

Abstract
The estrogen receptor (ER) is a hormone-regulated transcription factor that binds, as a dimer, to estrogens and to specific DNA sequences. To explore at a fundamental level the geometric and topological features of bivalent-ligand binding to the ER dimer, dimeric ER crystal structures were used to rationally design nonsteroidal bivalent estrogen ligands. Guided by this structure-based ligand design, we prepared two series of bivalent ligands (agonists and antagonists) tethered by flexible spacers of varying lengths (7–47Å) and evaluated their ER-binding affinities for the two ER subtypes and their biological activities in cell lines. Bivalent ligands based on the agonist diethylstilbestrol (DES) proved to be poor candidates, but bivalent ligands based on the antagonist hydroxytamoxifen (OHT) were well suited for intensive study. Binding affinities of the OHT-based bivalent ligands were related to spacer length in a distinctive fashion, reaching two maximum values at 14 and 29Å in both ER subtypes. These results demonstrate that the bivalent concept can operate in determining ER-ligand binding affinity and suggest that two distinct modes operate for the binding of bivalent estrogen ligands to the ER dimers, an intermolecular as well as an intramolecular mode. Our insights, particularly the possibility of intramolecular bivalent binding on a single ER monomer, may provide an alternative strategy to prepare more selective and active ER antagonists for endocrine therapy of breast cancer.
Keywords: Estrogen receptor, bivalent ligand, structure-activity relationship, drug design, tamoxifen
Introduction
The estrogen receptor (ER) is a nuclear receptor (NR) that binds various estrogens and regulates diverse physiological and pathological activities, largely at the level of gene transcription.1–3 Like other members of the NR superfamily, agonist binding causes a conformational change in the ER ligand binding domain (LBD) that induces receptor dimerization and results in the reorientation of the C-terminal helix (helix-12), completing the formation of a hydrophobic groove on the protein surface that is the docking site for the binding of coactivators, mediators of further transcriptional signaling.4,5 Because estrogen action can have both beneficial and detrimental health effects, major research efforts have been devoted to developing estrogens having selective activities.
The two human ER subtypes, ERα and ERβ, have different tissue distributions and distinct as well as overlapping regulatory functions,6 and thus provide interesting targets through which ER subtype-selective ligands might provide, for example, effective menopausal hormone replacement without increasing the risk of breast cancer.7–12 Ligands, such as tamoxifen and raloxifene (TAM and RAL, Figure 1), have been termed selective estrogen receptor modulators (SERMs) because they display tissue-selective pharmacology, protecting bone but blocking breast cancer, presumably by exploiting differences in cellular content of coregulator proteins in different target tissues. Hydroxytamoxifen (OHT, Figure 1), an active metabolite of tamoxifen, however, causes hot flashes and increases the risk of endometrial cancer.13
Figure 1.

The chemical structure of tamoxifen (TAM), hydroxytamoxifen (OHT), diethylstilbestrol (DES), raloxifene (RAL), and bivalent raloxifene ligands (BRLs).
Beyond ER subtype-selective ligands and SERMs, there are other, intriguing modes by which selective estrogens might be developed. Considerable work has gone into developing inhibitors of ER action that directly block coactivator-binding,14–16 although potent, selective agents have not yet been found. Finally, because ligand binding to the ER-LBD also induces receptor dimerization, which is essential for its transcriptional activity,17,18 an alternative strategy for regulating ER activity is to prepare bivalent estrogen ligands that can bridge between two ER-LBDs. Such bivalent ligands might bind especially well or in an unusual fashion that could affect the stability of the ER dimer and alter its transcriptional activity. Consequently, they might form the basis for the design of novel agents that could provide improved tissue selectivity or endocrine therapies.
During the last two decades, several groups have studied different bivalent estrogen ligands tethered with various types of spacers, such as flexible oligomers or rigid duplex DNA, to determine the optimal spacer length for two estrogen moieties to bind simultaneously to two ER-LBDs.19–25 Recently, we reported on bivalent raloxifene ligands (BRLs, Figure 1) tethered by oligoethylene glycol (OEG) spacers of varying length.26 BRLs tethered by long spacers bound more weakly to ER-LBD dimers than did those tethered by short spacers; the latter, short-spacer tethered BRLs, appeared to engage in intramolecular bivalent binding on the same ER-LBD monomer, rather than intermolecular binding between dimer pairs, a rather unexpected finding. Such a relationship between the architecture of other multivalent ligands and their ligand-protein binding mechanisms has also been observed.27,28
Herein, we report on bivalent ligands based on the agonist diethylstilbestrol (DES, Figure 1) and the antagonist OHT in studies designed to complement and extend our prior work on BRLs. The focus here was to examine how ER-binding affinities and cellular activities would be affected by the agonist versus antagonist nature of the ligand and the length and nature of the spacer. From our experimental and molecular modeling studies, we developed structure-activity relationships and obtained a number of insights from which we can draw instructive conclusions regarding further design of bivalent estrogen ligands. Our findings regarding the importance of ligand affinity, ER conformation, nature of the spacer, and especially our evidence that certain bivalent ligands bind to ER dimers in an intramolecular fashion—simultaneously in the cognate binding site and in the coactivator-binding groove of one ER monomer component—suggest a number of alternative strategies for preparing more selective and active ER ligands that might be useful in various endocrine therapies.
Results and Discussion
A thermodynamic approach to bivalent ligand design
The affinity (K°) of a binding event is characterized by the Gibbs free energy (ΔG°=−RT·lnK°), made up of enthalpic (ΔH°) and entropic components (T·ΔS°). Although free energy enhancement resulting from bivalent binding is attributed mostly to the entropic component,29–31 the influence of the binding moiety on the overall binding affinity of a bivalent ligand was unknown. Whereas the standard enthalpy ΔH° and entropy ΔS° can be considered to arise from changes in the intermolecular ligand-protein binding energies and the rearrangements undergone by water during the binding, respectively,32 one would expect that high-affinity estrogen agonist like DES or antagonist like OHT would have different effects on the enthalpic component, based on their distinctive patterns of hydrogen bonding and van der Waals interactions with ER; the interactions with these ligands would likely also be different from those of RAL, the ligand used our earlier study of BRLs. Thus, a comparison between agonist and antagonist (i.e., DES vs. OHT and RAL), which have very different conformations of binding to the ER,33–35 can help us parse out what components of bivalent ligands contribute to their binding affinity.
On the basis of thermodynamic considerations and to facilitate these comparisons, we constructed the new bivalent DES and OHT ligands using the same OEG spacers we had used for BRLs, ensuring that the spacers would have the same conformational entropic cost, so we could ascribe a decrease or increase in the Gibbs free energy for ER binding of our new bivalent ligands to changes in enthalpy rather than entropy. Because the nonsteroidal ligands contain a double bond, they provide us with both homo-bivalent (Z-Z or E-E isomers) and hetero-bivalent (Z-E isomer) ligands. Since double bond geometry affects ER-binding affinity, comparisons between homo- and hetero-bivalent ligands offer the potential to explore and interrogate at a more general level geometric and topological features of bivalent interactions.
Studies with DES-based bivalent ligands
DES-based ligand design
The structure of the ERα-LBD dimer bound to two DES molecules (PDB ID 3ERD) was modeled using PyMOL (Figure 2a). The closest distance between two DES molecules at carbon 1 of the stilbene structure is 30.0Å. As an ER agonist, DES binds deeply in the hydrophobic cognate binding site, and the direct path between ligands passes through portions of protein. Thus, a spacer would need to be longer than 30Å to adopt a non-linear pathway avoiding steric obstruction by the protein.
Figure 2.
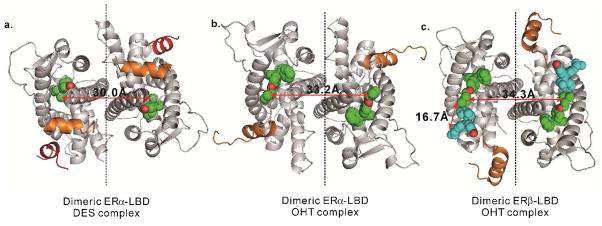
a) The distance between two DES molecules (spheres in green and red) bound to dimeric ERα-LBD (ribbon in gray, helix-12 in orange, coactivator in red, 3ERD) is 30.0Å. b) The distance between two OHT molecules in dimeric ERα-LBD (3ERT) is 33.2Å. c) The distance between two OHT molecules bound in two cognate binding sites of dimeric ERβ-LBD (2FSZ) is 34.3Å, between one OHT molecule bound in one cognate binding site and one OHT molecule (spheres in cyan and red) bound in the coactivator-binding groove is 16.7Å.
As previously, we chose OEG spacers of 39.5 to 46.7Å length (EG10 to EG12, approximately 10Å longer than the calculated distance) to tether two DES moieties via an amide link, producing bivalent ligands having the potential for intermolecular bivalent binding between two cognate binding sites in the ER dimer (Figure 3, 1–3). Moreover, to minimize the folding and conformational entropic cost of the OEG spacer, two carbon-hybrid OEG spacers and one biphenyl-hybrid OEG spacer, with extended lengths of 34.8 to 39.8Å, were chosen (4–6, Figure 3). In addition, a bivalent DES ligand (7) tethered by a much shorter 1,2-diethoxyethylene spacer (EG2, 10.8Å, Figure 3) and three monovalent DES ligands (8–10, Figure 3) were also prepared. Because the geometric isomers of DES bind with different affinities, separation of the low-binding Z-isomer from the desired high-binding E-isomer was performed.36
Figure 3.
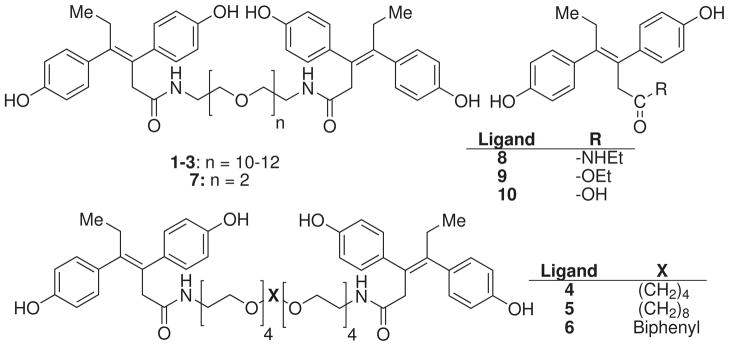
The chemical structure of E- and E-E isomers of mono- and bivalent DES ligands 1–10.
DES-based ligand ER-binding affinity
Binding affinities of E-E isomers of mono- and bivalent DES ligands (1–10) were evaluated by radiometric binding assays,37,38 using full-length human ERα and ERβ, with [3H]-E2 as tracer and E2 as standard. They are expressed as relative binding affinity (RBA) values, relative to that of E2 (RBA = 100%; Table 1). The affinities of ligands 1–10, except for DES itself, were considerably lower than E2. Among ligands 1–6, ligand 2 tethered by a 43.1Å OEG spacer had the highest affinity, which, however, was 10-fold lower than that of ligand 7, tethered by a 10.8Å spacer on both ER subtypes.
Table 1.
Spacer structures, maximum lengths, and relative binding affinity (RBA) values of mono- and bivalent DES ligands 1–10.
| Entry | Ligand | Spacer structure | Maximum length a [Å] | RBA for ERα [%] | RBA for ERβ [%] |
|---|---|---|---|---|---|
| 1. | 1 b | EG10 | 39.5 | 0.012 ± 0.001 | 0.039 ± 0.011 |
| 2. | 2 b | EG11 | 43.1 | 0.021 ± 0.005 | 0.048 ± 0.004 |
| 3. | 3 b | EG12 | 46.7 | 0.012 ± 0.003 | 0.023 ± 0.006 |
| 4. | 4 b | EG4C4EG4 | 34.8 | 0.006 ± 0.001 | 0.013 ± 0.004 |
| 5. | 5 b | EG4C8EG4 | 39.8 | 0.013 ± 0.002 | 0.022 ± 0.002 |
| 6. | 6 b | EG4Ph2EG4 | 39.0 | 0.008 ± 0.002 | 0.020 ± 0.006 |
| 7. | 6 c | EG4Ph2EG4 | 39.0 | 0.003 ± 0.001 | 0.008 ± 0.000 |
| 8. | 7 d | EG2 | 10.8 | 0.230 ± 0.005 | 0.247 ± 0.026 |
| 9. | 8 e | monovalent | - | 0.067 ± 0.004 | 0.159 ± 0.004 |
| 10. | 9 e | monovalent | - | 7.87 ± 1.9 | 41.98 ± 6.7 |
| 11. | 10 e | monovalent | - | 0.011 ± 0.002 | 0.051 ± 0.011 |
| 12. | DES | monovalent | - | 372 ± 106 f | 278 ± 54 f |
Extended spacer length between two nitrogen atoms measured by using PyMOL.
Pure E-E isomer was evaluated in assays.
Pure E-Z isomer was evaluated in assays.
The ER-binding affinity of bivalent DES ligand 7 was evaluated based on 60% E-E isomer.
The ER-binding affinities of monovalent ligands 8–10 were evaluated based on 80% E-isomer.
Reference 39.
The low affinities of bivalent DES ligands suggest two things: (1) Introduction of a polar group (carboxylic acid, ester, or amide, 8–10) onto the hydrophobic core of DES dramatically reduces ER-binding affinity, presumably because of the hydrophobic character of the binding pocket.12,39 (2) Agonist ligands, such as DES and E2, stabilize an ER-LBD conformation with ligand fully encapsulated by protein, with no obvious way for a tethering group to exit the fully enclosed pocket, making it particularly challenging to create bivalent estrogens based on agonist ligands.
DES-based ligand biological activity
Since RBA values do not reflect biological effects, the E-E isomers of bivalent DES ligands 1–7 were evaluated for their hormonal activities on ERα and ERβ-regulated reporter genes using U2OS cells transiently transfected with ER expression plasmids pSG5-ERα (U2OS/ERα) or pSG5-ERβ FL (U2OS/ERβ) and the reporter plasmid p(ERE)2-luc+ (Table 2).26 The intrinsic activity (IA) of each ligand was assayed at 10μM. Ligands 1–7 showed pure agonistic activity that was related to spacer length: Ligands 1 and 4–6 with spacer lengths of 34.8–39.8Å had less than 50% IA values on ERα, while on ERβ only ligands 1 and 5 have similar IA values. Cell density was used to evaluate cytotoxic effects, and none of these ligands influenced U2OS cell growth.
Table 2.
Hormonal activity and the growth inhibition of mono- and bivalent DES ligands (1–7).
| Entry | Ligand | Hormonal activity (U2OS Cells)a | Growth inhibition | ||||
|---|---|---|---|---|---|---|---|
| ERα | ERβ | IC50 after 96h [μM] | |||||
|
| |||||||
| IA b [%] | CD c [%] | IA b [%] | CD c [%] | MCF-7 | MDA-MB-231 | ||
| 1. | 1 d | 49 | 93 | 50 | 96 | 7.3 | 5.4 |
| 2. | 2 d | 79 | 95 | 104 | 92 | 9.0 | 9.1 |
| 3. | 3 d | 81 | 93 | 89 | 91 | 5.1 | 5.6 |
| 4. | 4 d | 22 | 86 | 58 | 86 | 5.5 | 4.9 |
| 5. | 5 d | 22 | 78 | 32 | 77 | 1.8 | 1.2 |
| 6. | 6 d | 33 | 88 | 89 | 85 | <0.63 | >20 |
| 7, | 6 e | −7 | 80 | 55 | 90 | <0.63 | >20 |
| 8. | 7 f | 90 | 93 | 95 | 92 | 6.5 | 5.6 |
| 9. | DES g | 105 | 103 | 111 | 90 | 5.9 | 4.3 |
Concentration of 1–7 was 10μM.
Intrinsic activity (IA).
Cell density (CD).
Pure Z-Z isomer.
Pure E-Z isomer.
The bivalent DES ligand 7 was evaluated based on 60% E-E isomer.
The concentration was 10nM.
In further evaluation of growth inhibitory effects in ER-positive (MCF-7) and ER-negative (MDA-MB-231) cell lines,26 IC50 values were calculated from concentration-dependent Treated/Controlcorr (T/Ccorr [%]) values after a 96h incubation (Table 2). ER agonists often show non-ER-mediated growth inhibition at high concentrations, exemplified by monovalent DES having an IC50 value of 5μM for both cell lines, and most bivalent ligands showed similar, non-ER dependent growth suppression. Ligand 6 (E-E isomer) was an exception, showing unusual cell line-dependent effects, being completely inactive in ER-negative cells (IC50 >20μM) but rather potent in ER-positive cells (IC50 <0.63μM). Remarkably, despite its low binding affinity (0.003%) for ERα, the E-Z isomer of ligand 6 showed identical growth suppression as its E-E isomer in both cells.
A possible explanation for the more potent growth suppression of ER-positive cells by ligand 6, lies in its unique spacer structure, the biphenyl segment; it might be interrupting ER signaling by a different mechanism, namely, a hydrophobic-driven interaction between the bis-arene and the coactivator-binding site of ERα. In this manner, ER signaling in support of cell proliferation would be inhibited in ER-positive but not ER-negative cells. Certain biphenyl systems are known to function as inhibitors of coactivator-binding, suggesting that this alternate mode of inhibition by ligand 6 is feasible.40 In summary, the effects of these ligands mirrored that of the parent DES ligand: they were mostly pure agonists and did not show any ER-dependent suppression of cell growth.
Studies with OHT-based bivalent ligands
OHT-based ligand design
Unlike ER agonists, the structures of ER bound to OHT are enlarged in the 11β direction to accommodate the bulky side chain. This local remodeling provides the OHT molecule access to the exterior of the ER, where the basic amino group forms a salt bridge with a surface aspartic acid (Asp351 ERα; Asp303 ERβ).34,35 This amino group, positioned at the protein surface, offers a promising site through which to tether the OHT moieties. To rationally design bivalent OHT ligands, computer modeling of dimeric ER-LBD crystal structures bound to OHT (PDB ID 3ERT for ERα and 2FSZ for ERβ) was performed using PyMOL. We found that the closest distance between two nitrogen atoms in the crystal structure is 33.2 and 34.3Å, for ERα and ERβ, respectively (Figure 2bc).
Notably, there are four OHT molecules in the dimeric ERβ-LBD crystal structure, two in the cognate binding sites and the other two in the hydrophobic coactivator-binding grooves on the protein surface.41 The distance between the nitrogen atom of one OHT molecule in the cognate binding site and the nitrogen atom of the other OHT molecule in the coactivator-binding groove on the same monomer component is much less, only 16.7Å.
To further understand the intra- vs. intermolecular bivalent binding relationship with bivalent OHT ligands, we prepared nine bivalent OHT ligands 11–19 (Figure 4) tethered through the amino groups by nine OEG spacers, with spacer lengths, 7.24–43.1Å, sufficient to span either intra- or intermolecular bivalent binding. Additionally, one monovalent OHT ligand 20 (Figure 4) with a side chain was prepared as a control. The stereochemistry of OHT molecules affects ER-ligand binding: Z-OHT is an antagonist with a 3-fold higher affinity than E2, while E-OHT is an agonist with only 5% the affinity of E2.42 As with DES ligands, geometric isomerization of OHT-based ligands means that separations need to be performed carefully.43
Figure 4.

The chemical structure of mono- and bivalent OHT ligands 11–20.
OHT-based ligand ER-binding affinity
The eight bivalent ligands (11–17 and 19) evaluated are of two types: (1) homo-bivalent ligands – both OHT moieties are high affinity Z-isomers, and (2) hetero-bivalent ligands – one Z-OHT moiety tethered to one low affinity E-OHT moiety. Because hetero-bivalent ligands (Z-E isomers) have essentially the same lipophilicity as homo-bivalent ligands (Z-Z isomers), the binding of hetero-bivalent ligands can be considered as bivalent controls for specific versus non-specific effects of binding to the second site. Both isomers of the monovalent control 20 were evaluated; binding affinities are expressed as RBA values (Table 3).
Table 3.
Spacer structures, maximum lengths, and relative binding affinity (RBA) values of mono- and bivalent OHT ligands (11–20).
| Entry | Ligand | Spacer structure | Maximum length a [Å] | Homo-bivalent ligand (Z-Z/Z isomer) | Hetero-bivalent ligand (Z-E/E isomer) | ||
|---|---|---|---|---|---|---|---|
| ERα | ERβ | ERα | ERβ | ||||
| 1. | 11 | EG1 | 7.24 | 13.8 ± 4.3 | 6.11 ± 1.4 | 6.60 ± 0.5 | 3.44 ± 0.7 |
| 2. | 12 | EG2 | 10.8 | 16.4 ± 2.1 | 5.55 ± 1.3 | 23.5 ± 5.2 | 6.25 ± 0.6 |
| 3. | 13 | EG3 | 14.4 | 37.2 ± 2.3 | 13.9 ± 3.4 | 8.16 ± 2.0 | 9.57 ± 1.8 |
| 4. | 14 | EG4 | 18.0 | 14.9 ± 4.0 | 12.2 ± 2.1 | 13.6 ± 1.3 | 4.77 ± 1.5 |
| 5. | 15 | EG5 | 21.6 | 20.8 ± 5.5 | 12.8 ± 2.0 | 10.9 ± 2.3 | 3.20 ± 1.0 |
| 6. | 16 | EG7 | 28.8 | 30.7 ± 2.8 | 20.7 ± 1.6 | 32.3 ± 8.1 | 9.48 ± 1.4 |
| 7. | 17 | EG9 | 35.9 | 28.1 ± 3.7 | 7.92 ± 1.5 | 23.9 ± 6.9 | 7.85 ± 2.0 |
| 8. | 18 b | EG10 | 39.5 | 17.2 ± 2.0 b | 9.53 ± 2.4 b | - | - |
| 9. | 19 | EG11 | 43.1 | 27.9 ± 4.0 | 7.17 ± 1.4 | 18.0 ± 5.0 | 7.60 ± 2.2 |
| 10. | 19 b | EG11 | 43.1 | 21.8 ± 3.1 b | 9.75 ± 0.57 b | - | - |
| 11. | 20 | monovalent | 19.2 c | 78.1 ± 12 | 34.2 ± 1.7 | 12.4 ± 2.2 | 8.03 ± 0.01 |
| 12. | OHT | monovalent | - | 285 d | 62 e | 5 d | - |
Extended spacer length between two nitrogen atoms measured by using PyMOL.
The ER-binding affinities for bivalent OHT ligands 18 and 19 were evaluated based on Z-Z:Z-E:E-E = 1:2:1 isomers according to analytical RP-HPLC analyses.
Maximum extended side chain length of EG5Me.
Reference 42.
Reference 44.
Generally, binding affinities of bivalent OHT ligands are only somewhat lower than that of E2, with ERα higher than ERβ. Thus, the amino link and the OEG spacer are tolerated by the cognate binding site. The monovalent ligand 20Z also had a much higher binding affinity than its E-isomer 20E. The Z-Z isomers (13–16) had overall stronger ER-binding affinities than the Z-E isomers on ERβ but not ERα, indicating that stereochemistry plays a variable role in second-site ER-binding.
Figure 5 illustrates the relationship between RBA values and spacer lengths of the bivalent OHT ligands: Affinities peaked at 14.4 and 28.8Å (EG3 and EG7, respectively) in both subtypes. This is not only consistent with our previous interpretation of intra- vs. intermolecular binding modes of BRLs,26 but also precise enough to provide spacer-length information for each binding mode. Interestingly, 11ZZ, tethered by a 7.24Å spacer (EG1), did not reach a maximum like the BRL with a 4.71Å spacer,26 while ligand 13ZZ, tethered by a longer 14.4Å spacer, peaked on both ER subtypes. This distinction suggests that the Z-OHT moiety binds to a different secondary site in ERα than did the RAL moiety. Thus, intramolecular bivalent binding can be achieved by the longer spacer of ligand 13ZZ. Moreover, the affinity difference between 13ZZ and 13ZE on ERα (37.2% vs. 8.16%) is greater than that on ERβ (13.9% vs. 9.57%), suggesting that the stereochemical tolerance of this secondary site on ERα is lower than on ERβ. Furthermore, ligand 12ZE tethered by a 10.8Å-long spacer (EG2) also reached a maximum binding peak on ERα, suggesting the E-OHT moiety can bind to a different secondary site, whereas the Z-OHT moiety cannot.
Figure 5.

The relationship between relative binding affinity (RBA) values of mono- and bivalent OHT ligands (11–17, 19, and 20) in both ER subtypes and their maximum spacer lengths.
A second maximum binding peak was reached by ligand 16 tethered by a 28.8Å-long spacer (EG7) on both subtypes, consistent with intermolecular bivalent binding between two ER cognate binding sites. It is challenging to rationalize why ER-binding affinities of 16ZZ and 16ZE on the ERα were nearly identical (30.7% vs. 32.3%), but the stereochemical tolerance at the second binding site might be rather low, so once one Z-OHT moiety binds in the cognate binding site of one monomer, the lipophilic nature of the second OHT moiety, rather than its Z/E stereochemistry, determines its ability either to bind to the second cognate binding site or elsewhere on the ERα surface. While the secondary site involved in intramolecular bivalent binding, the coactivator-binding groove, is precedented in the ERβ structure, the location of a non-cognate site for intermolecular bivalent binding cannot be ascertained with certainty.
In contrast to ERα, affinity differences between 16ZZ and 16ZE on ERβ (20.7% vs. 9.48%) were larger, suggesting that the ERβ subtype is more stereochemically discriminating in binding the second OHT moiety. Remarkably, a gradual decrease in affinity with increasing spacer length (16ZZ–18ZZ) on ERα but not on ERβ, revealed that intermolecular bivalent binding could still be achieved with spacers longer than 28.8Å on the ERα, whereas on the ERβ it peaked sharply with a 28.8Å spacer. This may reflect that the ERα cognate binding site is approximately 100Å3 larger than ERβ.12
OHT-based ligand biological activity
Because of cytotoxicity, we could not evaluate the biological activity of the OHT-based ligands (11–17, 19, and 20) in U2OS cells using the ER-regulated reporter genes used earlier for the DES ligands. Thus, their growth inhibitory effects were evaluated with MCF-7 and MDA-MB-231 cells (Table 4).
Table 4.
Hormonal activity and the growth inhibition of mono- and bivalent OHT ligands (11–17, 19, and 20).
| Entry | Ligand | Hormonal Activity (U2OS Cells) a | Growth inhibition | ||||
|---|---|---|---|---|---|---|---|
| ERα | ERβ | IC50 after 96h [μM] | |||||
|
| |||||||
| IA b [%] | CD c [%] | IA b [%] | CD c [%] | MCF7 | MDA-MB-231 | ||
| 1. | 11ZZ | −71 | 81 | −91 | 79 | 0.11 | >20 |
| 2. | 12ZZ | −64 | 80 | −89 | 78 | 0.07 | 1.84 |
| 3. | 13ZZ | −83 | 51 | −93 | 55 | 0.11 | 1.95 |
| 4. | 14ZZ | −85 | 61 | −96 | 59 | 0.06 | 1.23 |
| 5. | 15ZZ | −95 | 15 | −99 | 15 | 0.08 | 1.20 |
| 6. | 16ZZ | −100 | 10 | −99 | 9 | 0.10 | 0.69 |
| 7. | 17ZZ | −100 | 9 | −100 | 8 | 2.10 | 13.98 |
| 8. | 19ZZ | −100 | 8 | −100 | 7 | 0.01 | 0.32 |
| 9. | 19 d | −99 | 8 | −100 | 6 | 0.15 | 0.32 |
| 10. | 20Z | −42 | - | −77 | - | 0.37 | 2.17 |
| 11. | 20E | - | - | - | - | 0.40 | 1.94 |
| 12. | OHT e | 5 | 110 | −47 | 92 | 0.15 | 4.59 |
Concentration 10μM.
Intrinsic activity (IA).
Cell density (CD).
Cell assays were evaluated based on Z-Z:Z-E:E-E = 1:2:1 isomers.
The concentration was 1μM.
All the Z-Z isomers (except 17ZZ) were potent growth inhibitors of MCF-7 cells (IC50 ≤ 0.11μM). Notably, these bivalent ligands were more potent than the monovalent control 20Z (entry 10) or OHT itself (entry 12). The potency advantage of bivalent ligands compared to monovalent control 20Z and OHT is especially important, because it suggests that bivalent binding can significantly enhance activity. The growth inhibitory effects of all ligands is also higher in the ER-positive cells, with MDA-MB-231/MCF-7 potency ratios ranging from 7 (16ZZ, entry 6) to 32 (19ZZ, entry 8). The Z-Z isomer of ligand 19ZZ was the most potent in both cell lines, while the growth inhibition of its Z-E isomer (entry 9) was 15-fold weaker in ER-positive cells. By contrast, the monovalent controls 20Z and 20E had rather similar growth inhibitory potencies in both cell lines (entries 10 and 11).
While growth suppression of ER-positive cells can be attributed to the antagonist activity of bivalent OHT ligands, growth inhibition in ER-negative cells is significant and suggests that the cytotoxicity of the bivalent OHT ligands may be a combination of ER-dependent and ER-independent effects.
Computer modeling
ERβ complex with ligand 13ZZ and ligand 12ZE
The binding mode of the second OHT moiety in the ERβ coactivator-binding groove is known from a crystal structure (Figure 2c). Our binding mode for ligand 13ZZ, predicted by modeling, allows for interaction with the same residues as in the crystal structure (2FSZ), although the OHT moiety in the coactivator-binding groove adopts a reversed orientation, where the nitrogen atom connected to the spacer points towards the cognate binding site (Figure 6a), with the ethyl group protruding outward into solvent.
Figure 6.

a) Ligand 13ZZ (sticks in green and red) in complex with ERβ-LBD (ribbon in gray). b) Ligand 12ZE in complex with ERβ–LBD. c) Ligand 13ZZ in complex with ERα-LBD. d) Ligand 16ZZ in complex with ERα-LBD. e) Ligand 16ZZ in complex with ERβ-LBD. Results based on a 10ns simulation in explicit water. An overlay with OHT from crystal structure is shown in purple.
Ligand 12ZE is equally able to form interactions with the coactivator-binding groove, provided the spacer adopts an extended conformation, but the E-OHT moiety is less well accommodated in the coactivator-binding groove, because now the phenyl group from the outside OHT protrudes into solvent (Figure 6b). Thus, a better accommodation of this outside OHT moiety in the coactivator-binding groove may have been hindered by its short spacer, as is consistent with the RBA measurements.
ERα complex with ligand 13ZZ and ligand 12ZE
Because the topology of the coactivator-binding groove for the second OHT moiety is dependent on the position of helix-12, which in turn is dependent on the activation state of ERα, prediction of ERα binding modes at the coactivator-binding groove is complicated. Using structural knowledge on ERβ coactivator-site binding modes, we could obtain a stable binding mode for ligand 13ZZ that interacted with key residues of the groove and helix-12 (Figure 6c). The model suggests that helix-12 performs a slight shift due to interactions between the Z-OHT moiety and the coactivator-binding groove, while enclosing it as a binding site with multiple interactions. Interestingly, we could not find a stable binding mode for ligand 12ZE. The EG2 spacer is insufficient to enable good intramolecular binding with ERα, and the outside E-OHT moiety flipped over, forming non-specific interactions with other residues on the ERα surface.
ERα and ERβ complexes with ligand 16ZZ
By modeling ligand 16ZZ into the cognate binding sites of ERα and ERβ (Figure 6de), we found that it could bridge between both of them in an intermolecular bivalent binding fashion. The Z-OHT moieties were slightly shifted compared to the monovalent OHT in the crystal structure, possibly due to minor rearrangements during the simulation or constraints from the attached spacer that distort the ideal arrangement of the Z-OHT moieties in the cognate binding site.
Summary and Conclusion
Intermolecular and intramolecular modes of bivalent binding and unusual biological activity in cell
We have prepared two series of bivalent estrogen ligands tethered by OEG spacers of varying lengths (7.24 to 46.7Å) and evaluated their binding affinities and biological properties. Previously, several different types of spacers (e.g., flexible oligomers and rigid duplex DNA) were used to tether various estrogen binding moieties. Compared to rigid spacers, an oligomer like OEG has the advantage of high flexibility to avoid steric clashes with proteins. We found two peaks of binding affinity with bivalent OHT ligands tethered with 14.4 and 28.8Å spacers, which, based on our modeling efforts, correspond to the intra- and intermolecular bivalent ER-binding modes, respectively. Most notably, many bivalent OHT ligands are potent growth inhibitors of MCF-7 cells; they are more potent on ER-positive than ER-negative cells, and more potent than the monovalent control and OHT itself. Thus, bivalent OHT ligands exhibit some unusual biological activities in ER-positive cells, although it is less clear how growth inhibitory potency correlates with binding affinity and apparent bivalent binding behavior.
Global observations on bivalent ligand design
Based on our results and others,19–26 we can reach a number of conclusions regarding factors that influence bivalent ER-ligand interaction. First, an estrogen antagonist (e.g., RAL or OHT) induces an antagonistic ER-binding conformation, where the displacement of helix-12 creates a channel through which certain substituents can escape the confines of the cognate binding site in the direction of the ER dimer interface. By contrast, a substituent on an estrogen agonist has difficulty in getting access to the protein exterior unless it is at the 17α position of the steroidal structure;33,45 however, a spacer extending from this site is directed away from the dimer interface.23 Notably, the bivalent ethynyl estradiol (EE2) ligands prepared by LaFrate et al.23 had better ER-binding affinity than bivalent E2 ligands tethered elsewhere.21,22 Second, the functional group through which the spacer is linked to the ligand needs to be compatible with the environment of the bound ligand. Thus, introduction of a polar amide at C1 of the DES ligands dramatically reduced their affinity; by contrast, the tertiary amine in bivalent OHT ligands maintained salt bridge interactions with protein residues and gave them higher affinities. Third, a high-affinity estrogen antagonist like OHT gave bivalent ligands with higher affinity than BRLs studied earlier,26 attributed to a more negative enthalpy caused by OHT binding to the ER. Fourth, use of a rigid central element29,46 can help to minimize the conformational entropy loss and enhance the binding affinity, as noted by bivalent estrogens tethered by rigid duplex DNA, studied previously.25 While avoiding intramolecular hydrophobic interactions between spacer units like the biphenyl segment and DES moieties,47 it is also good to avoid additional mechanisms of activity, unless a coactivator-binding inhibition is desired. Finally, flexible spacers can have conformational biases; e.g., OEGs form helical coils because of their preferred gauche orientation.48 Spanning the same distance with more flexible oligomers, such as oligopropylene and oligobutylene glycols, might raise binding affinities.49
Consideration of heterofunctional bivalent ligand design
The good evidence for intramolecular ER-binding we provide suggests that even better bivalent ligands might be designed by explicitly optimizing their heterofunctional nature, i.e., one binding moiety optimized for the cognate binding site and the other for the coactivator-binding groove (or at other secondary sites on the same monomer). Presently most coactivator inhibitors designed on the basis of the LXXLL binding motif or found by high throughput screening have only micromolar activities, leaving a need for developing high-affinity ligands for this site.14–16,50 Nevertheless, our finding of the intramolecular bivalent binding mode suggests tantalizing new opportunities for the design of novel heterofunctional bivalent ligands as an alternative strategy to create more active and selective estrogens.
Methods
Chemical preparations, characterizations, biological evaluations, and computer modeling of mono-/bivalent ligands (1–20) are provided in Supporting Information. Cellular assays were performed according to the previous report.26
Supplementary Material
Acknowledgments
We thank the Deutsche Forschungsgemeinschaft (SFB 765 to R. Haag, M. Weber, and R. Gust) and the National Institutes of Health (PHS 5R37 DK015556 to J. A. Katzenellenbogen) for financial support. We thank T. Martin for help with binding assays.
Footnotes
Supporting Information Available: This material is free via the Internet at http://pubs.acs.org.
References
- 1.Avendaño C, Menéndez JC. Medicinal Chemistry of Anticancer Drugs. Elsevier; Amsterdam: 2008. Anticancer Drugs That Inhibit Hormone Action; pp. 53–92. [Google Scholar]
- 2.Weatherman RV. Untangling the Estrogen Receptor Web: Tools to Selectively Study Estrogen-binding Receptors. In: Ottow E, Weinmann H, editors. Nuclear Receptors as Drug Targets. Wiley-VCH; Weinheim: 2008. pp. 47–64. [Google Scholar]
- 3.Jordan VC, Furr BJA. Hormone Therapy in Breast and Prostate Cancer. Humana Press; Totowa: 2002. An Introduction to the Regulation of Sex Steroids for the Treatment of Cancer; pp. 1–14. [Google Scholar]
- 4.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The Nuclear Receptor Superfamily: The Second Decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olefsky JM. Nuclear receptor minireview series. J Biol Chem. 2001;276:36863–36864. doi: 10.1074/jbc.R100047200. [DOI] [PubMed] [Google Scholar]
- 6.Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS. Tissue distribution and quantitative analysis of estrogen receptor-α (ERα) and estrogen receptor-β (ERβ) messenger ribonucleic acid in the wild-type and ER alpha-knockout mouse. Endocrinology. 1997;138:4613–4621. doi: 10.1210/endo.138.11.5496. [DOI] [PubMed] [Google Scholar]
- 7.Grese TA, Sluka JP, Bryant HU, Cullinan GJ, Glasebrook AL, Jones CD, Matsumoto K, Palkowitz AD, Sato M, Termine JD, Winter MA, Yang NN, Dodge JA. Molecular determinants of tissue selectivity in estrogen receptor modulators. Proc Natl Acad Sci USA. 1997;94:14105–14110. doi: 10.1073/pnas.94.25.14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 1 Receptor interactions. J Med Chem. 2003;46:883–908. doi: 10.1021/jm020449y. [DOI] [PubMed] [Google Scholar]
- 9.Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 2 Clinical considerations and new agents. J Med Chem. 2003;46:1081–1111. doi: 10.1021/jm020450x. [DOI] [PubMed] [Google Scholar]
- 10.Minutolo F, Macchia M, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen Receptor β Ligands: Recent Advances and Biomedical Applications. Med Res Rev. 2011;31:364–442. doi: 10.1002/med.20186. [DOI] [PubMed] [Google Scholar]
- 11.Bertini S, De Cupertinis A, Granchi C, Bargagli B, Tuccinardi T, Martinelli A, Macchia M, Gunther JR, Carlson KE, Katzenellenbogen JA, Minutolo F. Selective and potent agonists for estrogen receptor beta derived from molecular refinements of salicylaldoximes. Eur J Med Chem. 2011;46:2453–2462. doi: 10.1016/j.ejmech.2011.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katzenellenbogen JA. The 2010 Philip S. Portoghese Medicinal Chemistry Lectureship: Addressing the “Core Issue” in the Design of Estrogen Receptor Ligands. J Med Chem. 2011;54:5271–5282. doi: 10.1021/jm200801h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, Tan-Chiu E, Ford L, Wolmark N. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90:1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 14.Parent AA, Gunther JR, Katzenellenbogen JA. Blocking Estrogen Signaling After the Hormone: Pyrimidine-Core Inhibitors of Estrogen Receptor-Coactivator Binding. J Med Chem. 2008;51:6512–6530. doi: 10.1021/jm800698b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gunther JR, Moore TW, Cottins ML, Katzenellenbogen JA. Amphipathic benzenes are designed inhibitors of the estrogen receptor α/steroid receptor coactivator interaction. ACS Chem Biol. 2008;3:282–286. doi: 10.1021/cb800056r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phillips C, Roberts LR, Schade M, Bazin R, Bent A, Davies NL, Moore R, Pannifer AD, Pickford AR, Prior SH, Read CM, Scott A, Brown DG, Xu B, Irving SL. Design and Structure of Stapled Peptides Binding to Estrogen Receptors. J Am Chem Soc. 2011;133:9696–9699. doi: 10.1021/ja202946k. [DOI] [PubMed] [Google Scholar]
- 17.Brandt ME, Vickery LE. Cooperativity and dimerization of recombinant human estrogen receptor hormone-binding domain. J Biol Chem. 1997;272:4843–4849. doi: 10.1074/jbc.272.8.4843. [DOI] [PubMed] [Google Scholar]
- 18.Tamrazi A, Carlson KE, Daniels JR, Hurth KM, Katzenellenbogen JA. Estrogen receptor dimerization: Ligand binding regulates dimer affinity and dimer dissociation rate. Mol Endocrinology. 2002;16:2706–2719. doi: 10.1210/me.2002-0250. [DOI] [PubMed] [Google Scholar]
- 19.Bergmann KE, Wooge CH, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. Bivalent Ligands as Probes of Estrogen-Receptor Action. J Steroid Biochem Mol Biology. 1994;49:139–152. doi: 10.1016/0960-0760(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 20.Groleau S, Nault J, Lepage M, Couture M, Dallaire N, Bérubé G, Gaudreault RC. Synthesis and preliminary in vitro cytotoxic activity of new triphenylethylene dimers. Bioorg Chem. 1999;27:383–394. [Google Scholar]
- 21.Rabouin D, Perron V, N’Zemba B, Gaudreault RC, Bérubé G. A Facile Synthesis of C2-Symmetric 17β-Estradiol Dimers. Bioorg Med Chem Lett. 2003;13:557–560. doi: 10.1016/s0960-894x(02)00987-3. [DOI] [PubMed] [Google Scholar]
- 22.Bérubé G, Rabouina D, Perron V, N’Zemba B, Gaudreault RC, Parenta S, Asselin EE. Synthesis of unique 17β-estradiol homo-dimers, estrogen receptors binding affinity evaluation and cytocidal activity on breast, intestinal and skin cancer cell lines. Steroids. 2006;71:911–921. doi: 10.1016/j.steroids.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 23.LaFrate AL, Carlson KE, Katzenellenbogen JA. Steroidal bivalent ligands for the estrogen receptor: Design, synthesis, characterization and binding affinities. Bioorg Med Chem. 2009;17:3528–3535. doi: 10.1016/j.bmc.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wendlandt AE, Yelton SM, Lou DY, Watt DS, Noonan DJ. Synthesis and functional analysis of novel bivalent estrogens. Steroids. 2010;75:825–833. doi: 10.1016/j.steroids.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abendroth F, Bujotzek A, Shan M, Haag R, Weber M, Seitz O. DNA-Controlled Bivalent Presentation of Ligands for the Estrogen Receptor. Angew Chem Int Edit. 2011;50:8592–8596. doi: 10.1002/anie.201101655. [DOI] [PubMed] [Google Scholar]
- 26.Shan M, Bujotzek A, Abendroth F, Wellner A, Gust R, Seitz O, Weber M, Haag R. Conformational Analysis of Bivalent Estrogen Receptor Ligands: From Intramolecular to Intermolecular Binding. Chem Bio Chem. 2011;12:2587–2598. doi: 10.1002/cbic.201100529. [DOI] [PubMed] [Google Scholar]
- 27.Bertozzi CR, Kiessling LL. Chemical Glycobiology. Science. 2001;291:2357–2364. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- 28.Gestwicki JE, Cairo CW, Strong LE, Oetjen KA, Kiessling LL. Influencing Receptor-Ligand Binding Mechanisms with Multivalent Ligand Architecture. J Am Chem Soc. 124:14922–14933. doi: 10.1021/ja027184x. [DOI] [PubMed] [Google Scholar]
- 29.Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Edit. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 30.Kiessling LL, Gestwicki JE, Strong LE. Synthetic Multivalent Ligands as Probes of Signal Transduction. Angew Chem Int Edit. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwefel D, Maierhofer C, Beck JG, Seeberger S, Diederichs K, Möller HM, Welte W, Wittmann V. Structural Basis of Multivalent Binding to Wheat Germ Agglutinin. J Am Chem Soc. 2010;132:8704–8719. doi: 10.1021/ja101646k. [DOI] [PubMed] [Google Scholar]
- 32.Testa B, Jenner P, Kilpatrick GJ, Eltayar N, Vandewaterbeemd H, Marsden CD. Do Thermodynamic Studies Provide Information on Both the Binding to and the Activation of Dopaminergic and Other Receptors. Biochem Pharmacol. 1987;36:4041–4046. doi: 10.1016/0006-2952(87)90559-4. [DOI] [PubMed] [Google Scholar]
- 33.Nettles KW, Bruning JB, Gil G, O’Neill EE, Nowak J, Guo Y, Kim Y, DeSombre ER, Dilis R, Hanson RN, Joachimiak A, Greene GL. Structural plasticity in the oestrogen receptor ligand-binding domain. EMBO Rep. 2007;8:563–568. doi: 10.1038/sj.embor.7400963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 35.Pike ACW, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson JK, Carlquist M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Winkler VW, Nyman MA, Egan RS. Diethylstilbestrol Cis-Trans Isomerization and Estrogen Activity of Diethylstilbestrol Isomers. Steroids. 1971;17:197–207. doi: 10.1016/s0039-128x(71)80124-1. [DOI] [PubMed] [Google Scholar]
- 37.Katzenellenbogen JA, Johnson HJ, Myers HN. Photoaffinity Labels for Estrogen Binding Proteins of Rat Uterus. Biochemistry. 1973;12:4085–4092. doi: 10.1021/bi00745a010. [DOI] [PubMed] [Google Scholar]
- 38.Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: Evidence that an open pocket conformation is required for ligand interaction. Biochemistry. 1997;36:14897–14905. doi: 10.1021/bi971746l. [DOI] [PubMed] [Google Scholar]
- 39.Waibel M, De Angelis M, Stossi F, Kieser KJ, Carlson KE, Katzenellenbogen BS, Katzenellenbogen JA. Bibenzyl- and stilbene-core compounds with non-polar linker atom substituents as selective ligands for estrogen receptor beta. Eur J Med Chem. 2009;44:3412–3424. doi: 10.1016/j.ejmech.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams AB, Weiser PT, Hanson RN, Gunther JR, Katzenellenbogen JA. Synthesis of Biphenyl Proteomimetics as Estrogen Receptor-α Coactivator Binding Inhibitors. Org Lett. 2009;11:5370–5373. doi: 10.1021/ol901999f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Chirgadze NY, Briggs SL, Khan S, Jensen EV, Burris TP. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor β. Proc Natl Acad Sci USA. 2006;103:9908–9911. doi: 10.1073/pnas.0510596103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robertson DW, Katzenellenbogen JA, Long DJ, Rorke EA, Katzenellenbogen BS. Tamoxifen Anti-Estrogens - a Comparison of the Activity, Pharmacokinetics, and Metabolic-Activation of the Cis-Isomer and Trans-Isomer of Tamoxifen. J Steroid Biochem Mol Biology. 1982;16:1–13. doi: 10.1016/0022-4731(82)90137-6. [DOI] [PubMed] [Google Scholar]
- 43.Katzenellenbogen JA, Carlson KE, Katzenellenbogen BS. Facile geometric isomerization of phenolic non-steroidal estrogens and antiestrogens: limitations to the interpretation of experiments characterizing the activity of individual isomers. J Steroid Biochem. 1985;22:589–596. doi: 10.1016/0022-4731(85)90210-9. [DOI] [PubMed] [Google Scholar]
- 44.Kim S, Katzenellenbogen JA. Triarylethylene Bisphenols with a Novel Cycle are Ligands for the Estrogen Receptor. Bioorg Med Chem. 2000;8:785–793. doi: 10.1016/s0968-0896(00)00016-x. [DOI] [PubMed] [Google Scholar]
- 45.Li MJ, Greenblatt HM, Dym O, Albeck S, Pais A, Gunanathan C, Milstein D, Degani H, Sussman JL. Structure of Estradiol Metal Chelate and Estrogen Receptor Complex: The Basis for Designing a New Class of Selective Estrogen Receptor Modulators. J Med Chem. 2011;54:3575–3580. doi: 10.1021/jm200192y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krishnamurthy VM, Estroff LA, Whitesides GM. Multivalency in Ligand Design. In: Jahnke W, Erlanson DA, editors. Fragment-based Approaches in Drug Discovery. Wiley-VCH; Weinheim: 2006. pp. 11–54. [Google Scholar]
- 47.Bujotzek A, Shan M, Haag R, Weber M. Towards a rational spacer design for bivalent inhibition of estrogen receptor. J Comput Aided Mol Des. 2011;25:253–262. doi: 10.1007/s10822-011-9417-1. [DOI] [PubMed] [Google Scholar]
- 48.Kjellander R, Florin E. Water Structure and Changes in Thermal Stability of the System Poly(ethylene oxide)-Water. J Chem Soc, Faraday Trans 1. 1981;77:2053–2077. [Google Scholar]
- 49.LaFrate AL. Synthesis and Biological Evaluation of Coactivator Binding Inhibitors and Bivalent Ligands for the Estrogen Receptor. University of Illinois at Urbana-Champaign; Urbana: 2008. Bivalent Ligands for the Estrogen Receptor; pp. 60–116. [Google Scholar]
- 50.Sun A, Moore TW, Gunther JR, Kim M, Rhoden E, Du Y, Fu H, Snyder JP, Katzenellenbogen JA. Discovering Small-Molecule Estrogen Receptor α/Coactivator Binding Inhibitors: High-Throughput Screening, Ligand Development, and Models for Enhanced Potency. Chem Med Chem. 2011;6:654–666. doi: 10.1002/cmdc.201000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.