

Abstract
The site- and stereo-selectivities of C-H oxidations of substituted cyclohexanes and trans-decalins by dimethyldioxirane (DMDO) were investigated computationally with quantum mechanical density functional theory (DFT). The multi-configuration CASPT2 method was employed on model systems to establish the preferred mechanism and transition state geometry. The reaction pathway involving a rebound step is established to account for the retention of stereochemistry. The oxidation of sclareolide with dioxirane reagents is reported, including the oxidation by the in situ generated tBu-TFDO, a new dioxirane that better discriminates between C-H bonds based on steric effects. The release of 1,3-diaxial strain in the transition state contributes to the site selectivity and enhanced axial reactivity for tertiary C-H bonds, a result of the lowering of distortion energy. In addition to this strain release factor, steric and inductive effects contribute to the rates of C-H oxidation by dioxiranes.
Introduction
The functionalization of unactivated sp3 C-H bonds is of considerable interest in contemporary synthetic organic and organometallic chemistry.1–3 Compared to traditional functional group manipulations and interconversions employed in synthesis, C-H bond functionalization offers a direct route avoiding pre-functionalization of substrates. However, differentiating between numerous C-H bonds and effecting site-specific and stereoselective chemical modifications is an ongoing challenge.4 A range of reagents are known for C-H to C-OH conversion, such as Fe, Cu, Ru, Cr and Pd based systems.5 The pioneering work of Murray6 and Curci7 pointed to the use of dimethyldioxirane (DMDO) and methyl(trifluoromethyl)dioxirane (TFDO) for sp3 C-H oxidation.8 The dioxiranes are potent oxidants that lead to conservation of stereochemistry but no intermediate was characterized.9 The mechanism is still under an ongoing debate and radical products are observed in some experiments.10–12 Due to their high reactivity, dioxiranes are sometimes generated in situ during synthesis.13
Recently, a two-phase strategy for terpene total synthesis was invented to holistically mimic the way such molecules are constructed in nature. In the first phase, the so-called “cyclase phase”, simple terpenes at low oxidation states are stitched together as a prelude to the “oxidase phase” wherein these molecules are systematically oxidized. In the case of the eudesmane terpene class this was done using both innate and guided C-H functionalization logic.4 The purpose of this strategy for retrosynthetic analysis is not to necessarily recapitulate biosynthesis but rather to uncover new basic reactivity principles and invent new reaction methodology.
The factors that contribute to site selectivity in C-H oxidation reactions has been a question of interest for some time and continues today.14 Terpenoids contain a host of cyclohexane derivatives, and the rationalization of reactivities in such systems traces back to Barton’s fundamental work in the fifties.15,16 Reactivity differences, such as observed for the oxidation of axial and equatorial secondary alcohols with chromic acid, were explained by steric effects. Eschenmoser interpreted the enhanced oxidation rates of axial alcohols as compared to their equatorial epimers as resulting from the release of 1,3-diaxial strain in the transition state of the oxidation of the axial epimers.17 Experimental measurements of the rates and regioselectivites of C-H oxidation of substituted cyclohexanes by dioxiranes were performed by Curci,18,19 and a preference for equatorial attack was observed. Furthermore, Curci8 and others20 delineated a variety of factors that contribute to site selectivity in oxidations by dioxiranes. Similar factors have also been reported by M. C. White in C-H oxidation reactions of complex molecules by a non-heme, iron-based system.21,22 This field has a storied history and has been reviewed extensively recently.23
In the context of Eschenmoser’s hypothesis based on strain release, a recent example of site-selective C-H oxidation of a substituted decalin from Baran’s laboratory has been rationalized.24 Of five unactivated tertiary C-H bonds in the substrate, only equatorial C-H1 is oxidized to an alcohol by the dioxirane reagent. (Scheme 1) Strain release in the transition state was proposed to account for the preference.25 Based on computational studies from the Houk26 and Bach27 labs, a flattening at the carbon undergoing oxidation is expected in the transition structure, which alleviates the 1,3-diaxial strain between two axial methyl groups in the transition state for H1 abstraction and lowers the activation energy for C-H1 oxidation.
Scheme 1.
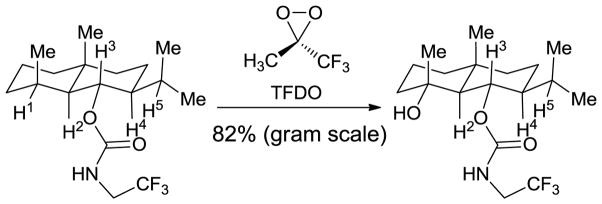
Selective C-H oxidation. Only one of the five tertiary C-H bonds is oxidized24
Given the intensive efforts underway to develop substrate controlled selective C-H activation processes, we undertook calculations at the density functional and multi-configurational ab initio levels of theory to gain quantitative insights into the exquisite selectivities reported by Baran. Here we describe computational studies of the transition structures for C-H oxidation of Baran’s eudesmane decalins and a variety of other simpler cyclohexanes by dioxirane reagents. We also analyze various substituted cyclohexyl substrates. For several such cases, we report transition structures and present explanations for the enhanced reactivity of equatorial C-H bonds in these strained systems. Additionally, we include a new synthesis of trifluoromethyl, tert-butyl ketone and its use as a reagent in C-H oxidation by in situ formation of the corresponding dioxirane, tBu-TFDO. A quantitative understanding of the factors contributing to selective C-H activation is provided, in the context of the distortion/interaction theory that has been successfully applied to other types of reactions.28–31
Computational Methods
All density functional theory (DFT) calculations were performed with Gaussian 09.32 Mimina and transition structures were optimized using the unrestricted DFT method, UB3LYP, with the 6–31G(d) basis set.32 Frequency analyses were carried out on stationary points to verify that they are minima or saddle points (transition structures - TSs). Energies were recalculated with UB3LYP/6–311++G(d, p) on these optimized geometries, unless otherwise stated. To ensure that the correct unrestricted wavefunctions were obtained, a stability test was carried out with Gaussian keyword stable=opt. Solvation corrections were calculated with the conductor-like polarizable continuum model (CPCM) using the UAHF atomic radii. Multi-determinant wavefunction theory, Complete Active Space with Second-order Perturbation Theory (CASPT2) calculations were conducted with MOLCAS 7.4.33 A 10-electron, 10-orbital active space was employed for CASPT2 calculations with a large cc-pVTZ basis set on the optimized geometries.
Results and Discussion
Modeling the Dioxirane Oxidation of C-H Bonds
Modeling of dioxirane oxidation has led to different conclusions about the TS geometries. Goddard34,35 and Bach36 investigated the electronic structure of dioxirane. The O-O bond lengths in both DMDO and TFDO are 1.51 Å, readily broken due to the ring strain. Early calculations show that a spiro geometry is preferred over a planar geometry for epoxidations.26,27,37 Houk explored the geometry and selectivities of dioxirane and cyanodioxirane, a model for TFDO, with restricted DFT calculations.26 Due to the complexity of the potential energy surface (PES), and possible intersystem crossing of the singlet and triplet PES, later studies proposed several different first-order saddle points, associated with three different transition structures.38,39 (Fig. 1) We have explored these in more detail in order to determine which is likely to be the most important transition structure for the reaction.
Figure 1.
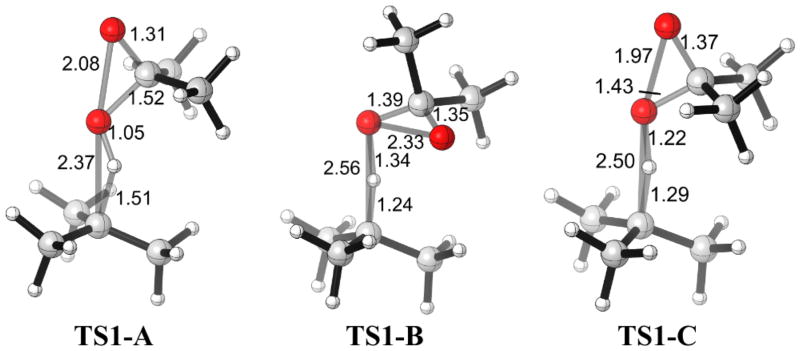
Transition state geometries for DMDO and isobutane reaction.
The first transition state, TS1-A, is a spiro transition state with all electrons paired, located by restricted DFT calculations.26,40 This corresponds to a concerted process in which the t-butyl alcohol and acetone are produced without the formation of an intermediate. A wavefunction stability test, however, reveals that this wavefunction is unstable with respect to an unrestricted wavefunction. That is, an open-shell wavefunction is lower in energy. The closed-shell treatment was further invalidated with the multi-determinant CASPT2, as strong diradical3 character is observed in the transition state, indicated by the HOMO/LUMO occupation number of 1.52/0.49. (Table 1)
Table 1.
Natural orbital occupancies of frontier orbitals, and activation energies in the transition statesa
| HOMO | LUMO | CASPT2 | UB3LYPb | |
|---|---|---|---|---|
| TS1-A | 1.42 | 0.58 | 21.5 | 19.9c |
| TS1-B | 1.24 | 0.76 | 22.5 | 15.8 |
| TS1-C | 1.64 | 0.37 | 19.5 | 20.0 |
CASPT2(10,10)/cc-pVTZ, energies in kcal/mol
UB3LYP/6–311++G(d, p)
24.2 kcal/mol with RB3LYP/6–311++G(d, p)
The second TS, TS1-B, has the O-O bond perpendicular to the breaking C-H bond. This is found to be a radical hydrogen abstraction process beginning from the 1,3-dioxy radical. The resulting radical intermediates could then recombine to transfer the OH group to form the products. For this mechanism, the rate limiting step is the dissociation of the DMDO O-O bond from the singlet diradical, which requires an activation enthalpy of 23.1 kcal/mol according to Cremer.41 The bond-dissociation rate determining step has a higher barrier than all three C-H activation barriers in Table 1, so it is disfavored under kinetic control.
The third TS, TS1-C, has the O-O bond aligned with the breaking C-H bond. We use stable=opt to obtain the correct wavefunction at the initial geometry, then perform each geometry optimization for transition state using the optimized wavefunction as an initial guess with the Gaussian keyword guess=read. To make sure the optimized geometries have the correct wavefunction, the same procedure was repeated on the optimized geometries. Both the UB3LYP and CASPT2 indicated slight diradical character for the transition state, with <S2> value = 0.5312 with B3LYP, and HOMO/LUMO occupation numbers 1.64/0.37 with CASPT2. Therefore TS1-C is the more reliable transition state. As CASPT2 becomes formidably expensive with larger systems, UB3LYP with the stable open-shell wavefunction was employed for further studies.
The mechanism involving TS1-C is analogous to the “oxygen rebound” mechanism common in iron-oxo oxidations.42,43 The complete reaction pathway was then explored and is summarized in Figure 2. The reactant complex goes through C-H activation transition state TS1 and forms a weakly-bound radical pair intermediate, which rebounds and forms the final product. The reaction can be tracked with the forming C-O bond length. As the two reactants approaches each other, the C-O bond distance decreases until it reaches a minimum at 2.50 Å in TS1, then increases back to 3.15 Å after hydrogen abstraction, leaving two radical centers in the intermediate. The C-O bond distance then decreases again, (“rebounds”) through 2.51 Å in TS2 to eventually 1.43 Å in the product. As the second transition state has essentially no barrier, the radical pair intermediate is expected to have too short a lifetime to escape from the solvent cage, leading to the retention of stereochemistry as observed in experiments.
Figure 2.

Reaction pathway in DMDO oxidations. Energies in kcal/mol.
The same conclusions hold for the more reactive oxidant, TFDO. The C-H activation free energy is around 5 kcal/mol lower with TFDO than DMDO, but the preferred transition state is again obtained with unrestricted and optimized wavefunctions. (Figure S3 for TS geometry and Table S1 for the natural orbital occupancies). The overall reaction pathway is similar and the rate-limiting step is again the C-H activation step with diradical character. (Figure S4 in the Supporting Information). Based on the similarity of the DMDO and TFDO reaction pathways, the in situ generated tBu-TFDO is expected to follow the same mechanism. The computational results are in agreement with Curci’s finding that TFDO is more reactive than DMDO without diminished selectivity, as an exception to the reactivity-selectivity rule.9
Oxidation of Equatorial and Axial C-H Bonds of Cyclohexane
The TSs for axial and equatorial C-H oxidation by DMDO are shown in Figure 3. No significant axial/equatorial preference is found. The activation energy for oxidizing the cyclohexane equatorial hydrogen is 21.6 kcal/mol, while for the axial hydrogen it is 21.4 kcal/mol.
Figure 3.

Optimized TSs for the reactions of DMDO with equatorial (2-eq-TS) and axial (2-ax-TS) C-H bonds. Energies in kcal/mol with respect to the corresponding reactants. Individual components of distortion energies (alkane distortion, dioxirane distortion) are given in parentheses.
To evaluate the factors governing relative reactivities of C-H bonds quantitatively, a distortion/interaction energy analysis was conducted. The distortion energy (ΔEd‡) is the energy required to distort reactants, hydrocarbon plus the oxidant, dimethyldioxirane (DMDO), into the transition state (activated) geometry. The interaction energy (ΔEi‡) is the energy lowering due to the interaction of the two distorted reactants. Distortion energies favor attack on the axial hydrogen by 1.1 kcal/mol, while interaction energies favor equatorial attack by 0.9 kcal/mol. These two components largely cancel out, leaving essentially no preference (0.2 kcal/mol). The nearly identical activation energies for 2-eq-TS and 2-ax-TS indicate very weak axial/equatorial selectivity, if any, as observed experimentally for C-H activations of different types.25
The distortion of the DMDO moiety is about the same for both transition states, and the difference in the distortion energies mainly comes from the cyclohexane distortion. Hyperconjugation is a likely factor to the 0.9 kcal/mol difference in interaction energies. (Fig. 3) In the 2-eq-TS, there are two C-C bonds anti to the breaking C-H bond, while there are two antiperiplanar C-H bonds in the 2-ax-TS. Since C-C bonds are generally better donors than C-H bonds, hyperconjugation is more effective at stabilizing the oxidation of equatorial C-H bonds due to the presence of better donors in the antiperplanar arrangement.20 A slight lengthening of the antiperiplanar bonds is observed in the optimized transition structures, indicative of this hyperconjugative interaction.
The distortion energy difference can be attributed to torsional interactions. The Newman projections of these two transition states are given in Fig. 3. The internal C-C-C-C dihedral angle is 56° in the 2-eq-TS, slightly more distorted than the one in cyclohexane (55°). This angle is 48° in 2-ax-TS, closer to the more relaxed angle in the cyclohexyl radical (44°). The relaxation is reflected as a 1.0 kcal/mol lowering in the distortion energy. Hydrogen-hydrogen repulsion also plays a part in the relative stability. As hydrogen is being abstracted from a tetrahedral sp3 carbon, the carbon flattens and becomes sp2 in character. As shown in the Newman projection for 2-eq-TS, this lead to slightly greater eclipsing of the vicinal C-H bonds, as indicated in the 45° dihedral angle, while that in 2-ax-TS is 50°. Therefore, distortion energy for 2-eq-TS is higher. This torsional effect favoring axial attack was observed in the case of nucleophilic attack on cyclohexanone (Felkin’s rule)44,45 and reactions of cyclohexyl radicals,46 in which the axial TS has a more staggered conformation than the equatorial TS. In the reactions studied here, the cancellation of distortion and interaction energy preferences leads to no intrinsic selectivity for the secondary C-H bonds of cyclohexane.
This type of analysis has been performed on a variety of cyclohexanes and decalins. The results are tabulated in Table 2.
Table 2.
Summary of activation energies and distortion/interaction energies (kcal/mol)
| Compound | C-H bond for oxidation | ΔH‡ | ΔE‡ | ΔEd‡ (alkane) | ΔEd‡ (DMDO) | ΔEd‡(total) | ΔEi‡ |
|---|---|---|---|---|---|---|---|
| 1 | tBu-H | 16.2 | 20.0 | 11.1 | 21.7 | 32.7 | −12.8 |
| 2-eq |
|
17.7 | 21.6 | 13.1 | 22.1 | 35.2 | −13.6 |
| 2-ax |
|
17.5 | 21.4 | 12.1 | 22.0 | 34.1 | −12.7 |
| 3-eq |
|
15.4 | 18.9 | 10.5 | 21.6 | 32.1 | −13.2 |
| 3-ax |
|
15.8 | 19.5 | 10.5 | 21.6 | 32.1 | −12.6 |
| 3′-ax |
|
16.1 | 19.6 | 10.5 | 21.6 | 32.2 | −12.6 |
| 4-eq |
|
15.3 | 18.7 | 10.2 | 21.5 | 31.7 | −13.0 |
| 4-ax |
|
15.8 | 19.2 | 10.2 | 21.5 | 31.7 | −12.5 |
| 4′-ax |
|
16.4 | 19.8 | 10.5 | 21.7 | 32.2 | −12.4 |
| 5-eq |
|
15.4 | 18.9 | 10.5 | 21.6 | 32.1 | −13.2 |
| 6 |
|
14.7 | 18.2 | 9.9 | 21.3 | 31.2 | −13.0 |
| 7 |
|
14.2 | 17.7 | 9.4 | 21.1 | 30.4 | −12.8 |
| 8a |

|
13.3 | 16.4 | 10.1 | 20.2 | 30.3 | −13.9 |
| 9a |
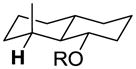
|
13.5 | 16.8 | 10.3 | 20.6 | 30.9 | −14.1 |
| 10-eq-C2 |
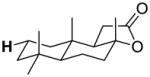
|
18.3 | 22.2 | 12.8 | 22.1 | 34.9 | −12.7 |
| 10-eq-C3 |
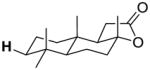
|
18.8 | 22.7 | 13.8 | 23.8 | 36.0 | −13.3 |
| 10-ax-C3 |
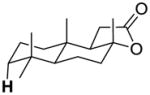
|
18.5 | 22.2 | 12.5 | 22.1 | 34.5 | −12.3 |
R = -C(O)NHCH2CF3, iPr on the decalin ring is omitted
Oxidation of Methylcyclohexane
The TSs for tertiary axial/equatorial C-H oxidation of cis-1,4-dimethylcyclohexane by DMDO are shown in Fig. 4a, as an eudesmane model system with both axial and equatorial tertiary C-H bonds. The activation energy for the reaction of the equatorial C-H bond in 3-eq-TS is 0.6 kcal/mol lower than for the axial C-H bond in 3-ax-TS. While interaction energies again favor equatorial hydrogen abstraction, the distortion energies are essentially the same. The axial/equatorial selectivity comes from the interaction energies, which are −13.2 kcal/mol for 3-eq-TS and −12.6 kcal/mol for 3-ax-TS. This preference, coming from interaction energies, is found again in 4-eq-TS and 4-ax-TS, where both axial and equatorial C-H bonds are present in the same molecule. (Fig. 4b)
Figure 4.

Optimized TSs for the reactions of DMDO with (a) cis-1,4-dimethylcyclohexanes at the equatorial (3-eq-TS) and axial C-H bonds (3-ax-TS); (b) cis-1,2-dimethylcyclohexanes at the equatorial (4-eq-TS) and axial (4-ax-TS) C-H bonds. Energies in kcal/mol with respect to the corresponding reactants. Individual components of distortion energies (alkane distortion, dioxirane distortion) are in parentheses.
The distortion energies are identical for the two transition states, while the interaction energies are −13.0 kcal/mol and −12.5 kcal/mol, respectively. Just as the case of secondary hydrocarbons, the lengthening of antiperiplanar C-C bonds is shown in the equatorial TS, leading to better hyper- conjugation. A more detailed study of how distortion energies change along the reaction coordinate is summarized in Figure S1 in the supporting information. Distortion energies are the same since the tertiary C-H oxidation transition states are earlier than the transition states for secondary C-H oxidation. With the same distortion energy and favored interaction energy for abstraction of equatorial hydrogens, the latter is slightly more susceptible to oxidation.
The stereochemistry of the remote methyl group has no effect on the reactivity, only the stereochemistry of the carbon on which hydrogen is abstracted matters. As in Table 2, axial C-H oxidations give almost identical distortion and interaction energies for cis-1,4-dimethylcyclohexane (3-ax) and trans-1,4-dimethylcyclohexane (3′-ax), leading to the same activation energy.
The stereochemistry of the methyl group α to the reacting center has little impact on the interaction energy, which is intrinsic whether an axial or equatorial hydrogen is abstracted. As in Table 2, oxidation of cis-1,2-dimethylcyclohexane (4-ax) and trans-1,2-dimethylcyclohexane (4′-ax) give virtually identical interaction energies. The distortion energy for 4′-ax is raised by 0.5 kcal/mol due to developing torsional strain in the transition state as the reacting site becomes more sp2 in character. Overall the activation energy for 4′-ax is 0.6 kcal/mol higher than that of 4-ax.
Compared with tertiary C-H bonds, activations of secondary C-H bonds require higher activation energies, which are around 21.5 kcal/mol in Fig. 3, due to the electrophilic nature of the oxidizing reagent, compared with those activation energies for tertiary C-H bonds, which are below 20.0 kcal/mol. This agrees with Curci’s experiments that tertiary C-H bonds are generally more reactive than secondary C-H bonds, and the usual greater stability of tertiary radicals.17 The differences in activation energies between secondary C-H oxidation (~18 kcal/mol) and tertiary C-H oxidation (15–16 kcal/mol) is comparable to, or slightly higher, than the difference between secondary and tertiary C-H bond dissociation enthalpies (BDEs) (95.6 and 98.1 kcal/mol, respectively47) because there is polarization and partial positive charge build up at the oxidized carbon in the transition states.
Influence of axial methyl substitution on reactivity
To systematically investigate the effects of 1,3-diaxial methyl-methyl interactions, reactivities of the tertiary equatorial C-H bonds reacting on axial methylcyclohexane (5), diaxial 1,3-dimethylcyclohexane (6), and triaxial 1,3,5-trimethylcyclohexane (7) were studied. The reactants and transition states are shown in Fig. 5. The methyl groups in 5, 6, 7 were axial rather than in the more stable equatorial position, to serve as models for rigid terpenes where the methyl groups are fixed to be axial, as in Baran’s selective oxidation of 1.24 The relative rate for oxidation of cis-1,3-dimethylcyclohexane (6) is slightly higher than the trans-1,3-dimethylcyclohexane, as strain release operates in the former isomer, in agreement with a ratio of 1.4–1.5:1 by TFDO oxidation generated in situ. The detailed experimental procedure is included in the supporting information.
Figure 5.

Optimized reactant and transition state geometries for the reaction of DMDO with substituted cyclohexanes. Energies are in kcal/mol. Individual components of distortion energies (alkane distortion, dioxirane distortion) are in parenthesis.
As more axial methyl substituents were added (5 to 6 to 7), the nearest H-H distance gets shorter, and more strain is imposed as the H-H closed shell repulsion increases. In the reactants, there is no significant H-H repulsion in 5, there is one H-H interaction with a 2.12 Å distance in 6, and there are three pairs of CH-CH interactions of 2.11 Å in 7. In the transition state for abstraction of equatorial hydrogens, the methyl bends away from the ring, releasing 1,3-diaxial strain. In 5, the H-H distances are above 2.4 Å, and no strain is released in the TS. In 6, the closest H-H bond distance increases from 2.12 Å in the reactant to 2.19 Å in TS, releasing strain. In 7, the two closest H-H distances increase from 2.11 Å to 2.20 Å and 2.21 Å, and further strain release is expected.
The trend towards lower activation energy was reflected in the decreasing distortion energies, from 32.1 (5-TS) to 31.2 (6-TS) to 30.4 kcal/mol (7-TS). The interaction energies are nearly the same, increasing by 0.2 kcal/mol (less negative) with each additional axial methyl group, but this change is smaller than the increase in distortion energies, which dominates the trend in reactivity. Most of the difference in distortion energy results from the hydrocarbon reactant. The steric acceleration proposed to result from strain release by Eschenmoser,17 is manifested in lower distortion energy in the TS. In other words, the reactants are pre-distorted towards the TS geometry, and the activation energy is lowered. We have referred to this phenomenon as distortion-acceleration.48 The higher reactivity along the series is also paralleled by slightly earlier transition states along the series, and so the distortion energy of the dioxirane decreases in the series.
Strain Release and Enhanced Reactivity towards Oxidations in Steroidal Systems
The strain release (or distortion-acceleration) effect has been taken advantage of by Baran24 to generate selectivity in C-H oxidations, as in Scheme 2. A 1:1 mixture of 8′ and 9′ gave the corresponding oxidation products in a ratio of 3:1. The strain release effect was proposed to account for the enhancement in reactivity of 8′ as compared with 9′. A similar trend in reactivity was observed in oxidation by ozone, which produced 8′ and 9′ in a 4:1 ratio.
Scheme 2.
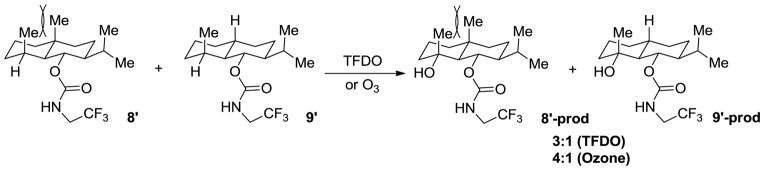
Reaction products in a competition reaction
We modeled the reaction with methyl(trifluoromethyl)dioxirane (TFDO) and dichloromethane as the solvent. The results are shown in Figure 6. A model study with DMDO and these substrates in the gas phase is summarized in Table 2.
Figure 6.

Optimized reactant and transition state geometries for the reaction of TFDO with (a) 8 and (b) 9. R = -C(O)NHCH2CF3. The iPr substituents on the decalin rings of 8′ and 9′ were omitted for simplicity. Energies are in kcal/mol. Individual components of distortion energies (alkane distortion, dioxirane distortion) are in parenthesis. Energies with dichloromethane solvation correction are in square brackets.
Reactants 8, 9, models for 8′ and 9′, and transition states for TFDO 8-TS and 9-TS to strong 1,3-diaxial strain, with the nearest H-H distance being 2.05 Å. This distance becomes 2.08 Å in 8-TS, which reduces the strain. The remaining H-H distances stay rather constant, contributing little to the activation energies due to strain release. The energy required to distort 8 into 8-TS is 26.3 kcal/mol, mainly from the dioxirane distortion, due to the stiffness of the ring.
The aforementioned methyl-methyl interaction in 8 is alleviated when the bridge methyl is replaced by a hydrogen atom for 9 in Fig. 6(b). Compared with the case of 8, there is less strain release. In other words, more energy was required to distort the reactant 9 into its transition states 9-TS, with distortion energy increased to 26.8 kcal/mol; the activation energy is 11.4 kcal/mol. The 0.5 kcal/mol difference in distortion energy, although rather small, dominates over the interaction energies, and lead to 0.3 kcal/mol lower activation energy for 8-TS than 9-TS. With solvent correction, there is 0.6 kcal/mol lower in activation enthalpy for 8-TS, in agreement with observed 3:1 experimental ratio. For model study with DMDO, predicted activation energy for 8-TS is 0.4 kcal/mol lower than 9-TS (16.4 kcal/mol and 16.8 kcal/mol, respectively). The axial methyl group gives only 0.4 kcal/mol rate enhancement for the substituted decalin, compared to 0.7 kcal/mol with substituted cyclohexane, which is likely due to the rigidity of the ring, as reflected in the distortion energies of the ring systems.
Selective Oxidations of Sclareolide
Sclareolide, 10, has five methylenes and ten distinct secondary hydrogens. Experimentally, 10 is oxidized at both the C2 and C3 positions (steroid numbering),49 and ratios of products are given in Table 3. Details of these new experimental results are given in the Supplemental Information.
Table 3.
Experimental and calculated ratios for C2/C3 products from 10. Energies in kcal/mol.
| oxidant | C2:C3 | ref | Calc | ΔE‡(C2) | ΔE‡(C3axial) | ΔE‡(C3equatorial) |
|---|---|---|---|---|---|---|
| DMDO | 1.5:1a | 49 | ~1:1b | 22.2b | 22.2b | 22.7b |
| TFDO | 2.1:1~1:2.0c | 49 | 2.7:1d | 10.2d | 12.1d | 10.9d |
| tBu-TFDO | 2.4:1e | 49 | 8.3:1d | 11.9d | 14.3d | 13.1d |
| Rh2(esp)2 f | Only C2 | 49 | ||||
| Mn(TPP)Clg | 7:1 | 50 | ||||
| Fe(PDP) | 1.4:1 | 21 |
1:1 acetone:DCM, <5% conversion
UB3LYP/6–311++G(d, p), gas phase
Ratios with a variety of solvents gave from 2.1:1 with hexafluoroisopropanol to 1:2.0 with acetonitrile.
UB3LYP/6–311++G(d, p), calculated ratio from activation energies in 298 K with CPCM/UAHF in acetonitrile
In acetonitrile solvent
Amination products
A ratio of 3:1 was observed for fluorination with Mn(TMP)Cl.
Electronically, oxidation of C3 is favored because it is remote from the electron-withdrawing lactone that deactivates the methylenes at C1, C5 and C6 and steric effects favor oxidation at C2. For reagents that behave like small reagents, such as dioxiranes and Fe(PDP) there is marginal selectivity between oxidation at the C2 and C3 positions. The use of a manganese porphyrin results in considerably higher site selectivity.50 In the case of the Rh-mediated amination employing a reagent that behaves like a large reagent, excellent C3 selectivity is observed.5n,24
Table 4 shows details of the computed activation barriers with different oxidants. The bulky tBu group increases the activation energies by about 2 kcal/mol, reflected in the distortion energies especially from the substrate 10. The interaction energies are essentially the same, since the methyl group in TFDO and tBu in tBu-TFDO have similar electronic effects. The reactants and transition states geometries are shown in Figure 7 for TFDO and Figure 8 for tBu-TFDO. With solvent correction (acetonitrile), the axial TS becomes disfavored probably due to a higher energy cost for the rearrangement of solvent molecules. Also a higher C2:C3 selectivity is expected. A thorough study of solvent effects is underway.
Table 4.
Analysis of site selectivities on 10 for TFDO vs tBu-TFDO. Energies in kcal/mol.
| oxidant | TS | ΔH‡(solv)a | ΔH‡ | ΔE‡ | ΔEd‡(10) | ΔEd‡(TFDO) | ΔEd‡(10+TFDO) | ΔEi‡ |
|---|---|---|---|---|---|---|---|---|
| TFDO | C2 | 6.7 | 12.2 | 15.7 | 8.7 | 20.2 | 28.9 | −13.2 |
| TFDO | C3-ax | 8.8 | 12.7 | 15.9 | 8.5 | 20.1 | 28.6 | −12.7 |
| TFDO | C3-eq | 7.5 | 12.8 | 16.1 | 9.2 | 20.3 | 29.5 | −13.4 |
| tBu-TFDO | C2 | 8.3 | 14.2 | 17.8 | 9.9 | 20.8 | 30.7 | −12.9 |
| tBu-TFDO | C3-ax | 10.7 | 15.1 | 18.7 | 10.3 | 20.8 | 31.1 | −12.4 |
| tBu-TFDO | C3-eq | 9.5 | 15.0 | 18.7 | 11.1 | 20.9 | 32.1 | −13.4 |
Calculated solvent with CPCM/UAHF in acetonitrile
Figure 7.

Optimized reactant and transition state geometries for the reaction of TFDO with sclareolide 10. Energies are in kcal/mol. Individual components of distortion energies (alkane distortion, dioxirane distortion) are in parenthesis. Energies with acetonitrile solvation correction are in square brackets.
Figure 8.

Optimized reactant and transition state geometries for the reaction of tBu-TFDO with sclareolide 10. Energies are in kcal/mol. Individual components of distortion energies (alkane distortion, dioxirane distortion) are in parenthesis. The prime labels on TS distinguish the transition states for tBu-TFDO oxidation with those with DMDO. Energies with acetonitrile solvation correction are in square brackets.
Figure 7 gives the geometries of sclareolide 10 and three transition states for C-H oxidations on the C2/C3 C-H bonds with TFDO. The methyl-hydrogen strain is released, as the bond lengths for nearest H-H pairs, which are 2.10 Å and 2.12 Å in 10, increased to 2.11 Å and 2.18 Å in 10-eq-C2-TS, but stays the same on 10-ax-C3-TS and 10-eq-C3-TS. Not surprisingly, the strain release effect is not as pronounced as is the case with the relief of a methyl-methyl 1,3 diaxial interaction. Accordingly, a mixture of products results.
Figure 8 gives the geometries of sclareolide 10 and three transition states for C-H oxidations on the C2/C3 C-H bonds with the bulkier oxidant tBu-TFDO. The site selectivity for oxidation on the C2 versus C3 position is predicted to be 0.9 kcal/mol. Experimentally a higher C2/C3 ratio was observed for tBu-TFDO than for TFDO. The steric effect of the tert-butyl group raises the activation barriers for tBu-TFDO oxidations compared with TFDO by about 2 kcal/mol. This energy difference is also consistent with the low yield and low levels of reactivity observed with tBu-TFDO compared with TFDO. The trace conversion affected by tBu-TFDO may also reflect the low concentration of this reagent in the reaction mixture, as it is formed in situ. The increased steric effects also render the equatorial hydrogen on C3 as susceptible to oxidation as the axial hydrogen at the same position.
Conclusions
The site selectivities and stereoselectivities of sp3 C-H activations in cyclohexane derivatives by dioxiranes were investigated computationally. CASPT2 calculations established the appropriate transition state for C-H oxidations by dioxiranes, which corresponds to that located using unrestricted DFT. The full pathway for dioxirane oxidation was established. The distortion/interaction model was used to understand the origins of selectivity in each case. In the absence of axial substitution of cyclohexanes there is no innate preference for either axial or equatorial C-H activation: eclipsing interactions slightly favor axial activation, but this is counterbalanced by the greater hyperconjugative stabilization in equatorial activation. The axial and equatorial C-H bonds of cyclohexane have similar reactivities.
However, with increasing axial substitution, computed activation barriers decrease, in accord with experiment. The enhancement in equatorial reactivity by geminal and diaxial substitution arises as a consequence of strain release in going to the transitions state.25 The distortion/interaction model allows for a quantification of the acceleration that arises as a result of strain release. The strain release effect is around 0.7 kcal/mol when the strain involving two 1,3-diaxial methyl groups is released. The eudesmane 8 is more reactive than 9 due to the strain release effect, but the C2/C3 site selectivity for sclareolide is dominated by steric effects and can be controlled by the bulkiness of the reagents.
Experimental Section
General Procedures
All reactions were carried out under an inert nitrogen atmosphere with dry solvents under anhydrous conditions unless otherwise stated. Dry acetonitrile (MeCN) was obtained by passing the previously degassed solvents through activated alumina columns. Product ratios were determined by analysis of the crude 1H NMR spectra. Yields refer to chromatographically and spectroscopically (1H-NMR) homogeneous materials, unless otherwise stated. Reagents were purchased at the highest commercial quality and used without further purification, unless otherwise stated. Reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25 mm E. Merck silica gel plates (60F-254 or RP-18 F254s) using UV light as the visualizing agent and an acidic solution of p-anisaldehyde, phosphomolybdic acid, cerric ammonium molybdate, Seebach’s stain (PMA and CAM), ninhydrin, or potassium permanganate and heat as developing agents. E. Merck silica gel (60, particle size 0.043–0.063 mm) was used for flash column chromatography
General Procedure for oxidation using TFDO generated in situ
The substrate (0.2 mmol) was dissolved in MeCN (6.0 mL, 0.03M) by stirring and sonication. To this solution was added aqueous sodium phosphate buffer (6.0 mL, pH = 7.5, 25 mM) and aqueous Na2EDTA (1.3 mL, 40 mM). The solution was then cooled to 4 °C (ambient temperature) and cool (4 °C) 1,1,1-trifluoroacetone (0.143 mL, 1.60 mmol, 8.0 equiv) was added. Oxone® • (KHSO5 0.5KHSO4 • 0.5K2SO4, 738 mg, 2.4 mmol, 12 equiv) was then added in a single portion and the suspension was rapidly stirred for 36 hours. Then, the mixture was warmed to room temperature and extracted with Et2O (3 × 15 mL). The combined organic layers were washed with brine (50 mL), dried over MgSO4, filtered and concentrated.
Similar conditions for in situ dioxirane formation for C-H oxidation have been developed:
Annese, C.; D’Accolti, L.; Fusco, C.; Curci, R. Org. Lett. 2011, 13, 2142.
The following references include spectral data for the sclareolide oxidation products:
Cambie, R. C.; Joblin, K. N.; McCallum, N. K. Aust. J. Chem. 1970, 23, 1439.
Choudhary, M. I.; Musharraf, S. G.; Sami, A.; Rahman, A.-u. Helv. Chim. Acta. 2004, 87, 2685.
Chen, M. S.; White, M. C. Science 2010, 327, 566.
The following reference includes data for the products from oxidation of 1,3-dimethylcyclohexanes:
Senda, Y.; Ishiyama, J.; Imaizumi, S. Tetrahedron 1975, 31, 1601.
t-butyl trifluoromethylketone could be synthesized according to: Wiedemann, J.; Heiner, T.; Mloston, G.; Prakash, G. K. S.; Olah, G. A. Angew. Chem. Int. Ed. 1998, 37, 820. Alternatively, CF3TMS could be added dropwise to a mixture of methyl pivalate and excess CsF at 0 ° C.
Supplementary Material
Acknowledgments
We are grateful to the Royal Commission for the Exhibition of 1851 for a Research Fellowship (RSP), National Institute of General Medical Sciences, National Institutes of Health (GM36700 to KNH and GM097444 to PSB), the TEVA Pharmaceuticals Scholars Grant (to PSB), and Bristol-Myers Squibb (graduate fellowship to TRN). Computations were performed using the UCLA Academic Technology Services Hoffman2 Beowulf cluster and the San Diego Supercomputer Center Thresher cluster. We thank Professor Yu Lan, Chongqing University, for helpful discussions.
Footnotes
Supporting Information Available: Optimized Cartesian coordinates of all stationary points and saddle points. Full reference 32 is also included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Yamaguchi J, Yamaguchi AD, Itami K. Angew Chem Int Ed. 2012;51:8960. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]
- 2.Doyle MP, Goldberg KI. Acc Chem Res. 2012;45:777. doi: 10.1021/ar300096z. [DOI] [PubMed] [Google Scholar]
- 3.Crabtree RH. Chem Rev. 2010;110:575. doi: 10.1021/cr900388d. [DOI] [PubMed] [Google Scholar]
- 4.Brueckl T, Baxter RD, Ishihara Y, Baran PS. Acc Chem Res. 2012;45:826. doi: 10.1021/ar200194b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For recent reviews, see: Davies HML, Beckwith REJ. Chem Rev. 2003;103:2861. doi: 10.1021/cr0200217.Punniyamurthy T, Velusamy S, Iqbal J. Chem Rev. 2005;105:2329. doi: 10.1021/cr050523v.Godula K, Sames D. Science. 2006;312:67. doi: 10.1126/science.1114731.Dick AR, Sanford MS. Tetrahedron. 2006;62:2439.Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485.Giri R, Shi BF, Engle KM, Maugel N, Yu JQ. Chem Soc Rev. 2009;38:3242. doi: 10.1039/b816707a.Daugulis O, Do HQ, Shabashov D. Acc Chem Res. 2009;42:1074. doi: 10.1021/ar9000058.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890. doi: 10.1021/cr900206p.Shul’pin GB. Org Biomol Chem. 2010;8:4217. doi: 10.1039/c004223d.Jazzar R, Hitce J, Renaudat A, Sofack-Kreutzer J, Baudoin O. Chem Eur J. 2010;16:2654. doi: 10.1002/chem.200902374.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e.Yu J-Q, Shi Z, editors. “C-H Activation”: Topics in Current Chemistry. Springer; Berlin: 2010. p. 292.Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem Rev. 2010;110:704. doi: 10.1021/cr900239n.Roizen JL, Harvey ME, Du Bois J. Acc Chem Res. 2012;45:911. doi: 10.1021/ar200318q.
- 6.Murray RW, Jeyaraman R. J Org Chem. 1985;50:2847. [Google Scholar]
- 7.Mello R, Fiorentino M, Sciacovelli O, Curci R. J Org Chem. 1988;53:3890. [Google Scholar]
- 8.Curci R, D’Accolti L, Fusco C. Acc Chem Res. 2006;39:1. doi: 10.1021/ar050163y. [DOI] [PubMed] [Google Scholar]
- 9.Curci R, Dinoi A, Rubino MF. Pure Appl Chem. 1995;67:811. [Google Scholar]
- 10.Bravo A, Bjorsvik HR, Fontana F, Minisci F, Serri A. J Org Chem. 1996;61:9409. [Google Scholar]
- 11.Curci R, Dinoi A, Fusco C, Lillo MA. Tetrahedron Lett. 1996;37:249. [Google Scholar]
- 12.Bravo A, Fontana F, Fronza G, Minisci F, Zhao LH. J Org Chem. 1998;63:254. [Google Scholar]
- 13.Romney DK, Miller SJ. Org Lett. 2012;14:1138. doi: 10.1021/ol3000712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newhouse T, Baran PS. Angew Chem Int Ed. 2011;50:3362. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barton DHR. J Chem Soc. 1953:1027. [Google Scholar]
- 16.Barton DHR, Beviere SD, Chavasiri W, Csuhai E, Doller D, Liu WG. J Am Chem Soc. 1992;114:2147. [Google Scholar]
- 17.Schreiber J, Eschenmoser A. Helv Chim Acta. 1955;38:1529. [Google Scholar]
- 18.D’Accolti L, Fiorentino M, Fusco C, Rosa AM, Curci R. Tetrahedron Lett. 1999;40:8023. [Google Scholar]
- 19.Mello R, Fiorentino M, Fusco C, Curci R. J Am Chem Soc. 1989;111:6749. [Google Scholar]
- 20.a) Gonzalez-Nunez ME, Castellano G, Andreu C, Royo J, Baguena M, Mello R, Asensio G. J Am Chem Soc. 2001;123:7487. doi: 10.1021/ja003667u. [DOI] [PubMed] [Google Scholar]; b) Gonzalez-Nunez ME, Royo J, Mello R, Baguena M, Ferrer JM, de Arellano CR, Asensio G, Prakash GKS. J Org Chem. 2005;70:7919. doi: 10.1021/jo0509511. [DOI] [PubMed] [Google Scholar]
- 21.Chen MS, White MC. Science. 2010;327:566. doi: 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]
- 22.Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]
- 23.Ishihara Y, Baran PS. Synlett. 2010:1733. [Google Scholar]
- 24.Chen K, Baran PS. Nature. 2009;459:824. doi: 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]
- 25.Chen K, Eschenmoser A, Baran PS. Angew Chem Int Ed. 2009;48:9705. doi: 10.1002/anie.200904474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du XH, Houk KN. J Org Chem. 1998;63:6480. doi: 10.1021/jo980760g. [DOI] [PubMed] [Google Scholar]
- 27.Glukhovtsev MN, Canepa C, Bach RD. J Am Chem Soc. 1998;120:10528. [Google Scholar]
- 28.Paton RS, Kim S, Ross AG, Danishefsky SJ, Houk KN. Angew Chem Int Ed. 2011;50:10366. doi: 10.1002/anie.201103998. [DOI] [PubMed] [Google Scholar]
- 29.Lan Y, Houk KN. J Am Chem Soc. 2010;132:17921. doi: 10.1021/ja108432b. [DOI] [PubMed] [Google Scholar]
- 30.Ess DH, Houk KN. J Am Chem Soc. 2008;130:10187. doi: 10.1021/ja800009z. [DOI] [PubMed] [Google Scholar]
- 31.Ess DH, Houk KN. J Am Chem Soc. 2007;129:10646. doi: 10.1021/ja0734086. [DOI] [PubMed] [Google Scholar]
- 32.Frisch, et al. Gaussian 09. B.01. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 33.Aquilante F, De Vico L, Ferre N, Ghigo G, Malmqvist PA, Neogrady P, Pedersen TB, Pitonak M, Reiher M, Roos BO, Serrano-Andres L, Urban M, Veryazov V, Lindh R. J Comput Chem. 2010;31:224. doi: 10.1002/jcc.21318. [DOI] [PubMed] [Google Scholar]
- 34.Harding LB, Goddard WA. J Am Chem Soc. 1978;100:7180. [Google Scholar]
- 35.Wadt WR, Goddard WA. J Am Chem Soc. 1975;97:3004. [Google Scholar]
- 36.Bach RD, Andres JL, Owensby AL, Schlegel HB, Mcdouall JJW. J Am Chem Soc. 1992;114:7207. [Google Scholar]
- 37.Shustov GV, Rauk A. J Org Chem. 1998;63:5413. [Google Scholar]
- 38.Freccero M, Gandolfi R, Sarzi-Amade M, Rastelli A. Tetrahedron Lett. 2001;42:2739. [Google Scholar]
- 39.Freccero M, Gandolfi R, Sarzi-Amade M, Rastelli A. J Org Chem. 2003;68:811. doi: 10.1021/jo0266184. [DOI] [PubMed] [Google Scholar]
- 40.Fokin AA, Tkachenko BA, Korshunov OI, Gunchenko PA, Schreiner PR. J Am Chem Soc. 2001;123:11248. doi: 10.1021/ja0158096. [DOI] [PubMed] [Google Scholar]
- 41.Cremer D, Kraka E, Szalay PG. Chem Phys Lett. 1998;292:97. [Google Scholar]
- 42.Ensing B, Buda F, Gribnau MCM, Baerends EJ. J Am Chem Soc. 2004;126:4355. doi: 10.1021/ja038865a. [DOI] [PubMed] [Google Scholar]
- 43.Kumar D, de Visser SP, Shaik S. J Am Chem Soc. 2003;125:13024. doi: 10.1021/ja036906x. [DOI] [PubMed] [Google Scholar]
- 44.Wu YD, Houk KN. J Am Chem Soc. 1987;109:908. [Google Scholar]
- 45.Wu YD, Houk KN, Trost BM. J Am Chem Soc. 1987;109:5560. [Google Scholar]
- 46.Damm W, Giese B, Hartung J, Hasskerl T, Houk KN, Zipse H. J Am Chem Soc. 1992;114:4067. [Google Scholar]
- 47.Luo YR. CRC Handbook of Chemistry and Physics. 91. CRC Press; Boca Raton, FL: 2010. [Google Scholar]
- 48.Gordon CG, Mackey JL, Jewett JC, Sletten EM, Houk KN, Bertozzi CR. J Am Chem Soc. 2012;134:9199. doi: 10.1021/ja3000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newhouse T. PhD thesis with Baran, P S. see SI for details. [Google Scholar]
- 50.(a) Liu W, Groves JT. J Am Chem Soc. 2010;132:12847. doi: 10.1021/ja105548x. [DOI] [PubMed] [Google Scholar]; (b) Liu W, Huang X, Cheng M-J, Nielsen RJ, Goddard WA, III, Groves JT. Science. 2012;337:1322. doi: 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.