

Abstract
Uncontrolled activation of tumor necrosis factor receptor-associated factor (TRAF) proteins may result in profound tissue injury by linking surface signals to cytokine release. Here we show that a ubiquitin E3 ligase component, Fbxo3, potently stimulates cytokine secretion from human inflammatory cells by destabilizing a sentinel TRAF inhibitor, Fbxl2. Fbxo3 and TRAF protein in circulation positively correlated with cytokine responses in septic subjects and we furthermore identified a hypofunctional Fbxo3 human polymorphism. A small molecule inhibitor targeting Fbxo3 was sufficient to lessen severity of cytokine-driven inflammation in several murine disease models. These studies identify a pathway of innate immunity that may characterize subjects with altered immune responses during critical illness or provide a basis for therapeutic intervention targeting TRAF protein abundance.
Tumor necrosis factor receptor associated factors (TRAF) are critically involved in inflammation, innate and adaptive immune responses, and programmed cell death 1. Six well-characterized TRAF proteins (TRAF1-6) exist and a newer homologue (TRAF7) has been identified 2. All TRAF members share a highly conserved carboxyl-terminal domain that mediates interactions with transmembrane tumor necrosis factor receptors (TNFR). TRAF members (with the exception of TRAF1) also contain an NH2-terminal RING finger domain. Identification of TRAF proteins has contributed significantly to the elucidation of the molecular mechanisms of signal transduction emanating from the TNFR superfamily and the Toll like/interleukin-1 receptor (TLR/IL-1R) family 1. TRAF family proteins also interact with CD40, RANK, I-TAC, and p75 NGF receptors 1. Specifically, TRAF2, TRAF5, and TRAF6 serve as adapter proteins that link cell surface receptors with downstream kinase cascades, which in turn activate key transcription factors, including nuclear factor κB (NFκB), which via canonical signaling results in cytokine gene expression. The execution of this cascade in some cases involves TRAF-mediated polyubiquitination of receptor interacting protein 1 (RIP1), which recruits and stimulates IkappaB kinase leading to NFκB activation 3. With an exaggerated immune response, TRAF-mediated cytokine release via this pathway leads to severe effects of edema, multi-organ failure, and shock 4,5. Hence, TRAF proteins have a central role as they mediate signal transduction to elicit transactivation of genes linked to a multitude of downstream cytokines that profoundly regulate host responses 6–10. These observations suggest that therapeutics designed to selectively down-regulate the abundance of TRAF proteins in cells could govern pro-inflammatory responses.
Protein ubiquitination is a universal, inducible, and reversible process in mammalian cells that regulates diverse processes by branding proteins for degradation, either by the 26S proteasome or the lysosome 11. Ubiquitin conjugation to a target protein is a multi-step process catalyzed by an E1 ubiquitin-activating enzyme, ubiquitin transfer from an E1-activating enzyme to an E2-conjugating enzyme, and last, generation of an isopeptide bond between the substrate’s ε-amino lysine and the carboxyl-terminus of ubiquitin catalyzed by a E3-ubiquitin ligase 12. One of the emerging families of E3 ligases is the Skp-Cullin1-F box (SCF) superfamily that regulates cell cycle progression, DNA repair, and cell survival 13. The SCF apparatus contains Rbx1 and Cullin1, which form a scaffold to link the E2 conjugating enzyme (Ubc) with a substrate specificity module 14–16. This substrate specificity module contains Skp1 binding to an F box protein, the latter of which targets many substrates through phosphospecific domain interactions 17. The F box proteins are sub classified as Fbxl or Fbxw proteins depending on the presence of a carboxyl-terminal leucine-rich repeat (LRR) motif or WD repeat motif, respectively, that are used for substrate recognition 16. A third subclass of F box proteins, however, contain other types of protein interaction domains or no recognizable domains, termed F box only proteins (Fbxo) 16. A new member of the Fbxl subclass, Fbxl2, was identified that regulates lipogenesis and the mitotic program 18,19.
In the current study we discovered that molecular interaction exists between two F box proteins that regulate cytokine secretion through TRAF protein stability. First, Fbxl2 acts as a crucial, pan-reactive inhibitor of TRAF function by mediating their ubiquitination and degradation in epithelia and human monocytes. Second, Fbxl2 itself is targeted for disposal in cells by Fbxo3, thereby upregulating TRAF protein levels to trigger cytokine-mediated inflammation. Fbxo3 and TRAFs were not only linked to elevated cytokine responses in the circulation of individuals with sepsis, but we also identified a subgroup harboring a hypomorphic Fbxo3 polymorphism with impaired cytokine production. These studies provide a new molecular model of regulating innate immunity, which for the first time, led to the development of a family of F box protein small molecule antagonists that exert potent anti-inflammatory activity.
RESULTS
Fbxl2 targets TRAFs for polyubiquitination
In the process of identifying substrates for Fbxl2, we observed that ectopic expression of this F box protein in a murine lung epithelial (MLE) cell line specifically reduced protein expression of TRAF1-6 and phospho-IκBα (Fig. 1a). Fbxl2 was also conditionally expressed in MLE cells using a doxycycline-inducible plasmid resulting in TRAF protein degradation in a time-dependent manner (Fig. 1b). In co-immunoprecipitation (co.i.p.) experiments where MLE cells were lysed and endogenous proteins were subjected to Fbxl2 immunoprecipitation (i.p.), all TRAF proteins were detected in Fbxl2 immunoprecipitates by immunoblotting (Fig. 1c). The results suggest that endogenous Fbxl2 interacts with TRAFs in epithelial cells. As a positive control, cyclin D3 was also found to precipitate with Fbxl2 20,21. Since Fbxl2 overexpression did not alter Bcl-2 and c-IAP1 protein, they served as negative controls not binding to Fbxl2 (Fig. 1a, c). Inclusion of purified Fbxl2 with the full complement of E1 and E2 enzymes plus ubiquitin (SCFFbxl2) was sufficient to generate polyubiquitinated TRAF species in vitro (Fig. 1d). Similarly, cyclin D3 was used as a positive control, and MAP2 (an Fbxl7 substrate, unpublished data) was used as a negative control in the in vitro SCFFbxl2 ubiquitination assay. Last, ectopic expression of Fbxl2 plasmid decreased TRAF protein half-life (Fig. 1e), but not their mRNA levels (data not shown).
Figure 1. Fbxl2 interacts with and targets TRAFs for polyubiquitination.
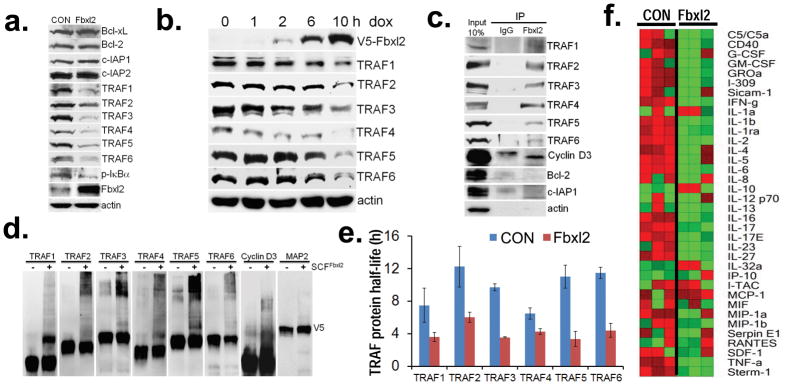
a. TRAF1-6 and control protein immunoblot of MLE cells after control (CON) plasmid or ectopic Fbxl2 plasmid expression. b. MLE cells were transfected with an inducible Fbxl2 plasmid under control of exogenous doxycycline. Cells were treated with doxycycline (500 ng/ml) for 1–10 h, cell lysates were analyzed for proteins by immunoblotting. c. Fbxl2 was immunoprecipitated from cells followed by TRAF 1-6 and control protein immunoblotting. d. In vitro ubiquitination assays. SCF complex components were incubated with individual V5-TRAFs and with (+) or without (−) Fbxl2 and the full complement of ubiquitination components (right lane in each pair). e. Half-life determination using cycloheximide of each TRAF protein with (red) or without (blue) Fbxl2 plasmid overexpression in MLE cells is shown (n=2± S.D.). Cells were exposed to cycloheximide (40 μg/ml) for up to 10 h. Cells were collected and assayed by immunoblotting. Bands corresponding to each TRAF protein were quantified using imageJ software and graphed to calculate a degradation curve and protein half-lives are shown. f. U937 cells were transfected with an empty plasmid or an Fbxl2 plasmid, followed by LPS exposure (100 ng/ml) for 24 h prior to assays for cytokine secretion using a human cytokine array prior to generation of a heat map (n=3). Green denotes relatively low-level cytokine levels and red represents relatively high-level expression. Panel (a) is representative of n=3 separate experiments, panels (b, d) represent at least n=2 separate experiments; panel c is from an individual experiment.
To identify a shared docking site within TRAF proteins that bind Fbxl2, we initially focused on molecular interaction between TRAF5 and Fbxl2. Wild-type (WT) and several TRAF5 deletional mutants were cloned into a pcDNA3.1D/V5-his vector (Supplementary Fig. 1a and b). Fbxl2 was immunoprecipitated from MLE cell lysates using Fbxl2 antibody and coupled to protein A/G beads. Fbxl2 beads were then incubated with in vitro synthesized his-V5-TRAF5 mutants (Supplementary Fig. 1b top). After washing, proteins were eluted and processed for V5-TRAF5 immunoblotting (Supplementary Fig. 1b bottom). We first determined that Fbxl2 binds within the c-terminal 403–421 residues of TRAF5 (Supplementary Fig. 1b). Sequence alignment of this region revealed conserved tryptophan and lysine residues within all TRAF proteins (Supplementary Fig. 1c). Next, we constructed c-terminal deletion mutants for other TRAF proteins. The TRAF c-terminal deletion mutants devoid of this conserved region lost the ability to bind Fbxl2 (Supplementary Fig. 1d–h). Last, in mutagenesis studies where the conserved tryptophan within TRAF1-6 proteins was substituted with alanine, we observed a significant decrease in interaction between Fbxl2 and TRAF1-6 mutants (Supplementary Fig. 1i). These binding studies are summarized in Supplementary Fig. 1j indicating that the tryptophan site within TRAF1-6 serves as a critical docking site for Fbxl2 interaction. Last, we identified putative ubiquitin acceptor sites within TRAF2-6 and observed that expression of TRAF protein variants containing mutations at these specific lysine residues confers greater protein stability in cycloheximide-based half-life studies (Supplementary Fig. 2).
To assess the biological function of Fbxl2, we monitored U937 human macrophage cytokine release upon lipopolysaccharide (LPS) challenge. Cells were first transfected with empty vector or Fbxl2 plasmid for 24 h before exposure to LPS (100 ng/ml) for an additional 24 h. Thirty six cytokines were measured using a human cytokine array. Fbxl2 cellular expression, compared to empty vector, down-regulated the vast majority of cytokine proteins released after LPS challenge (Fig. 1f). We recapitulated these findings in primary human peripheral blood mononuclear cells (PBMC) that were transfected with either empty vector or Fbxl2 plasmid for 12 h before exposure to LPS (2 μg/ml) for an additional 18 h. LPS treatment significantly increased TRAF protein levels, however Fbxl2 plasmid overexpression was able to variably suppress TRAF protein levels under unstimulated conditions and significantly block LPS-induction of TRAF1-6 proteins in PBMC (Supplementary Fig. 3a). As a complementary approach, when U937 cells were transfected with Fbxl2 short hairpin RNA (shRNA) before exposure to LPS, Fbxl2 knockdown increased several inflammatory cytokines including GM-CSF (3.2 fold), IL-6 (8.5 fold), IFN-γ (1.5 fold), IL-17 (3.5 fold), IL-23 (1.5 fold) and MIP-1β (8.7 fold) compared to LPS alone (data not shown). Fbxl2 knockdown increased TRAF1-6 protein in resting cells and generally augmented LPS-induction of TRAF proteins; these effects were associated with activation of key downstream signaling molecules including phospho-JNK and phospho-IKKα in both unstimulated and LPS stimulated U937 cells (Supplementary Fig. 3b). Thus, Fbxl2 may act as an inhibitor of TRAF protein activation by mediating their cellular disposal to modulate downstream kinase signaling. Because cytokine secretion from cells fluctuated with time, this raised the possibility that F box protein abundance also varied, we therefore investigated Fbxl2 proteolysis. Fbxl2 protein had a short lifespan (t½= 2.5 h), was K201 polyubiquitinated, and degraded in the proteasome (Supplementary Fig. 4).
Fbxl2 is phosphorylated and targeted by Fbxo3
SCF-based E3 ligases target phosphoproteins. Database analysis indicated many potential phosphorylation sites within the Fbxl2 (Fig. 2a). To confirm that Fbxl2 is a phosphoprotein, MLE cells were lysed and subjected to Fbxl2 i.p., and using phosphothreonine antibodies we were able to detect a band which migrated at the predicted size of Fbxl2 (Fig. 2b). To identify the potential kinase that targets Fbxl2 for phosphorylation, we performed co-i.p. experiments. MLE cells were lysed and subjected to Fbxl2 i.p. Of the seven kinases tested, glycogen synthase kinase (GSK3β) was the only protein detected in the Fbxl2 immunoprecipitates (Fig. 2c). GSK3β phosphorylated Fbxl2 in vitro (Fig. 2d).
Figure 2. Fbxl2 is phosphorylated and targeted by the SCF E3 ligase subunit Fbxo3.

a. Scheme of potential phosphorylation sites within Fbxl2 (GPS2.1 prediction). b. Endogenous Fbxl2 was immunoprecipitated from MLE cell lysates and followed by phosphothreonine (Phos-T) immunoblotting. HC=heavy, LC=light immunoglobulin chains. c. Endogenous Fbxl2 was immunoprecipitated from MLE cell lysates followed by immunoblotting for several candidate kinases. d. In vitro GSK3β kinase assay using Fbxl2 as a substrate. MEK1, a dual specificity kinase is shown as a negative control. *heat inactivated GSK3β. e. Endogenous Fbxl2 was immunoprecipitated from MLE cell lysates followed by Fbxo3 immunoblotting. f. In vitro ubiquitination assays. Purified SCFFbxo3 complex components were incubated with V5-Fbxl2 and the full complement of ubiquitination reaction components (right lane) showing polyubiquitinated Fbxl2 (arrows). g. Fbxl2 half-life upon Fbxo3 knockdown (top blot) or Fbxo3 plasmid overexpression (lower blot). MLE cells were either transfected with empty or Fbxo3 plasmid for 24 h, or transfected with scrambled RNA or Fbxo3 shRNA for 48 h. Cells were then exposed to cycloheximide (40 μg/ml) for up to 4.5 h prior to harvesting and processing for immunoblotting. h. U937 cells were transfected with either control shRNA or two sets of Fbxo3 shRNA. 48 h later, cells were exposed LPS (100 ng/ml) for an additional 18 h before harvesting and assaying for Fbxo3, TRAF and actin proteins by immunoblotting. Panels b,d,e,f, and g are representative of n=2 separate experiments, panel h represents n=3 separate experiments, and panel c is from an individual experiment.
Because SCF-based E3 ligases target phosphoproteins 17, we embarked on an unbiased screen randomly testing F box proteins that might mediate Fbxl2 ubiquitination and degradation. Upon overexpression of these proteins, only Fbxo3 was able to decrease immunoreactive levels of Fbxl2 (data not shown). To date, only one study has shown that Fbxo3 increases ubiquitination of a target protein (p300), and its authenticity as an SCF subunit and its substrates remain unknown 22. To confirm the specificity of Fbxo3 targeting Fbxl2, we performed co-i.p. experiments where Fbxo3 was detected in the Fbxl2 immunoprecipitates from MLE cells (Fig. 2e). Further, the SCFFbxo3 complex directly induced Fbxl2 polyubiquitination in vitro (Fig. 2f). Fbxo3 depletion increased Fbxl2 half-life whereas Fbxo3 plasmid overexpression destabilized Fbxl2 protein in MLE cells (Fig. 2g). Fbxo3 knockdown effectively decreased TRAF1-6 protein in both unstimulated and LPS stimulated U937 cells (Fig. 2h).
Using various Fbxl2 deletion mutants (Supplementary Fig. 5a), we performed mapping studies by transfecting cells with his-tagged Fbxl2 plasmid constructs followed by V5/his-pull down. Fbxo3 docked at the carboxyl-terminus (residues 390–410) of Fbxl2 (Supplementary Fig. 5b–d). To confirm that this region is important for Fbxl2 stability, WT Fbxl2 and several Fbxl2 carboxyl-terminal deletion or point mutants were tested for stability (Supplementary Fig. 5e and f). Notably, an Fbxl2 C390 deletion mutant exhibited a prolonged t½ compared to WT Fbxl2, suggesting that residues 390–423 were important for its stability. Consistent with the behavior of SCF based E3 ligase subunits to directly interact with phosphodegrons within their substrates, T404 within Fbxl2 served as a targeting molecular site for Fbxo3 interaction, as Fbxl2T404A lost the ability to bind Fbxo3 (Supplementary Fig. 5d), exhibited a prolonged half-life (Supplementary Fig. 5f), and displayed significant resistance to SCFFbxo3 in vitro ubiquitinylating activity (Supplementary Fig. 5g). To confirm that T404 is an authentic Fbxl2 phosphorylation site, cells transfected with either a WT Fbxl2 or a Fbxl2T404A plasmid were lysed and subjected to V5-Fbxl2 i.p., and immunoblotted using phospho-threonine antibodies; we were able to detect a significant decrease in Fbxl2 T404A protein phosphorylation levels (Supplementary Fig. 5h, lower blot). In summary, Fbxo3 uses Fbxl2T404 as a molecular signature to recruit the SCF complex to polyubiquitinate Fbxl2 at a K201 acceptor site (Supplementary Fig. 5i).
Fbxo3 contains a naturally occurring polymorphism at V221
Human SNP database analysis indicated a naturally occurring non-synonymous C/T polymorphism (rs1402954) within Fbxo3 (Val221Ile) in Caucasians with an allele frequency of 6.2% for European Caucasians (Table 1). To evaluate the biologic relevance of a V221I polymorphism within Fbxo3 in human cells, we analyzed PBMC from healthy Caucasian subjects. Genomic DNA was extracted from PBMC followed by genotyping using a specific primer and probe set. As expected, a total of eight PBMC were heterozygous for the Fbxo3V221I polymorphism among the sixty samples screened (Fig. 3a). In head-to-head studies with WT PBMC, the induction of IL-1β, tumor necrosis factor (TNF), and IL-6 were significantly suppressed in PBMC harboring the Fbxo3V221I polymorphism after LPS challenge (Fig. 3b). Further, these functional differences in Fbxo3V221I PBMC correlated with significantly reduced TRAF 1, TRAF2, TRAF 3, TRAF 4, and TRAF6 proteins and elevated Fbxl2 protein (Fig. 3c). Thus, a naturally occurring human Fbxo3V221I polymorphism might confer a reduced pro-inflammatory phenotype.
Table 1.
| Chr 11 pos | Sequence | AA change | ||
|---|---|---|---|---|
| 33777334(+) | GACATTGGTAGAAAA | V220I | ||
| C/ TTTCATTTTTGTTCG | ||||
| Sample Ascertainment | Alleles | |||
| ss# | Individual Group | Sample | C | T |
| ss24492449 | European | 48 | 0.875 | 0.13 |
| African American | 46 | 1 | ||
| Asian | 48 | 1 | ||
| ss38688029 | European | 220 | 0.945 | 0.06 |
| Asian | 90 | 1 | ||
| Asian | 88 | 1 | ||
| African | 120 | 1 | ||
| Italian | 174 | 0.885 | 0.12 | |
| ss69314691 | European | 120 | 0.95 | 0.05 |
| Asian | 90 | 1 | ||
| Asian | 90 | 1 | ||
| African | 120 | 1 | ||
SNP analysis of Fbxl2 protein indicating a V221I polymorphism (non-coding strand is shown http://www.ncbi.nlm.nih.gov/projects/SNP).
Figure 3. Fbxo3 contains a naturally occurring polymorphism at V221.

a. Genomic DNA was extracted from human PBMC from sixty healthy Caucasian donors followed by SNP genotyping using the TaqMan® SNP probes with real-time PCR. TaqMan® SNP genotyping was performed using probes containing VIC™ dye linked to the 5′ end of the wild-type (WT) probe, and a FAM™ dye linked to the 5′ end of the mutant probe. An increase in the VIC-dye fluorescence signal generated by real-time PCR indicates homozygosity for allele 1(+/+), whereas an increased FAM-dye fluorescence indicates homozygosity for allele 2 (−/−); an increase in both VIC-dye and FAM-dye fluorescence signal intensity indicates allele 1-allele 2 Fbxo3 heterozygosity (+/−). Shown are relative numbers of WT (blue, ++) or heterozygotes (red, +/−) identified. b–c. Eight WT human PBMC and eight PBMC containing the heterozygous V221I polymorphism were treated with LPS (2 μg/ml) for 24 h before assays for cytokine release (b) or TRAFs, Fbxl2, and Fbxo3 immunoblotting (c). Bands corresponding to indicated proteins on immunoblots were quantified using ImageJ software and results displayed graphically (c).
Fbxo3V221I: a loss-of-function F box protein polymorphism
To further assess the behavior of Fbxo3V221I, we measured ubiquitinylating activity of the F box protein. Compared to WT Fbxo3, the SCFFbxo3V221I complex displayed markedly reduced ability to polyubiquitinate Fbxl2 with most of the substrate intact (Fig. 4a). By using U937 cells, which adopt the morphology and many characteristics of mature macrophages, Fbxo3 plasmid overexpression decreased Fbxl2 protein, yet increased the amount of all six TRAF proteins (Fig. 4b), but not their steady-state mRNA levels (data not shown). However, overexpression of Fbxo3V221I (Fbxo3 V/I) plasmid in U937 cells resulted in no or only modest increases in TRAF proteins (Fig. 4b). As a control we identified another Fbxo3 mutation (Fbxo3P109R) that when overexpressed was able to increase TRAF protein comparable to WT Fbxo3 plasmid (data not shown). Thus P109R is not a loss of function mutation of Fbxo3. We further monitored U937 cell cytokine release by array upon LPS challenge by transfecting cells with LacZ, Fbxo3, or Fbxo3V221I plasmids for 24 h before exposure to LPS. Fbxo3 transfection stimulated elevated secretion of the majority of cytokines tested following LPS challenge. In contrast, Fbxo3V221I plasmid expression did not dramatically alter cytokine release from cells compared to the LacZ control (Supplementary Fig. 6a). Hence, the in vitro studies suggest that Fbxo3V221I is a loss-of-function polymorphism of Fbxo3. As an acute cytokine storm contributes to tissue injury, the results raise the possibility that expression of this polymorphism in vivo might alter host inflammatory responses or vulnerability to inflammatory injury.
Figure 4. Fbxo3V221I is a loss-of-function polymorphism of Fbxo3 in vitro.

a. In vitro ubiquitination assays. Purified SCFFbxo3 (WT, left two lanes) or SCFFbxo3V221I variant complex (V221I, right two lanes) components were incubated with V5-Fbxl2 and the full complement of ubiquitination reaction components showing levels of polyubiquitinated Fbxl2 (arrows). b. U937 cells were transfected with V5-WT Fbxo3 or the V5-Fbxo3V221I plasmids, followed by immunoblotting for V5, Fbxl2, and TRAF proteins. c–e. C57BL/6J mice were administered diluent (control, CON), intratracheal (i.t.) lenti-LacZ, lenti-Fbxo3, or lenti-Fbxo3V221I (107 pfu/mouse, n=4–6 mice/group) for 120 h, prior to inoculation with P. aeruginosa (PA103, 104 cfu/mouse) for 24 h. Mice were then euthanized, lungs were lavaged with 3 ml saline, harvested, and then homogenized; lavage protein (c), cell counts (d), and cytokine secretion (e) were determined. f. Survival studies of mice administered i.t. PA103 (105 cfu/mouse, n= 7 mice/group) was determined. Mice were carefully monitored over time; moribund, preterminal animals were immediately euthanized and recorded as deceased. Kaplan-Meier survival curves were generated using Prism software (P1: Fbxo3 V/I versus Fbxo3, P2: LacZ versus Fbxo3). g. H&E staining was performed on lung samples in (c–d). *P<0.05 versus Fbxo3. #P<0.05 versus LacZ or CON. Panel a is representative of n=2 separate experiments, panel b represents n=3 separate experiments.
To extend the above observations in vivo, mice were infected with an empty lentivirus, or lentivirus encoding either Fbxo3 or Fbxo3V221I genes for 120 h (107 pfu/mouse, intratracheally [i.t.]). Mice were then challenged with Pseudomonas aeruginosa (strain PA103,104 cfu/mouse, i.t.) for an additional 24 h. Mice were then euthanized prior to analysis of parameters of inflammatory injury. WT Fbxo3 plasmid expression, but not Fbxo3V221I, augmented PA103 induced lung injury. Specifically, Fbxo3 gene transfer significantly increased lavage protein concentration and cell counts (Fig. 4c,d). In addition, Fbxo3 plasmid overexpression in vivo significantly increased lavage cytokine levels compared to mice receiving an empty vector with PA103 infection (Fig. 4e). Mice ectopically expressing Fbxo3 also had significantly reduced survival after PA103 infection compared to mice infected with PA103 alone (Fig. 4f). Lastly, Fbxo3 plasmid overexpression significantly increases lung cell infiltrates in mice (Fig. 4g). These biologic effects were less pronounced after gene transfer of the Fbxo3V221I plasmid, further supporting that Fbxo3V221I is a loss-of-function polymorphism of an F box protein. Thus, Fbxo3 triggers pro-inflammatory effects in a model of Pseudomonas pneumonia.
Fbxo3 knockdown ameliorates pseudomonas lung injury
To assess a requirement of Fbxo3 in pneumonia, we pursued in vivo knockdown studies where mice were infected with lentivirus encoding empty shRNA or Fbxo3 shRNA for 120 h (107 pfu/mouse, i.t). Mice were then challenged with PA103 for an additional 24 h. Fbxo3 knockdown significantly ameliorated adverse effects of PA103 on lung mechanics as depletion of the Fbxo3 led to increased compliance, and decreased lung resistance with reduced elastance (Fig. 5a–d). Fbxo3 knockdown also decreased lavage protein concentration, lavage cell counts, and cell infiltrates (Fig. 5e–g). Further, mice receiving Fbxo3 silencing plasmid had decreased lavage cytokine levels after PA103 infection (Fig. 5h) and had increased survival (Fig. 5i). These in vivo studies suggest that endogenous Fbxo3 plays an essential role in mediating cytokine-driven inflammation during microbial infection.
Figure 5. Fbxo3 knockdown ameliorates pseudomonas-induced lung injury.

C57BL/6J mice were administered diluent (control, CON), lentivirus encoding control shRNA (CON shRNA) or lenti-Fbxo3 shRNA (107 pfu/mouse) (intratracheally [i.t.], n=5 mice/group) for 120 h, and mice were then inoculated with PA103 (104 cfu/mouse) for 24 h. Mice were monitored on FlexiVent to measure lung mechanics (a–d). Mice were then euthanized and lungs lavaged with saline, harvested, and then homogenized. Lavage protein, cell counts, and cytokine secretion were measured in (e, f, h). g. H&E staining representative 1 of 4 samples from (a) was performed on lung tissue. i. Survival studies of mice administered PA103 (i.t. 105 cfu/mouse, n=6 mice per group) was determined (time=hours). Mice were carefully monitored over time; moribund, preterminal animals were immediately euthanized and recorded as deceased. Kaplan-Meier survival curves were generated using Prism software. *P<0.05 versus CON shRNA and #P<0.05 versus CON.
To further assess specificity of our model, we used Pam3CSK4 and TNF to stimulate U937 cells after Fbxo3 depletion. Briefly, U937 cells were first transfected with either control shRNA or Fbxo3 shRNA for 48 h, cells were then exposed to either Pam3CSK4 (500 ng/ml) (Supplementary Fig. 6b) or TNF (20 ng/ml) (Supplementary Fig. 6c) for an additional 18 h. Cell free medium was collected and assayed for mediator secretion. Compared to LPS stimulation, Pam3CSK4 and TNF were less potent in triggering release of a broad range of cytokines (data not shown). However, of the pro-inflammatory products induced by Pam3CSK4, Fbxo3 knockdown was able to suppress release of CCL1 (I-309), IFN-γ, IL-8, CCL5 (RANTES) and TNF (Supplementary Fig. 6b). Moreover, of the proteins induced by TNF, Fbxo3 knockdown was able to suppress CD40L, CCL1, IL-1α, IL1-β, IL-23 and serpin E1 (Supplementary Fig. 6c). Thus, depletion of endogenous Fbxo3 also appears to reduce, in part, certain pro-inflammatory mediators released by stimuli other than endotoxin such as Pam3CSK4 and TNF.
Fbxo3-TRAF pathway in subjects with sepsis
Pro-inflammatory cytokines were assayed in plasma from control subjects (n=7) including two patient controls mechanically ventilated without acute lung injury, and septic subjects (n=16). All septic subjects met the definition evidenced by the presence of a systemic inflammatory response associated with infection confirmed by culture (or strongly suspected) with or without signs of hypoperfusion or organ dysfunction 23 (Supplementary Table 1). Four subjects with sepsis had positive blood cultures and the majority of the remaining subjects had evidence of localizing pulmonary, gastrointestinal, or urinary infectious sources. Subjects with sepsis had significantly elevated plasma levels of TNF, IL-1β, and IL-6 (Fig. 6a). We analyzed TRAF1-6, Fbxl2, and Fbxo3 protein content by immunoblotting in circulating white blood cells from control subjects (n=6) and septic subjects (n=10) with available leukocytes. Septic subjects had a significant increase in TRAF2, TRAF4, TRAF5, TRAF6 and Fbxo3 proteins and decreased immunoreactive Fbxl2 protein in cells versus control subjects (Fig. 6b). We also extracted genomic DNA from septic subjects (n=16) for genotyping; in this group we identified two subjects harboring an Fbxo3V221I polymorphism (Het). In accordance with its putative hypofunction, the Fbxo3V221I subjects expressed lower plasma TNF, IL-1β, and IL-6 protein compared to subjects harboring WT Fbxo3 (Fig. 6a, inset). These results suggest that the Fbxo3-TRAF pathway is functional in individuals with systemic inflammation.
Figure 6. F box and TRAF proteins in subjects with sepsis.

a. Plasma from control (CON) subjects (n=7) and septic subjects (n=16) were assayed for TNF, IL-1β, and IL-6 using a cytokine ELISA kit. Inset shows levels of cytokines in heterozygotic (Het) subjects harboring a Fbxo3V221I (n=2, m±SD) polymorphism and subjects harboring wild type Fbxo3 (n=14). b. Leukocytes from control subjects (n=6) and septic subjects (n=10) were assayed for TRAFs, Fbxl2 and Fbxo3 protein by immunoblotting and results quantified using ImageJ software and data displayed graphically.
Small molecule inhibition of Fbxo3
Fbxo3 harbors a 125 amino acid bacterial-like ApaG domain within its carboxyl-terminus, the function of which is unknown. Structural analysis from different ApaG proteins shows a fold of several beta-sheets. Deletional mapping indicated that the Fbxo3-ApaG signature was required for Fbxl2 ubiquitination (data not shown). Based on molecular docking and best-fit analysis of suitable Fbxo3 ligands, we selected benzathine as a backbone to develop a series of small molecules (Fig. 7a). One such molecule, termed BC-1215, scored high on docking studies with Fbxo3-ApaG, and it exhibited a low half maximal inhibitory concentration (IC50) (0.9 μg/ml) with regard to IL-1β release and a high lethal concentration, 50% (LC50) (87 μg/ml) in vitro (Fig. 7b). BC-1215 decreased Fbxo3-Fbxl2 interaction in a dose-dependent manner and prevented SCFFbxo3 catalyzed Fbxl2 ubiquitination (data not shown). Furthermore, BC-1215 effectively reduced TRAF 1-6 protein levels (Supplementary Fig. 7a) without altering TRAF steady-state mRNA levels (data not shown). BC-1215 not only decreased TRAF1-6 protein under resting conditions, but also blocked LPS induction of TRAF1-6 proteins in PBMC (Supplementary Fig. 7b). BC-1215 also blunted induction of TRAF1-6 proteins after ectopic expression of Fbxo3 plasmid in U937 cells (Supplementary Fig. 7c). Thus, BC-1215 inhibits the Fbxo3-TRAF activation pathway by destabilizing TRAF1-6. Last, BC-1215 inhibited LPS-induced secretion of a broad spectrum of TH1 panel cytokines in human PBMC (Supplementary Fig. 7d).
Figure 7. Fbxo3 inhibitor BC-1215 reduces bacterial-induced inflammation.

a. Structures of a base compound benzathine and BC-1215. b. PBMC (0.6 ml at 1.5 × 106/ml) were treated with LPS (2 μg/ml) for 16 h with BC-1215 and cytokine release was monitored by ELISA. The drug concentrations that effectively reduced cytokine release were used to calculate the IC50. In separate studies, U937 cells (0.6 ml at 1.5 × 106/ml) were treated with BC-1215 for 16 h. Cells were then stained with trypan blue to identify dead cells to calculate the LC50 (or LD50). c. Mice were anesthetized prior to administration of BC-1215 (100 μg) through an intraperitoneal (i.p.) injection immediately prior to cecal ligation and puncture (CLP) 43. 6 h later, mice were euthanized and blood collected for assays of IL-6, TNF, and IL1-β levels. d. Peritoneal fluid was obtained for determination of bacterial counts after CLP in mice. e–h. BC-1215 (100 μg) was administered to mice though an i.p. injection, and mice were immediately challenged with P. aeruginosa (strain PA103, 104 cfu/mouse, i.t.) or without (control, CON) for an additional 18 h. Mice were then euthanized and lungs were lavaged with saline, harvested, and then homogenized. Lavage protein (e), cell counts (f) and cytokine secretion (h) were measured. g. H&E staining was performed on lung samples. The data in (c, d) represent n=4–5 mice/group, *P<0.05 BC-1215 with CLP versus CLP and **P<0.001 versus SHAM. The data in e–h are representative of data from n=4–6 mice/group, *P<0.05 versus vehicle, ** P<0.05 versus CON.
Anti-inflammatory activity of Fbxo3 inhibitor
To assess in vivo anti-inflammatory activity, we first tested effects of administration of BC-1215 in a model of a cecal ligation and puncture (CLP)-induced sepsis. Mice with CLP had significantly increased cytokine release compared to sham treated mice. BC-1215 treatment significantly attenuated CLP-induced secretion of all three circulating pro-inflammatory cytokines in mice (Fig. 7c). BC-1215 also decreased bacterial counts in the CLP model (Fig. 7d) and modestly produced inhibition of bacterial growth using Kirby Bauer testing (data not shown). Finally, BC-1215 was administered in a pneumonia model where it significantly ameliorated adverse effects of PA103 on lung mechanics (data not shown). BC-1215 also substantially decreased lavage protein concentration, lavage cell counts, and reduced cell infiltrates after PA103 infection (Fig. 7e–g). These beneficial effects of the agent were associated with significantly decreased lavage pro-inflammatory cytokine levels in PA103 infected mice (Fig. 7h). Similar effects of BC-1215 were observed in an H1N1 pneumonia model (data not shown). Hence, small molecule targeting of the Fbxo3→TRAF pathway reduces severity of cytokine-driven inflammation in preclinical models (Fig. 8).
Figure 8. Molecular regulation of pro-inflammatory cytokines mediated by F box proteins.

Microbial infection or stimuli can robustly activate a variety of cell surface receptors linked to TRAF proteins that serve as critical intermediary signaling proteins to mediate cytokine synthesis and release. The F box protein Fbxl2 serves as a sentinel inhibitor of TRAFs by mediating their polyubiquitination (red circles) and proteasomal degradation in cells. Fbxl2 specifically targets at a conserved tryptophan domain within all TRAF 1-6. During microbial infection, another F box protein, Fbxo3, targets Fbxl2 for its ubiquitination and degradation at K201; this process is facilitated by glycogen synthase kinase (GSK3β) phosphorylation (green circle) of Fbxl2 at T404. WT Fbxo3 in this pathway potently activates cytokine driven inflammation, whereas a naturally occurring Fbxo3V221I polymorphism is hypofunctional. A small molecule Fbxo3 inhibitor, BC-1215, reduces inflammation by antagonizing actions of Fbxo3 on TRAF–cytokine signaling.
DISCUSSION
Cytokine driven inflammation is a hallmark of severe infectious and noninfectious disorders that can contribute significantly to tissue injury. Here we provide the first evidence for two indispensible molecular inputs, Fbxo3 and Fbxl2, into TRAF mediated signaling for cytokine gene expression. The data authenticate Fbxo3 as an E3 ligase subunit that triggers ubiquitination and degradation of another E3 ligase subunit, Fbxl2, thereby increasing levels of TRAF proteins. In essence, the SCFFbxl2 complex appears to act as a counter-regulatory repressor of inflammation by destabilizing TRAF proteins. Specifically, Fbxl2 targets six TRAF family proteins (TRAF1-6) for their ubiquitination and degradation using a conserved molecular signature leading to reduced cytokine secretion in inflammatory cells. In turn, Fbxo3 opposes Fbxl2 activity by specifically targeting it for ubiquitination and degradation to stimulate cytokine release. Targeting is facilitated by GSK3β phosphorylation of Fbxl2, this provides a phosphodegron molecular signal for Fbxo3 recruitment to Fbxl2. In addition, our data reveal the presence of a naturally occurring hypofunctional Fbxo3V221I polymorphism. Human monocytes expressing this polymorphism showed reduced sensitivity to endotoxin stimulation. These data may be clinically relevant as subjects with sepsis exhibited activation of the Fbxo3-TRAF pathway within circulating leukocytes that correlated with elevated pro-inflammatory cytokines in plasma. The discovery of Fbxo3 as a stimulator of TRAF signaling provided the mechanistic platform that led to the design, synthesis, and biological evaluation of an F box small molecule inhibitor that exerted robust anti-inflammatory activity by impairing cytokine release in vivo. The effectiveness of this inhibitor underscores the physiological importance of the Fbxo3-Fbxl2 pathway for regulating inflammation.
Individual TRAF proteins may exert very distinct functions and F box protein regulation of this family might lead to divergent biological responses affecting long-term adaptive immunity or the innate immune system depending on the cell type, the surface receptor, or the predominant TRAF protein that is modulated by any given stimulus 24. For example, overexpression of TRAF2 and TRAF5 can activate NF-κB canonical signaling 25. TRAF3 is highly conserved with TRAF2 and TRAF5, however, TRAF3 overexpression does not activate the NF-κB pathway and it negatively regulates TLR-stimulated pro-inflammatory cytokine release 26–28. Splice variants with altered Zn2+ finger domains of TRAF3 do activate the NF-κB pathway 29. Thus, the ability of F box proteins to elicit specific innate or adaptive effector functions will depend on the differential availability of these intermediary adaptors within any given cell population.
Aside from TRAF1, TRAFs themselves function as ubiquitin E3 ligases through their RING dimerization interface that mediate K63 polyubiquitination of adaptor molecules; TRAFs are also prone to ubiquitination by a HECT-type E3 Smad ubiquitination regulation factor 1 and Triad3A 30,31,32. An E3 ligase can target multiple proteins for ubiquitination and different E3 ligases can target the same substrate 33. Here one E3 ligase subunit (Fbxl2) within the SCF E3 ligase complex ubiquitinates several TRAF proteins. Thus, given the common Fbxl2 docking site identified within TRAF proteins, our data are not inconsistent with the promiscuity of some F box proteins and highlights a potentially fundamental role for this F box protein in regulating the innate immune response during agonist-induced inflammation.
The molecular behavior of Fbxo3 appears to be unique by virtue of its ability to target Fbxl2 for ubiquitination through its prokaryotic ApaG domain. Some components within the SCF machine are targeted by the ubiquitin ligase anaphase-promoting complex or by auto-ubiquitination within their own SCF complex 34,35. It is likely that Fbxo3 modulates the TRAF-cytokine axis through Fbxl2, however, the results do not rule out other intermediary proteins that link Fbxo3 with TRAF proteins. Importantly, the Fbxo3 ApaG domain served as the mechanistic centerpiece and structural basis for our drug design testing actions of F box inhibitors. This domain is present in various species of bacteria such as Xanthomonas, Vibrio, and Salmonella where it is postulated to interact with pyrophosphate, nucleotide phosphates, or heavy metals 36. The ApaG domain to date has been identified only within some F box proteins and PDIP38, a protein that interacts with proliferating cell nuclear antigen 37,38. By adding a pyridine group to the benzathine backbone generating BC-1215, the small molecule generated here optimally interacted with ApaG to profoundly inhibit secretion of a broad spectrum of TH1 panel cytokines from human PBMC. Indeed, structure-activity analysis led to synthesis of several modified Fbxo3 antagonists that displayed potent anti-inflammatory activities (data not shown).
Our model of Pseudomonas induced pneumonia in mice was recapitulated, in part, with observations in humans with sepsis underscoring the biologic relevance of the Fbxo3-TRAF pathway for cytokine secretion. Functional characterization of eight human PBMC harboring Fbxo3V221I polymorphisms unexpectedly showed that these cells do not fully retain cytokine responses to LPS-stimulation. Because valine and isoleucine are similarly branched chain hydrophobic residues, the prediction is that such naturally occurring polymorphisms would not result in profound functional consequences. Interestingly, similar amino acid polymorphisms within ubiquitin-specific protease 26 are linked to human phenotypes 39. All subjects identified in our studies were heterozygotes who had diminished immunoreactive Fbxo3 levels, lower TRAF proteins, and cytokine responses coupled with higher Fbxl2 protein levels. However, in these heterozygotic subjects, TRAF5 levels were not altered versus wild-type subjects that may be attributed to the experimental design and kinetics of TRAF5 expression or its unique molecular regulation. It is possible that heterozygotic Fbxo3V221I polymorphisms confer a selective biological advantage in silent carriers after microbial infection by limiting host inflammation that significantly contributes to end-organ damage. This heterozygotic advantage within Fbxo3 would mimic other genetic mutations such as sickle cell anemia 40–42. The inability to identify homozygotic Fbxo3 subjects might be due to its genetic lethality with marked inability of subjects to recover from severe infection as innate immunity might be significantly disarmed. Future studies will evaluate a larger cohort of subjects with sepsis and other immunological disorders and correlate TRAFs, F box proteins, and inflammatory responses to additional clinical endpoints. Ultimately the heterogeneity of cytokine responses across individuals that stem from Fbxo3-TRAF signaling might lend itself to personalized therapy using Fbxo3 inhibitors in subjects with inflammatory illness.
ONLINE METHODS
Materials
Cells lines were obtained from ATCC. Purified SCFFbxo3 complex was purchased from Abnova. Purified ubiquitin, E1, E2, MG132, and cycloheximide were purchased from Calbiochem. Molecular reagents were from Invitrogen. Lentivirus kit was from Clontech. PBMC were from Sanguine Life Sciences. The F box proteins cDNA, scrambled shRNA, Fbxo3 and Fbxl2 shRNA sets were purchased from OpenBiosystems. MG132 was from Roche. The Fbxl2 antibodies were from Aviva Biosciences and Santa Cruz. Fbxo3 and all TRAFs antibodies were from Santa Cruz. IL1β, TNFα, IL6, human cytokine array were from R&D systems. Pam3GSK4 was from InvivoGen.
Human Samples
This study was approved by the University of Pittsburgh Institutional Review Board. After obtaining informed consent from the subjects, we collected blood from subjects diagnosed with sepsis (clinical diagnosis and/or confirmed with blood culture) and healthy donors. Blood was further processed for the plasma and leukocyte fraction. Plasma samples were processed for cytokine assays. Cell pellets were assayed for proteins by immunoblotting.
Cell culture
MLE cells were cultured in Dulbecco’s Modified Eagle Medium-F12 (Gibco) supplemented with 2–10% fetal bovine serum (DMEM-2 or 10). U937 cells and PBMC were cultured in RPMI medium supplemented with 10% fetal bovine serum. For Fbxl2 plasmid was overexpressed in PBMCs using Fugene6HD; cells were treated with LPS at 2 μg/ml for an additional 18 h. For Fbxo3 or Fbxl2 knockdown studies in U937 cells, Hilymax reagent was used to transfect cells for 48 h. U937 cells were then treated with LPS at 100 ng/ml, Pam3CSK4 at 500 ng/ml or TNFα at 20 ng/ml for an additional 18 h. For drug treatment, compounds were solubilized either in acetic acid or ethanol before adding to the cells for up to 18 h. For half-life studies, MLE cells were exposed to cycloheximide (40 μg/ml) for up to 10 h.
In vitro binding assays
Fbxl2 was immunoprecipitated from 1 mg of MLE cell lysate using Fbxl2 antibody (goat) and coupled to protein A/G beads. Fbxl2 beads were then incubated with in vitro synthesized products (50μl) expressing his-V5-TRAF5 mutants. After washing, proteins were eluted and processed for V5-TRAF immunoblotting. Similarly, Fbxo3 was immunoprecipitated from 1 mg MLE cell lysate using Fbxo3 antibody (rabbit) and coupled to protein A/G beads. Fbxo3 beads were then incubated with in vitro synthesized products (50 μl) expressing his-V5-Fbxl2 mutants. After washing, proteins were eluted and processed for V5-Fbxl2 immunoblotting.
In vitro ubiquitin conjugation assays
The ubiquitination of substrates were performed in a volume of 25 μl containing 50 mM Tris pH 7.6, 5 mM MgCl2, 0.6 mM DTT, 2 mM ATP, 1.5 ng/μl E1, 10 ng/μl Ubc5, 10 ng/μl Ubc7, 1μg/μl ubiquitin (Calbiochem), 1 μM ubiquitin aldehyde, 4–16μl of purified Cullin1, Skp1, Rbx1, and in vitro synthesized Fbxl2 or Fbxo3. Reaction products were processed for V5 immunoblotting.
Molecular docking studies and compound design
The docking experiments were carried out by using software from Discovery studio 3.1. A library containing 6507 approved or experimental drugs were first used to screen potential ligands for Fbxo3. Based on the docking and best-fit analysis of suitable ligands, benzathine was selected as a backbone to develop a series of new small molecules, including BC-1215.
Quantitative RT–PCR, cloning, and mutagenesis
All mutant plasmid constructs were generated using PCR-based approaches using appropriate primers and subcloned into a pcDNA3.1D/V5-His vector.
Lentivirus construction
To generate lentivirus encoding Fbxo3, Plvx-Fbxo3 plasmid was co-transfected with Lenti-X HTX packaging plasmids into 293FT cells following the manufacturers’ instructions. 72 h later, virus was collected and titered using a p24 Rapid Titration Kit (Clontech).
Cytokine arrays
Cytokine array panel A (R&D) was used to profile thirty-six cytokines in cell free medium or murine lavage. Each cytokine signal value was quantified using imageJ software based on the relative signal intensities in each group and converted into a heatmap (Heatmap Builder).
SNP genotyping
Genomic DNAs were extracted from PBMC or leukocyte samples. 10 ng genomic DNA (11.25 μl), 12.5 μl of Taqman Universal PCR master mix and 1.25 μl of SNP genotyping assay were added to optical reaction plates followed by real-time PCR and results were analyzed using CFX manager software (BioRad). TaqMan® SNP genotyping was performed using a specific primer and probe set. Each TaqMan probe contains a VIC™ dye linked to the 5′ end of the wild-type (WT) probe, a FAM™ dye linked to the 5′ end of the mutant probe, and a nonfluorescent quencher (NFQ) at the 3′ end of each probe. The fluorescence signal generated by real-time PCR system indicates which alleles are present in the sample.
Animal studies
C57BL6 mice were anesthetized and the larynx was visualized before endotracheal intubation with a 24-gaugeplastic catheter. 107 CFU of lentivirus encoding genes was instilled intratracheally (i.t.) for 120 h before administration of P. aeruginosa (strain PA103, 104CFU/mouse, i.t.) for 18 h, after which animals were analyzed. Pulmonary function analysis was performed using Flexivent. Bronchoalveolar lavage (BAL) fluid from animals was collected by flushing the lung using 3 ml of sterile PBS. Survival studies of mice were performed on mice that were administered P. aeruginosa (strain PA103, 105 CFU/mouse, i.t. 7 mice/group). For drug studies, BC-1215 (100 μg) was administered to mice through an i.p. injection. For CLP, a 3-cm midline laparotomy was made first through the skin and then the cecum with the adjoining intestine was exteriorized and ligated at 0.5 cm from its end with a 3.0 silk. The ligated cecum was punctured with an 18-gauge needle, allowing entrapped fecal material to leak into the normally sterile peritoneal cavity. The abdomen was closed. Sham-operated animals received laparotomy only. After 6 h, plasma was collected. For pneumonia studies, PA103, was instilled i.t. with administration of BC-1215 (100 μg, intraperitoneal [i.p.]) for 18 h, after which animals were analyzed. All procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Statistical Analysis
Statistical comparisons were performed with the Prism program, version 4.03 (GraphPad Software, Inc., San Diego, CA) using an ANOVA 1 or an unpaired 2 t-test with p<0.05 indicative of significance.
Supplementary Material
Acknowledgments
The authors thank Alex Lagneaux and Suchitra Barge for assistance with studies. This material is based upon work supported, in part, by the US Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. This work was supported by a Merit Review Award from the US Department of Veterans Affairs and National Institutes of Health R01 grants HL096376, HL097376 and HL098174 (to R.K.M.), HL116472 (to B.B.C.) HL01916 (to Y.Z.), P50HL084948 (F.C.S.), and American Heart Association (AHA) awards 12SDG9050005 (J.Z.), 12SDG12040330 (C.Z.), and AHA Grant-in Aid 12GRNT11820019 (B.J.M.). The contents presented do not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
AUTHOR CONTRIBUTIONS
R.K.M. and B.B.C. were jointly responsible for oversight of the studies. B.B.C. designed the study, performed experiments, analyzed the data and wrote the manuscript; T.A.C., J.R.G., J.Z, Y.Z. and C.Z. performed experiments and assisted with animal experiments; B.J.M., B.E., F.S. F.C.S. and Y.Z. assisted with human studies. R.K.M. revised the manuscript and directed the study.
COMPETING FINANCIAL INTERESTS
A provisional patent application (US 61/657, 423) covering Fbxo3 inhibitors and additional modifications was filed jointly through the United States Department of Veterans Affairs and the University of Pittsburgh. The authors declare no competing financial interests.
References
- 1.Inoue J, et al. Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp Cell Res. 2000;254:14–24. doi: 10.1006/excr.1999.4733. [DOI] [PubMed] [Google Scholar]
- 2.Xu LG, Li LY, Shu HB. TRAF7 potentiates MEKK3-induced AP1 and CHOP activation and induces apoptosis. J Biol Chem. 2004;279:17278–82. doi: 10.1074/jbc.C400063200. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez SE, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–8. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.London NR, et al. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med. 2010;2:23ra19. doi: 10.1126/scitranslmed.3000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrison C. Sepsis: calming the cytokine storm. Nat Rev Drug Discov. 2010;9:360–1. doi: 10.1038/nrd3162. [DOI] [PubMed] [Google Scholar]
- 6.Lutgens E, et al. Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med. 2010;207:391–404. doi: 10.1084/jem.20091293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu LJ, et al. Suppression of tumor necrosis factor receptor associated factor (TRAF)-2 attenuates the proinflammatory and proliferative effect of aggregated IgG on rat renal mesangial cells. Cytokine. 2010;49:201–8. doi: 10.1016/j.cyto.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Yamashita M, et al. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell. 2008;31:918–24. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamothe B, et al. The RING domain and first zinc finger of TRAF6 coordinate signaling by interleukin-1, lipopolysaccharide, and RANKL. J Biol Chem. 2008;283:24871–80. doi: 10.1074/jbc.M802749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ha H, Han D, Choi Y. TRAF-mediated TNFR-family signaling. Curr Protoc Immunol. 2009;Chapter 11(Unit 11):9D. doi: 10.1002/0471142735.im1109ds87. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka Y, et al. c-Cbl-dependent monoubiquitination and lysosomal degradation of gp130. Mol Cell Biol. 2008;28:4805–18. doi: 10.1128/MCB.01784-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hochstrasser M. Biochemistry. All in the ubiquitin family. Science. 2000;289:563–4. doi: 10.1126/science.289.5479.563. [DOI] [PubMed] [Google Scholar]
- 13.Tyers M, Willems AR. One ring to rule a superfamily of E3 ubiquitin ligases. Science. 1999;284:601, 603–4. doi: 10.1126/science.284.5414.601. [DOI] [PubMed] [Google Scholar]
- 14.Zheng N, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–9. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 15.Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–51. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- 16.Jin J, et al. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18:2573–80. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cenciarelli C, et al. Identification of a family of human F-box proteins. Curr Biol. 1999;9:1177–9. doi: 10.1016/S0960-9822(00)80020-2. [DOI] [PubMed] [Google Scholar]
- 18.Chen BB, Coon TA, Glasser JR, Mallampalli RK. Calmodulin antagonizes a calcium-activated SCF ubiquitin E3 ligase subunit, FBXL2, to regulate surfactant homeostasis. Mol Cell Biol. 2011;31:1905–20. doi: 10.1128/MCB.00723-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen BB, et al. F box protein FBXL2 targets cyclin D2 for ubiquitination and degradation to inhibit leukemic cell proliferation. Blood. 2012;119:3132–41. doi: 10.1182/blood-2011-06-358911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen BB, Glasser JR, Coon TA, Mallampalli RK. F-box protein FBXL2 exerts human lung tumor suppressor-like activity by ubiquitin-mediated degradation of cyclin D3 resulting in cell cycle arrest. Oncogene. 2011 doi: 10.1038/onc.2011.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen BB, Glasser JR, Coon TA, Mallampalli RK. FBXL2 is a ubiquitin E3 ligase subunit that triggers mitotic arrest. Cell Cycle. 2011;10:3487–94. doi: 10.4161/cc.10.20.17742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shima Y, et al. PML activates transcription by protecting HIPK2 and p300 from SCFFbx3-mediated degradation. Mol Cell Biol. 2008;28:7126–38. doi: 10.1128/MCB.00897-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levy MM, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250–6. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- 24.Hildebrand JM, et al. Roles of tumor necrosis factor receptor associated factor 3 (TRAF3) and TRAF5 in immune cell functions. Immunol Rev. 2011;244:55–74. doi: 10.1111/j.1600-065X.2011.01055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishida TK, et al. TRAF5, a novel tumor necrosis factor receptor-associated factor family protein, mediates CD40 signaling. Proc Natl Acad Sci U S A. 1996;93:9437–42. doi: 10.1073/pnas.93.18.9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vallabhapurapu S, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol. 2008;9:1364–70. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tseng PH, et al. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol. 2010;11:70–5. doi: 10.1038/ni.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie P, Stunz LL, Larison KD, Yang B, Bishop GA. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity. 2007;27:253–67. doi: 10.1016/j.immuni.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Eyndhoven WG, Gamper CJ, Cho E, Mackus WJ, Lederman S. TRAF-3 mRNA splice-deletion variants encode isoforms that induce NF-kappaB activation. Mol Immunol. 1999;36:647–58. doi: 10.1016/s0161-5890(99)00079-6. [DOI] [PubMed] [Google Scholar]
- 30.Habelhah H. Emerging complexity of protein ubiquitination in the NF-kappaB pathway. Genes Cancer. 2010;1:735–747. doi: 10.1177/1947601910382900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li S, et al. Ubiquitin ligase Smurf1 targets TRAF family proteins for ubiquitination and degradation. Mol Cell Biochem. 2010;338:11–7. doi: 10.1007/s11010-009-0315-y. [DOI] [PubMed] [Google Scholar]
- 32.Nakhaei P, et al. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 2009;5:e1000650. doi: 10.1371/journal.ppat.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skaar JR, D’Angiolella V, Pagan JK, Pagano M. SnapShot: F Box Proteins II. Cell. 2009;137:1358, 1358 e1. doi: 10.1016/j.cell.2009.05.040. [DOI] [PubMed] [Google Scholar]
- 34.Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature. 2004;428:190–3. doi: 10.1038/nature02330. [DOI] [PubMed] [Google Scholar]
- 35.Galan JM, Peter M. Ubiquitin-dependent degradation of multiple F-box proteins by an autocatalytic mechanism. Proc Natl Acad Sci U S A. 1999;96:9124–9. doi: 10.1073/pnas.96.16.9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chin KH, et al. Preparation, crystallization and preliminary X-ray analysis of XC2382, an ApaG protein of unknown structure from Xanthomonas campestris. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:700–2. doi: 10.1107/S1744309105018956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ilyin GP, Rialland M, Pigeon C, Guguen-Guillouzo C. cDNA cloning and expression analysis of new members of the mammalian F-box protein family. Genomics. 2000;67:40–7. doi: 10.1006/geno.2000.6211. [DOI] [PubMed] [Google Scholar]
- 38.Liu L, Rodriguez-Belmonte EM, Mazloum N, Xie B, Lee MY. Identification of a novel protein, PDIP38, that interacts with the p50 subunit of DNA polymerase delta and proliferating cell nuclear antigen. J Biol Chem. 2003;278:10041–7. doi: 10.1074/jbc.M208694200. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, et al. Novel mutations in ubiquitin-specific protease 26 gene might cause spermatogenesis impairment and male infertility. Asian J Androl. 2007;9:809–14. doi: 10.1111/j.1745-7262.2007.00305.x. [DOI] [PubMed] [Google Scholar]
- 40.Month SR, et al. Analysis of 5′ flanking regions of the gamma globin genes from major African haplotype backgrounds associated with sickle cell disease. J Clin Invest. 1990;85:364–70. doi: 10.1172/JCI114447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geva A, et al. Hemoglobin Jamaica plain--a sickling hemoglobin with reduced oxygen affinity. N Engl J Med. 2004;351:1532–8. doi: 10.1056/NEJMoa040771. [DOI] [PubMed] [Google Scholar]
- 42.Dodge JA, Burton L. Letter: Heterozygote advantage in cystic fibrosis. Lancet. 1975;1:572–3. doi: 10.1016/s0140-6736(75)91587-1. [DOI] [PubMed] [Google Scholar]
- 43.Ebong SJ, et al. Immunopathologic responses to non-lethal sepsis. Shock. 1999;12:118–26. doi: 10.1097/00024382-199908000-00005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.