

Abstract
Endoplasmic reticulum (ER) quality control is a conserved process by which misfolded or unassembled proteins are selectively retained in the endoplasmic reticulum (ER). Failure in oligomerization of multisubunit membrane proteins is one of the events that triggers ER quality control. The transmembrane domains (TMDs) of unassembled subunits are determinants of ER retention in many cases, although the mechanism of the TMD-mediated sorting of unassembled subunits remains elusive. We studied a yeast iron transporter complex on the cell surface as a new model system for ER quality control. When Fet3p, a transmembrane subunit, is not assembled with the other membrane subunit, Ftr1p, unassembled Fet3p is exclusively localized to the ER at steady state. The TMD of Fet3p contains a determinant for this process. However, pulse-chase analysis and in vitro budding assays indicate that unassembled Fet3p rapidly escapes from the ER. Furthermore, Rer1p, a retrieval receptor for ER-resident membrane proteins in the Golgi, is responsible for the TMD-dependent ER retrieval of unassembled Fet3p. These findings provide clear evidence that the ER quality control of unassembled membrane proteins can be achieved by retrieval from the Golgi and that Rer1p serves as a specific sorting receptor in this process.
INTRODUCTION
The eukaryotic secretory pathway initiates from the endoplasmic reticulum (ER), where newly synthesized proteins are assisted in their folding and oligomerization by molecular chaperones. The ER has a surveillance system termed ER quality control (ERQC), which prevents transport of immature proteins beyond the ER (Ellgaard et al., 1999; Ellgaard and Helenius, 2003). ERQC is generally observed in yeast and higher eukaryotes and is now known to relate to a number of diseases, including cystic fibrosis and α1-antitrypsin deficiency (Ward et al., 1995; Teckman and Perlmutter, 1996). Molecules recognized by ERQC are often subject to rapid degradation by the ER-associated degradation (ERAD) mechanism, in which proteins are dislocated from the ER and targeted for ubiquitin-proteasome-dependent degradation (Brodsky and McCracken, 1999; Tsai et al., 2002). On the other hand, some misfolded proteins such as a variant of vesicular stomatitis virus G protein (VSV-G) tsO45 are rather stably localized in the ER for a prolonged period (Doms et al., 1987; Loayza et al., 1998). ER-resident chaperones and glycosylation events are also involved in recognition of ERQC substrates (Ellgaard et al., 1999; Ellgaard and Helenius, 2003).
Compared with the ERAD and chaperone-dependent mechanisms, how the substrates of ERQC are retained in the ER is poorly understood in terms of protein transport. ER localization of ERQC substrates could occur either by static retention in the ER or retrieval from post-ER compartments. Retrograde transport from the Golgi to the ER, which is mediated by COPI-coated vesicles, plays a critical role in correct localization of authentic ER-resident proteins. Protein sorting in the early-Golgi is accomplished by multiple mechanisms. Erd2p recognizes the C-terminal HDEL (KDEL in mammals) sequence of lumenal proteins and recruits them into COPI-vesicles (Semenza et al., 1990). The C-terminal di-lysine motif of type I transmembrane proteins also acts as a retrieval signal through its direct binding to the COPI complex (Jackson et al., 1993; Letourneur et al., 1994). In addition to these mechanisms, we and others have elucidated the Rer1p-dependent retrieval pathway (Nishikawa and Nakano, 1993; Boehm et al., 1994; Sato et al., 1995). Rer1p is a Golgi-resident protein with four transmembrane domains (TMDs) that is required for the ER localization of a variety of ER membrane proteins (Sato et al., 1997; Massaad et al., 1999). Rer1p recognizes polar amino acid residues specifically arranged in the TMD of its ligands and returns them to the ER via COPI vesicles (Sato et al.,1996, 1997, 2001). Recently, there were reports in yeast showing that retrieval from the Golgi is involved in ERQC of mutant forms of soluble proteins but not in that of misfolded membrane proteins (Vashist et al., 2001; Haynes et al., 2002). In mammalian cells, the static retention mechanism was suggested for several membrane proteins arrested by ERQC, including VSV-G tsO45 and the unassembled major histo-compatibility complex (MHC) class I molecule (Nehls et al., 2000; Spiliotis et al., 2002). Thus, the current idea is that soluble ERQC substrates travel to the Golgi but transmembrane substrates are statically retained in the ER. Molecular mechanisms of these sorting events are not understood.
Here, we focus our attention to the sorting of a membrane protein subunit missing its intrinsic partner. Failure in oligomerization is known to be a general cause of ERQC for multisubunit membrane proteins. An interesting feature of this type of ERQC in mammalian cells is that the TMDs of unassembled subunits are the determinants of ER retention in many cases (Bonifacino et al., 1990, 1991; Cosson et al., 1991; Reth et al., 1991; Letourneur et al., 1995). Although such a specific region has been identified as an ER localization signal, it remains unknown how this TMD-mediated molecular sorting of unassembled proteins is accomplished. To address this question, we chose an iron transporter complex of Saccharomyces cerevisiae as a new model system for ERQC of unassembled membrane proteins. Two subunits, Fet3p and Ftr1p, constitute a high-affinity iron transporter on the plasma membrane (PM) (Figure 1A) (Askwith et al., 1994; Dancis et al., 1994). Ftr1p is an iron permease with multiple TMDs, whereas Fet3p is a type I transmembrane protein containing a large lumenal (extracellular) domain. The lumenal domain of Fet3p exhibits similarity to human ceruloplasmin and possesses a multicopper oxidase activity essential for iron transport across membranes. This complex and related factors have an important role in iron homeostasis, which is associated with many disease processes (Dancis et al., 1994; Yuan et al., 1995; Stearman et al., 1996). A previous study suggested that Ftr1p and Fet3p form a complex in the ER and that this is prerequisite for exit from the ER (Stearman et al., 1996). In the absence of Fet3p, Ftr1p is not transported to the PM but is retained in the ER. On the other hand, Ftr1p expression is required for Fet3p to be loaded with copper, which takes place in the Golgi or a post-Golgi compartment, suggesting that in the absence of Ftr1p Fet3p is retained in an earlier compartment. Thus, Fet3p and Ftr1p depend on each other, and the lack of the partner seems to result in a typical ERQC case of unassembled subunits.
Figure 1.

Unassembled Fet3p is localized to the ER. (A) Schematic representation of a yeast high-affinity iron transporter complex on the PM. (B) Rer1p-dependent ER localization of unassembled Fet3p-GFP. FET3-GFP was integrated into the FET3 locus of the wild-type (SMY502; a), Δpep4 (SMY501; b), Δftr1 Δpep4 (SMY531; c), or Δftr1 Δrer1 Δpep4 (SMY541; d). The GFP signal was observed in living cells by confocal microscopy. The right panels in the pair show Nomarski images in the same field. (C) Immunoblotting of Fet3-3HAp. FET3-3HA was integrated into the FET3 locus of the wild-type (YPH500; lane 1), Δpep4 (SMY501; lane 2), Δftr1 (SMY53; lane 3), Δftr1 Δpep4 (SMY531; lane 4), Δftr1 Δrer1 (SMY54; lane 5), or Δftr1 Δrer1 Δpep4 (SMY541; lane 6). Total cell lysates were prepared and analyzed by immunoblotting by using the anti-HA antibody.
To reveal mechanisms underlying the sorting events of unassembled membrane protein, we analyzed the fate of Fet3p in Δftr1 cells by using morphological and biochemical approaches. We confirmed morphologically that, in the cells lacking Ftr1p, Fet3p is exclusively localized to the ER at steady state. However, our in vivo and in vitro analysis demonstrated that unassembled Fet3p is rapidly exported from the ER and reaches the early-Golgi compartment. We further showed that the ER localization of orphan Fet3p strongly depends on its TMD and that Rer1p, the sorting receptor in the early-Golgi, is required for the retrieval of Fet3p to the ER by recognizing a specific residue in the TMD. These results indicate that Rer1p-dependent retrieval is a molecular mechanism responsible for TMD-mediated sorting of an unassembled membrane protein. We suggest that the ER localization of ERQC substrates is fulfilled by multiple mechanisms depending upon the substrate.
MATERIALS AND METHODS
Plasmids Construction
The FET3 gene was amplified by polymerase chain reaction (PCR) by using primers that correspond to 1 kb upstream and 0.7 kb downstream of the FET3 open reading frame and cloned into the SacI and XhoI sites of pRS306 (Sikorski and Hieter, 1989). A SpeI site was created just before the stop codon and the fragments encoding 3HA or green fluorescent protein (GFP) were inserted into this SpeI site to produce pFET3-42 and pFET3-43, respectively. For integration of FET3-3HA and FET3-GFP into the FET3 locus, pFET3-42 and pFET3-43 were digested with SacI and BsiWI, blunted and self-ligated to yield pFET3-52 and pFET3-53 that contain only the 3′ halves of the tagged FET3 genes.
The TMD mutants were made as follows. In pFET3-42, NheI and AflII sites were inserted after the 560th and 584th codons of FET3, respectively (pFET3-802NA). The NheI to AflII region corresponding to the fragment encoding the TMD was replaced with the DNA fragment encoding 24 leucines (pFET3-802L24). Silent mutations were introduced at the 596th and 597th codons to create a BglII site (GAT CTG) in pFET3-42 (pFET3-42B). The PCR-based site-directed mutagenesis was performed to change each polar residue in the TMD to leucine, and these fragments were inserted into the BamHI (intrinsic)-BglII site of pFET3-42B. FET3-GFP versions of these mutants were constructed by the same strategy by using pFET3-43 instead of pFET3-42. To integrate these constructs into the ura3 locus, plasmids were linearized by the use of NspV in the URA3 gene of pRS306 and used for transformation.
The open reading frame and the downstream sequence of FET3-3HA and its mutant forms (L24 and S567L) were cloned into the EcoRI and XhoI sites of pTU1, which contains the TDH3 promoter on a single-copy plasmid pRS316. GFP-RER1 under the TDH3 promoter (pSKY5-RER1) was described previously (Sato et al., 2001).
Strains and Culture Conditions
The Δfet3::kanMX and Δftr1::kanMX4 loci were amplified by PCR by using the genomic DNA of the Δymr058w and Δyer145c strains from EUROSCARF as templates. SNY9 (the wild-type) (Nishikawa and Nakano, 1993) and SKY7 (Δrer1) (Sato et al., 1995) were transformed with these DNA fragments, and transformants were screened on YPD plates containing 0.2 mg/ml G418. The correct disruption was checked both by PCR and by the sensitivity to a Fe(II) chelator, bathophenanthroline sulfonate (Sigma-Aldrich, St. Louis, MO). They are crossed with the wild-type YPH499 (Sikorski and Hieter, 1989), and MATα Δfet3 (SMY51), MATα Δftr1 (SMY53), MATα Δftr1 Δrer1 (SMY54), and MATα Δrer1 (SMY55) were selected after tetrad dissection. The Δfet3 and Δftr1 Δrer1 cells were crossed, and MATα Δfet3 Δftr1 (SMY60) and MATα Δfet3 Δftr1 Δrer1 (SMY61) were selected from nonparental ditype asci of tetrad dissection. In YPH500 (Sikorski and Hieter, 1989), SMY51, SMY53, SMY54, SMY55, SMY60, and SMY61, PEP4 was disrupted by the ADE2 marker (resulting in SMY501, SMY511, SMY531, SMY541, SMY631, SMY601, and SMY611, respectively), or the ADE2 gene was reintroduced into the ade2 locus (SMY502, SMY512, SMY532, SMY542, SMY632, SMY602, and SMY612, respectively).
To replace the FET3 locus with FET3-3HA, yeast cells were transformed with pFET3-52 digested with BamHI. The correct transformants were streaked onto plates containing 5′-fluoroorotic acid to loop out URA3 (Guthrie and Fink, 1991), and the cells expressing FET3-3HA were screened by immunoblotting using the anti-hemagglutinin (HA) antibody. FET3-GFP was also integrated into the FET3 locus by using pFET3-53 after BamHI digestion and correct integration was checked by PCR.
Yeast cells were grown in SC (0.67% yeast nitrogen base without amino acids [Difco, Detroit, MI], 2% glucose, and 0.2% Drop-out mix) (Kaiser et al., 1994) supplemented with 10 μM FeSO4 to keep the expression level of FET3 constant.
Immunoblotting and Cross-linking Experiment
Cells were cultured at 30°C to the middle logarithmic phase. Total cell lysates (60 μg) were prepared by agitation with glass beads in 62.5 mM Tris-HCl (pH 6.8), 2% SDS, 10% (wt/vol) glycerol, and 1 mM phenylmethylsulfonyl fluoride and boiling at 70°C for 10 min. They were subjected to immunoblotting by using the anti-HA monoclonal antibody (mAb) (16B12; Covance Research Products, Berkeley, CA) and visualized by the ECL-plus chemiluminescent detection system (Amersham Biosciences, Piscataway, NJ).
Chemical cross-linking between Rer1p and Fet3-3HAp was performed using the thiol-cleavable linker dithiobis-(succinimidylpropionate) (DSP) as described previously (Sato et al., 2001). SMY611 expressing GFP-Rer1p and Fet3-3HAp (or its mutant form) under the TDH3 promoter on a single-copy plasmid (8 × 107 cells) was lysed and divided. An aliquot was treated with 5 mM DSP at 20°C for 30 min. Reactions were terminated by the addition of 50 mM Tris-HCl (pH 8.0), and subjected to the immunoprecipitation with the anti-GFP polyclonal antibody (Sato et al., 2001). The immunoprecipitates were treated with 5% β-mercaptoethanol to cleave DSP and analyzed by immunoblotting with the anti-GFP mAb (BD Biosciences Clontech, Palo Alto, CA) and the anti-HA mAb (16B12). By using the other aliquots, total Fet3-HAp was immunoprecipitated with the anti-HA mAb (16B12) and detected by the anti-HA polyclonal antibody (Y11; Santa Cruz Biotechnology, Santa Cruz, CA).
Confocal Laser Microscopy
GFP fluorescence was visualized under an Olympus BX-60 fluorescence microscope equipped with a confocal laser scanner unit CSU10 (Yokogawa Electronic, Tokyo, Japan). Images were acquired by a high-resolution digital charge-coupled device camera (ORCA-ER; Hamamatsu Photonics, Hamamatsu, Japan), and processed by the IPLab software (Scanalytics, Fairfax, VA).
Pulse-Chase Analysis
Cells grown in SC to the early logarithmic phase were washed and preincubated in SC without methionine and cysteine for 1 h at 30°C. Cells were pulse-labeled with Redivue Promix [35S] (Amersham Biosciences) for 15 min, centrifuged, and resuspended in SC supplemented methionine and cysteine. All medium contained 10 μM FeSO4. Cells were harvested at appropriate time points, and cell lysates were prepared as described previously (Sato et al., 1999) and subjected to immunoprecipitation with the anti-HA mAb (16B12). The first immunoprecipitates were divided into three aliquots and subjected to the second immunoprecipitation with either the anti-HA antibody, the anti-α1,6 mannose antiserum or the anti-α1,3 mannose antiserum (Sato et al., 1996). Radioimages were observed and quantified with a Fuji Film image analyzer BAS-1000.
In Vitro Vesicle Budding Assay
Microsomes and cytosol were prepared as described previously (Wuestehube and Schekman, 1992). Microsomes (60 μg of protein) were incubated in B88 with 150 μg of wild-type cytosol, 0.2 mM GTP, 1 mM ATP, and an ATP degeneration system at 25°C for 45 min. The reactions were placed on ice for 5 min, and the vesicle fractions were separated by centrifugation at 15,000 × g for 5 min and analyzed by immunoblotting. Polyclonal antibodies against Sec22p and Sec61p were described previously (Stirling et al., 1992; Bednarek et al., 1995).
RESULTS
Fet3p Is Localized to the ER in Δftr1 Cells
It has been suggested that the oligomerization of Fet3p and Ftr1p is required for the correct surface targeting of the iron transporter complex and that failure of oligomerization results in the ER localization of the both subunits (Stearman et al., 1996). Because Fet3p acquires N-linked oligosaccharide modification, which serves as an indicator of intracellular transport, but Ftr1p does not (Yuan et al., 1995), we decided to select Fet3p as a marker to study the ERQC of this complex. To follow the intracellular behavior of Fet3p, a GFP- or 3HA-tag was fused to the C terminus of Fet3p. Expression of either fusion from a single-copy plasmid fully remedied the enhanced sensitivity of Δfet3 to an Fe(II) chelator, bathophenanthroline sulfonate (Askwith et al., 1994), indicating that these tagged versions of Fet3p are functional (our unpublished data). We found that imbalanced expression of FET3 and FTR1 resulted in the ER localization of the excess protein (our unpublished data). For the subsequent experiments, the tagged FET3 gene was integrated into the genomic FET3 locus by replacing the authentic gene to maintain the appropriate expression level.
Using the Fet3p-GFP fusion, the subcellular localization of Fet3p was directly examined in wild-type and Δftr1 strains (Figure 1B). To detect a possible Fet3p signal in the vacuole, we used a Δpep4 background in which most vacuolar proteolytic activity was inactivated (Zubenko et al., 1983). In wild-type cells, the majority of Fet3p-GFP was localized to the PM, although faint intracellular GFP fluorescence was observed. When FTR1 was deleted, Fet3p-GFP clearly showed the typical ER pattern consisting of the nuclear membrane and the peripheral ER. A similar FTR1-dependent change of subcellular localization was observed by immunofluorescence microscopy of Fet3-3HAp (our unpublished data). These results showed that unassembled Fet3p was retained in the ER. Next, the protein level of Fet3-3HAp was examined by immunoblotting with an anti-HA antibody (Figure 1C). In wild-type cells, Fet3-3HAp was detected as a smear band around 120-145 kDa due to elongation of sugar chains in the Golgi as commonly observed for glycoproteins passing through the secretory pathway. The amount of high-molecular-weight Fet3p was increased in the Δpep4 background, suggesting that a significant fraction of Fet3p is normally turned over in the vacuole. In the Δftr1 strain, a substantial amount of Fet3-3HAp was detected, but it occurred as a sharp 120-kDa band, implying that Fet3-3HAp was not converted to the Golgi form in the absence of Ftr1p.
The fate of Fet3-3HAp was further analyzed in a pulse-chase experiment. In the FTR1 Δpep4 strain, Fet3-3HAp was first detected as a 120-kDa band corresponding to the core-glycosylated ER form. Then, the molecular mass increased to 127-145 kDa in a time-dependent manner (Figure 2A). In yeast, carbohydrate modifications with α1,6 and α1,3 mannosyl linkages occur in the early- and late-Golgi compartments, respectively (Orlean, 1997). We confirmed that Fet3p with high molecular weight contained both α1,6 and α1,3 mannose modifications (Figure 2B). This indicates that the mobility shift in Fet3p reflects transport of Fet3-3HAp through the Golgi where Fet3-3HAp receives further mannose modifications. When FTR1 was intact, most of Fet3-3HAp became fully glycosylated within 30 min of the chase. In contrast, in Δftr1 cells, Fet3-3HAp was not converted to the high-molecular-weight species even after a 60-min chase (Figure 2A). This result is consistent with our morphological findings showing that Fet3p-GFP or Fet3-3HAp missing Ftr1p is not found at the PM but is localized to the ER.
Figure 2.

Intracellular transport of Fet3-3HAp. (A) Pulse-chase analysis of Fet3-3HAp in Δpep4 (SMY501), Δftr1 (SMY53), Δftr1 Δpep4 (SMY531), and Δftr1 Δrer1 Δpep4 (SMY541) strains. Cells were pulse labeled for 15 min and chased for 60 min. Immunoprecipitates with the anti-HA antibody were separated by SDS-PAGE and analyzed by autoradiography. (B) Unassembled Fet3-3HAp receives mannosyl modification in the early-Golgi. The immunoprecipitates were prepared as described in A, divided into three aliquots, and subjected to second immunoprecipitation with antibodies against HA, α1,6 mannosyl linkages (α1,6) and α1,3 mannosyl linkages (α1,3).
Unassembled Fet3p Is Exported from the ER
To examine whether unassembled Fet3p is statically retained in the ER or recycles between the ER and post-ER compartments, we analyzed carbohydrate modifications in more detail by a second immunoprecipitation by using anti-α1,6 or anti-α1,3 mannose antibodies. As shown in Figure 2B, in Δftr1 cells, a significant part of Fet3-3HAp received α1,6 mannose, suggesting that Fet3-3HAp travels to the early-Golgi. Strikingly, the rate of α1,6 modification to Fet3-3HAp in Δftr1 was comparable with that in wild type. In spite of this, transport of Fet3p beyond the early-Golgi seemed to be impaired because α1,3 modification was significantly lower than in wild-type cells.
As an alternative strategy to prove ER exit of Fet3-3HAp, we performed an in vitro budding assay from the ER (Wuestehube and Schekman, 1992). Microsomes were prepared from Δftr1 cells expressing FET3-3HA and let react with wild-type cytosol. The vesicles released from the microsomes were separated by centrifugation and analyzed by immunoblotting. Figure 3 demonstrates that Fet3-3HAp was incorporated into the vesicle fraction as efficiently as the established cargo protein Sec22p. The negative control, Sec61p, was not detected in the vesicle fraction in this condition, showing that this reaction is selective. Together with the exclusive ER localization of Fet3p-GFP at the steady state, both in vivo and in vitro results suggest that unassembled Fet3-3HAp is exported from the ER but selectively retrieved from the early-Golgi back to the ER.
Figure 3.
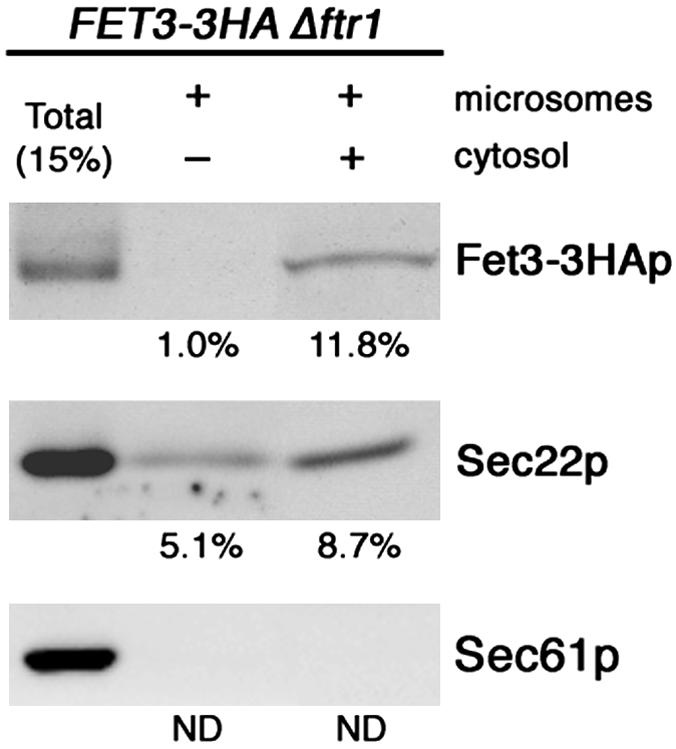
Unassembled Fet3-3HAp is incorporated into the ER-derived vesicles. In vitro budding assay was performed with microsomes prepared from Δftr1 Δpep4 (SMY531) expressing FET3-3HA. Fifteen percent of total reaction and budded vesicle fractions were analyzed by immunoblotting with anti-HA, anti-Sec22p, and anti-Sec61p antibodies. Sec22p and Sec61p represent a positive and negative control, respectively.
TMD Confers the ER Localization of Unassembled Fet3p
We examined whether any particular sequence within the TMD is required for the ER localization of unassembled Fet3p as is the case for ERQC of unassembled membrane proteins in mammalian cells (Figure 4). First, the whole TMD of Fet3p-GFP corresponding to the 561-584 residues of Fet3p was replaced by a stretch of 24 leucine residues (L24; Figure 4A) and the localization of this mutant Fet3p was observed in Δftr1 cells (Figure 4B). L24 was no longer localized in the ER, and most of the GFP signal was observed in the vacuole, demonstrating that the specific sequence of the Fet3p TMD is required for the ER localization in the absence of Ftr1p. Next, we looked at the four polar residues in the Fet3p TMD. Each residue was mutated to leucine (Figure 4A). First, the effect on the subcellular localization of unassembled Fet3p-GFP was examined (Figure 4B). Among four mutants, S567L lost its ER localization almost completely and resulted in redistribution of Fet3p to the vacuole. The other mutations (T565L, T577L, and Y581L) showed only minor defects in the ER localization of Fet3p-GFP in Δftr1 cells. These results were confirmed for all mutant forms of Fet3p expressed in the context of Fet3-3HAp in Δftr1 and Δftr1 Δpep4 cells (Figure 4C). Wild-type Fet3-3HAp was detected as a sharp band of 120 kDa either in Δftr1 and Δftr1 Δpep4 cells. In contrast, slow migrating bands corresponding to the Golgi-modified form of Fet3-3HAp were obvious in L24 and S567L mutants. These bands became faint in the PEP4+ background, suggesting that these mutants are transported through the Golgi to the vacuole and degraded there. As for T565L, T577L, and Y581L mutants, most of the protein remained in the 120-kDa band, which is consistent with the morphological observation by using GFP fusions. Thus, the TMD of Fet3p, especially the S567 residue within the TMD, is important for the ER localization of unassembled Fet3p.
Figure 4.

Fet3p TMD is required for ER localization of Fet3p in Δftr1. (A) Sequences of the wild-type Fet3p TMD and its mutant forms. (B) Subcellular localization of Fet3p-GFP and its mutant forms. Wild-type FET3-GFP or indicated mutants were integrated into the ura3 locus of Δfet3 Δftr1 Δpep4 (SMY611) and observed by confocal microscopy. Fluorescence (left panel in the pair) and Nomarski (right panel in the pair) images are shown. (C) PEP4-dependent degradation of mutant Fet3p. Wild-type FET3-3HA and indicated mutants were integrated into the ura3 locus of either Δfet3 Δftr1 (SMY60) or Δfet3 Δftr1 Δpep4 (SMY611) and detected by the anti-HA antibody.
Rer1p Is Responsible for the ER Localization of Fet3p in Δftr1
Our data suggest that the ER localization of unassembled Fet3p is achieved by retrieval from the Golgi. Among the known retrieval pathways, the Rer1p-dependent pathway has a unique feature in that it retrieves a variety of ER-resident membrane proteins from the early-Golgi in a TMD-dependent manner (Sato et al., 2001). Although these ligands show no apparent similarity in amino acid sequence, Rer1p recognizes their TMDs, which contain polar residues in particular positions (Sato et al., 1996; Sato et al., 2003). Because the ER localization of unassembled Fet3p depends on the TMD containing a polar residue essential for ERQC, we tested whether Rer1p is involved in the retrieval process of unassembled Fet3p. First, subcellular localization of Fet3p-GFP was observed in a Δftr1 Δrer1 Δpep4 strain (Figure 1B, d). In these cells, unassembled Fet3p-GFP was clearly localized to the vacuole, indicating that the ER retention of unassembled Fet3p-GFP depends strongly upon Rer1p. It should be noted that, in Δrer1, most unassembled Fet3p-GFP is transported to the vacuole instead of the PM, the normal destination of the Fet3p-Frt1p complex. This implies the existence of a second quality control system that targets unassembled Fet3p-GFP escaping ER retention to the vacuole. In the vacuole, GFP signal was observed both on the limiting membrane and in the lumen. The luminal GFP signal may be due to internalization of Fet3p-GFP via the multivesicular body sorting pathway.
We further analyzed the effect of the deletion of RER1 by immunoblotting (Figure 1C) and pulse-chase analysis (Figure 2). As shown in Figure 1C, in the absence of Rer1p, unassembled Fet3-3HAp was degraded in a PEP4-dependent manner, supporting vacuolar targeting and PEP4-dependent degradation of Fet3-3HAp. Pulse-chase analysis also demonstrated that, when RER1 was disrupted, unassembled Fet3-3HAp was converted to the high-molecular-weight Golgi form containing α1,3 mannose linkage like the assembled Fet3-3HAp. Importantly, the kinetics of the α1,6 mannosyl modification was not affected by the deletion of RER1, suggesting that Rer1p is involved in the sorting event in the early-Golgi compartment.
A chemical cross-linking experiment was performed to prove physical interaction between Rer1p and unassembled Fet3p. Because physical interaction between Rer1p and its ligands is expected to be transient, Fet3-3HAp and GFP-Rer1p were overexpressed in Δftr1 to increase the chances of detecting their physical interaction. Cell lysates were prepared from these cells and reacted with a thiol-cleavable linker DSP. Cross-linked products were immunoprecipitated with an anti-GFP antibody, treated with β-mercaptoethanol to cleave the linker, and subjected to immunoblotting with the anti-HA antibody. Figure 5 demonstrates that Fet3-3HAp was cross-linked with GFP-Rer1p in Δftr1 cell lysates. This physical interaction strongly suggests that Rer1p directly recognizes unassembled Fet3p. Moreover, we corroborated that the TMD of Fet3p is required for the interaction between Rer1p and Fet3p. Fet3-3HA with a L24 or S567L mutation was expressed in Δftr1 cells and cross-linking was carried out by the same procedure. As shown in Figure 5, L24 and S567L were no longer cross-linked with Rer1p. These results suggest that S567 is critical for recognition by Rer1p, which confers ER localization to unassembled Fet3p.
Figure 5.

Physical interaction of Rer1p and unassembled Fet3p. GFP-Rer1p and Fet3-3HAp or its mutant forms (L24 and S567L) were co-overproduced under the TDH3 promoter in Δfet3 Δftr1 Δrer1 (SMY611). Total cell lysates were subjected to chemical cross-linking with DSP. The immunoprecipitates with the anti-GFP antibody were examined by immunoblotting with the anti-GFP and anti-HA antibodies. Total Fet3-3HAp was also immunoprecipitated from an aliquot of cell lysates by using the anti-HA antibody and detected by the anti-HA antibody.
Finally, we asked whether Rer1p-dependent recycling is also involved in oligomerization of Fet3p and Ftr1p in the wild-type cells. When RER1 was deleted in the presence of FTR1, the steady-state amount of Fet3-3HAp was markedly decreased compared with that in RER1 FTR1 cells (Figure 6). The amount of Fet3-3HAp was partially restored by disruption of PEP4, suggesting that degradation of Fet3-3HAp in the vacuole is enhanced in FTR1 Δrer1 cells. One possible explanation is that in the absence of Rer1p, the Fet3p-Frt1p complex is not formed efficiently, and unassembled Fet3p is susceptible of degradation in the vacuole and other compartments.
Figure 6.
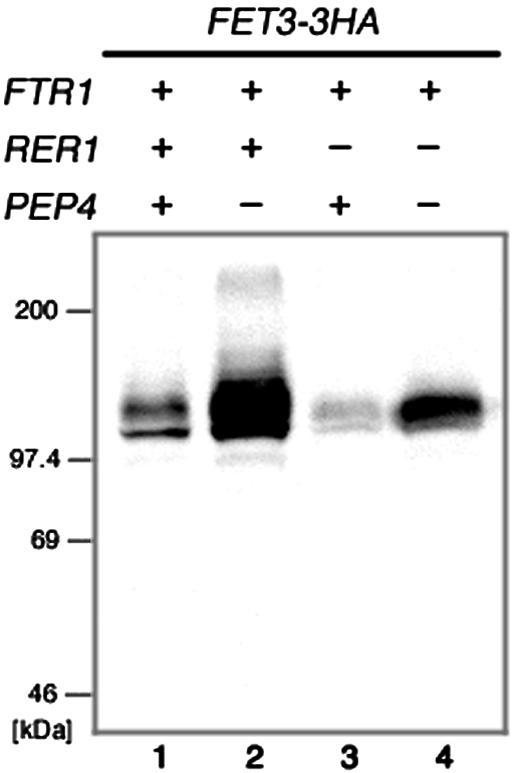
Deletion of RER1 causes enhanced degradation of Fet3-3HAp in the presence of Ftr1p. FET3-3HA was integrated into the FET3 locus of the wild-type (YPH500; lane 1), Δpep4 (SMY501; lane 2), Δrer1 (SMY632; lane 3), and Δrer1 Δpep4 (SMY631; lane 4). Total cell lysates were prepared and analyzed by immunoblotting by using the anti-HA antibody.
DISCUSSION
ERQC is a process of conformation-dependent molecular sorting in the ER, leading to the selective ER localization of misfolded or unassembled proteins and eventual degradation (Ellgaard et al., 1999; Ellgaard and Helenius, 2003). In contrast to the degradation step, molecular mechanisms are still elusive that should explain the selective ER localization of ERQC substrates in terms of protein transport.
In this article, we studied the Fet3p-Ftr1p complex, the iron transporter on the PM, as a new model of ERQC of incompletely assembled transmembrane subunits. Failure in complex formation has been known as a general trigger of ERQC in mammalian systems. In many cases, including T-cell receptor, CD8, and B-cell receptor, TMDs contain determinants of ER localization of incompletely assembled subunits, and a common feature of such determinants is the presence of one or several hydrophilic or polar residues within the TMD (Bonifacino et al., 1990, 1991; Cosson et al., 1991; Reth et al., 1991; Hennecke and Cosson, 1993; Letourneur et al., 1995). In yeast, we demonstrated by using tagged Fet3p that the absence of Ftr1p causes relocalization of Fet3p to the ER. Similar to previous studies of mammalian systems, the TMD of Fet3p, particularly a single polar residue in the TMD, is critical for this ER localization. Interestingly, in vivo pulse-chase analysis monitoring the processing of sugar chains indicates that unassembled Fet3p is exported from the ER and modified in the early-Golgi in spite of its extensive ER localization at steady state. ER exit of Fet3p was further supported by the in vitro budding assay. Furthermore, Rer1p, a retrieval receptor for ER-resident membrane proteins (Sato et al., 2001), turns out to be responsible for the ER localization of unassembled Fet3p. This finding not only demonstrates that ER retention of unassembled Fet3p is achieved by continuous retrieval from the Golgi but also provides a new insight into the molecular mechanism underlying ER localization of ERQC substrates.
Contribution of the retrieval pathway to ERQC is still controversial. Recent studies suggest the static retention mechanism for transmembrane substrates, including VSV-G tsO45 and unassembled MHC class I molecule in mammalian cells (Nehls et al., 2000; Spiliotis et al., 2002) and Ste6-166p in yeast (Vashist et al., 2001). On the other hand, several lines of evidence showed involvement of the retrieval pathway from the post-ER compartments. It was reported that, under a certain experimental condition, VSV-G tsO45 and unassembled MHC class I are recycled between the ER and the intermediate compartment (Hsu et al., 1991; Hammond and Helenius, 1994). As for soluble misfolded proteins, a more positive role of the retrieval pathway was reported especially for the ERAD step in yeast. CPY* (mutant version of carboxypeptidase Y) and KHN (simian virus 5 hemagglutinin neuraminidase with the Kar2p signal sequence), well-characterized soluble substrates of ERQC, are transported to the Golgi before degradation. This transport is an obligatory step for efficient degradation by ERAD, although the exit rate from the ER is lower than that of normal cargo proteins (Vashist et al., 2001; Haynes et al., 2002). There are also morphological studies in which post-ER localization was demonstrated for important ERQC components such as UDP-glucose:glycoprotein glucosyltransferase and endo-α-mannosidase (Zuber et al., 2000, 2001). Thus, at least a part of ERQC substrates should be retrieved from the post-ER compartment. Our study provides clear evidence that the retrieval pathway is involved in ERQC of an unassembled transmembrane protein. Fet3p in Δftr1 cells is exported from the ER as efficiently as in wild-type cells. Furthermore, analysis of mannosyl modification indicates that protein sorting of unassembled Fet3p takes place mainly in the early-Golgi. Unassembled Fet3p could represent a novel type of ERQC substrate whose ER localization strongly depends on retrieval from the Golgi. It should be noted that Fet3p itself is intact in this system and is potentially competent to form a complex with Ftr1p. One possibility is that even unassembled Fet3p is exported from the ER due to a presumed ER export signal in its cytosolic region. If it is the case, retrieval from the Golgi would restrict transport of the immature protein to further destinations and increase the chance of complex formation. A more positive role of retrieval might be that recycling between the ER and Golgi prevents rapid degradation of Fet3p by ERAD. Unlike the case of the mutant Ste6-166 protein that is rapidly degraded by ERAD, unassembled Fet3p is relatively stable in RER1 Δftr1 cells. Recycling might ensure enough time to oligomerize with Ftr1p in wild-type cells (also discussed below). Multiple mechanisms of ERQC must operate depending on the status of substrates, and misfolded and unassembled proteins could follow different pathways.
An important finding is that Rer1p plays a central role in the sorting of the unassembled Fet3p in the Golgi. Rer1p is a conserved protein originally identified as a Golgi membrane protein required for correct ER localization of Sec12p, a type II transmembrane protein essential for budding of COPII vesicles from the ER (Sato et al., 1995, 1999; Fullekrug et al., 1997). Subsequent studies uncovered that Rer1p recognizes a variety of ER membrane proteins including type III and polytopic membrane proteins and sends them back to the ER via COPI vesicles (Sato et al., 1997; Massaad et al., 1999). When RER1 was deleted, most unassembled Fet3p-GFP was relocalized to the vacuole. This effect is direct because the physical interaction between Rer1p and unassembled Fet3p was shown by chemical cross-linking experiments. This finding not only reinforces the idea that ER retention of unassembled Fet3p is dependent on retrograde transport but also gives a mechanical insight into sorting and localization of unassembled proteins. A previous report suggested the involvement of Rer1p in a particular type of ERQC (Letourneur and Cosson, 1998). Glycosylphosphatidylinositol (GPI)-anchored proteins are synthesized as a precursor with a membrane-anchoring domain. In the ER, this precursor is cleaved at the luminal site adjacent to the membrane anchor and attached to a GPI-anchor. When the cleavage and subsequent attachment of GPI is inhibited by a mutation of the cleavage site, this mutant protein is retained in the ER in Rer1p- and COPI-dependent manner. Rer1p-dependent retrieval seems to contribute commonly to ERQC of a subset of membrane proteins.
Rer1p recognizes the TMDs of its ligands that contain polar residues (Sato et al., 1996, 2003). This holds true for unassembled Fet3p because L24 and S567L mutations in the TMD diminish binding to Rer1p and cause vacuolar localization. It is likely that the complex formation with Ftr1p masks S567 in the TMD, which releases the assembled complex from Rer1p-dependent recycling between the ER and the Golgi. Even in wild-type cells, Rer1p could prevent transport of premature Fet3p beyond the Golgi and return it to the ER to provide a next chance of complex formation, which improves the efficiency of correct complex formation. Supporting this possibility, the deletion of RER1 remarkably reduced the stability of Fet3-3HAp even in the presence of Ftr1p, implying that inefficient complex formation in Δrer1 results in degradation of unassembled Fet3p in the vacuole and other compartment. In this context, the function of Rer1p could be similar to that of molecular chaperons. Rer1p may conceal polar residues exposed in the lipid bilayer and stabilizes premature proteins in a hydrophobic environment. The deletion of RER1 causes the sensitivity to hygromycin B, an inhibitor of protein synthesis (Sato, unpublished observation). This might be explained if the efficiency of complex formation is generally reduced in Δrer1, which causes shortage of functional proteins when protein synthesis is also inhibited. As mentioned above, TMD-mediated ERQC in mammalian cells requires hydrophilic or polar residues within the TMD, which is similar to the Rer1p-dependent retrieval signal in yeast. We would speculate that Rer1p is also involved in ERQC of unassembled membrane proteins in higher eukaryotes.
A previous study suggested another quality control mechanism in the late-Golgi (Hong et al., 1996). It showed that a soluble misfolded protein is captured by Vps10p at the Golgi and selectively targeted to the vacuole. In the present study, we found that unassembled Fet3p, which escapes ERQC either by the deletion of RER1 or by mutations in the TMD, is transported not to the PM but to the vacuole, implying that unassembled Fet3p is subjected to second quality control in the late secretory pathway. Multiple quality control systems through the secretory pathway may monitor the folding status of proteins to prevent potentially toxic effects caused by accumulation of unfolded proteins.
Acknowledgments
We are grateful to Randy Schekman and Hiroshi Abe for antibodies, Jerry Kaplan for a plasmid, Barth Grant for a critical reading of the manuscript, and the members of the Nakano laboratory for helpful discussions. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, Culture and Technology of Japan and by grants from the Bioarchitect Research Project of RIKEN. M.S. was a Special Postdoctoral Researcher of RIKEN Discovery Research Institute.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E03-10-0765. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E03-10-0765.
Abbreviations used: ERAD, endoplasmic reticulum-associated degradation; ERQC, endoplasmic reticulum quality control; GFP, green fluorescent protein; PM, plasma membrane; TMD, transmembrane domain; VSV-G, vesicular stomatitis virus G protein.
References
- Askwith, C., Eide, D., Van Ho, A., Bernard, P.S., Li, L., Davis-Kaplan, S., Sipe, D.M., and Kaplan, J. (1994). The FET3 gene of S. cerevisiae encodes a multi-copper oxidase required for ferrous iron uptake. Cell 76, 403-410. [DOI] [PubMed] [Google Scholar]
- Bednarek, S.Y., Ravazzola, M., Hosobuchi, M., Amherdt, M., Perrelet, A., Schekman, R., and Orci, L. (1995). COPI- and COPII-coated vesicles bud directly from the endoplasmic reticulum in yeast. Cell 83, 1183-1196. [DOI] [PubMed] [Google Scholar]
- Boehm, J., Ulrich, H.D., Ossig, R., and Schmitt, H.D. (1994). Kex2-dependent invertase secretion as a tool to study the targeting of transmembrane proteins which are involved in ER-Golgi transport in yeast. EMBO J. 13, 3696-3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino, J.S., Cosson, P., and Klausner, R.D. (1990). Colocalized transmembrane determinants for ER degradation and subunit assembly explain the intracellular fate of TCR chains. Cell 63, 503-513. [DOI] [PubMed] [Google Scholar]
- Bonifacino, J.S., Cosson, P., Shah, N., and Klausner, R.D. (1991). Role of potentially charged transmembrane residues in targeting proteins for retention and degradation within the endoplasmic reticulum. EMBO J. 10, 2783-2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky, J.L., and McCracken, A.A. (1999). ER protein quality control and proteasome-mediated protein degradation. Semin. Cell Dev. Biol. 10, 507-513. [DOI] [PubMed] [Google Scholar]
- Cosson, P., Lankford, S.P., Bonifacino, J.S., and Klausner, R.D. (1991). Membrane protein association by potential intramembrane charge pairs. Nature 351, 414-416. [DOI] [PubMed] [Google Scholar]
- Dancis, A., Yuan, D.S., Haile, D., Askwith, C., Eide, D., Moehle, C., Kaplan, J., and Klausner, R.D. (1994). Molecular characterization of a copper transport protein in S. cerevisiae: an unexpected role for copper in iron transport. Cell 76, 393-402. [DOI] [PubMed] [Google Scholar]
- Doms, R.W., Keller, D.S., Helenius, A., and Balch, W.E. (1987). Role for adenosine triphosphate in regulating the assembly and transport of vesicular stomatitis virus G protein trimers. J. Cell Biol. 105, 1957-1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard, L., and Helenius, A. (2003). Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell. Biol. 4, 181-191. [DOI] [PubMed] [Google Scholar]
- Ellgaard, L., Molinari, M., and Helenius, A. (1999). Setting the standards: quality control in the secretory pathway. Science 286, 1882-1888. [DOI] [PubMed] [Google Scholar]
- Fullekrug, J., Boehm, J., Rottger, S., Nilsson, T., Mieskes, G., and Schmitt, H.D. (1997). Human Rer1 is localized to the Golgi apparatus and complements the deletion of the homologous Rer1 protein of Saccharomyces cerevisiae. Eur. J. Cell Biol. 74, 31-40. [PubMed] [Google Scholar]
- Guthrie, C., and Fink., G. (1991). Guide to yeast genetics and molecular biology. Methods Enzymol. 194, 182-187. [PubMed] [Google Scholar]
- Hammond, C., and Helenius, A. (1994). Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J. Cell Biol. 126, 41-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes, C.M., Caldwell, S., and Cooper, A.A. (2002). An HRD/DER-independent ER quality control mechanism involves Rsp5p-dependent ubiquitination and ER-Golgi transport. J. Cell Biol. 158, 91-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennecke, S., and Cosson, P. (1993). Role of transmembrane domains in assembly and intracellular transport of the CD8 molecule. J. Biol. Chem. 268, 26607-26612. [PubMed] [Google Scholar]
- Hong, E., Davidson, A.R., and Kaiser, C.A. (1996). A pathway for targeting soluble misfolded proteins to the yeast vacuole. J. Cell Biol. 135, 623-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, V.W., Yuan, L.C., Nuchtern, J.G., Lippincott-Schwartz, J., Hammerling, G.J., and Klausner, R.D. (1991). A recycling pathway between the endoplasmic reticulum and the Golgi apparatus for retention of unassembled MHC class I molecules. Nature 352, 441-444. [DOI] [PubMed] [Google Scholar]
- Jackson, M.R., Nillson, T., and Peterson, P.A. (1993). Retrieval of transmembrane proteins to the endoplasmic reticulum. J. Cell Biol. 121, 317-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser, C., Michaelis, S., and Mitchell, A. (1994). Methods in Yeast Genetics, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- Letourneur, F., and Cosson, P. (1998). Targeting to the endoplasmic reticulum in yeast cells by determinants present in transmembrane domains. J. Biol. Chem. 273, 33273-33278. [DOI] [PubMed] [Google Scholar]
- Letourneur, F., Gaynor, E.C., Hennecke, S., Demolliere, C., Duden, R., Emr, S.D., Riezman, H., and Cosson, P. (1994). Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell 79, 1199-1207. [DOI] [PubMed] [Google Scholar]
- Letourneur, F., Hennecke, S., Demolliere, C., and Cosson, P. (1995). Steric masking of a dilysine endoplasmic reticulum retention motif during assembly of the human high affinity receptor for immunoglobulin E. J. Cell Biol. 129, 971-978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loayza, D., Tam, A., Schmidt, W.K., and Michaelis, S. (1998). Ste6p mutants defective in exit from the endoplasmic reticulum (ER) reveal aspects of an ER quality control pathway in Saccharomyces cerevisiae. Mol. Biol. Cell 9, 2767-2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaad, M.J., Franzusoff, A., and Herscovics, A. (1999). The processing alpha1, 2-mannosidase of Saccharomyces cerevisiae depends on Rer1p for its localization in the endoplasmic reticulum. Eur. J. Cell Biol. 78, 435-440. [DOI] [PubMed] [Google Scholar]
- Nehls, S., Snapp, E.L., Cole, N.B., Zaal, K.J., Kenworthy, A.K., Roberts, T.H., Ellenberg, J., Presley, J.F., Siggia, E., and Lippincott-Schwartz, J. (2000). Dynamics and retention of misfolded proteins in native ER membranes. Nat. Cell Biol. 2, 288-295. [DOI] [PubMed] [Google Scholar]
- Nishikawa, S., and Nakano, A. (1993). Identification of a gene required for membrane protein retention in the early secretory pathway. Proc. Natl. Acad. Sci. USA 90, 8179-8183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlean, P. (1997). Biogenesis of yeast wall and surface components. In: The Molecular and Cellular Biology of the Yeast Saccharomyces cerevisiae, vol 3, ed. J.R. Pringle, J.R. Broach, and E.W. Jones, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 229-362. [Google Scholar]
- Reth, M., Hombach, J., Wienands, J., Campbell, K.S., Chien, N., Justement, L.B., and Cambier, J.C. (1991). The B-cell antigen receptor complex. Immunol. Today 12, 196-201. [DOI] [PubMed] [Google Scholar]
- Sato, K., Nishikawa, S., and Nakano, A. (1995). Membrane protein retrieval from the Golgi apparatus to the endoplasmic reticulum (ER): characterization of the RER1 gene products as a component involved in ER localization of Sec12p. Mol. Biol. Cell 6, 1459-1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, M., Sato, K., and Nakano, A. (1996). Endoplasmic reticulum localization of Sec12p is achieved by two mechanisms: Rer1p-dependent retrieval that requires the transmembrane domain and Rer1p-independent retention that involves the cytoplasmic domain. J. Cell Biol. 134, 279-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, K., Sato, M., and Nakano, A. (1997). Rer1p as common machinery for the endoplasmic reticulum localization of membrane proteins. Proc. Natl. Acad. Sci. USA 94, 9693-9698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, K., Sato, M., and Nakano, A. (2001). Rer1p, a retrieval receptor for endoplasmic reticulum membrane proteins, is dynamically localized to the Golgi apparatus by coatomer. J. Cell Biol. 152, 935-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, K., Sato, M., and Nakano, A. (2003). Rer1p, a retrieval receptor for ER membrane proteins, recognizes transmembrane domains in multiple modes. Mol. Biol. Cell 14, 3605-3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, K., Ueda, T., and Nakano, A. (1999). The Arabidopsis thaliana RER1 gene family: its potential role in the endoplasmic reticulum localization of membrane proteins. Plant Mol. Biol. 41, 815-824. [DOI] [PubMed] [Google Scholar]
- Semenza, J.C., Hardwick, K.G., Dean, N., and Pelham, H.R. (1990). ERD2, a yeast gene required for the receptor-mediated retrieval of luminal ER proteins from the secretory pathway. Cell 61, 1349-1357. [DOI] [PubMed] [Google Scholar]
- Sikorski, R.S., and Hieter, P. (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiliotis, E.T., Pentcheva, T., and Edidin, M. (2002). Probing for membrane domains in the endoplasmic reticulum: retention and degradation of unassembled MHC class I molecules. Mol. Biol. Cell 13, 1566-1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearman, R., Yuan, D.S., Yamaguchi-Iwai, Y., Klausner, R.D., and Dancis, A. (1996). A permease-oxidase complex involved in high-affinity iron uptake in yeast. Science 271, 1552-1557. [DOI] [PubMed] [Google Scholar]
- Stirling, C.J., Rothblatt, J., Hosobuchi, M., Deshaies, R., and Schekman, R. (1992). Protein translocation mutants defective in the insertion of integral membrane proteins into the endoplasmic reticulum. Mol. Biol. Cell 3, 129-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teckman, J.H., and Perlmutter, D.H. (1996). The endoplasmic reticulum degradation pathway for mutant secretory proteins alpha1-antitrypsin Z and S is distinct from that for an unassembled membrane protein. J. Biol. Chem. 271, 13215-13220. [DOI] [PubMed] [Google Scholar]
- Tsai, B., Ye, Y., and Rapoport, T.A. (2002). Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell. Biol. 3, 246-255. [DOI] [PubMed] [Google Scholar]
- Vashist, S., Kim, W., Belden, W.J., Spear, E.D., Barlowe, C., and Ng, D.T. (2001). Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding. J. Cell Biol. 155, 355-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, C.L., Omura, S., and Kopito, R.R. (1995). Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121-127. [DOI] [PubMed] [Google Scholar]
- Wuestehube, L.J., and Schekman, R.W. (1992). Reconstitution of transport from endoplasmic reticulum to Golgi complex using endoplasmic reticulum-enriched membrane fraction from yeast. Methods Enzymol. 219, 124-136. [DOI] [PubMed] [Google Scholar]
- Yuan, D.S., Stearman, R., Dancis, A., Dunn, T., Beeler, T., and Klausner, R.D. (1995). The Menkes/Wilson disease gene homologue in yeast provides copper to a ceruloplasmin-like oxidase required for iron uptake. Proc. Natl. Acad. Sci. USA 92, 2632-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubenko, G.S., Park, F.J., and Jones, E.W. (1983). Mutations in PEP4 locus of Saccharomyces cerevisiae block final step in maturation of two vacuolar hydrolases. Proc. Natl. Acad. Sci. USA 80, 510-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber, C., Fan, J.Y., Guhl, B., Parodi, A., Fessler, J.H., Parker, C., and Roth, J. (2001). Immunolocalization of UDP-glucose:glycoprotein glucosyltransferase indicates involvement of pre-Golgi intermediates in protein quality control. Proc. Natl. Acad. Sci. USA 98, 10710-10715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber, C., Spiro, M.J., Guhl, B., Spiro, R.G., and Roth, J. (2000). Golgi apparatus immunolocalization of endomannosidase suggests post-endoplasmic reticulum glucose trimming: implications for quality control. Mol. Biol. Cell 11, 4227-4240. [DOI] [PMC free article] [PubMed] [Google Scholar]