

Abstract
We introduce Pathifier, an algorithm that infers pathway deregulation scores for each tumor sample on the basis of expression data. This score is determined, in a context-specific manner, for every particular dataset and type of cancer that is being investigated. The algorithm transforms gene-level information into pathway-level information, generating a compact and biologically relevant representation of each sample. We demonstrate the algorithm’s performance on three colorectal cancer datasets and two glioblastoma multiforme datasets and show that our multipathway-based representation is reproducible, preserves much of the original information, and allows inference of complex biologically significant information. We discovered several pathways that were significantly associated with survival of glioblastoma patients and two whose scores are predictive of survival in colorectal cancer: CXCR3-mediated signaling and oxidative phosphorylation. We also identified a subclass of proneural and neural glioblastoma with significantly better survival, and an EGF receptor-deregulated subclass of colon cancers.
Keywords: computational biology, systems biology, oncogenomics, principal curve
The operation of many important pathways is altered during cancer initiation and progression. Identifying the involved pathways and quantifying their deregulation is a very important step toward understanding the malignancy process (1–5). Because advanced therapies target specific pathways, pathway-level understanding is a key step also for developing personalized cancer treatments. Indeed, many methods, such as those described in refs. 5–10, were developed for pathway analysis of high-throughput data. Nearly all methods characterize a pathway’s activity for an entire sample set and do not provide information on its deregulation in a particular tumor. One prominent exception is Pathway Recognition Algorithm using Data Integration on Genomic Models (PARADIGM) (11), a tool that deduces for each pathway and sample a score using the pathway’s known connectivity and functional structure. Hence, it may not work well for many complex pathways that play significant roles in cancer, for which either the mechanism of pathway activity is not well known or essential relevant data (such as protein abundance and phosphorylation status) are unavailable.
We introduce a method to calculate, independently for every pathway, a score that represents the extent to which the pathway is deregulated in every individual sample. We quantify the level of deregulation of a pathway in a sample by measuring the deviation of the sample from normal behavior. We do not need detailed reliable knowledge of the network or wiring diagram that underlies the pathway’s activity. Hence, our estimates of pathway deregulation in a given sample are not restricted to only simple pathways. The method is knowledge-based, because we use generally well-known external information on the identity of the genes that belong to each pathway. Since the detailed interactions in each pathway are largely unknown and are context-dependent, we derive our deregulation scores in a “phenomenological” context-specific manner. Because at every stage the analysis is performed in a relatively low-dimensional space, we evade the “curse of dimensionality” stemming from using a small number of data points in a larger dimensional space. Hence, our results are more robust to perturbations such as removing some of the pathways or samples from the analysis.
To demonstrate that the pathway deregulation scores obtained this way indeed capture biologically and clinically relevant information in a sensible manner, and the validity and usefulness of the method, we apply it on many pathways to generate a pathway-level representation of every sample and show that our representation generates clinically relevant stratifications and outcome predictors for glioblastoma and colorectal cancer. Pathifier is available at www.weizmann.ac.il/pathifier/.
Results
Brief Outline of Pathifier.
Pathifier analyzes NP pathways, one at a time, and assigns to each sample i and pathway P a score DP(i), which estimates the extent to which the behavior of pathway P deviates, in sample i, from normal. To determine this pathway deregulation score (PDS), we use the expression levels of those dP genes that belong to P, for example, using databases such as those described in refs. 12–15. Each sample i is a point in this dP dimensional space; the entire set of samples forms a cloud of points, and we calculate the (nonlinear) “principal curve” (16) that captures the variation of this cloud. Next, we project each sample onto this curve; the PDS is defined as the distance DP(i), measured along the curve, of the projection of sample i, from the projection of the normal samples (Fig. 1). The variance of the PDS of the normals is much lower than that of the tumors (SI Appendix, Fig. S1). On the basis of genome-wide gene-level expression data we generate a pathway-level, biologically relevant NP-dimensional representation of each sample and mine this representation for insights.
Fig. 1.

The principal curve learned for the apoptosis pathway [as defined by the Kyoto Encyclopedia of Genes and Genomes (KEGG)] on the colorectal dataset of Sheffer et al. (30). The data points (representing samples of different tissue types, colored accordingly) and the principal curve are projected onto the three leading principal components. (A) The principal curve (in blue) going through the cloud of samples. The curve is directed so that normal samples are near the beginning of the curve (Methods). (B) The samples projected onto the curve. Each point carries its color from A.
PDSs Capture Biologically Relevant Information in Glioblastoma.
Pathifier was applied to expression data from 445 glioblastoma multiforme (GBM) and 10 normal brain samples from The Cancer Genome Atlas (TCGA) (17). The PDS for 548 pathways from Kyoto Encyclopedia of Genes and Genomes (KEGG) (12, 13), BioCarta (14) and the National Cancer Institute–Nature Pathway Interaction Database (15) are presented in a 548 × 455 table (Fig. 2A), representing the deregulation score of each pathway in every sample and summarized in Fig. 2B. For 135 of the 445 tumors, TCGA identified point mutations in key genes. Ten genes (listed in Fig. 2C) were mutated in more than 5% of the samples; 96 of the 135 sequenced samples had mutations in one or more of these genes. Ninety-four pathways are significantly related to a mutation [Mann–Whitney, false discovery rate (FDR) < 1%]. These 94 pathways are partitioned, on the basis of their PDS, into three clusters (Fig. 2C and SI Appendix, Table S1). Pathways of cluster P1 are deregulated mostly in samples of cluster S2, which comprises tumors with IDH1 mutation. All 32 pathways in P2 are activated by EGF. Indeed, they are highly deregulated on sample cluster S5, which includes almost all patients with EGFR mutations. The fact that our scores capture the deregulation of EGF signaling pathways, expected in samples with oncogenic EGF mutations (18), is reassuring and indicates that Pathifier indeed captures relevant biological information. Also note cluster P3, which contains many pathways with high PDS in tumors with NF1 mutations (mostly in sample cluster S4) and low PDS in tumors with IDH1 mutation (mostly in S2). Another indication of the biological relevance of the PDS is the observed correlation of the scores of many cell death-related pathways with the necrosis levels of GBM samples (SI Appendix, Table S2).
Fig. 2.

(A) Pathway deregulation scores (PDSs) of the TCGA glioblastoma (GBM) dataset (17). Each row corresponds to a pathway and each column to a sample. Pathways and samples are clustered according to PDS. Blue color represents low score (“no deregulation”) and red high. The bottom bar represents the GBM subtype. Notice that pathway-based clustering captures the subtypes well and identifies a secondary substratification. (B) Summary of clustered PDS for the TCGA (Left) and REMBRANDT (19) (Right) GBM datasets. Each row corresponds to a pathway cluster and each column to a sample cluster, displaying the median value of deregulation for each pair of clusters. Arrows connect between pathway clusters that match (that is, the pathways in the clusters have significant overlap). When several matches are significant (as for ReP9 and ReP10) all are shown in dashed arrows, except for the extremely significant ones (P < 10−5). Some of the neurals/proneurals are mostly not deregulated, and some are deregulated on TgP1–TgP3 or matching ReP1/ReP2/ReP7. Classical tumors are deregulated on TgP4/TgP5 and possibly TgP6/TgP7 as well as matching ReP10 (and unmatched ReP6/ReP7). Mesenchymal samples are highly deregulated on TgP8–TgP16 as well as matching ReP8–10/ReP3/ReP4 (and unmatchable ReP5). The classical-mesenchymal cluster TgS4 matches ReS8, and indeed they are both deregulated on TgP4/TgP5/TgP10–12/TgP14/TgP15 and matching ReP8–10/ReP3 (as well as unmatchable ReP5). (C) Normalized PDS of 94 pathways correlated with mutations. The bottom bars display the mutation status, each bar for one gene (samples with mutation are marked in black). Cluster S1 corresponds to normal samples, S2 mostly to samples with IDH1 mutations, S4 mostly to samples with NF1 mutations, and S5 mostly to samples with EGFR mutations. Notice pathway cluster P2, which consist mostly of EGF-activated pathways, and is highly deregulated on the EGFR mutated samples. (D) Normalized PDS of the REMBRANDT GBM dataset. As in A, the pathway clusters correspond to the known subtypes but offer additional substratifications.
PDS-Based Stratification of Glioblastoma.
Hierarchical average-linkage clustering according to PDS of the TCGA data (Fig. 2A) generates (i) sample clusters, which are consistent with known classification and extend it, and (ii) pathway clusters, with related biological functions. The normal samples form cluster TgS7 (sample cluster 7 of the TCGA dataset); mesenchymal form TgS1–3 and TgS11; classical cancers are in TgS8–9, and neurals and proneurals are in TgS12–16. (Fig. 2A and Dataset S1). A concise representation of the characteristic deregulation profiles of the sample types over the pathway clusters (Fig. 2B), reveals further, more subtle, substratification of the tumors. Mesenchymal samples are mostly deregulated in TgP8–TgP16 (pathway clusters 8–16 of TCGA). The main differences between the subtypes are (i) no deregulation of TgP7–TgP9 pathways in the samples of TgS2 and (ii) deregulation of TgP2 in TgS3 and (iii) of TgP4 pathways in TgS11. TgS11 and TgS4 contain classical-like mesenchymals and mesenchymal-like classicals; these intermediate tumor types are deregulated on both the typical mesenchymal pathway clusters TgP8–TgP15 and the characteristic classical TgP4 and TgP5. The emergence of this “subclass” might be due to heterogeneous samples containing both types of cells, or to a new subtype with classical and mesenchymal features. The neural and proneural samples appear mostly in TgS12–TgS16. Some neural/proneural tumors, of TgS13 and TgS15, are normal-like and are not deregulated on most pathways. To validate these results we used data from the Repository of Molecular Brain Neoplasia Data (REMBRANDT) (19) (Fig. 2D and Dataset S1). Fisher’s exact test (P < 0.05) identified correspondence between pathway clusters found in the TCGA data and in REMBRANDT (marked RePi, where i is the cluster number); the main PDS-based features and results are mostly reproduced (Fig. 2B and SI Appendix).
Pathway-Based Substratification of GBM Has Important Clinical Implications.
Neural and proneural samples are thought to have better prognosis (20, 21); the pathway-based substratification reveals, however, that this notion is due to a subset of better survivors (logrank P value < 0.05). In the TCGA data, patients of clusters TgS15 and TgS13, which have relatively few deregulated pathways, survive significantly longer than other neural and proneural samples (P = 0.009 for TgS15 and P = 0.015 for TgS13), whereas patients from TgS12 have worse prognosis (P = 0.003, Fig. 3A). If this group of good survivors is removed from the neural and proneural samples, the remaining patients of these classes do no better than patients with mesenchymal and classical tumors. The separation between survival of the patients of TgS12, 13, and 15 remains significant even if the comparison is made only for the proneural samples.
Fig. 3.
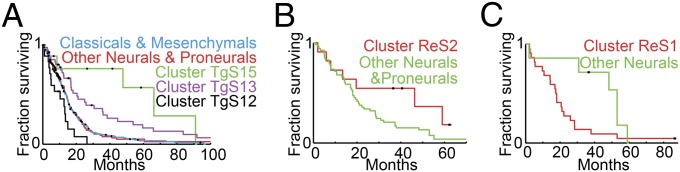
Kaplan–Meier plots for neural and proneural substratification. (A) Patients in TCGA clusters TgS13 and TgS15 have better prognosis. Neural and proneural tumors were divided into three groups, cluster TgS12 (in black), TgS13 (in purple), and TgS15 (in green), and all “others” (in red). Kaplan–Meier plots show clear separation between the four, where cluster TgS15 patients survive the longest (P = 0.009) and cluster TgS13 a little less, but still better compared with the others (P = 0.015); those in TgS12 survive less than the others (P = 0.003). The prognosis of the other neural and proneural tumors is similar to classical and mesenchymal tumors (blue). (B) In the REMBRANDT dataset, neural and proneural tumors were divided into two groups: those in cluster ReS2 (in red) and all others (in green). Kaplan–Meier plots show clearly better survival of the ReS2 patients (P = 0.066). (C) In the REMBRANDT dataset, cluster ReS1 contains only normal samples and normal-like neural samples. Interestingly, these neural patients (in red) have significantly worse prognosis (P = 0.032) than other neurals (in green).
These results are reproduced on the REMBRANDT data as well: Patients with neural or proneural tumors in cluster ReS2 (REMBRANDT sample cluster 2), the one for which only few pathways are deregulated, have better prognosis than other neurals and proneurals (P = 0.066, Fig. 3B). Cluster ReS1 contains only normals and normal-like neural samples. Interestingly, these normal-like neural tumors have worse prognosis than other neurals (P = 0.032, Fig. 3C).
Pathways Associated with Survival in Glioblastoma.
Seventy-seven pathways are significantly related to survival on the TCGA data, and 187 on the REMBRANDT data (FDR < 10%, from Kaplan–Meier analysis, comparing the top one-third of deregulated samples to the bottom one-third, logrank P value). Thirty-seven of these pathways overlap, constituting a significant intersection (P = 0.005). Higher PDSs were associated with bad prognosis on both datasets for all but two pathways. Many of the other 35 pathways (SI Appendix, Table S3) make biological sense: Some are related to angiogenesis, critical to glioblastoma progression (such as VEGF signaling, Fibrinolysis, PDGFRβ signaling, α4β1 integrin signaling, and hypoxia-inducible factor 2-α pathway); many are known key players in glioblastoma and cancer, have a prognostic value, and are promising drug targets [such as MAP kinase (22, 23), Insulin signaling and its components (24), RET tyrosine kinase (25), EGFR/ERBB signaling (26), PDGF signaling (27), and integrins (28)]. SI Appendix gives the full list of other survival-related pathways and their roles in glioblastoma.
Pathway Deregulation in Colorectal Cancer Is Associated with Chromosomal and Microsatellite Instability.
Two kinds of genetic instabilities were identified in colon cancer: chromosomal instability (CIN) and microsatellite instability (MSI-high). CIN tumors (85% of colon cancers) exhibit abnormal numbers of chromosomes, deletions and amplifications of smaller genomic regions, and translocations, and tend to have p53 mutations (29, 30). MSI-high tumors (15% of colon cancers) have highly varying lengths of short sequences of nucleotides, caused by dysfunctional mismatch-repair genes, and usually display no large-scale deletions and amplifications. High-CIN tumors are usually microsatellite-stable (MSS). We applied Pathifier to the data of Sheffer et al. (30), 313 samples of normal colon, polyps, primary carcinoma, and metastases, and validated the results on datasets of Sveen et al. (31) and Kogo et al. (32), 89 and 141 samples, respectively (SI Appendix). Notably, for many relevant pathways the PDS reflected disease progression (Figs. 1 and 4). One hundred six pathways showed increased deregulation with disease progression, that is, significant (Mann–Whitney, 5% FDR) and consistent in all three transitions tested: from normals to polyps, polyps to primary, and primary tumors to metastasis (SI Appendix, Table S4). The deregulation scores of many pathways are correlated with the level of chromosomal instability within the tumor, as measured by the CIN index. Eighty-four pathways show increasing deregulation with increase of the CIN index in all three datasets (SI Appendix, Table S5). This level of overlap is highly significant (P < 4 × 10−4 for every dataset pair), indicating that the correlation found is a robust, fundamental aspect of colorectal cancer. These 84 pathways belong to many biological functions, as expected, considering that many genes are affected by the diverse chromosomal aberrations that characterize colon cancer. Many pathways are differentially deregulated between MSS and MSI-high tumors (325 pathways in the Sheffer data, with significant overlap to those in the Sveen data, P = 1.92 × 10−5 for MSS deregulated pathways, P = 0.022 for MSI-high, Fisher’s exact test; SI Appendix, Tables S6 and S7). The pathways that were highly deregulated in MSI-high tumors, in both datasets, included the mismatch-repair pathway and pathways related to immune response. This is reassuring, because MSI-high tumors have defective mismatch repair (33) and higher levels of inflammation and tumor-infiltrating lymphocytes (34). We also observe that pathways downstream of p53 were highly deregulated in MSS tumors, where p53 mutations are frequent, whereas in MSI-high tumors many pathways upstream of p53 are deregulated.
Fig. 4.

(A) Clustered normalized PDS of the Sheffer dataset. Pathways and samples are clustered according to PDS. For most pathways the PDS of the normal samples are minimal (dark blue), and hence the higher the PDS are the more deregulated the pathway is. For a few pathways (mostly in ShP2) tumors deviate from normals in both directions; PDSs of normal samples have PDS ∼ 0 (green); both highly positive PDS (dark red) and highly negative PDS (dark blue) correspond to pathway deregulation, but in different directions. The color bars at the bottom correspond to the sample type (met denotes metastasis), the MSI status [normal, low, high, MSS, and not available (NA)], p53 mutation status, anatomic location of the tumor, and the CIN index (equally distributed into 20 bins). (B) Summary of clustered pathway scores for the Sheffer dataset. Each row corresponds to a pathway cluster and each column to a sample cluster, displaying the median value of deregulation for each pair of clusters. The color bar indicates the major groups of samples. (C) Oxidative phosphorylation pathway is associated with survival. Kaplan–Meier plots for groups defined by the deregulation scores of oxidative phosphorylation in the Sheffer dataset. The primary tumor samples were divided into three equal groups, based on their level of deregulation (high, medium, and low). Low deregulation scores are associated with better prognosis. (D) CXCR3 pathway is associated with survival. Kaplan–Meier plots for the deregulation scores of CXCR3 pathway in the Sheffer dataset. The primary tumor samples were divided into three equal groups, based on their level of deregulation (high, medium, and low). High deregulation scores are associated with better prognosis.
PDS-Based Stratification of Colorectal Cancer.
Clustering analysis of the PDSs of the Sheffer data identified 11 pathway clusters (denoted by ShP1–ShP11) and 12 sample clusters (ShS1–ShS12) (Fig. 4A, Dataset S1). The normal samples comprise cluster ShS1; the polyps ShS2 (and ShS12), and metastatic samples belong mainly to ShS3. High-CIN primary tumors belong to clusters ShS3–ShS7 and ShS11. These samples are located mainly at the distal part of the colon and are mostly MSS. Clusters ShS8–ShS10 are associated with lower CIN levels, showing mixed locations. Clusters ShS9 and ShS10 include most of the low-CIN, MSI-high samples. A concise coarse-grained representation of the characteristic deregulation profiles is shown in Fig. 4B. Clusters ShP1 and ShP2 (immune response-related) are deregulated on a subset of the polyps. Interestingly, normals have a midlevel score on ShP2: Tumors (and polyps) can deviate along two distinct routes from the normals. Samples in ShS10–12 have lower-than-normal (negative) scores, whereas those of ShS3–ShS5 get positive PDS. Negative PDS correspond to high expression levels of HLA class II molecules and T- and B-cell receptors, which are responsible for activation of the immune response, and hence are indicative of high levels of tumor-infiltrating lymphocytes (similar results are shown for Sveen and Kogo datasets, SI Appendix, Fig. S2).
We discovered a unique class of colon cancer, characterized by high deregulation of cluster ShP3, which contains EGF signaling pathways. This cluster is markedly deregulated in ShS8 and is composed of nine tumors and two polyps. The main cause of the deregulation is overexpression of EGF; no prior identification of such a subgroup has been made, even though there are some reports of EGFR mutations in colon cancer. Apparently about 5% of the tumors belong to this class; hence, relatively large cohorts are needed to observe them. Identification of mutations or amplifications associated with these tumors may provide a new therapeutic strategy that targets the EGF pathway. Clusters ShP4 and ShP5, containing pathways known to play a role in cell migration and invasion (35), show marked deregulation in most samples of ShS10, a unique subgroup of low-CIN tumors that contains both MSS and MSI-high samples, and hence is probably independent of the MSI status; the survival rates of this cluster are not different from those of the high-CIN groups. Clusters ShP6, ShP7, ShP9, and ShP10 show high deregulation in the high-CIN clusters ShS3–4. Many of the pathways that show increase of PDS with progression of the disease (see above) belong to ShP8–10, suggestive of their role in cancer initiation and development.
Repeating the analysis for the Sveen and Kogo datasets (SI Appendix, Figs. S3 and S4), some of the pathway clusters significantly overlap in their pathway content among the three datasets (pairwise Fisher’s exact test, FDR < 1%; SI Appendix, Fig. S5 and Dataset S2). We validated several aspects of the tumor stratification of the Sheffer dataset. The pathway content of the immune cluster ShP2 matches clusters SvP4 (Sveen Pathway cluster 4) and KoP1 (Kogo Pathway cluster 1); all three show consistent bimodal deregulation. ShP5 (migration, inflammation, and angiogenesis) matches SvP9 and KoP10. These clusters have high PDS in low-CIN tumors in all three datasets. The cluster ShP8 (cAMP-dependent signaling) matches SvP10 and KoP7, and ShP11 (cell cycle) matches SvP14 and KoP15.
Survival Analysis of Pathways in Colorectal Cancer.
Thirteen pathways are significantly related to survival in the Sheffer data (FDR < 10%, comparing primary tumors with the top one-third deregulation scores to the bottom one-third, logrank P value). For three of these pathways the same comparison yields significant association (logrank P value < 0.01) with disease-free survival in the Sveen dataset (no survival information was given by Kogo et al.). The first is oxidative phosphorylation (logrank P value < 1.98 × 10−3 for Sheffer, P < 0.027 for Sveen; Fig. 4C and SI Appendix, Fig. S3B). Indeed, deletions of genomic regions enriched by oxidative phosphorylation genes were associated with survival and progression (30), consistent with the Warburg effect and with HIF1 activation that leads to angiogenesis, invasion, and metastasis. A significant finding is the prognostic value of the CXCR3 pathway (P < 3.28 × 10−5 for Sheffer, P < 1.13 × 10−3 for Sveen; Fig. 4D and SI Appendix, Fig. S3C); this is a chemokine receptor expressed by activated T cells and natural killer (NK) cells (36). In both datasets, deregulation of the CXCR3 pathway is governed by the expression levels of four chemokine ligands: CXCL9, CXCL10, CXCL11, and CXCL13, all located at chromosome 4q21. CXCL9–11 bind CXCR3 and show involvement in immune cell recruitment and antiangiogenesis (36). CXCL13 is a chemokine that binds CXCR5, and high CXCL13 expression levels showed improved outcome in early HER2-positive breast cancer (37). In the Sheffer and Sveen datasets, these genes are down-regulated in tumors with poor outcome, suggesting that higher expression of these ligands is associated with recruitment of T cells and NK cells and angiogenesis inhibition that lead to better prognosis of the disease. The third is the IL22BP pathway, which may play a role in anti-inflammation (P < 1.41 × 10−3 for Sheffer, P < 0.014 for Sveen). Notably, both oxidative phosphorylation and CXCR3 remained significant in both datasets, even when we considered only MSS and MSI-low tumors (CXCR3: P < 2.5 × 10−4 Sheffer, P < 0.043 Sveen; oxidative phosphorylation: P < 7.34 × 10−3 Sheffer, P < 0.035 Sveen), suggesting that these pathways are related to survival in colorectal cancer independently of microsatellite stability.
Discussion
Pathifier performs pathway-level analysis of an expression dataset of tumors and determines for each sample a set of PDSs. These PDSs are calculated separately for each pathway using genes that are known to take part in its functioning. The approach can be used with any other kind of data with known pathway assignments (not just mRNA). The approach is data-based: For each pathway we construct a principal curve that captures the variation of the data. All samples are projected onto this curve, and for each sample the distance between this projection and that of the normal samples is measured along the curve. This distance represents the level of deregulation of the pathway. The method copes successfully with the biggest challenges of expression-based pathway analysis: (i) knowledge of biological pathways is partial, (ii) pathway deregulation is context-specific, and (iii) available data contain only part of the relevant information. Using expression values of the genes that were labeled by different studies as belonging to a pathway, measured on the very tissues we wish to study, we are able to define a context-specific PDS. This is accomplished without relying on (incompletely known) underlying network connectivity and function. Even though having more information can only improve the inference made, in most cases one must deal with the absence of relevant information (e.g., posttranslational modifications and protein localization); we do this by projecting the very complex (and unavailable) parameterization of the “biological state” onto expression space, where deregulation is defined by the deviation from the signature of normal samples, measured along a trajectory that reflects context-specific deregulation.
The conceptual aim of defining these scores is to incorporate a large body of prior biological knowledge (in the form of assignment of genes to pathways) to allow further analysis on a “higher” (pathway) level, instead of analyzing the expression levels of thousands of genes, in a brute-force, “ignorance-based” manner. Because this high dimensionality is the cause of many of the challenges and failures of predicting prognosis and response to therapy (38–40), we believe that switching to pathway representation is of key importance for development of reliable clinically relevant prognostic and predictive methods.
We showed for glioblastoma and colorectal cancer that the PDSs successfully reflect deregulation of pathways and constitute a compact and biologically relevant representation of the samples, that retains most of the essential information present in the original data. We stratified tumors into subtypes, interpretable in terms of biologically meaningful and relevant pathways. Whereas the resulting tumor groups were consistent with previously identified clinical classes of glioblastoma and colon cancer, we also identified subclasses with important clinical characteristics.
For glioblastoma we found a clinically relevant substratification of neural and proneural samples, separating them into poor and good survivors, as well as a robust substratification of the mesenchymal subtype. We showed that the method is robust and validated the results on an additional dataset. We showed that important recurrent mutations in glioblastoma have a clear impact on the deregulation scores of the relevant pathways. Some samples without the mutation exhibit deregulation profiles similar to those of the mutated ones, suggesting alternative equivalent deregulation mechanisms. We find 35 pathways whose deregulation score is significantly correlated with survival, with higher levels of deregulation indicative of poor survival in both datasets. This high overlap [in contrast with the low overlap between prognostic gene lists derived using gene-level analyses (38–40)] demonstrates the robustness of the pathway-level findings. Some of these pathways (such as MAP kinase) were previously known to be associated with survival in glioblastoma patients, whereas several others constitute unique findings (such as PDGFRβ and WNT signaling), that may serve as hypotheses for glioblastoma research.
For colorectal cancer we showed that CXCR3-mediated signaling and oxidative phosphorylation pathways are significantly predictive of survival in two different datasets. Furthermore, we suggest a classification of tumors based on their CIN status, high and low, which is broader than the known partition into MSS and MSI-high. Many of the pathways show differential deregulation between these two CIN-based classes of tumors, which cannot be explained solely by their MSI status, emphasizing the important effect of CIN on tumor development. Within the class of low-CIN tumors we found a subgroup, composed of both MSI-high and MSS, that show high deregulation of pathways related to migration, inflammation, and angiogenesis, and indeed these tumors have survival rates similar to those of the group of high-CIN tumors. We also discovered a subclass of tumors related to aberrant EGF signaling that comprise about 5% of the patients.
Methods
Scoring a Gene Set.
Denote by SP the dP-dimensional space, where each coordinate is the expression level of a gene that belongs to a given pathway P, and represent each sample by a point in this space. We look for a one-dimensional curve in SP (or in a subspace of SP) that best describes the variability (e.g., due to disease progression) of the samples across SP (16). That is, we look for a curve that passes through the “middle of the cloud” of samples, and we assume that any two points (samples) that have proximal projections onto the curve also share similar pathway functionality.
Variance stabilization.
Because for some genes we can observe large variation of expression, whereas for others a similar effect on a pathway’s functionality may be induced by a smaller variation, we do not use the absolute expression values. Rather, we divide a gene’s expression values by the SD of its expression in some normalization set of samples (such as normal samples, or all the samples). To avoid genes whose entire variation is due to noise, we keep the 5,000 genes of the highest variance over all samples.
Correlations.
Many of the genes in the gene set of a pathway might be highly correlated, conveying the same information, whereas some other important information might reside in a single gene in the set. To counter this effect, and to improve the running time, we do not actually search for a curve in SP, but in a space SP′ of smaller dimensionality k, identified as follows. First, we perform principal component (PC) analysis and keep only PCs along which the variance exceeds by more than 10% that of the normalization set. The number of such PCs is k and the entire ensuing computation is done in the space SP′ spanned by these components.
Principal curve.
We use Hastie and Stuetzle’s algorithm (16) to find a principal curve in SP′ (Fig. 1). After such a curve is found, we project each point xi, that represents sample i in SP′, onto fi, its closest point on the curve. The deregulation score DP(i) of sample i is defined as the distance along the curve between fi and a reference point r, defined as the centroid of some reference set of samples. The reference set is used also to define the curve’s direction, by making sure the point representing the median coordinates of the reference set is closer to the beginning of the curve (flip the curve’s direction otherwise). In this study, the reference set is composed of the healthy samples from the same tissue (henceforth “normal samples”), which indeed tend to concentrate on one side of the curve, due to the high similarity among normal samples and the large difference from tumor samples. The distance DP(i) provides a measure of the extent to which the expression levels of the genes associated with pathway P were perturbed in sample i by the disease.
In some cases the normal samples fall roughly in the middle of the curve. When this happens, the curve captures two different kinds of deregulation, with tumors moving away from the normal samples along two distinct paths. In principle one can use other (than normal) samples as reference, although doing this makes sense only in cases when the inner variability of the new reference set is considerably smaller than the overall variability.
Stability and Robustness of the PDS.
Finding a stable gene set.
Often some of the genes in the gene set are noisy (in the sense that their variation does not reflect information relevant to the biology we are trying to capture; Discussion), and we would rather omit them. Because we work in SP′ and not in SP, we actually omit metagenes (linear combinations of genes), but similar considerations imply that some of the metagenes might be noisy and should be omitted. This is partly taken care of by omitting genes and metagenes that do not vary much, but some of the noise might be due to highly varying metagenes, where most of the variation is unrelated to the biological information captured by the gene set.
To find out which metagenes should be omitted, we select, one at a time, those along which the samples are farthest from the curve, as expected for noisy metagenes, and find after each omission the new corresponding principal curve. To assess which curve is the best, we check the sensitivity of the gene set’s scores to sampling noise (the variance over 100 repeats of 80% of the samples selected randomly each time). If there is a significant improvement in the stability, we omit the metagene whose omission yields the most stable curve and continue in a greedy fashion. If the improvement is not significant (or stability actually becomes worse), we stop.
Stability against dilution of the normal samples.
To test for sensitivity of the PDS with respect to the reference used (the mean of the normal) we excluded from the analysis 20% (selected at random, 10 times) of the normal samples and recalculated the PDS. For a large majority (but not all) of the pathways and for nearly all of the samples the correlations between the new scores and the old ones are high, indicating robustness of the PDS against such dilution of the data (SI Appendix, Fig. S6).
Comparison with Alternative Methods.
We compared our method with three simpler ones and with PARADIGM. The first, by Segal et al. (3), was not designed to provide individual pathway deregulation scores. Nevertheless, such scores were calculated by Segal et al. as interim results, derived by counting the number of significantly up- and down-regulated genes and performing gene set enrichment analysis (separately for the two gene groups), assigning scores of ±1 or 0 to each pathway and sample. These scores have low correlation with those of Pathifier (SI Appendix, Figs. S7 and S8). This method detects only extreme changes; the resulting deregulation score matrix (SI Appendix, Fig. S9A) is very sparse and most of our observations reported above are missed: Only two pathways were found to be related to gene mutations in glioblastoma, known subtypes were not well separated by the scores, and no subtle substratification was identified (except, perhaps, for mesenchymals). No pathway score is correlated with survival for any of the datasets of glioblastoma, and only two survival-related pathways were found, for only one of the colorectal cancer datasets (at FDR < 10%).
The second method is a linear version of Pathifier, with scores defined as the Euclidean distance of a sample to the normals (without first projecting to a principal curve). These scores have a wide dynamic range and separate known GBM subtypes well (SI Appendix, Fig. S9B). In fact, the scores are quite correlated to those of Pathifier (SI Appendix, Figs. S7 and S8); however, Pathifier captures more subtle changes, and, importantly, captures clinical relevance better. When the linear scores are used, none of the pathways is found to be related to survival (at FDR < 10%) for the GBM TCGA dataset. On the REMBRANDT dataset Pathifier discovered 187 survival-related pathways and the linear version only 54 (8 of which were not identified by Pathifier). In the colorectal Sheffer dataset no pathway related to survival was discovered (at 10% FDR) by the linear scores.
The third method we tested was the MAPPFinder module of GenMAPP (6), which we adapted to provide a score for every pathway in each sample (this algorithm was also designed to yield a single score per pathway for an entire cohort). This was done by calling differentially expressed genes for every sample by Z-test compared with the normal samples (instead of t test over the entire cohort). The average correlation of the pathway profiles with those of Pathifier is around 0.30 (SI Appendix, Figs. S7 and S8). For GBM (SI Appendix, Fig. S9C), no pathways having significant correlations with mutations were found (at FDR < 1%), the scores do not separate the clinical subtypes well, and no pathway is found to have significant correlation with survival (at FDR < 10%). We found only one pathway whose MAPPFinder scores were positively correlated with the chromosomal instability index in all three colon cancer datasets; one pathway showed correlation with survival in the Sheffer dataset (at FDR < 10%), but it was not significant in the Sveen dataset and no pathway was correlated with progression (at FDR < 5%).
Despite the differences in the approaches, we also compared the results of Pathifier to those of PARADIGM for TCGA GBM (SI Appendix, Text and Fig. S10). Some of the results of the two methods were in agreement, but some findings of Pathifier were not detected by PARADIGM.
Supplementary Material
Acknowledgments
We thank Shlomo Urbach for his part in a preliminary study of similar aims and Dr. Charles Vaske and Dr. Joshua Stuart of University of California, Santa Cruz for their assistance with PARADIGM and supplying their raw results. This work was supported by the Leir Charitable Foundation, the Israel Science Foundation, and a Germany–Israel Cooperative Research grant (Department of International Programs).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1219651110/-/DCSupplemental.
References
- 1.Markert EK, Mizuno H, Vazquez A, Levine AJ. Molecular classification of prostate cancer using curated expression signatures. Proc Natl Acad Sci USA. 2011;108(52):21276–21281. doi: 10.1073/pnas.1117029108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas DC, et al. American Association for Cancer Research Approaches to complex pathways in molecular epidemiology: Summary of a special conference of the American Association for Cancer Research. Cancer Res. 2008;68(24):10028–10030. doi: 10.1158/0008-5472.CAN-08-1690. [DOI] [PubMed] [Google Scholar]
- 3.Segal E, Friedman N, Koller D, Regev A. A module map showing conditional activity of expression modules in cancer. Nat Genet. 2004;36(10):1090–1098. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- 4.Bild AH, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439(7074):353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 5.Chin L, Hahn WC, Getz G, Meyerson M. Making sense of cancer genomic data. Genes Dev. 2011;25(6):534–555. doi: 10.1101/gad.2017311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doniger SW, et al. MAPPFinder: Using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol. 2003;4(1):R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cary MP, Bader GD, Sander C. Pathway information for systems biology. FEBS Lett. 2005;579(8):1815–1820. doi: 10.1016/j.febslet.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Efroni S, Schaefer CF, Buetow KH. Identification of key processes underlying cancer phenotypes using biologic pathway analysis. PLoS ONE. 2007;2(5):e425. doi: 10.1371/journal.pone.0000425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas DC, et al. Use of pathway information in molecular epidemiology. Hum Genomics. 2009;4(1):21–42. doi: 10.1186/1479-7364-4-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emmert-Streib F, Glazko GV. Pathway analysis of expression data: deciphering functional building blocks of complex diseases. PLOS Comput Biol. 2011;7(5):e1002053. doi: 10.1371/journal.pcbi.1002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaske CJ, et al. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;26(12):i237–i245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(Database issue):D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishimura D. BioCarta. Biotech Software Internet Report. 2001;2(3):117–120. [Google Scholar]
- 15.Schaefer CF, et al. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009;37(Database issue):D674–D679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hastie T, Stuetzle W. Principal curves. J Am Stat Assoc. 1989;84(406):502–516. [Google Scholar]
- 17.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JC, et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006;3(12):e485. doi: 10.1371/journal.pmed.0030485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Madhavan S, et al. Rembrandt: Helping personalized medicine become a reality through integrative translational research. Mol Cancer Res. 2009;7(2):157–167. doi: 10.1158/1541-7786.MCR-08-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phillips HS, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 21.Verhaak RG, et al. Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mawrin C, et al. Prognostic relevance of MAPK expression in glioblastoma multiforme. Int J Oncol. 2003;23(3):641–648. [PubMed] [Google Scholar]
- 23.Mason WP, et al. A phase II study of the Ras-MAPK signaling pathway inhibitor TLN-4601 in patients with glioblastoma at first progression. J Neurooncol. 2012;107(2):343–349. doi: 10.1007/s11060-011-0747-6. [DOI] [PubMed] [Google Scholar]
- 24.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441(7092):424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 25.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353(2):172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 26.Andersson U, et al. Epidermal growth factor receptor family (EGFR, ErbB2-4) in gliomas and meningiomas. Acta Neuropathol. 2004;108(2):135–142. doi: 10.1007/s00401-004-0875-6. [DOI] [PubMed] [Google Scholar]
- 27.Dong Y, et al. Selective inhibition of PDGFR by imatinib elicits the sustained activation of ERK and downstream receptor signaling in malignant glioma cells. Int J Oncol. 2011;38(2):555–569. doi: 10.3892/ijo.2010.861. [DOI] [PubMed] [Google Scholar]
- 28.Desgrosellier JS, Cheresh DA. Integrins in cancer: Biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396(6712):643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 30.Sheffer M, et al. Association of survival and disease progression with chromosomal instability: A genomic exploration of colorectal cancer. Proc Natl Acad Sci USA. 2009;106(17):7131–7136. doi: 10.1073/pnas.0902232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sveen A, et al. Transcriptome instability in colorectal cancer identified by exon microarray analyses: Associations with splicing factor expression levels and patient survival. Genome Med. 2011;3(5):32. doi: 10.1186/gm248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kogo R, et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011;71(20):6320–6326. doi: 10.1158/0008-5472.CAN-11-1021. [DOI] [PubMed] [Google Scholar]
- 33.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363(6429):558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 34.Smyrk TC, Watson P, Kaul K, Lynch HT. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer. 2001;91(12):2417–2422. [PubMed] [Google Scholar]
- 35.Na KY, Bacchini P, Bertoni F, Kim YW, Park YK. Syndecan-4 and fibronectin in osteosarcoma. Pathology. 2012;44(4):325–330. doi: 10.1097/PAT.0b013e328353447b. [DOI] [PubMed] [Google Scholar]
- 36.Lacotte S, Brun S, Muller S, Dumortier H. CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci. 2009;1173:310–317. doi: 10.1111/j.1749-6632.2009.04813.x. [DOI] [PubMed] [Google Scholar]
- 37.Razis E, et al. Improved outcome of high-risk early HER2 positive breast cancer with high CXCL13-CXCR5 messenger RNA expression. Clin Breast Cancer. 2012;12(3):183–193. doi: 10.1016/j.clbc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 38.Ein-Dor L, Kela I, Getz G, Givol D, Domany E. Outcome signature genes in breast cancer: Is there a unique set? Bioinformatics. 2005;21(2):171–178. doi: 10.1093/bioinformatics/bth469. [DOI] [PubMed] [Google Scholar]
- 39.Drier Y, Domany E. Do two machine-learning based prognostic signatures for breast cancer capture the same biological processes? PLoS ONE. 2011;6(3):e17795. doi: 10.1371/journal.pone.0017795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simon R. Lost in translation: Problems and pitfalls in translating laboratory observations to clinical utility. Eur J Cancer. 2008;44(18):2707–2713. doi: 10.1016/j.ejca.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.