insulin (0.75 U/kg body weight) and blood glucose was quantified tail vein blood at the indicated time points.
Statistics
Data are represented as mean ± SEM. Statistical differences between the groups were evaluated by one-way ANOVA, post-hoc LSD or Bonferroni test and paired t-test where applicable. A p-value of ≤0.05 was considered as statistically significant. Analyses were performed with SPSS, version 17 (SPSS, Chicago, IL, USA) and GraphPad Prism Software (GraphPAd Software, LaJolla, CA). Ingenuity™ systems pathway analysis (Ingenuity, Redwood City, CA) was used to analyze genearray results.
Results
Loss of hepatocyte KLF6 protects mice from diet-induced steatohepatitis and improves insulin response
To assess the potential impact of KLF6 on hepatic lipid and glucose homeostasis in vivo, we generated mice with hepatocyte-specific knockdown of KLF6 using a Cre-lox strategy in which mice expressing Cre driven by the albumin promoter were crossed with animals containing two floxed KLF6 alleles and further backcrossed into the C57BL/6 background [8, 17]. These albumin-Cre X KLF6LoxP/LoxP mice (termed ‘DeltaHepKlf6 mice’) had a normal phenotype when fed standard chow, with body and liver weights, serum transaminases and blood lipids that were no different from controls (KLF6LoxP/LoxP) after eight and sixteen weeks [8, 18] (supplementary tbl. S1).
We explored the impact of a high fat diet (HFD) on DeltaHepKlf6 mice. Because histologic features of NASH are well characterized [20], we compared hepatic histology between DeltaHepKlf6 mice and controls for macro- and microvesicular steatosis, ballooning, portal- and lobular inflammation and necrosis in a blinded fashion. After eight weeks of HFD, DeltaHepKlf6 mice had fewer ballooned hepatocytes and significantly fewer features of steatohepatitis, assessed by a cumulative score comprised of steatosis, ballooning and inflammation (mNAS) (Fig. 1A, B, supplementary fig. 1). After 16 weeks of HFD, both groups had similar amounts of steatosis, but in controls, fat was predominantly macrovesicular. Correspondingly, lipid droplets as assessed by oil red-O staining had smaller diameters in DeltaHepKlf6 mice, whole body fat content and liver triglyceride content was lower in DeltaHepKLF6 mice on HFD compared to controls on the same diet (Fig. 1C, D, supplementary fig. 1). The trend for both ALT and AST was lower after 16 weeks of HFD in DeltaHepKlf6 mice, compared to controls, consistent with attenuated hepatic inflammation and necrosis (Tab. 1).
Figure 1. Less steatosis in DeltaHepKlf6 mice after eight weeks of high fat diet.

Representative liver sections show in H&E staining more ballooned hepatocytes and a significantly higher cumulative mNAS score in controls vs. DeltaHepKlf6 (A,B). Accordingly, a larger diameter and quantity of lipid droplets are present in controls vs. DeltaHepKlf6 in Oil Red O staining (C,D). In support of our model, we observed less KLF6 positive hepatocytes in DeltaHepKLF6 mice compared to controls in KLF6 immunohistochemistry (E,F). Interestingly, the KLF6 staining in non-parenchymal cells appeared weaker in controls compared to DeltaHepKLF6 livers.
Table 1.
Basic characteristics HFD mice
| Characteristics | 8 weeks HFD control | 8 weeks HFD DeltaKLF6 | Sig. | 16 weeks HFD control | 16 weeks HFD DeltaKLF6 | Sig. |
|---|---|---|---|---|---|---|
| Weight (g) | 24.86 ± 0.74 | 25.66 ± 0.52 | n.s. | 35.55 ± 1.19 | 34.55 ± 2.26 | n.s. |
| liver/body ratio | 3.3 ± 0.09 | 3.53 ± 0.1 | n.s. | 3.86 ± 0.19 | 3.59 ± 0.2 | n.s. |
| AST (U/l) | 144.0 ± 43.47 | 115.71 ± 28.04 | n.s. | 151 ± 61.61 | 66 ± 6.93 | n.s. |
| ALT (U/l) | 16.0 ± 4.81 | 20.43 ± 3.63 | n.s. | 106 ± 52.83 | 23 ± 5.57 | n.s. |
| Fasting Blood Glucose (mg/dl) | 152.46 ± 8.48 | 155.11 ± 5.67 | n.s. | 138.71 ±9.09 | 182.5 ± 9.09 | p<0.05 |
| Fasting Serum Insulin (mU/l) | 10.42 ± 2.74 | 8.96 ± 2.59 | n.s. | 27.16 ± 8.42 | 20.39 ± 7.14 | n.s. |
| HOMA-IR | 2.5 ± 0.87 | 1.9 ± 0.58 | n.s. | 8.3 ± 2.54 | 9.09 ± 3.22 | n.s. |
| Serum Triglycerides (mg/dl) | 37.5 ± 4.5 | 51.0 ± 6.93 | n.s. | 51 ± 11.36 | 56 ± 13.23 | n.s. |
| Serum Cholesterol (mg/dl) | 111.17 ± 26.1 | 138.14 ± 10.46 | n.s. | 224 ± 42.51 | 144 ± 21.07 | n.s. |
In DeltaHepKlf6 hepatocytes, knockdown of KLF6 protein occurred progressively with aging (Fig. 1E, F; Fig. 2A, B, supplementary fig. 2), associated with progressive Cre activity [21].
Figure 2. Improved glucose metabolism with KLF6 knockdown.

KLF6 whole liver protein as quantified by western blots was lower in DeltahepKlf6 mice in both chow (A) and HFD (B) at 8 and 16 weeks. AUC of glucose tolerance tests (GTTs) at 8 weeks of diets shows significantly improved GTT results in DeltaHepKlf6 compared to controls (C), while their response to Insulin tolerance test (ITT) remained unchanged (D). While in the 16 week diet groups, we did not observe a significant benefit of KLF6 knockdown in the GTT (E), their response to ITT was significantly better (F) [*: p<0.05].
Fasting glucose levels were similar in HFD-fed controls and DeltaHepKlf6 mice after 8 weeks, but were significantly increased after 16 weeks of HFD. In DeltaHepKlf6 mice, administration of glucose was followed by a blunted rise in blood glucose levels compared to controls following 8 weeks of diet (Fig. 2B, E). As we recently reported [8], insulin levels and HOMA-IR were comparable between the groups when fed a chow diet (supplementary tab. S1). Similarly, whereas there were no significant differences in response to insulin tolerance test (ITT) after eight weeks of HFD, DeltaHepKlf6 mice had a significantly improved response to an insulin bolus after 16 weeks of HFD, compared to controls (Fig. 2D, F). Data from euglycemic insulin-clamp studies and mice that were heterozygous for the KLF6 gene in all tissues (Klf6 +/−) support the observation that KLF6 depletion improves systemic insulin response, protects from steatosis and attenuates hepatic gluconeogenesis (supplementary fig. 1, 5).
KLF6 post-transcriptionally activates PPARα protein
In order to determine potential pathways accounting for these differences in glucose and lipid homeostasis, we compared transcriptomic profiles of liver tissue between 3 controls and 3 DeltaHepKlf6 mice on HFD. Based on Ingenuity™ pathway analysis (Ingenuity, Redwood City, CA), PPARα signaling-related genes were among the most affected by KLF6 knockdown (supplementary tbl. 2). Based on these findings, we assessed PPARα protein and mRNA expression in whole liver. Whereas PPARα protein was decreased in DeltaHepKlf6 and Klf6 +/− mice, Pparα mRNA was not affected (Fig. 3A–E). Similarly, PPARα protein and transcriptional activity assessed using a PPRE luciferase reporter were significantly up-regulated in HepG2cells stably overexpressing KLF6 (Fig. 3F), whereas knockdown by KLF6 siRNA decreased PPARα protein (supplementary fig. 3).
Figure 3. Loss of PPARα protein but not RNA in hepatocytes of DeltaHepKlf6 mice.
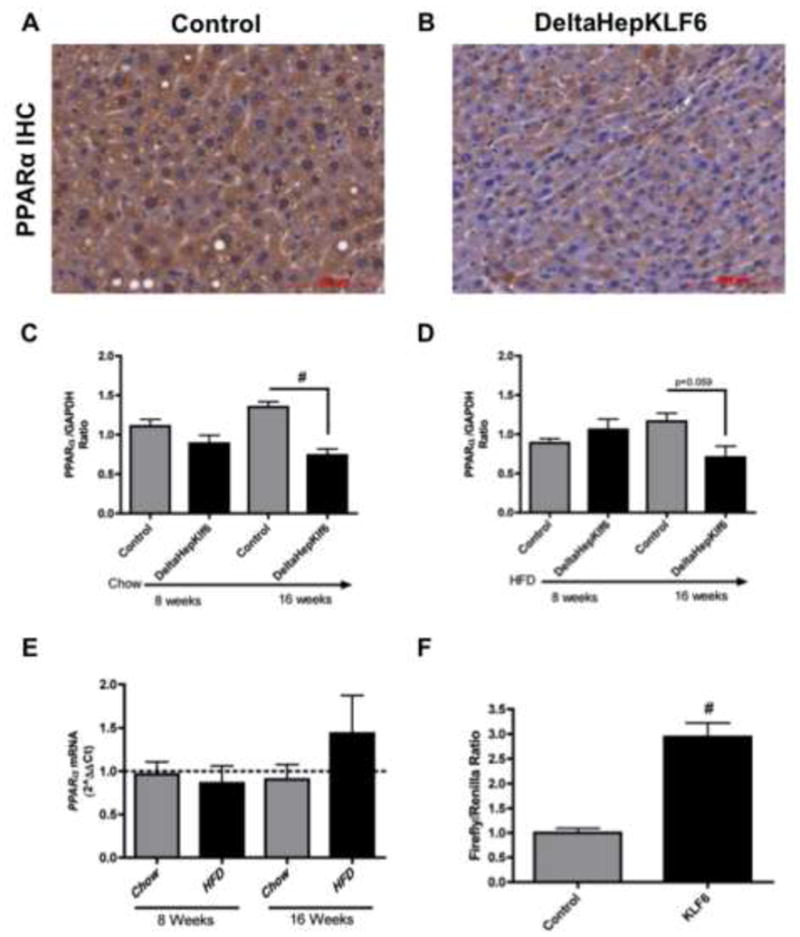
PPARα protein expression is decreased in DeltaHepKLF6 mice compared to controls (representative slides) in IHC (A, B) as well as whole liver tissue western blots (C,D). Interestingly, PPARα mRNA expression remains unchanged in DeltaHepKLF6 mice compared to controls (dotted line) during the feeding periods (E). KLF6 expression in HepG2 cells leads to a significant induction of PPAR responsive elements as quantified by luciferase reporter assay (F) [*: p<0.05; #: p<0.01].
KLF6 induces PPARα downstream targets in mice and human NAFLD
PPARα transcriptionally activates TRB3, which inhibits insulin signaling by binding to AKT, thereby preventing its phosphorylation and activation [22]. In Klf6 deficient mice, Trb3 mRNA expression was reduced compared to controls (Fig. 4A). In NAFLD patients, KLF6 mRNA was associated with TRB3 mRNA expression (Fig. 4B). Because TRB3 inhibits Ser-phosphorylation of AKT, we also quantified AKT- phosphorylation by western blot and observed a trend towards increased AKT-phosphorylation after 16 weeks of a high fat diet. In HepG2 cells over-expressing KLF6, AKT-phosphorylation was decreased, whereas siKLF6 increased AKT-phosphorylation (supplementary fig. 3). Phospho-AKT inhibits PEPCK expression via FOXO1 translocation. In DeltaHepKlf6 mice, Pepck was down-regulated under some treatment conditions and in NAFLD patients, KLF6 mRNA expression was significantly correlated with PEPCK expression (Fig. 4C, supplementary fig. 2).
Figure 4. KLF6 expression is associated with expression of PPARα downstream targets and repression of miRNA 10b.

In DeltaHepKlf6 compared to controls (dotted line), mRNA expression of the PPARα downstream target Trb3 was down-regulated (A). In 28 NAFLD patients, we observed a significant association between KLF6 mRNA expression and TRB3 mRNA (B) as well as its downstream target PEPCK (C). We did not observe a negative association between KLF6 mRNA and miRNA-10b expression in the NAFLD cohort. However, when divided into patients with or without histological features of inflammation, we observed an increase of KLF6, TRB3 and PEPCK mRNA in those individuals with inflammation, while miRNA10b was significantly repressed (D). Since miRNA 10b has been identified, to repress PPARα protein, but not mRNA expression, similar to our in vivo observations, we quantified miRNA-10b expression in whole liver lysate of DeltaHepKlf6 vs. controls (dotted line) and observed a significant upregulation in 8-week fed mice. As miRNA-10b is a miRtron of HoxD4, we quantified HoxD4 mRNA expression and found a significant upregulation in DeltaHepKlf6 mice vs. controls (dotted line) (E). A luciferase assay, in which luciferase activity is inversely correlated with miRNA-10b activation, showed a significant repression of miRNA-10b activation with KLF6 expression and an induction with SiKLF6 transfection in three independent experiments (C) [*: p<0.05; #: p<0.01].
KLF6 mediated induction of PPARα protein expression is not due to induction of known co-activators, but rather through miRNA 10b repression
KLF6 knockdown did not reduce, and actually induced expression of the known PPARα co-activator PGC1α as well as FXR (supplementary fig. 2). The substantial differences in PPARα protein but not mRNA expression implicated a post-transcriptional, rather than a direct transcriptional effect of KLF6 on PPARα. Because miRNA 10b [13] has previously been characterized as a repressor of PPARα, we assessed whether it contributes to reduce PPARα protein in DeltaHepKlf6 livers. In fact, miRNA 10b was significantly induced in DeltaHepKLF6 mice after eight weeks of diets (Fig. 4E). To directly link KLF6 to miR10b activity, we performed a functional luciferase assay (pMIR-Luc™, Signosis, Sunnyvale, CA), in which luciferase expression is reduced with increasing miRNA 10b activation. As predicted, siRNA knockdown of KLF6 led to a significant decrease in luciferase activity, indicating induction of miRNA 10b transcription, whereas KLF6 over-expression led to the opposite effect (Fig. 4F).
In order to characterize the interplay between KLF6 and miRNA 10b, we evaluated the miRNA 10b coding sequence. In contrast to the canonical activation typical of most microRNAs via Drosha mediated cleavage of a micro-RNA hairpin-derived pri-microRNA, miRNA 10b is encoded within intron 1 of the homeobox gene HoxD4, and directly transcribed into pre-microRNA [23]. This pattern of activation (termed ‘miRtron’) depends upon transcriptional activation of the host gene, in this case HoxD3 and HoxD4. While HoxD3 was not expressed in mouse liver, HoxD4 mRNA was increased in DeltaHepKlf6 mice compared to controls (Fig. 4E).
In our cohort of NAFLD patients, there was neither an overall inverse correlation between KLF6 mRNA and miRNA 10b expression, nor an effect of steatosis on the expression of miRNA 10b and PPARα related targets (supplementary fig. 4). However, when only patients with histological features of inflammation (Brunt criteria) were analyzed, there was increased expression of KLF6 mRNA and protein, PPARα protein, TRB3 and PEPCK mRNA and a significant decrease in expression of miRNA 10b (Fig. 4D, supplementary fig. 4).
Interestingly, while expression of fatty acid oxidation related targets remained unchanged or partially reduced, the key lipogenic gene Fasn was elevated in DeltaHepKlf6 mice (supplementary fig. 2, 3). Among other fatty acids, FASN produces 16:0/18:1-GPC, an endogenous and physiologically relevant agonist of PPARα [24]. In chow-fed mice, Fasn was unchanged between controls and DeltaHepKlf6 mice. However, Fasn was progressively and significantly induced in DeltaHepKlf6 mice following 16 weeks of HFD. FASN is also transcriptionally modified by FXR. Other nuclear receptors, particularly PPARγ, which are reportedly repressed by KLF6 [25] might also account for these alterations in lipid metabolism related genes, although their mRNAs were not altered. PPARγ protein concentration remained unchanged in most groups; however, a significant decrease was observed in 16-week chow-fed mice (supplementary fig. 2).
Discussion
Our findings substantially clarify clinical findings implicating KLF6 in the pathogenesis of NAFLD [7, 8] by demonstrating in cultured cells, mouse models and human samples that KLF6 mediates glucose tolerance and insulin responses in fatty liver disease. This phenotype is accompanied by KLF6-mediated induction of PPARα signaling and its downstream target Trb3. Furthermore, in DeltaHepKlf6 mice, HFD-induced hepatic steatohepatitis is attenuated compared to controls, which is associated with altered expression of key genes regulating fatty acid metabolism. Our data suggests that in KLF6 deficient mice, miRNA 10b represses PPARα protein, which reduces Trb3 and Pepck, thereby impairing hepatic gluconeogenesis and improving glucose tolerance (supplementary fig. 6). Loss of wild type KLF6 improves hepatic insulin resistance in humans, and KLF6 is positively correlated with TRB3 and PEPCK in NAFLD patients [8]. In humans, KLF6 is progressively increased in the progression of NAFLD to NASH [7].
Our findings also link KLF6 to the emerging interdependence between hepatic steatosis, hepatic lipid and glucose metabolism, inflammation and carcinogenesis [26]. KLF6 deficiency contributes to carcinogenesis in HCC in part by transcriptional de-repression of Mdm2, which leads to loss of p53 [19]. Apoptosis and cellular stress, which are key features of NAFLD, are also closely linked to p53 activity, and thus p53 is an increasing focus in NAFLD research [27]. On a regulatory level, glycogen synthase kinase (GSK3) activation promotes hepatocyte lipoapoptosis, a key feature of NAFLD, and we previously demonstrated that GSK3β directly phosphorylates KLF6, and thus increases its growth suppressor properties in vitro [28, 29]. PPARα, which we identify here as a KLF6-regulated protein, is critical for the development of hepatic steatosis associated with hepatitis C infection, a major risk factor for hepatocellular carcinoma (HCC) [30].
Based on the observation that PPARα protein expression and downstream targets were reduced in DeltaHepKlf6 mice, yet PPARα mRNA expression remained unchanged, we sought a post-transcriptional effect of KLF6 on PPARα rather than a direct transcriptional effect. This supposition was based in part on a recent study implicating miRNA 10b in the development of NAFLD through repression of PPARα protein expression, but not transcription [13]. Indeed, miRNA 10b expression was negatively correlated with KLF6 expression in our models. Interestingly, the transcription pattern of miRNA 10b differs from the canonical transcription of micro RNAs by Drosha mediated splicing from pri-miRNA, since miRNA 10b is directly spliced out of a shared sequence with the first introns of the homeobox related genes HoxD3 and HoxD4, and is thus termed a ‘miRtron’ [23]. While HoxD3 is not expressed in adult mouse liver, HoxD4 was up-regulated in KLF6 deficient mice and controlled by KLF6 in culture.
KLF6 contributes to a network of interactions that also converge on TRB3. PPARα activation transactivates Trb3, which is further augmented by PGC1α co-activation [15, 31]. Consistent with our findings that the KLF6 genotype correlates with fasting glucose and insulin resistance, TRB3 mRNA expression is up-regulated in insulin-resistant patients [8, 31]. Furthermore, mice administrated a TRB3-RNAi vector have improved GTTs [15]. TRB3 affects glucose metabolism by inhibition of Ser-phosphorylation of Akt and thus TRB3 disrupts insulin signaling [22]. Several studies favor TRB3-mediated repression of AKT-signaling and its downstream targets glycogen synthase and PEPCK to account for its role in glucose metabolism, which is also supported by our data [12, 22, 31]. However, in vivo knockdown of Trb3 by antisense oligonucleotides improves insulin signaling, but does not affect AKT activity [16]. Interestingly, since PPARγ expression was up-regulated in this study, the authors concluded that TRB3 mediates glucose metabolism by repression of PPARγ. Since KLF6 represses PPARγ expression in vitro [6], and the PPARγ downstream target Fasn was up-regulated in HFD-fed DeltaHepKlf6 mice, we quantified PPARγ protein expression in HFD-fed DeltaHepKlf6 mice, which remained unchanged.
In NAFLD patients, we found a clear correlation between KLF6 and PEPCK mRNA expression. Utilizing a global and a hepatocyte specific knockout model for PEPCK, She et al. identified a key metabolic role for PEPCK in vivo, linking hepatic lipid and glucose metabolism, exceeding its established role in gluconeogenesis [32]. Liver specific deletion of PEPCK led to marked hepatic steatosis and increased lipid accumulation. Yang et al. identified PEPCK to be essential in the susceptibility of fatty livers to acute injury, as ob/ob mice show diminished regeneration, which was attributed to lack of PEPCK expression [33]. The finding of elevated fasting glucose levels in HFD-fed DeltaHepKlf6 mice compared to controls is consistent with features of hepatocyte-specific PEPCK knockout mice, in which extrahepatic gluconeoenesis accounts for this pattern [34]. In euglycemic clamp studies, we observed a higher glucose disposal rate with KLF6 knockdown, which suggests an improved whole body insulin tolerance, compared to controls. However, we did not specifically examine extra-hepatic insulin sensitivity. The reduced features of steatohepatitis following HFD in both DeltaHepKlf6 and Klf6+/− mice might be sufficiently explained by improved insulin sensitivity secondary to loss of TRB3 leading to altered AKT-phosphorylation and PEPCK expression. However, we also observed changes in expression genes regulating lipid-metabolism. In particular, fatty acid transporters Cd36/fat and Got2 were reduced in DeltaHepKlf6 mice at earlier time points (supplementary fig. 2). Expression of fatty acid transporters is not only associated with fatty acid uptake, which was up-regulated in our model [10], but also with hepatocyte apoptosis, a hallmark of NASH [35]. On the other hand, the lipogenic gene Fasn was induced in HFD-fed Delta-hepKlf6 mice. Interestingly, an important role for Fasn in PPARα biology has recently been described, since it produces a specific fatty acid (16:0/18:1-GPC), which acts as a potent PPARα ligand, that accounts for a negative feedback-loop [24]. PPARα is an important nuclear receptor in the regulation of fatty acid oxidation [9]. However, neither the Gene array nor qRT PCR analyses of relevant fatty acid oxidation related targets revealed significant alterations in any of these genes. In fact, carnitine palmitoyltransferase 1 (CPT1), a key gene in mitochondrial uptake of long-chain fatty acids, was down-regulated with KLF6 knockdown only under HFD. Interestingly, CPT1 activity is known to be regulated by a p53-dependant manner [36]. We have previously identified KLF6 as an important mediator of p53 stability via transcriptional repression of Mdm2, which is lost when KLF6 is depleted in HCC patients and in a mouse model [19]. However, in the current study, we did not explore a role of KLF6 in p53-associated steatosis or fatty acid oxidation. Our observation that CPT1 expression was down-regulated under some treatment conditions in KLF6-deficient mice suggests a complex regulatory network beyond this single target. Further studies are warranted to fully elucidate the role of KLF6 in NAFLD, beyond the observed effects of KLF6 depletion on hepatic insulin sensitivity.
We have previously linked KLF6 to hepatic glucose metabolism by identifying KLF6 as a transcriptional activator of hepatic glucokinase, and here we link KLF6 to PPARα via repression of miRNA 10b. In aggregate, our data implicate KLF6 as a nexus for glucose- and lipid homeostasis and nuclear receptor signaling in fatty liver disease. Presumably, the role of KLF6 in the pathogenesis of NAFLD cannot be entirely explained by transcriptional control of glucokinase and posttranscriptional regulation of PPARα alone. Rather KLF6 is likely to act as a central integrator of fatty liver, encompassing direct transcriptional regulation of metabolic genes, as well as a broader interaction with nuclear receptors, growth suppressors and apoptosis related targets. These additional possibilities merit further evaluation in future studies.
Supplementary Material
Acknowledgments
Financial support:
Supported by grants from IFORES program of the University of Essen and the European Association for the Study of the Liver (EASL) Sheila Sherlock Short-Term Fellowship (LPB); Swiss National Fund (DV); The European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement Health-F2-2009-241762, for the project FLIP (HLR, GLP); National Institutes of Health (NIH): DK37340, DK56621 and AA017067 (SLF); Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST Program) of the Japan Society for the Promotion of Science (JSPS, RN); Takeda Science Foundation (TS); NIH/ NIDDK DK-52401(PDB); NIH DK 20541, Einstein Diabetes Research and Training Center (GS); Deutsche Forschungsgemeinschaft, DFG (PK, YML)
Footnotes
Disclosure: There is no conflict of interest to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Feldstein AE. Novel insights into the pathophysiology of nonalcoholic fatty liver disease. Semin Liver Dis. 2010;30(4):391–401. doi: 10.1055/s-0030-1267539. [DOI] [PubMed] [Google Scholar]
- 2.Narla G, Heath KE, Reeves HL, Li D, Giono LE, Kimmelman AC, et al. KLF6, a candidate tumor suppressor gene mutated in prostate cancer. Science. 2001;294(5551):2563–2566. doi: 10.1126/science.1066326. [DOI] [PubMed] [Google Scholar]
- 3.Yea S, Narla G, Zhao X, Garg R, Tal-Kremer S, Hod E, et al. Ras promotes growth by alternative splicing-mediated inactivation of the KLF6 tumor suppressor in hepatocellular carcinoma. Gastroenterology. 2008;134(5):1521–1531. doi: 10.1053/j.gastro.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki T, Yamamoto T, Kurabayashi M, Nagai R, Yazaki Y, Horikoshi M. Isolation and initial characterization of GBF, a novel DNA-binding zinc finger protein that binds to the GC-rich binding sites of the HIV-1 promoter. J Biochem. 1998;124(2):389–395. doi: 10.1093/oxfordjournals.jbchem.a022124. [DOI] [PubMed] [Google Scholar]
- 5.Li D, Yea S, Li S, Chen Z, Narla G, Banck M, et al. Kruppel-like factor-6 promotes preadipocyte differentiation through histone deacetylase 3-dependent repression of DLK1. J Biol Chem. 2005;280(29):26941–26952. doi: 10.1074/jbc.M500463200. [DOI] [PubMed] [Google Scholar]
- 6.Qi W, Chen X, Holian J, Tan CY, Kelly DJ, Pollock CA. Transcription factors Kruppel-like factor 6 and peroxisome proliferator-activated receptor-{gamma} mediate high glucose-induced thioredoxin-interacting protein. Am J Pathol. 2009;175(5):1858–1867. doi: 10.2353/ajpath.2009.090263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miele L, Beale G, Patman G, Nobili V, Leathart J, Grieco A, et al. The Kruppel-like factor 6 genotype is associated with fibrosis in nonalcoholic fatty liver disease. Gastroenterology. 2008;135(1):282–291. e281. doi: 10.1053/j.gastro.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bechmann LP, Gastaldelli A, Vetter D, Patman GL, Pascoe L, Hannivoort RA, et al. Glucokinase links Kruppel-like factor 6 to the regulation of hepatic insulin sensitivity in nonalcoholic fatty liver disease. Hepatology. 2012;55(4):1083–1093. doi: 10.1002/hep.24793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol. 2012;56(4):952–964. doi: 10.1016/j.jhep.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 10.Berk PD. Regulatable fatty acid transport mechanisms are central to the pathophysiology of obesity, fatty liver, and metabolic syndrome. Hepatology. 2008;48(5):1362–1376. doi: 10.1002/hep.22632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cha DR, Han JY, Su DM, Zhang Y, Fan X, Breyer MD, et al. Peroxisome proliferator-activated receptor-alpha deficiency protects aged mice from insulin resistance induced by high-fat diet. Am J Nephrol. 2007;27(5):479–482. doi: 10.1159/000106485. [DOI] [PubMed] [Google Scholar]
- 12.Stayrook KR, Bramlett KS, Savkur RS, Ficorilli J, Cook T, Christe ME, et al. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology. 2005;146(3):984–991. doi: 10.1210/en.2004-0965. [DOI] [PubMed] [Google Scholar]
- 13.Zheng L, Lv GC, Sheng J, Yang YD. Effect of miRNA-10b in regulating cellular steatosis level by targeting PPAR-alpha expression, a novel mechanism for the pathogenesis of NAFLD. J Gastroenterol Hepatol. 2010;25(1):156–163. doi: 10.1111/j.1440-1746.2009.05949.x. [DOI] [PubMed] [Google Scholar]
- 14.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449(7163):682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 15.Koo SH, Satoh H, Herzig S, Lee CH, Hedrick S, Kulkarni R, et al. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat Med. 2004;10(5):530–534. doi: 10.1038/nm1044. [DOI] [PubMed] [Google Scholar]
- 16.Weismann D, Erion DM, Ignatova-Todorava I, Nagai Y, Stark R, Hsiao JJ, et al. Knockdown of the gene encoding Drosophila tribbles homologue 3 (Trib3) improves insulin sensitivity through peroxisome proliferator-activated receptor-gamma (PPAR-gamma) activation in a rat model of insulin resistance. Diabetologia. 2011;54(4):935–944. doi: 10.1007/s00125-010-1984-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leow CC, Wang BE, Ross J, Chan SM, Zha J, Carano RA, et al. Prostate-specific Klf6 inactivation impairs anterior prostate branching morphogenesis through increased activation of the Shh pathway. J Biol Chem. 2009;284(31):21057–21065. doi: 10.1074/jbc.M109.001776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vetter D, Cohen-Naftaly M, Villanueva A, Lee YA, Kocabayoglu P, Hannivoort R, et al. Enhanced hepatocarcinogenesis in mouse models and human hepatocellular carcinoma by coordinate KLF6 depletion and increased messenger RNA splicing. Hepatology. 2012 doi: 10.1002/hep.25810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarocchi M, Hannivoort R, Hoshida Y, Lee UE, Vetter D, Narla G, et al. Carcinogen-induced hepatic tumors in KLF6+/− mice recapitulate aggressive human hepatocellular carcinoma associated with p53 pathway deregulation. Hepatology. 2011;54(2):522–531. doi: 10.1002/hep.24413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 21.Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis. 2000;26(2):149–150. doi: 10.1002/(sici)1526-968x(200002)26:2<149::aid-gene16>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 22.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300(5625):1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- 23.Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448(7149):83–86. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chakravarthy MV, Lodhi IJ, Yin L, Malapaka RR, Xu HE, Turk J, et al. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell. 2009;138(3):476–488. doi: 10.1016/j.cell.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qi W, Holian J, Tan CY, Kelly DJ, Chen XM, Pollock CA. The roles of Kruppel-like factor 6 and peroxisome proliferator-activated receptor-gamma in the regulation of macrophage inflammatory protein-3alpha at early onset of diabetes. Int J Biochem Cell Biol. 2010;43(3):383–92. doi: 10.1016/j.biocel.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 26.Hirsch HA, Iliopoulos D, Joshi A, Zhang Y, Jaeger SA, Bulyk M, et al. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell. 2010;17(4):348–361. doi: 10.1016/j.ccr.2010.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomita K, Teratani T, Suzuki T, Oshikawa T, Yokoyama H, Shimamura K, et al. p53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J Hepatol. 2012;57(4):837–843. doi: 10.1016/j.jhep.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 28.Ibrahim SH, Akazawa Y, Cazanave SC, Bronk SF, Elmi NA, Werneburg NW, et al. Glycogen synthase kinase-3 (GSK-3) inhibition attenuates hepatocyte lipoapoptosis. J Hepatol. 2011;54(4):765–772. doi: 10.1016/j.jhep.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lang UE, Kocabayoglu P, Cheng GZ, Ghiassi-Nejad Z, Munoz U, Vetter D, et al. GSK3beta phosphorylation of the KLF6 tumor suppressor promotes its transactivation of p21. Oncogene. 2012 doi: 10.1038/onc.2012.457. (E-pub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanaka N, Moriya K, Kiyosawa K, Koike K, Gonzalez FJ, Aoyama T. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J Clin Invest. 2008;118(2):683–694. doi: 10.1172/JCI33594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oberkofler H, Pfeifenberger A, Soyal S, Felder T, Hahne P, Miller K, et al. Aberrant hepatic TRIB3 gene expression in insulin-resistant obese humans. Diabetologia. 2010;53(9):1971–1975. doi: 10.1007/s00125-010-1772-2. [DOI] [PubMed] [Google Scholar]
- 32.She P, Shiota M, Shelton KD, Chalkley R, Postic C, Magnuson MA. Phosphoenolpyruvate carboxykinase is necessary for the integration of hepatic energy metabolism. Mol Cell Biol. 2000;20(17):6508–6517. doi: 10.1128/mcb.20.17.6508-6517.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang SQ, Lin HZ, Mandal AK, Huang J, Diehl AM. Disrupted signaling and inhibited regeneration in obese mice with fatty livers: implications for nonalcoholic fatty liver disease pathophysiology. Hepatology. 2001;34(4 Pt 1):694–706. doi: 10.1053/jhep.2001.28054. [DOI] [PubMed] [Google Scholar]
- 34.She P, Burgess SC, Shiota M, Flakoll P, Donahue EP, Malloy CR, et al. Mechanisms by which liver-specific PEPCK knockout mice preserve euglycemia during starvation. Diabetes. 2003;52(7):1649–1654. doi: 10.2337/diabetes.52.7.1649. [DOI] [PubMed] [Google Scholar]
- 35.Bechmann LP, Gieseler RK, Sowa JP, Kahraman A, Erhard J, Wedemeyer I, et al. Apoptosis is associated with CD36/fatty acid translocase upregulation in non-alcoholic steatohepatitis. Liver Int. 2010;30(6):850–859. doi: 10.1111/j.1478-3231.2010.02248.x. [DOI] [PubMed] [Google Scholar]
- 36.Derdak Z, Villegas KA, Harb R, Wu AM, Sousa A, Wands JR. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J Hepatol. 2012 doi: 10.1016/j.jhep.2012.11.042. (E-pub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.