

Abstract
Acid-sensing ion channels (ASICs), a novel family of proton-gated amiloride-sensitive cation channels, are expressed primarily in neurons of peripheral sensory and central nervous systems. Recent studies have shown that activation of ASICs, particularly the ASIC1a channels, plays a critical role in neuronal injury associated with neurological disorders such as brain ischemia, multiple sclerosis, and spinal cord injury, etc. In normal conditions in vitro, ASIC1a channels desensitize rapidly in the presence of a continuous acidosis or following a pre-exposure to minor pH drop, raising doubt for their contributions to the acidosis-mediated neuronal injury. It is now known that the properties of ASICs can be dramatically modulated by signaling molecules or biochemical changes associated with pathological conditions. Modulation of ASICs by these molecules can lead to dramatically enhanced and/or prolonged activities of these channels thus promoting their pathological functions. Understanding of how ASICs behave in pathological conditions may help define new strategies for the treatment and/or prevention of neuronal injury associated with various neurological disorders.
Keywords: Acid-sensing ion channel, acidosis, ischemia, neuron, modulation
Introduction
In the normal condition, extracellular pH (pHo) is maintained at ~7.4 through various H+ transporting mechanisms (Chesler 1990). In pathological conditions such as tissue inflammation, ischemic stroke, traumatic brain injury, and epileptic seizure, a marked reduction of pHo, a condition termed acidosis, takes place (Ljunggren et al. 1974;Nedergaard et al. 1991;Rehncrona 1985;Revici et al. 1949;Siesjo 1988;Siesjo et al. 1996;Sutherland et al. 2000;Tombaugh and Sapolsky 1993). In brain ischemia, for example, the shortage of oxygen, due to the lack of blood supply, results in increased anaerobic glycolysis which leads to lactic acid accumulation (Siesjo et al. 1996;Tombaugh and Sapolsky 1993). Accumulation of lactic acid, along with H+ release from ATP hydrolysis, results in a dramatic decrease in tissue pH. In addition, cessation of local circulation results in carbon dioxide accumulation and carbonic acid build up, which also contribute to the decrease of brain pH (Siesjo 1988).
For many years, acidosis has been known to be associated with neuronal injury (Siesjo 1988;Siesjo et al. 1996;Tombaugh and Sapolsky 1993). However, the underlying mechanism was not entirely clear. Low tissue pH may cause cell injury through non-selective denaturation of proteins and nucleic acids (Kalimo et al. 1981); hinderance of postischemic metabolic recovery by inhibiting mitochondrial energy metabolism; impairment of postischemic blood flow via vascular edema (Hillered et al. 1985); stimulation of pathologic free radical formation (Rehncrona et al. 1989); and inhibition of astrocytic glutamate uptake which may contribute to excitatory neuronal injury (Swanson et al. 1995). Recent finding that acidosis, at the level commonly seen in neurological disorders, can induce neuronal injury by activating a distinct family of ligand-gated cation channels, the acid-sensing ion channels (ASICs), has dramatically changed the view of acid signaling and provided a novel therapeutic target for neuroprotection (Benveniste and Dingledine 2005;Huang and McNamara 2004;Voilley 2004;Wemmie et al. 2006;Xiong et al. 2004;Xiong et al. 2007;Yermolaieva et al. 2004).
Following the cloning and characterization of the first ASIC subunit (ASIC1a) in 1997 (Waldmann et al. 1997), six additional subunits of ASICs (1b1, 1b2, 2a, 2b, 3, and 4) have been identified (Wemmie et al. 2006). ASICs are voltage-independent, amiloride-sensitive, and Na+-selective cation channels belonging to the degenerin/epithelial Na+ channel (DEG/ENaC) superfamily (Alvarez et al. 2000). Expression of 1a, 2a, 2b, and 4 has been demonstrated in CNS neurons, while all other ASICs, except ASIC4, are expressed in peripheral sensory neurons. Non-neuronal tissues such as vascular smooth muscle cells (Grifoni et al. 2008), some glial cells (Feldman et al. 2008;Huang et al. 2010), and bone (Jahr et al. 2005) have been shown to express ASICs.
Each ASIC subunit consists of two transmembrane domains (TM1 and TM2) and a large cysteine rich extracellular loop, with the pre-TM2 region essential for ion permeability and the gating of these channels (Bassler et al. 2001;Krishtal 2003;Waldmann et al. 1997). Although a tetrameric assembly was initially proposed for functional ASICs, the recent success of crystal structure of chicken ASIC1a channels has revealed a trimeric assembly (Jasti et al. 2007). Among all ASICs, the homomeric ASIC1a channel is of particular interest, because of its wide distribution, high sensitivity to acid, and permeability to Ca2+ (Waldmann et al. 1997;Xiong et al. 2004;Yermolaieva et al. 2004).
In peripheral sensory neurons, ASICs are implicated in nociception (Benson et al. 1999;Bevan and Yeats 1991;Bohlen et al. 2011;Chen et al. 2002;Deval et al. 2008;Krishtal and Pidoplichko 1981;Mazzuca et al. 2007;Sluka et al. 2003;Ugawa et al. 2002;Wu et al. 2004), mechanosensation (Page et al. 2005;Price et al. 2000;Price et al. 2001), and taste transduction (Lin et al. 2002;Ugawa et al. 2003;Ugawa 2003). In CNS, ASIC1a is involved in synaptic plasticity, learning/memory, fear conditioning (Wemmie et al. 2002;Wemmie et al. 2003), and retinal physiology (Ettaiche et al. 2006). A number of studies have demonstrated that activation or sensitization of Ca2+-permeable ASIC1a channels is involved in acidosis-mediated neuronal injury (Friese et al. 2007;Gao et al. 2005;Li et al. 2011;Pignataro et al. 2011;Xiong et al. 2004), disclosing a novel neuroprotective target (Xiong et al. 2008).
Under experimental conditions (e.g. in patch-clamp recordings), ASICs are activated only by rapid pH drops and the currents of most ASIC subtypes, particularly the homomeric ASIC1a channels, desensitize rapidly in the continuous presence of acidic pH (Fig. 1A). Pre-exposure of ASIC1a channels to very small pH decrease (e.g. from 7.4 to 7.2) that does not activate the channel itself also suppresses the channel activity in response to subsequent large pH drops (Fig. 1B). Thus, whether a significant amount of ASIC current can be activated in the pathological conditions and whether the effects of ASIC activation could be long-lasting are important questions crucial in determining the functional significance of these channels. In this regard, recent findings showing that endogenous signaling molecules and biochemical changes associated with various pathological conditions can dramatically alter the properties of ASICs have provided desirable answer to the questions. Understanding the modulation of ASICs by molecules or biochemical changes associated with pathological conditions will help with understanding the pathological functions of these channels. It will also aid the development of new and effective strategies for the treatment and/or prevention of neuronal injury associated with various neurological disorders.
Figure 1.

Desensitization of ASIC current in cultured mouse cortical neurons. (A) ASIC current was activated by lowering the extracellular pH from 7.4 to 6.5 or 6.0. The current decays rapidly in the continuous presence of reduced pH. (B) ASIC current was activated by lowering the extracellular pH to 6.0 from a preset pH of 7.4 or 7.2. Pre-incubation of cells at pH 7.2 dramatically inhibited the ASIC current activated at pH 6.0.
Lactate
In the normal brain, the concentration of lactate is at ~1 mM. Following ischemia, anaerobic metabolism of glucose leads to increased production of lactate. A concentration of lactate between 12–20 mM has been reported (Schurr 2002;Schurr and Rigor 1998). Increased concentration of lactate correlates well with increased degree of brain injury (Siesjo et al. 1996), though the detailed mechanism was unclear. The finding that lactate enhances the ASIC current provided a new interpretation (Immke and McCleskey 2001).
In sensory neurons that innervate the heart, Immke & McCleskey first demonstrated that the addition of 15 mM lactate dramatically increased the amplitude the ASIC current activated by a moderate pH drop to ~7.0 (Immke and McCleskey 2001). Applications of the same concentration of lactate at pH values that do not activate ASICs (e.g. pH 8.0 or pH 7.4) caused no response. Thus, lactate acts by potentiating but not activating the ASICs. In COS-7 transfected with different subunit of ASICs, both ASIC3 and ASIC1a currents were potentiated by lactate (Immke and McCleskey 2001). The effect of lactate on ASIC current persists in excised cell-free membrane patches suggesting that no second messenger or signaling cascade is involved. Since lactate has the ability to chelate the divalent cations including Ca2+ and Mg2+, which have a modulatory role on various membrane receptors and ion channels (Hess et al. 1986;Xiong and MacDonald 1999;Zhou and Jones 1995), the authors hypothesized that potentiation of the ASIC currents may be due to the chelation of divalent cations by lactate. Indeed, adjusting the concentrations of Ca2+ and Mg2+ eliminated the effect of lactate. Whereas, reducing the divalent concentrations mimicked the effect of lactate (Immke and McCleskey 2001). Other monocarboxylic acids which have divalent cation chelation ability also potentiated the ASIC current. Similar to the cardiac sensory neurons, potentiation of the ASIC current by lactate has been reported in other neurons such as cerebellar Purkinje neurons (Allen and Attwell 2002).
Ca2+
Ca2+ is one of the most important modulators of various voltage-gated and ligand-gated ion channels including ASICs. Co-application of Ca2+ with acidic solution reduces ASIC currents (Gao et al. 2004;Immke and McCleskey 2003;Paukert et al. 2004;Waldmann et al. 1997). Similarly, pretreatment followed by continuous presence of the extracellular Ca2+ also inhibits the ASIC currents (de Weille and Bassilana 2001;Wang et al. 2006). Investigation into the mechanisms underlying Ca2+ modulation of ASICs has led to the finding that Ca2+ decreases the affinity of ASICs for H+ (Babini et al. 2002;Immke and McCleskey 2003). It has been proposed that, at a pH of 7.4, ASICs (e.g. ASIC3 channels) are closed because of the Ca2+ blockade. As the pHo is decreased, binding of H+ to the channel displaces Ca2+ from its binding site, leading to opening of the channel (Immke and McCleskey 2003). For ASIC1a channels, different models have been proposed (Paukert et al. 2004;Zhang et al. 2006). Paukert and colleagues showed that two negatively charged residues near the entrance of the channel pore, E425 and D432, are crucial for Ca2+ blockade of the ASIC1a channel (Paukert et al. 2004). They proposed that there are more than one Ca2+ binding site, one that mediates blocking and one mediates modulation, exist on the ASIC1a channel. In pathological conditions such as brain ischemia, significant decrease of extracellular Ca2+ concentration ([Ca2+]e) takes place (Ekholm et al. 1995;Hansen and Zeuthen 1981). Reduction in [Ca2+]e is expected to relieve the inhibition of ASICs thus increasing the activity and pathological function of these channels.
Arachidonic acid
Arachidonic acid (AA) is a polyunsaturated fatty acid present in the phospholipids of all cell membranes. It is one of the most abundant fatty acids in the brain. In addition to being involved in cellular signaling as a lipid second messenger involved in the regulation of signaling enzymes such as PLC and PKC (Farooqui and Horrocks 2006;Sang and Chen 2006), AA plays important roles in various pathological conditions. For example, it is a key intermediate in inflammatory responses. In addition, the role of AA in the pathophysiology of neurological disorders including ischemic brain injury has been well documented (Farooqui et al. 2006;Farooqui and Horrocks 2006;Muralikrishna and Hatcher 2006;Siesjo and Katsura 1992). Following brain ischemia, the rise of [Ca2+]i leads to the activation of phospholipase A2, resulting in increased production of lipid mediators including AA (Farooqui and Horrocks 2006;Muralikrishna and Hatcher 2006;Rehncrona et al. 1982). High concentrations of lipid mediators cause neurotoxicity (Farooqui and Horrocks 2006).
Earlier studies have shown that AA has effects on a variety of voltage-gated and ligand-gated ion channels (Angelova and Muller 2006;Hu et al. 2006;Keros and McBain 1997;Mignen et al. 2005;Miller et al. 1992;Nagano et al. 1995;Paoletti and Ascher 1994). Particularly, it potentiates the opening of NMDA-gated channels (Casado and Ascher 1998;Miller et al. 1992;Paoletti and Ascher 1994), which might contribute to its neurotoxicity.
Allen and Attwell were the first to show that AA also enhances the activity of ASICs (Allen and Attwell 2002). In rat cerebellar Purkinje cells, bath perfusion of solution with 5 or 10 μM arachidonic acid produced a large increase in the amplitude of ASIC current. In addition to the peak current, application of AA enhanced or induced a sustained component of the ASIC current (Allen and Attwell 2002). The potentiation of the ASIC current by AA appears to be independent of its derivatives, since an agent known to block the breakdown of AA did not affect its capacity to potentiate the ASIC current (Allen and Attwell 2002). The molecular mechanism for AA potentiation of ASICs is not clear. One possibility is that, insertion of AA into the membrane induces membrane stretch and that the ASICs are stretch-sensitive, as are NMDA channels (Casado and Ascher 1998). This explanation is supported by the finding that perfusion of neurons with hypotonic saline, which causes cell swelling and membrane stretch, mimicked the potentiation of ASIC currents by AA (Allen and Attwell 2002). Subsequent studies by Smith et al also suggested that AA potentiates ASIC activation by a direct mechanism (Smith et al. 2007). They confirmed that inhibition of AA metabolism had no effect on the potentiation of ASIC1a. However, the potentiation of single ASIC2a channels by AA could also be observed in cell-free patches, suggesting that membrane stretch is not required.
Spermine
Spermine is a polyvalent cation whose extracellular concentration fluctuates significantly within the nervous system. Although spermine is involved in various physiological processes, high concentration of spermine can induce neuronal depolarization and cytoplasmic Ca2+ overload, which might lead to neuronal damage (Toninello et al. 2004). Following ischemia, the activity of ornithine decarboxylase (ODC), a rate-limiting enzyme responsible for polyamine synthesis is enhanced, leading to increased concentration of spermine and neuronal injury (Kindy et al. 1994).
The mechanism underlying increased neuronal injury by spermine has not been entirely clear. Spermine can modulate NMDA receptor function (Benveniste and Mayer 1993;Rock and Macdonald 1995). Thus, several studies have attempted to link its neurotoxicity to increased activation of NMDA receptors. However, these studies have yielded inconsistent results (Johnson 1998;Li et al. 2007). Additional mechanisms are likely involved in spermine-mediated neurotoxicity.
Babini and colleagues were the first to demonstrate that spermine potentiates the activities of ASICs (Babini et al. 2002). Subsequent studies by Duan et al showed that extracellular spermine exacerbated ischemic neuronal injury through sensitization of ASIC1a channels to extracellular acidosis (Duan et al. 2011). Pharmacological blockade of ASIC1a or deletion of the ASIC1 gene reduced the enhancing effect of spermine on ischemic neuronal damage, both in cultured neurons and in a mouse model of focal ischemia. Spermine also reduced desensitization of ASIC1a in the open state and accelerated recovery from desensitization in response to repeated acid stimulation. Enhanced channel activity was accompanied by increased acid-induced neuronal depolarization and cytoplasmic Ca2+ overload, which may explain the exacerbation of neuronal damage caused by spermine. Thus, extracellular spermine contributes to ischemic neuronal injury, at least in part, by enhancing the activity of ASIC1a channels (Duan et al. 2011).
Proteases
Brain injury is accompanied by increased protease activity (Gingrich and Traynelis 2000). Blood-derived proteases such as thrombin, tissue plasminogen activator, and plasmin can gain access to CNS interstitial spaces due to a compromised blood-brain barrier (Gingrich and Traynelis 2000;Vivien and Buisson 2000). Previous studies have demonstrated that proteases modulate the activities of various ion channels including ENaC, which belongs to the same superfamily as ASICs (Chraibi et al. 1998;Holt et al. 2001;Nicole et al. 2001), suggesting that ASICs could be a target. Indeed, subsequent studies by Poirot and colleagues showed that the activity of ASIC1a is modulated by serine proteases (Poirot et al. 2004). Exposure of CHO cells stably expressing ASIC1a channels to trypsin or other serine proteases (e.g. proteinase K and chymotrypsin) shifted the pH dependence of activation and steady-state inactivation of the ASIC1a channels to more acidic pH values. As a consequence, protease exposure leads to a decrease in ASIC1a activity when currents are activated by a pH drop from 7.4. Interestingly, if the channel is activated from a basal pH of 7, a condition relevant to ischemia, protease exposure increases rather than decreases the ASIC1a activity. In addition, protease treatment dramatically accelerates the recovery rate of ASIC1a channels from desensitization (Poirot et al. 2004). The effects of proteases on ASICs involves proteolysis of the channel protein, as the capacity of trypsin to modulate the ASIC1a channels was eliminated with soybean trypsin inhibitor or modification of trypsin’s catalytic site with TLCK, a reagent that irreversibly modifies a histidine residue in the catalytic site of trypsin. Cleavage of the channel protein was confirmed by Western blot showing reduction of a 64-kDa ASIC protein band to a lower molecular weight band of 49 kDa (Poirot et al. 2004). Further studies demonstrated that trypsin cleaves ASIC1a subunits at Arg-145 in the N-terminal part of the extracellular loop. The cleavage site is between a highly conserved sequence and a sequence that is critical for ASIC1a inhibition by PcTx1 (Vukicevic et al. 2006). Since activation of ASIC1a is involved in acidosis-mediated ischemic brain injury [30,33], modulation of ASIC1a by proteases could be relevant to its pathological function in brain ischemia.
CaMKII
Ca2+/calmodulin (CaM)-dependent protein kinase II (CaMKII) is the most abundant kinase isoform in brain, particularly enriched in neurons. It is a multifunctional protein kinase that regulates normal neuronal function. It is regulated by multi-site phosphorylation, which can alter the enzyme activity and targeting to cellular microdomains through interactions with binding proteins. CaMKII is a major mediator of the function of excitatory glutamate receptors. Activation of glutamate receptors triggers an increase of intracellular Ca2+ and an autophosphorylation of CaMKII at T286. This process makes the kinase active independent from Ca2+ stimulation, a transition required for synaptic plasticity. Increased CaMKII activity was also implicated in the regulation of neuronal death, though the detailed mechanism was unclear.
Recent studies by Gao et al suggest that during ischemia, coupling between the NMDARs/CaMKII cascade and the ASIC1a channel contributes significantly to acidotoxic neuronal death (Gao et al. 2005). They demonstrated that global brain ischemia in rats results in an increased phosphorylation of ASIC1a by CaMKII at Ser478 and Ser479. This phosphorylation sensitizes the channel to low pH, exacerbating cell death by allowing increased calcium conductance. The phosphorylation is a result of the activation of NR2B-containing NMDA receptors and an increase of intracellular Ca2+ during ischemia.
Consistent with the previous study (Xiong et al. 2004), they observed an enhancement of ASIC currents by oxygen glucose deprivation (OGD). Inhibition of CaMKII with KN-93 or CaMKIINtide abolished the enhancement of ASIC currents, indicating an involvement of CaMKII. Consistent with the involvement of CaMKII, they showed an increased phosphorylation of ASIC1a after transient global ischemia which can be blocked by intracerebroventricular administration of CaMKII inhibitor KN-93 or CaMKIINtide. In addition, there was an increased co-immunoprecipitation of ASIC1a with CaMKIIα after global ischemia, which can be inhibited by KN-93 or CaMKIINtide. Pharmacological inhibition of CaMKII phosphorylation of ASIC1a with KN-93, or mutation of ASIC1a at Ser478 and Ser479, produced neuroprotection. Thus, phosphorylation of ASIC1a by CaMKII at the site of Ser478 and Ser479 plays an essential role in ischemia-induced cell death.
Nitric oxide
Nitric oxide (NO) is an important reactive oxygen/nitrogen species which has a variety of physiological and pathological functions (Star 1993). Endogenous synthesis of NO is catalyzed by nitric oxide synthase, which is Ca2+ dependent. During ischemia, over-activation of the glutamate receptors and subsequent increases in intracellular Ca2+ leads to activation of the Ca2+-dependent neuronal form of nitric oxide synthase (nNOS), resulting in an increased production of NO (Bolanos and Almeida 1999;Schulz et al. 1997). NO can also be released by activated microglia (Boje and Arora 1992). NO regulates the protein function by two main pathways: an indirect mechanism that involves the production of cGMP and the activation of protein kinase G, and a direct mechanism that involves modification of the tertiary structure of proteins by S-nitrosylating the thiol side-chains of cysteine residues (Jaffrey et al. 2001). Increased NO production is known to increase neuronal injury (Boje and Arora 1992). Although the formation of a strong oxidant of peroxynitrite is likely involved in cell injury, other mechanisms cannot be excluded. Cadiou and colleagues were the first to report that ASICs are a target of NO. NO donor S-nitroso-N-acetylpenicillamine (SNAP) potentiates proton-gated currents in DRG neurons and in CHO cells expressing each of the ASIC subunits. Modulators of the cGMP/PKG pathway had no effect on the potentiation, but in excised patches from CHO cells expressing ASIC2a, the potentiation could be reversed by externally applying reducing agents. NO therefore has a direct external effect on ASICs, probably through oxidization of cysteine residues (Cadiou et al. 2007).
Consistent with the potentiation of ASIC currents by NO, subsequent studies by Jett et al demonstrated that acid-induced cell injury is potentiated by NO donor (Jetti et al. 2010). They showed that, at a pH of 6.1, death rates of Neuro2A cells expressing ASIC1 channels were significantly higher than the cells that do not express ASICs. Amiloride, a blocker of ASICs, protected the cell from acid-injury, suggesting that acid injury is mediated by activating ASICs. Sodium nitroprusside, a potent NO donor, not only increased the ASIC mediated currents but also increased acid-induced cell death.
Dynorphins
Dynorphins are endogenous opioid neuropeptides abundantly expressed in the CNS. They are involved in a variety of physiologic functions including antinociception and neuroendocrine signaling, and may be protective to neurons and oligodendroglia via their opioid receptor-mediated effects. However, under pathophysiological conditions where dynorphin levels are substantially elevated, these peptides are excitotoxic, partially through actions at glutamate receptors (Hauser et al. 1999). The excitotoxic actions of dynorphins require supraphysiological concentrations or prolonged tissue exposure. Thus, dynorphins can have either a protective or destructive action in neurons and glia, and the net effect may depend upon the distribution of receptors in a particular region and the amount of dynorphins released.
Recently Sherwood and Askwith reported that, at high concentrations dynorphin A and big dynorphin potentiate acid-activated currents in cortical neurons and in CHO cells expressing homomeric ASIC1a subunits (Sherwood and Askwith 2009). The potentiation of the ASIC1a activity was mediated through a limitation of steady-state desensitization of the channel. The potentiation of ASIC1a activity by dynorphin was not mediated by opioid receptor activation but through a direct interaction with ASIC1a. Alteration of steady-state desensitization by dynorphins enhanced ASIC1a-triggered neuronal injury during prolonged acidosis. Thus, ASIC1a is a new non-opioid receptor target for dynorphins, and that dynorphins can enhance ischemic brain injury by preventing steady-state desensitization of ASIC1a channels.
FMRFamide
FMRFamide and structurally related peptides are abundant in invertebrate nervous systems where they function as neurotransmitters and neuromodulators. Although FMRFamide itself has not been isolated in mammals, several FMRFamide-related peptides exist in the mammalian nervous system. FMRFamide and related peptides are generally thought to exert their physiological roles through G-protein coupled receptors (Lingueglia et al. 2006). However, two ionotropic receptors involved in the function of these peptides have recently been identified. FMRFamide-gated Na+ channel (FaNaC), which is a neuronal Na+ channel in invertebrates, is directly activated by micromolar concentrations of FMRFamide and RFamide-related peptides (RFRPs) (Lingueglia et al. 2006). In addition, ASICs, which share significant structure and sequence homology with FaNaC in the mammalian nervous system, can be modulated by FMRFamide and RFRPs. FMRFamide, and RFRPs such as neuropeptide FF are incapable of generating any ASIC currents on their own, but significantly potentiate ASIC currents in sensory neurons and in heterologous expression systems (Askwith et al. 2000;Catarsi et al. 2001;Xie et al. 2003). In addition to their effects on the amplitude of ASIC currents, FMRFamide and RFRPs also reduce the rate of current desensitization (Askwith et al. 2000;Catarsi et al. 2001;Xie et al. 2003).
Insulin
Insulin and insulin receptors are expressed at high levels in discrete regions within the CNS, and insulin is released by depolarization in cultured CNS neurons (Wozniak et al. 1993). In addition to its conventional role in glucose uptake, insulin has been shown to act as a neuromodulator of many brain functions, such as food intake, neuronal growth and maturation. It has also been shown that insulin administration protects neurologic function in cerebral ischemia in non-diabetic rats (LeMay et al. 1988). The exact mechanism was not clear. Almost 15 years ago, Wan and colleagues first demonstrated that insulin promotes surface expression of inhibitory GABAA receptors thereby potentiating GABAA-receptor-mediated synaptic inhibition. This effect may partially explain the neuroprotective effect of insulin in non-diabetic animals.
Very recently, Chai and colleagues demonstrated that insulin plays an important role in regulating the level of surface expression of ASIC1a channels (Chai et al. 2010). This finding provided an alternative explanation for the protective effect of insulin. In CHO cells expressing ASIC1a subunit, serum depletion, a condition mimicking ischemia, induced a significant increase in the level of ASIC1a surface expression. As a result, the ASIC1a current was dramatically enhanced. Among the components of serum, insulin was identified as the key factor that maintains a low level of ASIC1a expression on the plasma membrane. Removing insulin from culture medium increased the trafficking of the ASIC1a channels to the surface membrane. Similarly, neurons subjected to insulin depletion increased the surface expression of ASIC1a subunit with resultant potentiation of ASIC1a currents. Thus, in the normal condition where insulin is present, ASIC1a is predominantly localized to the endoplasmic reticulum. Under conditions of substrate depletion (e.g. following brain ischemia), the lack of insulin may stimulate translocation of the ASIC1a channels to the cell surface, thus increasing the function of these channels (Chai et al. 2010). Since activation of ASIC1a channels plays a critical role in acidosis-mediated ischemic brain injury, inhibition of ASIC1a expression by insulin may account partially for its neuroprotective effect.
Conclusion
ASICs represent new biological components in neurons. Increasing evidence indicates the involvement of these channels in both physiological and pathological processes such as nociception, mechanosensation, taste transduction, synaptic plasticity, learning/memory, and acidosis-mediated neurodegeneration. Although in most electrophysiological recordings ASIC responses appear to be transient in nature, various biochemical changes, largely occur in pathological conditions, dramatically enhance the amplitude and reduce the desensitization of the ASIC current (Figure 2), or increase the recovery of ASICs from desensitization. Knowing the changes of ASIC property and the mechanisms of modulation in pathological conditions is, with no doubt, critical for understanding the pathological functions of these channels and for establishing effective therapeutic interventions.
Figure 2.
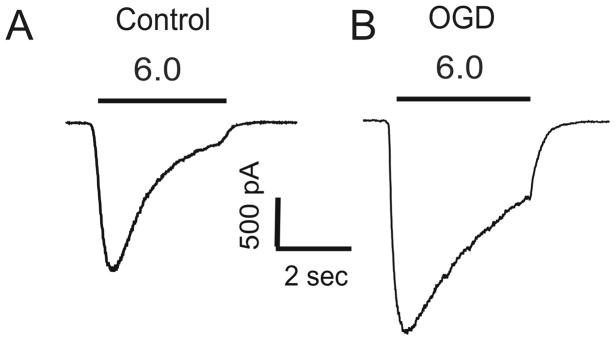
Oxygen glucose deprivation (OGD), an in vitro model of ischemia, increases the amplitude but reduces the desensitization of ASIC current in mouse cortical neurons.
Acknowledgments
XPC is supported by American Heart Association Scientist Development Grant 0735092N, University of Missouri Research Board and University of Missouri-Kansas City School of Medicine Start-up Funding. The work in ZGX’s lab is supported in part by NIH R01NS047506, R01NS066027, UL1 RR025008, U54 RR026137, AHA 0840132N, and ALZ IIRG-10-173350.
References
- Allen NJ, Attwell D. Modulation of ASIC channels in rat cerebellar Purkinje neurons by ischemia-related signals. J Physiol (Lond) 2002;543:521–529. doi: 10.1113/jphysiol.2002.020297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez dlR, Canessa CM, Fyfe GK, Zhang P. Structure and regulation of amiloride-sensitive sodium channels. Annu Rev Physiol. 2000;62:573–594. doi: 10.1146/annurev.physiol.62.1.573. [DOI] [PubMed] [Google Scholar]
- Angelova P, Muller W. Oxidative modulation of the transient potassium current IA by intracellular arachidonic acid in rat CA1 pyramidal neurons. Eur J Neurosci. 2006;23:2375–2384. doi: 10.1111/j.1460-9568.2006.04767.x. [DOI] [PubMed] [Google Scholar]
- Askwith CC, Cheng C, Ikuma M, Benson C, Price MP, Welsh MJ. Neuropeptide FF and FMRFamide potentiate acid-evoked currents from sensory neurons and proton-gated DEG/ENaC channels. Neuron. 2000;26:133–141. doi: 10.1016/s0896-6273(00)81144-7. [DOI] [PubMed] [Google Scholar]
- Babini E, Paukert M, Geisler HS, Grunder S. Alternative splicing and interaction with di-and polyvalent cations control the dynamic range of acid-sensing ion channel 1 (ASIC1) J Biol Chem. 2002;277:41597–41603. doi: 10.1074/jbc.M205877200. [DOI] [PubMed] [Google Scholar]
- Bassler EL, Ngo-Anh TJ, Geisler HS, Ruppersberg JP, Grunder S. Molecular and functional characterization of acid-sensing ion channel (ASIC) 1b. J Biol Chem. 2001;276:33782–33787. doi: 10.1074/jbc.M104030200. [DOI] [PubMed] [Google Scholar]
- Benson CJ, Eckert SP, McCleskey EW. Acid-evoked currents in cardiac sensory neurons: A possible mediator of myocardial ischemic sensation. Circ Res. 1999;84:921–928. doi: 10.1161/01.res.84.8.921. [DOI] [PubMed] [Google Scholar]
- Benveniste M, Dingledine R. Limiting stroke-induced damage by targeting an acid channel. N Engl J Med. 2005;352:85–86. doi: 10.1056/NEJMcibr045010. [DOI] [PubMed] [Google Scholar]
- Benveniste M, Mayer ML. Multiple effects of spermine on N-methyl-D-aspartic acid receptor responses of rat cultured hippocampal neurones. J Physiol. 1993;464:131–163. doi: 10.1113/jphysiol.1993.sp019627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan S, Yeats J. Protons activate a cation conductance in a sub-population of rat dorsal root ganglion neurones. J Physiol (Lond ) 1991;433:145–161. doi: 10.1113/jphysiol.1991.sp018419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlen CJ, Chesler AT, Sharif-Naeini R, Medzihradszky KF, Zhou S, King D, Sanchez EE, Burlingame AL, Basbaum AI, Julius D. A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature. 2011;479:410–414. doi: 10.1038/nature10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- Bolanos JP, Almeida A. Roles of nitric oxide in brain hypoxia-ischemia. Biochim Biophys Acta. 1999;1411:415–436. doi: 10.1016/s0005-2728(99)00030-4. [DOI] [PubMed] [Google Scholar]
- Cadiou H, Studer M, Jones NG, Smith ES, Ballard A, McMahon SB, McNaughton PA. Modulation of acid-sensing ion channel activity by nitric oxide. J Neurosci. 2007;27:13251–13260. doi: 10.1523/JNEUROSCI.2135-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado M, Ascher P. Opposite modulation of NMDA receptors by lysophospholipids and arachidonic acid: common features with mechanosensitivity [In Process Citation] J Physiol (Lond ) 1998;513:317–330. doi: 10.1111/j.1469-7793.1998.317bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catarsi S, Babinski K, Seguela P. Selective modulation of heteromeric ASIC proton-gated channels by neuropeptide FF. Neuropharmacology. 2001;41:592–600. doi: 10.1016/s0028-3908(01)00107-1. [DOI] [PubMed] [Google Scholar]
- Chai S, Li M, Branigan D, Xiong ZG, Simon RP. Activation of acid-sensing ion channel 1a (ASIC1a) by surface trafficking. J Biol Chem. 2010;285:13002–13011. doi: 10.1074/jbc.M109.086041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Zimmer A, Sun WH, Hall J, Brownstein MJ, Zimmer A. A role for ASIC3 in the modulation of high-intensity pain stimuli. Proc Natl Acad Sci U S A. 2002;99:8992–8997. doi: 10.1073/pnas.122245999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M. The regulation and modulation of pH in the nervous system. Prog Neurobiol. 1990;34:401–427. doi: 10.1016/0301-0082(90)90034-e. [DOI] [PubMed] [Google Scholar]
- Chraibi A, Vallet V, Firsov D, Hess SK, Horisberger JD. Protease modulation of the activity of the epithelial sodium channel expressed in Xenopus oocytes. J Gen Physiol. 1998;111:127–138. doi: 10.1085/jgp.111.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Weille J, Bassilana F. Dependence of the acid-sensitive ion channel, ASIC1a, on extracellular Ca(2+) ions. Brain Res. 2001;900:277–281. doi: 10.1016/s0006-8993(01)02345-9. [DOI] [PubMed] [Google Scholar]
- Deval E, Noel J, Lay N, Alloui A, Diochot S, Friend V, Jodar M, Lazdunski M, Lingueglia E. ASIC3, a sensor of acidic and primary inflammatory pain. EMBO J. 2008;27:3047–3055. doi: 10.1038/emboj.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan B, Wang YZ, Yang T, Chu XP, Yu Y, Huang Y, Cao H, Hansen J, Simon RP, Zhu MX, Xiong ZG, Xu TL. Extracellular spermine exacerbates ischemic neuronal injury through sensitization of ASIC1a channels to extracellular acidosis. J Neurosci. 2011;31:2101–2112. doi: 10.1523/JNEUROSCI.4351-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekholm A, Kristian T, Siesjo BK. Influence of hyperglycemia and of hypercapnia on cellular calcium transients during reversible brain ischemia. Exp Brain Res. 1995;104:462–466. doi: 10.1007/BF00231980. [DOI] [PubMed] [Google Scholar]
- Ettaiche M, Deval E, Cougnon M, Lazdunski M, Voilley N. Silencing acid-sensing ion channel 1a alters cone-mediated retinal function. J Neurosci. 2006;26:5800–5809. doi: 10.1523/JNEUROSCI.0344-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqui AA, Horrocks LA. Phospholipase A2-generated lipid mediators in the brain: the good, the bad, and the ugly. Neuroscientist. 2006;12:245–260. doi: 10.1177/1073858405285923. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Ong WY, Horrocks LA. Inhibitors of brain phospholipase A2 activity: their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol Rev. 2006;58:591–620. doi: 10.1124/pr.58.3.7. [DOI] [PubMed] [Google Scholar]
- Feldman DH, Horiuchi M, Keachie K, McCauley E, Bannerman P, Itoh A, Itoh T, Pleasure D. Characterization of acid-sensing ion channel expression in oligodendrocyte-lineage cells. Glia. 2008 doi: 10.1002/glia.20693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJ, Vincent A, Fugger L. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007;13:1483–1489. doi: 10.1038/nm1668. [DOI] [PubMed] [Google Scholar]
- Gao J, Duan B, Wang DG, Deng XH, Zhang GY, Xu L, Xu TL. Coupling between NMDA receptor and acid-sensing ion channel contributes to ischemic neuronal death. Neuron. 2005;48:635–646. doi: 10.1016/j.neuron.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Gao J, Wu LJ, Xu L, Xu TL. Properties of the proton-evoked currents and their modulation by Ca2+ and Zn2+ in the acutely dissociated hippocampus CA1 neurons. Brain Res. 2004;1017:197–207. doi: 10.1016/j.brainres.2004.05.046. [DOI] [PubMed] [Google Scholar]
- Gingrich MB, Traynelis SF. Serine proteases and brain damage - is there a link? Trends Neurosci. 2000;23:399–407. doi: 10.1016/s0166-2236(00)01617-9. [DOI] [PubMed] [Google Scholar]
- Grifoni SC, Jernigan NL, Hamilton G, Drummond HA. ASIC proteins regulate smooth muscle cell migration. Microvasc Res. 2008;75:202–210. doi: 10.1016/j.mvr.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AJ, Zeuthen T. Extracellular ion concentrations during spreading depression and ischemia in the rat brain cortex. Acta Physiol Scand. 1981;113:437–445. doi: 10.1111/j.1748-1716.1981.tb06920.x. [DOI] [PubMed] [Google Scholar]
- Hauser KF, Foldes JK, Turbek CS. Dynorphin A (1-13) neurotoxicity in vitro: opioid and non-opioid mechanisms in mouse spinal cord neurons. Exp Neurol. 1999;160:361–375. doi: 10.1006/exnr.1999.7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess P, Lansman JB, Tsien RW. Calcium channel selectivity for divalent and monovalent cations. Voltage and concentration dependence of single channel current in ventricular heart cells. J Gen Physiol. 1986;88:293–319. doi: 10.1085/jgp.88.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillered L, Smith ML, Siesjo BK. Lactic acidosis and recovery of mitochondrial function following forebrain ischemia in the rat. J Cereb Blood Flow Metab. 1985;5:259–266. doi: 10.1038/jcbfm.1985.33. [DOI] [PubMed] [Google Scholar]
- Holt JC, Lioudyno M, Athas G, Garcia MM, Perin P, Guth PS. The effect of proteolytic enzymes on the alpha9-nicotinic receptor-mediated response in isolated frog vestibular hair cells. Hear Res. 2001;152:25–42. doi: 10.1016/s0378-5955(00)00225-2. [DOI] [PubMed] [Google Scholar]
- Hu HZ, Xiao R, Wang C, Gao N, Colton CK, Wood JD, Zhu MX. Potentiation of TRPV3 channel function by unsaturated fatty acids. J Cell Physiol. 2006;208:201–212. doi: 10.1002/jcp.20648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Hu ZL, Wu WN, Yu DF, Xiong QJ, Song JR, Shu Q, Fu H, Wang F, Chen JG. Existence and distinction of acid-evoked currents in rat astrocytes. Glia. 2010;58:1415–1424. doi: 10.1002/glia.21017. [DOI] [PubMed] [Google Scholar]
- Huang Y, McNamara JO. Ischemic stroke: “acidotoxicity” is a perpetrator. Cell. 2004;118:665–666. doi: 10.1016/j.cell.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Immke DC, McCleskey EW. Lactate enhances the acid-sensing Na+ channel on ischemia-sensing neurons. Nat Neurosci. 2001;4:869–870. doi: 10.1038/nn0901-869. [DOI] [PubMed] [Google Scholar]
- Immke DC, McCleskey EW. Protons open acid-sensing ion channels by catalyzing relief of Ca2+ blockade. Neuron. 2003;37:75–84. doi: 10.1016/s0896-6273(02)01130-3. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Jahr H, van Driel M, van Osch GJ, Weinans H, van Leeuwen JP. Identification of acid-sensing ion channels in bone. Biochem Biophys Res Commun. 2005;337:349–354. doi: 10.1016/j.bbrc.2005.09.054. [DOI] [PubMed] [Google Scholar]
- Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature. 2007;449:316–323. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- Jetti SK, Swain SM, Majumder S, Chatterjee S, Poornima V, Bera AK. Evaluation of the role of nitric oxide in acid sensing ion channel mediated cell death. Nitric Oxide. 2010;22:213–219. doi: 10.1016/j.niox.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Johnson TD. Polyamines and cerebral ischemia. Prog Drug Res. 1998;50:193–258. doi: 10.1007/978-3-0348-8833-2_5. [DOI] [PubMed] [Google Scholar]
- Kalimo H, Rehncrona S, Soderfeldt B, Olsson Y, Siesjo BK. Brain lactic acidosis and ischemic cell damage: 2. Histopathology. J Cereb Blood Flow Metab. 1981;1:313–327. doi: 10.1038/jcbfm.1981.35. [DOI] [PubMed] [Google Scholar]
- Keros S, McBain CJ. Arachidonic acid inhibits transient potassium currents and broadens action potentials during electrographic seizures in hippocampal pyramidal and inhibitory interneurons. J Neurosci. 1997;17:3476–3487. doi: 10.1523/JNEUROSCI.17-10-03476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindy MS, Hu Y, Dempsey RJ. Blockade of ornithine decarboxylase enzyme protects against ischemic brain damage. J Cereb Blood Flow Metab. 1994;14:1040–1045. doi: 10.1038/jcbfm.1994.136. [DOI] [PubMed] [Google Scholar]
- Krishtal O. The ASICs: signaling molecules? Modulators? Trends Neurosci. 2003;26:477–483. doi: 10.1016/S0166-2236(03)00210-8. [DOI] [PubMed] [Google Scholar]
- Krishtal OA, Pidoplichko VI. A receptor for protons in the membrane of sensory neurons may participate in nociception. Neuroscience. 1981;6:2599–2601. doi: 10.1016/0306-4522(81)90105-6. [DOI] [PubMed] [Google Scholar]
- LeMay DR, Gehua L, Zelenock GB, D’Alecy LG. Insulin administration protects neurologic function in cerebral ischemia in rats. Stroke. 1988;19:1411–1419. doi: 10.1161/01.str.19.11.1411. [DOI] [PubMed] [Google Scholar]
- Li J, Doyle KM, Tatlisumak T. Polyamines in the brain: distribution, biological interactions, and their potential therapeutic role in brain ischaemia. Curr Med Chem. 2007;14:1807–1813. doi: 10.2174/092986707781058841. [DOI] [PubMed] [Google Scholar]
- Li MH, Inoue K, Si HF, Xiong ZG. Calcium-permeable ion channels involved in glutamate receptor-independent ischemic brain injury. Acta Pharmacol Sin. 2011;32:734–740. doi: 10.1038/aps.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Ogura T, Kinnamon SC. Acid-activated cation currents in rat vallate taste receptor cells. J Neurophysiol. 2002;88:133–141. doi: 10.1152/jn.2002.88.1.133. [DOI] [PubMed] [Google Scholar]
- Lingueglia E, Deval E, Lazdunski M. FMRFamide-gated sodium channel and ASIC channels: a new class of ionotropic receptors for FMRFamide and related peptides. Peptides. 2006;27:1138–1152. doi: 10.1016/j.peptides.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Ljunggren B, Norberg K, Siesjo BK. Influence of tissue acidosis upon restitution of brain energy metabolism following total ischemia. Brain Res. 1974;77:173–186. doi: 10.1016/0006-8993(74)90782-3. [DOI] [PubMed] [Google Scholar]
- Mazzuca M, Heurteaux C, Alloui A, Diochot S, Baron A, Voilley N, Blondeau N, Escoubas P, Gelot A, Cupo A, Zimmer A, Zimmer AM, Eschalier A, Lazdunski M. A tarantula peptide against pain via ASIC1a channels and opioid mechanisms. Nat Neurosci. 2007;10:943–945. doi: 10.1038/nn1940. [DOI] [PubMed] [Google Scholar]
- Mignen O, Thompson JL, Shuttleworth TJ. Arachidonate-regulated Ca2+-selective (ARC) channel activity is modulated by phosphorylation and involves an A-kinase anchoring protein. J Physiol. 2005;567:787–798. doi: 10.1113/jphysiol.2005.090209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B, Sarantis M, Traynelis SF, Attwell D. Potentiation of NMDA receptor currents by arachidonic acid. Nature. 1992;355:722–725. doi: 10.1038/355722a0. [DOI] [PubMed] [Google Scholar]
- Muralikrishna AR, Hatcher JF. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic Biol Med. 2006;40:376–387. doi: 10.1016/j.freeradbiomed.2005.08.044. [DOI] [PubMed] [Google Scholar]
- Nagano N, Imaizumi Y, Watanabe M. Modulation of calcium channel currents by arachidonic acid in single smooth muscle cells from vas deferens of the guinea-pig. Br J Pharmacol. 1995;116:1887–1893. doi: 10.1111/j.1476-5381.1995.tb16678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Kraig RP, Tanabe J, Pulsinelli WA. Dynamics of interstitial and intracellular pH in evolving brain infarct. Am J Physiol. 1991;260:R581–R588. doi: 10.1152/ajpregu.1991.260.3.R581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med. 2001;7:59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- Page AJ, Brierley SM, Martin CM, Price MP, Symonds E, Butler R, Wemmie JA, Blackshaw LA. Different contributions of ASIC channels 1a, 2, and 3 in gastrointestinal mechanosensory function. Gut. 2005;54:1408–1415. doi: 10.1136/gut.2005.071084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Ascher P. Mechanosensitivity of NMDA receptors in cultured mouse central neurons. Neuron. 1994;13:645–655. doi: 10.1016/0896-6273(94)90032-9. [DOI] [PubMed] [Google Scholar]
- Paukert M, Babini E, Pusch M, Grunder S. Identification of the Ca2+ blocking site of acid-sensing ion channel (ASIC) 1: implications for channel gating. J Gen Physiol. 2004;124:383–394. doi: 10.1085/jgp.200308973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignataro G, Cuomo O, Esposito E, Sirabella R, Di Renzo G, Annunziato L. ASIC1a contributes to neuroprotection elicited by ischemic preconditioning and postconditioning. Int J Physiol Pathophysiol Pharmacol. 2011;3:1–8. [PMC free article] [PubMed] [Google Scholar]
- Poirot O, Vukicevic M, Boesch A, Kellenberger S. Selective regulation of acid-sensing ion channel 1 by serine proteases. J Biol Chem. 2004;279:38448–38457. doi: 10.1074/jbc.M407381200. [DOI] [PubMed] [Google Scholar]
- Price MP, Lewin GR, McIlwrath SL, Cheng C, Xie J, Heppenstall PA, Stucky CL, Mannsfeldt AG, Brennan TJ, Drummond HA, Qiao J, Benson CJ, Tarr DE, Hrstka RF, Yang B, Williamson RA, Welsh MJ. The mammalian sodium channel BNC1 is required for normal touch sensation. Nature. 2000;407:1007–1011. doi: 10.1038/35039512. [DOI] [PubMed] [Google Scholar]
- Price MP, McIlwrath SL, Xie J, Cheng C, Qiao J, Tarr DE, Sluka KA, Brennan TJ, Lewin GR, Welsh MJ. The DRASIC cation channel contributes to the detection of cutaneous touch and acid stimuli in mice. Neuron. 2001;32:1071–1083. doi: 10.1016/s0896-6273(01)00547-5. [DOI] [PubMed] [Google Scholar]
- Rehncrona S. Brain acidosis. Ann Emerg Med. 1985;14:770–776. doi: 10.1016/s0196-0644(85)80055-x. [DOI] [PubMed] [Google Scholar]
- Rehncrona S, Hauge HN, Siesjo BK. Enhancement of iron-catalyzed free radical formation by acidosis in brain homogenates: differences in effect by lactic acid and CO2. J Cereb Blood Flow Metab. 1989;9:65–70. doi: 10.1038/jcbfm.1989.9. [DOI] [PubMed] [Google Scholar]
- Rehncrona S, Westerberg E, Akesson B, Siesjo BK. Brain cortical fatty acids and phospholipids during and following complete and severe incomplete ischemia. J Neurochem. 1982;38:84–93. doi: 10.1111/j.1471-4159.1982.tb10857.x. [DOI] [PubMed] [Google Scholar]
- Revici E, Stoopen E, Frenk E, Ravich RA. The painful focus. II. The relation of pain to local physico-chemical changes. Bull Inst Appl Biol. 1949;1:21. [Google Scholar]
- Rock DM, Macdonald RL. Polyamine regulation of N-methyl-D-aspartate receptor channels. Annu Rev Pharmacol Toxicol. 1995;35:463–482. doi: 10.1146/annurev.pa.35.040195.002335. [DOI] [PubMed] [Google Scholar]
- Sang N, Chen C. Lipid signaling and synaptic plasticity. Neuroscientist. 2006;12:425–434. doi: 10.1177/1073858406290794. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Matthews RT, Klockgether T, Dichgans J, Beal MF. The role of mitochondrial dysfunction and neuronal nitric oxide in animal models of neurodegenerative diseases. Mol Cell Biochem. 1997;174:193–197. [PubMed] [Google Scholar]
- Schurr A. Lactate, glucose and energy metabolism in the ischemic brain (Review) Int J Mol Med. 2002;10:131–136. [PubMed] [Google Scholar]
- Schurr A, Rigor BM. Brain anaerobic lactate production: a suicide note or a survival kit? Dev Neurosci. 1998;20:348–357. doi: 10.1159/000017330. [DOI] [PubMed] [Google Scholar]
- Sherwood TW, Askwith CC. Dynorphin opioid peptides enhance acid-sensing ion channel 1a activity and acidosis-induced neuronal death. J Neurosci. 2009;29:14371–14380. doi: 10.1523/JNEUROSCI.2186-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siesjo BK. Acidosis and ischemic brain damage. Neurochem Pathol. 1988;9:31–88. doi: 10.1007/BF03160355. [DOI] [PubMed] [Google Scholar]
- Siesjo BK, Katsura K. Ischemic brain damage: focus on lipids and lipid mediators. Adv Exp Med Biol. 1992;318:41–56. doi: 10.1007/978-1-4615-3426-6_5. [DOI] [PubMed] [Google Scholar]
- Siesjo BK, Katsura K, Kristian T. Acidosis-related damage. Adv Neurol. 1996;71:209–233. [PubMed] [Google Scholar]
- Sluka KA, Price MP, Breese NM, Stucky CL, Wemmie JA, Welsh MJ. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain. 2003;106:229–239. doi: 10.1016/S0304-3959(03)00269-0. [DOI] [PubMed] [Google Scholar]
- Smith ES, Cadiou H, McNaughton PA. Arachidonic acid potentiates acid-sensing ion channels in rat sensory neurons by a direct action. Neuroscience. 2007;145:686–698. doi: 10.1016/j.neuroscience.2006.12.024. [DOI] [PubMed] [Google Scholar]
- Star RA. Nitric oxide. Am J Med Sci. 1993;306:348–358. doi: 10.1097/00000441-199311000-00015. [DOI] [PubMed] [Google Scholar]
- Sutherland SP, Cook SP, McCleskey EW. Chemical mediators of pain due to tissue damage and ischemia. Prog Brain Res. 2000;129:21–38. doi: 10.1016/S0079-6123(00)29003-1. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Farrell K, Simon RP. Acidosis causes failure of astrocyte glutamate uptake during hypoxia. J Cereb Blood Flow Metab. 1995;15:417–424. doi: 10.1038/jcbfm.1995.52. [DOI] [PubMed] [Google Scholar]
- Tombaugh GC, Sapolsky RM. Evolving concepts about the role of acidosis in ischemic neuropathology. J Neurochem. 1993;61:793–803. doi: 10.1111/j.1471-4159.1993.tb03589.x. [DOI] [PubMed] [Google Scholar]
- Toninello A, Salvi M, Mondovi B. Interaction of biologically active amines with mitochondria and their role in the mitochondrial-mediated pathway of apoptosis. Curr Med Chem. 2004;11:2349–2374. doi: 10.2174/0929867043364559. [DOI] [PubMed] [Google Scholar]
- Ugawa S. Identification of sour-taste receptor genes. Anat Sci Int. 2003;78:205–210. doi: 10.1046/j.0022-7722.2003.00062.x. [DOI] [PubMed] [Google Scholar]
- Ugawa S, Ueda T, Ishida Y, Nishigaki M, Shibata Y, Shimada S. Amiloride-blockable acid-sensing ion channels are leading acid sensors expressed in human nociceptors. J Clin Invest. 2002;110:1185–1190. doi: 10.1172/JCI15709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugawa S, Yamamoto T, Ueda T, Ishida Y, Inagaki A, Nishigaki M, Shimada S. Amiloride-insensitive currents of the acid-sensing ion channel-2a (ASIC2a)/ASIC2b heteromeric sour-taste receptor channel. J Neurosci. 2003;23:3616–3622. doi: 10.1523/JNEUROSCI.23-09-03616.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivien D, Buisson A. Serine protease inhibitors: novel therapeutic targets for stroke? J Cereb Blood Flow Metab. 2000;20:755–764. doi: 10.1097/00004647-200005000-00001. [DOI] [PubMed] [Google Scholar]
- Voilley N. Acid-sensing ion channels (ASICs): new targets for the analgesic effects of non-steroid anti-Inflammatory drugs (NSAIDs) Curr Drug Targets Inflamm Allergy. 2004;3:71–79. doi: 10.2174/1568010043483980. [DOI] [PubMed] [Google Scholar]
- Vukicevic M, Weder G, Boillat A, Boesch A, Kellenberger S. Trypsin cleaves acid-sensing ion channel 1a in a domain that is critical for channel gating. J Biol Chem. 2006;281:714–722. doi: 10.1074/jbc.M510472200. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386:173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- Wang W, Duan B, Xu H, Xu L, Xu TL. Calcium-permeable acid-sensing ion channel is a molecular target of the neurotoxic metal ion lead. J Biol Chem. 2006;281:2497–2505. doi: 10.1074/jbc.M507123200. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Askwith CC, Lamani E, Cassell MD, Freeman JH, Jr, Welsh MJ. Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J Neurosci. 2003;23:5496–5502. doi: 10.1523/JNEUROSCI.23-13-05496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E, Hoshi T, Freeman JH, Welsh MJ. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron. 2002;34:463–477. doi: 10.1016/s0896-6273(02)00661-x. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Price MP, Welsh MJ. Acid-sensing ion channels: advances, questions and therapeutic opportunities. Trends Neurosci. 2006;29:578–586. doi: 10.1016/j.tins.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Wozniak M, Rydzewski B, Baker SP, Raizada MK. The cellular and physiological actions of insulin in the central nervous system. Neurochem Int. 1993;22:1–10. doi: 10.1016/0197-0186(93)90062-a. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Duan B, Mei YD, Gao J, Chen JG, Zhuo M, Xu L, Wu M, Xu TL. Characterization of acid-sensing ion channels in dorsal horn neurons of rat spinal cord. J Biol Chem. 2004;279:43716–43724. doi: 10.1074/jbc.M403557200. [DOI] [PubMed] [Google Scholar]
- Xie J, Price MP, Wemmie JA, Askwith CC, Welsh MJ. ASIC3 and ASIC1 mediate FMRFamide-related peptide enhancement of H+-gated currents in cultured dorsal root ganglion neurons. J Neurophysiol. 2003;89:2459–2465. doi: 10.1152/jn.00707.2002. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Chu XP, Simon RP. Acid sensing ion channels--novel therapeutic targets for ischemic brain injury. Front Biosci. 2007;12:1376–1386. doi: 10.2741/2154. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, MacDonald JF. Sensing of extracellular calcium by neurones. Can J Physiol Pharmacol. 1999;77:715–721. [PubMed] [Google Scholar]
- Xiong ZG, Pignataro G, Li M, Chang SY, Simon RP. Acid-sensing ion channels (ASICs) as pharmacological targets for neurodegenerative diseases. Curr Opin Pharmacol. 2008;8:25–32. doi: 10.1016/j.coph.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable Acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci U S A. 2004;101:6752–6757. doi: 10.1073/pnas.0308636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Sigworth FJ, Canessa CM. Gating of acid-sensitive ion channel-1: release of Ca2+ block vs. allosteric mechanism. J Gen Physiol. 2006;127:109–117. doi: 10.1085/jgp.200509396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Jones SW. Surface charge and calcium channel saturation in bullfrog sympathetic neurons. J Gen Physiol. 1995;105:441–462. doi: 10.1085/jgp.105.4.441. [DOI] [PMC free article] [PubMed] [Google Scholar]