

Abstract
Whereas models of cancer disparities and variation in cancer burden within population groups now specify multiple levels of action from biologic processes to individual risk factors and social and physical contextual factors, approaches to estimating the preventable proportion of cancer use more traditional direct models often from single exposures to cancer at specific organ sites. These approaches are reviewed, and the strengths and limitations are presented. The need for additional multilevel data and approaches to estimation of preventability are identified. International or regional variation in cancer may offer the most integrated exposure assessment over the life course. For the four leading cancers, which account for 50% of incidence and mortality, biologic, social, and physical environments play differing roles in etiology and potential prevention. Better understanding of the interactions and contributions across these levels will help refine prevention strategies.
Keywords: chemoprevention, obesity, activity, built environment, tobacco
INTRODUCTION
Fifty to sixty percent of cancer deaths can be prevented (32). Such estimates draw on data from several sources demonstrating international variation in cancer incidence and mortality; changes in risk observed in migrant studies; and etiologic studies with data on individual participants, such as prospective cohort studies and reduction in risk of smoking-related cancers after stopping smoking. Recently, a small number of randomized controlled trials of prevention strategies including vaccination (64), drugs (71), and dietary supplements (7) have added to the evidence base. Given that most cancer can be prevented with what we already know, public health authorities, health care providers, and individuals have responded by adopting prevention targets and strategies. To achieve prevention goals, we need multipronged approaches through health care providers, regulatory changes, and individual and community behaviors (4). Examples include provider counseling for risk reduction and early detection through screening; implementation of regulations to enforce health-related protections (e.g., SunSmart (75); and global public health campaigns to impact personal, community, and corporate decisions that improve the public’s lifestyle (98). Identifying the relative contributions of biologic, social, and physical environments on cancer incidence and mortality will clarify the importance of these factors for prevention and help set priorities, informing goals and realistic time lines for achieving reduction in the cancer burden.
Interventions through the health care system such as early detection and effective treatment of diagnosed cancer cases are critical to improving quality of life for individuals with cancer and to decreasing cancer-related deaths (36, 48). Although some treatment advances have reduced cancer mortality (6), the overall impact of the health care system through advances in therapy appears limited to date. Extensive investment towards genome-based therapies for cancer remain the focus and future therapeutic potential (53). Because breast and prostate cancer are two of the top four cancer diagnoses and the majority of cases die from causes other than their primary cancer, much remains to be done to implement lifestyle changes that will reduce the burden of chronic disease mortality among these survivors (108). Thus issues of access to care, adherence to therapy, lifestyle patterns, and genetics all interconnect among those with diagnosed cancer to determine, in part, their survival and the overall number of cancer deaths. To address the interplay of biologic, social, and physical environments in driving the cancer burden, we therefore focus primarily on cancer incidence. Considering the interplay of age, timing of exposure, dose and duration necessary to increase/decrease risk, and the corresponding changes that must be sustained to reduce risk informs our understanding of the contributions of factors to the burden of cancer in society (24,26).
Tarlov & St. Peter defined the determinants of population health as genes and biology, health behaviors, medical care, social/societal characteristics, and total ecology (96). They define social/structural determinants of population health as including poverty and income inequality, education, employment, housing, mobility, transportation, pollution, and nutrition. To date, many of these components are not well characterized in relation to cancer incidence and mortality. This type of model can be expanded to a multilevel approach where factors are nested with the social environment and genetic factors interact with environmental determinants of cancer (50, 100). Such models typically lead to downstream interventions that may be disease specific but can include upsream interventions decreasing social isolation, promoting early detection, integrating social indicators into clinical care, and community partnerships combining research and social change in disease prevention. To address the biologic, social, and physical environmental determinants of cancer, we group factors that may traditionally be considered as components of the total preventable burden of disease but acknowledge that the boundaries are somewhat arbitrary.
BURDEN OF CANCER
Taking a global perspective, the number of cancer deaths is expected to grow from 12 million new cases and 7 million deaths from cancer in 2008 to a projected 26 million new cases and 11.4 million deaths in 2030 (57). The leading causes of cancer mortality in the world are lung (1.4 million deaths per year), stomach (866,000), colon (677,000), and breast (548,000). Approximately 72% of cancer deaths occurred in low- and middle-income countries in 2007, where the leading causes of cancer mortality are lung, stomach, liver, colon and rectum, and cervix. Increasing cancer death rates can be attributed, in part, to the aging population and also to the epidemic of tobacco use in the developing world (83), with additional increases in female cancers due to changing reproductive patterns (66, 67). Within the United States, as is typical of other established market economies, the leading cancers are breast, prostate, colon, and lung, which together account for at least half of all new cases and all deaths (see Table 1).
Table 1.
Cancer incidence mortality and change in mortality, United States. For 5 leading cancer sites and population total. Total numbers are total cancer cases all casues not jsu the 5 leading cuases called out in the table
| Incidence cases (% of total) | Mortality cases (% of total) | Annual change in mortality 1997–2006 | |
|---|---|---|---|
| Male | |||
| Lung | 116,750 (14.8) | 86,220 (28.8) | −2.0 |
| Colorectal | 71,090 (9) | 26,580 (8.9) | −2.9 |
| Prostate | 217,730 (27.6) | 32,050 (10.7) | −4.1 |
| Pancreas | 21,370 (2.7) | 18,770 (6.3) | 0.2 |
| Esophagus | 13,130 (1.7) | 11,650 (3.9) | 0.4 |
| TOTAL | 789,620 | 299,200 | −1.8 |
| Female | |||
| Breast | 207,090 (28.0) | 39,840 (14.7) | −2.0 |
| Lung | 105,770 (14.3) | 71,080 (26.3) | −0.1 |
| Colorectal | 70,480 (9.5) | 24,790 (9.2) | −2.7 |
| Pancreas | 21,770 (2.9) | 18,030 (6.7) | 0.1 |
| Ovary | 21,880 (2.9) | 13,850 (5.1) | −0.4 |
| TOTAL | 739,940 | 270,290 | −1.1 |
Source: Edwards, ref 43
We now consider the three approaches to identify their strengths and limitations for quantifying the preventable burden of cancer. These approaches range from contrasting international or regional variation in cancer risk (including migrant studies), to modeling individual risk factors and interpreting the potential for prevention, and to a more classic approach of estimating the population attributable risk (PAR) for specific cancer risk factors. Although we consider these approaches separately, some mixing of methods is possible.
INTERNATIONAL/REGIONAL VARIATION
Drawing on epidemiologic surveillance data, Doll & Peto (41) compared the rates of different cancer types in high- and low-incidence populations and estimated the proportion of cancers that could be attributed to nongenetic factors that vary across these populations. The ratio of highest rate to lowest rate of cancer was as high as 100-fold or more. Connecticut was used as the reference population for the United States because it has a longstanding population-based cancer registry dating back to the early 1940s. On the basis of comparisons of high- and low-incidence regions, Doll & Peto concluded that 75–80% of cancers diagnosed in the United States in 1970 theoretically could have been avoided. What made the U.S. population different from low-risk populations? The environmental (nongenetic) factors that differ between the United States and low-risk populations are many and diverse and are experienced across the life course. They include factors such as birth weight; age at puberty; lifelong patterns of diet, weight gain, alcohol consumption, use of tobacco, and use of pharmacological agents; and reproductive factors. This conclusion was provocative because, at the time, only limited data from rigorously performed epidemiological studies related diet, obesity, and alcohol intake to cancer risk. Other commonly studied environmental exposures, such as differences in air, water, and food contamination between the United States and other populations, were also thought to be involved in determining cancer risk, but to a lesser extent than previously assumed.
Many investigators criticized the Doll & Peto studies, indicating that they placed too much emphasis on lifestyle factors—for example, smoking and diet—with too little emphasis on involuntary exposures such as occupational and environmental carcinogens that clearly covary with the geography and level of development of countries (44). Extensive epidemiologic data documented the carcinogenic hazards of workplace exposures, including asbestos, benzene, arsenic, nickel, polycyclic hydrocarbons, and vinyl chloride (9). As a result, the Occupational Safety and Health Administration was established in 1970 to ensure safe and healthy working conditions. Over time, the agency has had a positive impact, decreasing cancer risk among industrial workers through reduced exposure to carcinogens (105). However, as U.S. workplaces have become less carcinogenic, the hazards of production for products such as steel—which is associated with exposure to crystalline silica, polycyclic aromatic hydrocarbons, and various other carcinogenic chemicals—have been largely exported to countries that have cheaper labor and lower production costs (112). Although fewer U.S. workers are exposed to occupational carcinogens, and industrial exposures have only a minor contribution to cancer in the United States (74), it is still important to understand the cancer risk associated with occupational exposures. Recent reviews show a continuing small proportion of cancer due to these exposures in industrialized countries despite regulations on workplace exposures (10, 11). Boffetta (9) completed a rigorous review of evidence and concluded that occupational and environmental exposures account for 1–2% of cancers. Here exposure means carcinogens contaminating the environment, not the built environment, and social influences of smoking, diet, and physical activity (see below). By studying occupational as well as lifestyle factors that contribute to cancer incidence, it becomes possible to find ways to prevent cancer on many fronts worldwide.
Doll & Peto acknowledged that their estimate that cancer rates could be reduced by 75–80% was a theoretical maximum and that it was unlikely that society could change enough—even over many years—to decrease cancer incidence by this amount. Their analysis, however, provided an important starting point for subsequent studies of cancer causes and strategies for cancer prevention (87, 106, 109) and has even led to some strictly defined timelines for reducing cancer incidence in the United States (13). In the past 30 years, many subsequent epidemiologic studies have confirmed the contribution of specific lifestyle factors to the etiology of cancer (27, 32) and have expanded the list of cancer causes to include obesity and lack of physical activity (55, 89). Specifically, achievable changes in the preventable causes of cancer now account for an estimated more than 60% of all cancer cases in the United States (27, 32, 107). Thus the estimates by Doll & Peto have proven in large part to be correct in overall magnitude with some rebalancing of the contributions of different lifestyle components, greater roles for obesity and lack of physical activity, and a lesser role for diet.
STATISTICAL MODELS INFORMING PREVENTABLE CANCER
Models of cancer incidence can build on the epidemiologic data accumulated through detailed studies over the past decades and also integrate biologic understanding of cell division, repair, and other defense mechanisms.
Modeling Incidence
Two distinct classes of mathematical models have been used in cancer epidemiology. Statistical models draw on established mathematical structures (including linear and logistic regression) to evaluate relationships between risk factors and cancer incidence. Biomathematical models are derived by translating into mathematical terms a series of hypotheses about the biologic process involved in carcinogenesis (58). One of the best known models developed by Armitage & Doll (2) lays the foundation for a long history of applying mathematical models to cancer incidence rates and with extension can relate epidemiologic risk factors to cancer incidence to provide a structure to view the process of carcinogenesis and estimate the preventability of cancer (12, 21, 103). Drawing on cancer mortality data from the 1940s, a time when morality directly reflected incidence data because treatment had little impact on cancer outcomes, Fisher & Hollomon (46) used stomach cancer statistics and Nordling (81) combined all cancer sites to note that for ages 25–74 years the logarithm of the death rate increased in direct proportion to the logarithm of age. Armitage & Doll then built on this work to evaluate cancer mortality in the United Kingdom in men and women in 1950 and 1951. They noted that a gradient of 6 to 1 (i.e., 6 units increase in the logarithm of the death rate per unit increase in the logarithm of age) was more or less consistent across 17 cancer sites and concluded that the theory that cancer is the end result of several successive cellular changes is supported by cancers of the esophagus, stomach, colon, rectum, and pancreas in men and stomach, colon, rectum, and pancreas in women. Furthermore, a slowing in the rate of increase in mortality from breast, ovary, and cervical cancer in women beyond age 50 was noted by Armitage & Doll. They attributed this to a reduction during midlife in the rate of production of one of the later changes in the process of carcinogenesis (2). Through this work, they set forth a multistage model of carcinogenesis long before laboratory or biologic understanding was established.
These types of mathematical models can also summarize the impact of multiple variables that may modify the incidence rates and so can identify areas of research that require more study (76). They may also allow for refinement and improved precision in risk estimation and ultimately produce better tools for clinical risk assessment and decision-making regarding the use of chemopreventive agents (49). These same models have also been applied to estimate the preventability of cancer, perhaps best represented in lung cancer, where the relative benefits of interventions in smoking initiation and cessation were presented in 1987 (12). Doll & Peto (39) applied this multistage cancer incidence model to lung cancer within the British Doctor’s Study and observed that incidence is proportional to (dose +6)2. (age – 22.5)4.5, where dose equals cigarettes per day. This calculation was consistent with the multistage model of carcinogenesis and generated coefficients for the components of the model that are not readily interpretable beyond comparing their magnitude and the power function that approximates the number of stages in the model. However, in this and similar models, incidence is proportional to the fourth to sixth power of time, suggesting four to six independent steps are necessary for cancer development. Such extrapolations have been confirmed by Vogelstein and colleagues, who documented that more than four genetic alterations are necessary for colon cancer development (99). Mechanistic implications of this work for lung cancer indicated that more than one of the stages of lung carcinogenesis was strongly affected by smoking (12, 54). Extensive application of the Armitage & Doll model to radiation exposure also attests to its utility (38, 69).
Moolgavkar and colleagues (77) modeled stages of clonal expansion from preinitiation on through malignancy. They consider loss of tumor suppressor at initiation and clonal expansion as a two-stage process and then fit three- and four-stage clonal expansion models to colorectal cancer and pancreatic cancer (72). This approach builds on the application of the Knudson two-stage model of cancer (63) and provides insight to the timescales for progression of common malignancies.
The underlying Armitage & Doll approach to modeling has also been the basis for better understanding of breast cancer incidence where Pike (86) then Rosner & Colditz (28, 90) built on this approach. Pike took the lung cancer models of Peto & Doll and applied them to breast cancer, which gave a framework for breast tissue aging and risk accumulation over the life course that was modified by reproductive exposures (menarche, parity, and menopause). Rosner & Colditz (90, 91) expanded this approach to include number and timing of births and menopause, family history of breast cancer (31), benign breast disease, alcohol intake, height and obesity, type of menopause (bilateral oophorectomy versus natural), and use of postmenopausal hormones (28). This model then allows estimation of the contribution of individual risk factors to cumulative risk, an approximation of the population burden of breast cancer.
The model has been applied to Chinese data where incidence trends have changed rapidly. We show how reproductive factors, weight, and alcohol together predict a doubling in the age-specific rates of breast cancer by 2021 (68).
CLASSIC PAR APPROACH FOR INDIVIDUAL EXPOSURES
Estimating the proportion of cancer due to specific exposures (tobacco, diet, obesity) often draws on data from individual studies or a synthesis of such data. This approach was used in a quantitative review of risk factors for specific cancers by Danaei and colleagues (37). This approach is far more conservative than the earlier approach of Doll & Peto (40), who estimated the overall proportion of cancer that could be avoided mainly by comparing high-and low-risk populations. Danaei and colleagues limit their prevention estimates to lifestyle changes that could reasonably be achieved and base these estimates on a very limited number of cancers for which their consensus approach leads to a causal inference. This approach requires extensive epidemiologic data measured on the exposure and time frame of life to come to a causal inference. Estimates for smoking are comparable in both reports because, like Doll & Peto, Danaei et al. (40) used the American Cancer Society (ACS) Cancer Prevention Study 1 mortality data (for cancers of the lung, mouth, larynx, esophagus, bladder, pancreas) and estimates for other cancers (such as kidney and liver) to derive their estimate of 30% of cancer mortality.
However, for obesity, for example, the ACS data indicate that 14–20% of cancer mortality is attributable to obesity (14), whereas the Danaei estimate is limited to only breast, colon, and uterus (37), omitting causal evidence for esophagus [as classified by the International Agency for Research on Cancer (IARC) report (55)] and for other cancers (liver, pancreas, multiple myeloma, and non-Hodgkin lymphoma) identified as having significant excess mortality among overweight and obese participants in the ACS cohort. These additional cancers are confirmed as causally related to obesity through the rigorous systematic review and meta-analysis reported by Renehan (89). Thus, in contrast with the ACS mortality data, Danaei and colleagues use a more conservative consensus approach to causal inference and attribute only 3% of cancers in high-income countries to overweight and obesity. The disagreement pertaining to the importance of obesity to cancer burden is almost an order of magnitude different and clearly misses the evidence accrued prior to their review.
Furthermore, it is noteworthy that the choice of exposures to consider can limit the final estimate for potential preventability of cancer. For example, Danaei and colleagues omit drugs such as postmenopausal hormones as causes of breast and endometrial cancers, as noted above accounting for 10% of postmenopausal breast cancer. They also omit the contribution of chemoprevention and vaccination programs against infectious agents. For these preventive strategeies we might summarize data estimating the proportion of disese prevented (72a). Although we may debate whether these are biologic or social exposures, because peer pressure may be one influence on interest in taking drugs and subsequent adherence to therapy, biologic agents appear to be a fitting classification for hormones, vaccines, and other chemopreventive agents. We included these in our review.
TRADE-OFFS FROM DIFFERENT APPROACHES
The various approaches to estimating contributions to prevention have strengths and limitations. We summarize these in Table 2. International or regional variation gives an integration of exposure over the life course that includes lifestyle patterns, be it low smoking, low red meat consumption, and low alcohol intake (such as Utah in the United States), or patterns of growth, activity, and energy balance (such as Asia before industrialization). Although the actual exposures driving incidence may not be known precisely, an integrated summary of the impact of lifestyle factors is provided by these incidence data. However, separating out components of exposure may be difficult. Some societies, however, show substantial variation in population-level exposures such as smoking, which is historically low in the Mormon population in Utah. Migrant studies add precision to the age at exposure or age at change in exposure as well as timing of subsequent exposure. The level of exposure may still be imprecisely quantified. Individual disease models provide opportunities to integrate a range of initiating factors as well as progression or promoters and evaluate rates of change in risk in biologically plausible ways (70). However, these models deal with one cancer site at a time and may be limited by available measures needed across the life course. One limitation of the individual-level data approach is the reliance on measures of exposure in the assumed time frame for etiology of cancer or its prevention (104) and lack of broader social ecologic framework. PAR estimates may thus be biased by measures of exposure at etiologic times that are not relevant to disease progression.
Table 2.
Strengths and limitations of approaches to estimating preventability of cancer.
| Strengths | Limitations | |
|---|---|---|
| International/regional variation | Integration of exposure across life course | Separating out component exposures is difficult |
| Migrant studies | Defines timing of change in “exposure” and change in risk | -Separating out component exposures is difficuly for many cancers |
| Individual models | Integrates range of factors as initiator and promoters Evaluates change in biologically plausible way |
Requires input data to have necessary measures Precision and timing of measures within biologic pathway may vary |
| Population attributable risk | Apparent precision | Choice of disease conditions, quality of classification and documentation of cancer incidence or mortality Choice of exposures and crieria for including exposure, evidence included in deriving summary level of relative risk Timing of exposure may not reflect etiologic role Mis-specified preventive potential |
Data from international variation subsequently supported by individual-level data from cohort studies indicates that in the United States, tobacco use accounts for some 30% of all cancer cases, alcohol consumption for 4% of cancer cases, and poor diet for a variable proportion of cases. In addition to these lifestyle factors, Doll & Peto (41) proposed a link between use of estrogen and endometrial cancer, and they speculated that estrogens might also increase breast cancer risk. Substantial additional data have accumulated to confirm these causal relations due to exogenous hormone therapy (a biologic agent) (8, 20, 35, 92). The decline in incidence after reduction in use of combination estrogen plus progestin therapy was observed within a health maintenance organization population, then in U.S. SEER (Surveillance Epidemiology and End Results) rates, and in other countries (23, 61, 88). These data indicate that use of hormone therapy may account for 10% of postmenopausal breast cancer (25).
Summing Up the Biologic, Social, and Physical Environment as Causes of Cancer
The somewhat arbitrary classification of biologic, social, and physical environments is best exemplified by tobacco, a known source of carcinogens and so a biologically active agent: Its uptake is a socially learned response, addiction is a biologic phenomenon, and long-term exposure to known carcinogens is both biologic and, through secondhand smoke, a social/ecologic exposure. Maintenance of clean indoor air may be a prevention strategy by regulating the physical environment. Unarguably, together these factors operate for tobacco exposure to account for at least one-third of cancers that are completely preventable. Subdividing the overall impact of tobacco into these three component areas would not be based on data.
Established as the primary cause of cancer-related deaths and considered the single largest preventable cause of cancer in the world (85), the impact of tobacco on international health is hugely detrimental. Tobacco smoking causes bladder, cervical, esophageal, kidney, laryngeal, lung, oral, pancreatic, and stomach cancers and acute myeloid leukemia (97). In the United States alone, smoking causes at least 30% of cancer deaths annually; globally, tobacco will kill more than five million people per year. Risk increases with daily consumption as well as duration of smoking. Secondhand smoke poses significant risk as well, which makes tobacco the only legal consumer product that can harm everyone exposed to it. Furthermore, the reduction in mortality from tobacco-related cancer after cessation from smoking is substantial, attaining mortality rates of never smokers in 20 to 30 years (60).
At the other end of the interface among the determinants, social and physical environments appear intertwined around obesity, physical activity, diet, and alcohol. Sun exposure, a strong physical environmental carcinogen, includes social behavioral determinants from tanning and related exposures. As noted above under the PAR discussion, obesity is a preventable cause of at least 15% of cancers;- weight gain is a result of excess energy intake over energy expenditure (107). Is this a consequence of the social norms around eating and recreation, of our built or physical environment, or of an interaction between these factors and genetics? Lack of physical activity clearly causes cancers of the breast and colon independent of obesity (55, 109), yet again the role of social and physical environments is intertwined. Lack of physical activity causes 5% of cancers, and the built environment contributes to both development of obesity and physical activity patterns (42). A detailed analysis of Behavioral Risk Factor Surveillance data from 2007 shows how poverty and race are directly related to obesity, and the local food economy is negatively related (95). If we accept that diet is a socially determined behavior, we may allocate red meat consumption, lack of folate, and higher alcohol intakes to social environment causes. The lack of these habits in some populations adds further support for this classification. Evidence for these components is summarized below.
Biologic Agents
Infections, drugs, and chemopreventive agents may be most clearly allocated to biologic agents. Together these factors cause the majority of cancers in developing countries (infection) and in established market economies may account for 10% of breast cancer (postmenopausal hormones). Agents such as aspirin clearly prevent colon cancer with established latency (93, 94), and calcium can also reduce colon cancer incidence as shown through randomized controlled trial evidence and corresponding observational data (7, 22).
The etiology of some 18% of cancers worldwide can be linked to chronic infections such as hepatitis B (HBV), hepatitis C (HCV), human papillomavirus, Epstein-Barr virus, HIV, human herpes virus 8, Helicobacter pylori, and Schistosoma haematobium (82). The current burden of cancer in the developing world, aside from smoking-related cancers, is dominated by infection. Of 5 million new cases of cancer worldwide, those caused by infection are divided among several sites that contribute to the total global burden and are attributable to chronic infection: stomach (H. pylori, 5.5%); cervix, ano-genital, mouth, and pharynx (HPV, 5.2%); liver (HBV HCV, 4.9%); and other less common sites. More than 25% of cancer incidence in developing countries could be avoided if infectious causes of cancer were prevented (82). These biologic agents can be interrupted through vaccination programs, which have the potential to reduce cancer incidence and mortality; for example, Taiwan’s HBV vaccination program was initiated in 1984. High coverage rates (up to 97% in 2004) have led to a consistent decline in hepatocellular cancer rates (16). However, two recent reports by Chang et al. (17, 18) underscore the importance of a multipronged approach. Although Taiwan has seen a decrease in the incidence of hepatocellular cancer in children and adolescents since initiation of the vaccination program (from 0.54 to 0.20 per 100,000 before and after the program), vertical transmission, vaccine failure, and the lack of hepatitis B immunoglobulin injection has affected program effectiveness and public health impact (17, 18).
Medication use is widespread in the high-income countries and limited in low- and middle-income countries. Strong evidence indicates several medications as either causing cancer (e.g., postmenopausal hormone therapy with estrogen plus progestin) (25) or reducing cancer [e.g., oral contraceptives and ovarian cancer (34); aspirin and colon cancer (15, 93, 94)]. For combination estrogen plus progestin, the IARC has now classified this combination therapy as carcinogenic in humans (56), and estimates indicate that the reduction in use of hormones after the widespread publicity of the results of the Women’s Health Initiative (stopped early due to excess breast cancer) accounts for a ~10% decline in incidence among women 40–70 years of age (25). Thus, for this combination therapy, evidence shows that risk rises with duration of use and that, acting as a late promoter, removal of the drug leads to a rapid decline in incidence (25); however, among women with longer durations of use, risk may not return to that of women who have never used combination therapy (29). Unopposed estrogen, particularly when used from menopause, also significantly increases risk of breast cancer (8). Other less widespread drugs may also contribute to cancer risk (e.g., Diethylstilbestrol - DES), but the population impact will be substantially smaller than the examples based on much more widespread use described above.
Chemoprevention Represents the Use of Biologic Agents to Reduce Cancer Incidence and Mortality
Use of oral contraceptives (OCs) for five years halves a woman’s risk of ovarian cancer and substantially reduces risk of endometrial cancer (34). The protection is long lasting, and in high-income countries, rates of use approach 80%. Adverse effects are largely limited to increased risk of breast cancer and stroke while women are currently using OCs. Because these side effects are strongly age-dependent, use of OCs during late teens and early twenties could be widened for greater reduction in ovarian and endometrial cancers and overall net health benefit (59) and be an integral component of cost-saving preventive health services.
Aspirin has been extensively studied in observational epidemiologic settings that address duration of use, dose, and magnitude of risk reduction. The observational evidence is consistent with evidence from randomized primary prevention trials showing that intake of at least 300 mg of aspirin per day for at least 5 years is effective in preventing colon cancer, reducing risk by ~25% (47). A latency of ~10 years is observed. Five years of use reduced the 20-year risk of colon cancer by 25%, and benefit was seen for 75mg per day as well as higher doses (94). Combined analysis of individual patient data from 8 randomized controlled trials with more than 25,000 participants and 674 cancer deaths showed an overall reduction in cancer mortality by 20% at 5 years and greater reductions with longer use of aspirin. Latency of 5 years was observed for esophageal, pancreatic, and lung cancer mortality with longer latency for colorectal, stomach, and prostate cancers (93). Like all chemoprevention strategies, risks and benefits must be balanced (51). To date, the risk-benefit considerations of cardiovascular disease, bleeding complications, stomach pain, and heart burn have precluded recommendations for aspirin use as a widespread cancer prevention strategy (52).
Selective estrogen receptor modulators (SERMs) such as tamoxifen and raloxifene have been shown in randomized controlled prevention trials to reduce risk of preinvasive and invasive breast cancer (45, 71). Whereas tamoxifen increases risk of uterine cancer, raloxifene does not, and the risk profile for raloxifene looks considerably safer (19). On this basis, we have estimated the potential for risk reduction among women over age 50 who are postmenopausal. Our estimates indicate that if a trade-off of excess adverse events versus cases of breast cancer prevented must be less than 1, then ~30% of the 27 million women between ages 50 and 69 in the United States have benefits exceeding risks and would achieve a 50% reduction in the burden of breast cancer by taking a SERM. This figure represents a population benefit of 42,900 fewer cases of invasive breast cancer among the more than 7 million women with sufficiently high risk to justify chemoprevention (19). The reduction in risk observed in the chemoprevention randomized trials is rapid; within two years of patients beginning therapy, incidence curves have clearly separated. This result is consistent with the pharmacologic action of the agents inhibiting estrogen receptors. These agents show protection against estrogen receptor–positive breast cancers (risk reduction up to 76%) and no protection against receptor-negative cancers (71). Although models to classify risk of breast cancer have been developed and validated, to date prediction of receptor-positive tumors is no more accurate than prediction of risk overall (30). Refining risk stratification and developing tools to aid women in considering trade-offs of risks and benefits of chemoprevention therapy are necessary next steps to widespread use of these promising strategies for women at elevated risk of breast cancer (101).
Although infection (8%), medication use (5%), and reproductive factors (5%) operate through direct biologic actions accounting for 18% of preventable cancer, the potential is much greater for prevention through elimination of some drugs and increase in use of others. Likewise, the role of infection varies greatly around the world.
Physical Environment
The physical or biophysical environment includes the physical and biologic factors along with their chemical interactions that affect an organism. A component of the physical environment is the built environment, the constructed surroundings that provide the setting for human activity. As the American population has moved from predominantly rural population in 1900 (60.4%) to a small minority remaining rural in 2010 (20.78%), the built environment, transportation, and urban structure have modified our energy expenditure in occupational settings, commuting, and recreation. As noted above, this change over more than 100 years also alters access to fresh fruit and vegetables and modern postindustrial foods, contributing to energy imbalance, weight gain, and obesity (78). The physical environment also includes chemical agents introduced with industrialization, in occupations, and as contaminants of our environment. Solar radiation, another carcinogen, also has varied with changing cultural norms around tanning, surfing, and outdoor recreational activities.
Environmental and occupational exposures account for 1–4% of cancers (9). Occupational exposures such as asbestos, arsenic in drinking water, food contaminants such as aflatoxins and pesticides, and radiation exposure are classified as environmental carcinogens; however, in countries with established market economies, exposure is now largely limited by regulation to reduce harm. International agencies have responded by identifying carcinogens (e.g., IARC classification of carcinogenic compounds) and regulating use, exposure, and protection for employees in the case of occupational hazards.
The WHO has identified legislative enforcement of identification and elimination/reduction approaches, government-driven dissemination of information and awareness-raising activities, and increased access to information as effective strategies to combat carcinogenic environmental exposures (111). An end result is that, in some cases, production has been exported to countries with more lenient requirements for environmental exposure and contaminants, thereby not eliminating, but shifting, the cancer risk from an international scope. Despite regulatory changes in many countries, exposure to asbestos, for example, continues through occupations such as construction, ship work, and asbestos mining. Given the long lag between exposure and lung and pleural cancers, mortality from asbestos-related disease is estimated to remain at 90,000 per year (110). Successful enforcement of approaches to reduce exposure to known carcinogens in both the work place and the home is necessary to achieve successful cancer prevention.
Together, these components and consequences of the physical environment account for at least 26% of preventable cancers (obesity accounts for 15% or more of cancer cases, physical inactivity accounts for 5% of cases, radiation and sun exposures account for 2%, and occupation and environment account for 4%). Figure 1 summarizes the contributions.
Figure 1.

Biological, social, and physical environmental causes of cancer.
Integrated Summary
Tobacco use is the dominant cause of cancer and shows ease of measurement in level of exposure, timing of exposure, duration of exposure, and time of cessation. The addictive component adds to ease of epidemiologic assessment. Other major causes of cancer and preventive strategies are not so easily quantified. Furthermore, when imprecise measures are added for neighborhood education and socioeconomic status, the relation to cancer risk may appear to be mediated by lifestyle factors such as healthy diet (62).
If we integrate these social, biologic, and physical environmental factors in disease models, such as that developed by Wei et al. (103), we see that the combined effect on colon cancer in U.S. adults is substantial. For example, combining data on family history of colon cancer, age, height, body mass index, past and current use of postmenopausal hormones, red meat consumption, folate intake, physical activity, smoking (pack years before age 30), aspirin use, and screening history, a high-risk profile (defined by smoking 10 pack-years before age 30, consistently high relative body weight, physical activity of 2 MET-hours per week, 1 serving of red or processed meat per day, never having been screened, and folate intake of 150 μg per day) gave a relative risk of 3.8 [95%, CI (confidence interval) 1.61–9.16] compared with a low-risk profile (nonsmoker, consistently lean, physical activity of 21 MET-hours/week, no red or processed meat consumption, never having been screened, and folate intake of 400 μg/day). Although screening significantly reduces risk of colon cancer, the modifiable factors persist as important even after screening. See Figure 2.
Figure 2.
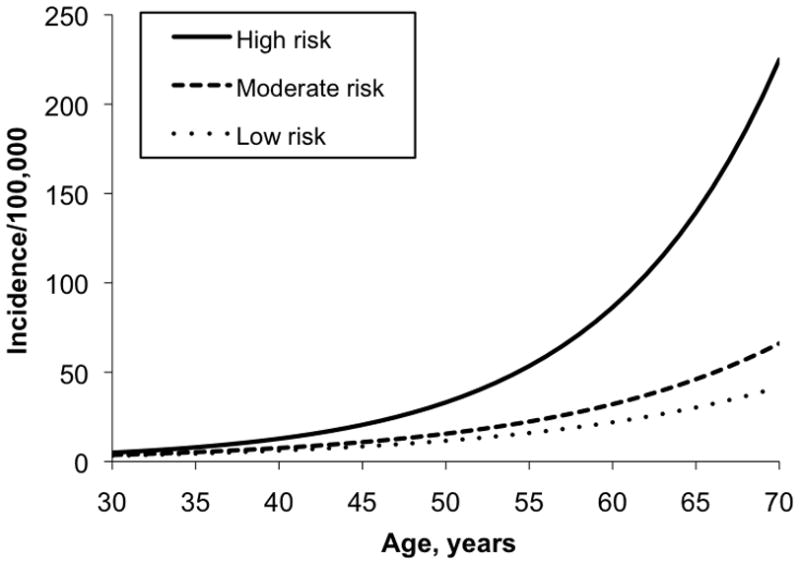
Age-specific incidence per 100,000 of colon cancer comparing high-, moderate, and low-risk women within the Nurses’ Health Study cohort.
A parallel estimate by Edwards et al. (43) evaluates contributors to the 26% decline on colorectal cancer mortality from 1975 through 2000. That analysis suggests lifestyle changes account for more than one-third of the reduction in mortality, screening accounts for half the reduction, and treatment accounts of just over 10% of the overall decline in mortality. See Figure 3.
Figure 3.
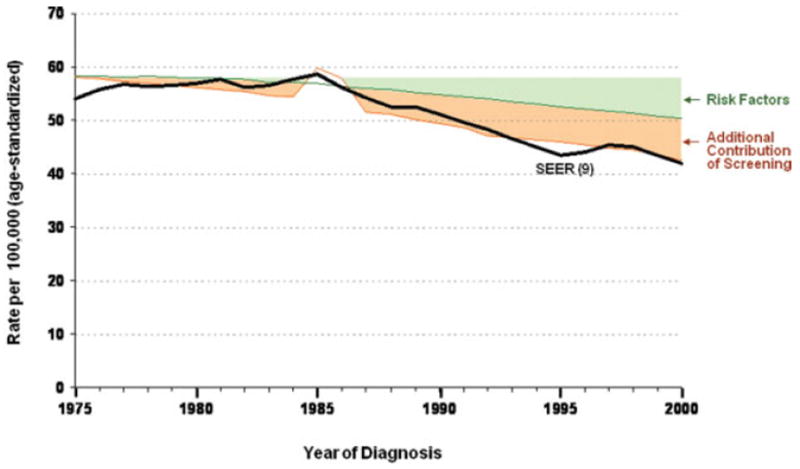
Decline in colorectal cancer incidence partitioned between changing risk factors and screening. From Edwards et al. 2010 (43).
Even these summary measures underestimate possible time-related exposures that integrate across the life course. Recent advances in genomic sciences allowed Yachida and colleagues (113) to estimate genomic evolution of pancreatic cancer. They estimate the carcinogenesis spans some 18–25 years. Tumorgenesis begins with an initiating mutation transforming a normal epithelium cell to have growth advantage. After successive clonal expansion over some 12 or so years, the development of pancreatic intraepithelial neoplasia is assumed to have occurred, which is followed by a parental clone expanding as an infiltrating carcinoma (some 7 years later) and eventual metastatic spread (113). Because diagnosis usually occurs after metastatic spread, preventive inquiry for pancreatic cancer should focus on exposures 20 or more years before diagnosis. Such exposures may inhibit or promote the progression to cancer. Alas, the vast majority of epidemiologic data has focused on lifestyle only months or up to two years before diagnosis, likely of no relevance to the etiology of this malignancy. We set forth in Figure 4 a summary of the time line for several major cancers to refine further the focus on timing of exposure for cancer prevention.
Figure 4.
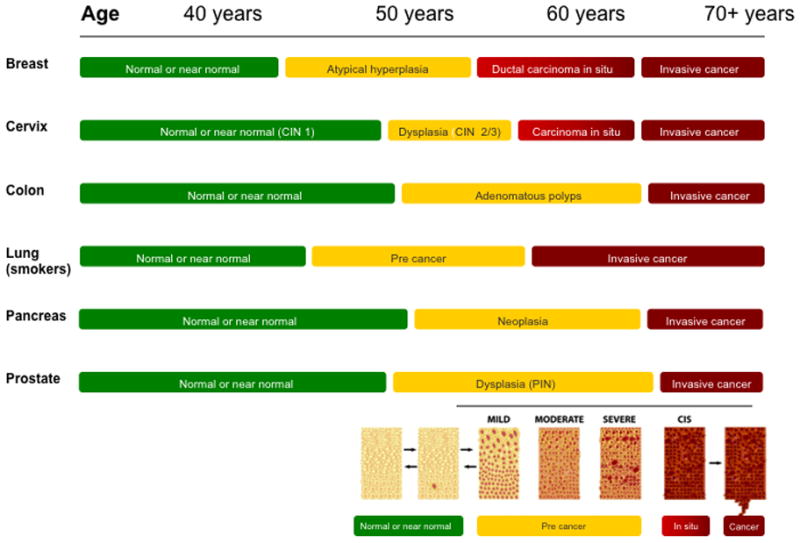
Average time line for normal tissue to invasive cancer.
In this context, one important omission from estimates of the proportion of cancer that can be prevented is a detailed understanding of the time course of risk reduction for many of the behaviors that cause cancer as well as consideration of achievable or sustainable change in exposure to the causes. Except in rare settings, such as cessation from smoking cigarettes, the time course to achieve reduction in the cancer burden is not quantified. Cigarette smoking does, however, offer strong evidence on the change in cancer risk after stopping smoking at the individual level (84). Additional evidence on the importance of timing of exposure is reported by Lee et al. (65), who studied both colon polyps (precursor lesions) and colon cancer in the Health Professionals Follow-Up Study. They noted that total folate intake 12–16 years before diagnosis was associated with a significant 30% reduced risk of colorectal cancer comparing ≥800 μg per day compared with <250 μg per day, but recent intake was not related to cancer risk. On the other hand, long- and short-term intakes of folate were associated with reduced risk of colorectal adenoma. Finally, the relationship between body size and cancer risk is another example of the complex relationship between the time course of exposure and disease that is missing from causal models but is captured in international variation. Although adult obesity is associated with an increased risk of a wide range of cancers (107), emerging evidence supports an even earlier window for prevention based on body size during childhood, adolescence, and even birth. For breast cancer, higher body fatness at ages 5, 10, and 20 was associated with a reduced risk of both pre- and postmenopausal breast cancer, independent of adult body size. The association was also stronger among those with lower birth weights (<8.5 lbs versus >8.5 lbs) (5). For colon cancer, evidence supports an inverse U-shaped association between birth weight and later adult risk of colon cancer (102). In addition, body size at ages 5 and 10 have been associated with the development of colorectal adenoma, the precursor to colon cancer (80). Body size [measured as body mass index (BMI)] at age 18 has also been associated with risk of colon cancer, independent of current BMI. The potential effect of interventions that target early-life obesity on later risk of cancer needs to be quantified; our ability to intervene successfully and prevent cancer depends on a complete understanding of these windows of risk because they represent multiple prevention opportunities. Thus correct specification of timing of exposure is imperative to quantify the potential for prevention.
The impact of change in level of exposure to other causes of cancer, or following the implementation of prevention strategies, is less well supported by rigorous evaluation. Several important aspects of the time course of prevention interventions must be considered. These are (a) the ability of interventions to change the exposure sufficiently, (b) the timing in the process of carcinogenesis, or the development of cancer, and (c) how sustained the behavior change is over time (114). This last issue, in particular, plagues randomized trials of prevention strategies, which may be more accurately assessed through observational studies (33).
In sum, the time course to achieve reduction in cancer incidence through active prevention programs may vary substantially. The timing of the intervention in the time course of carcinogenesis and the ability of individuals or populations to maintain the lifestyle changes necessary to reduce the cancer burden both contribute to the ultimate benefit of the active prevention intervention. One population that shows how much reduction can be achieved through long-term adherence to a cancer-reducing lifestyle is the members of the Seventh Day Adventist church in the United States. This population avoids smoking, alcohol, and consumption of meat, being largely lacto-ovo-vegetarian, and shows an overall 27% lower cancer mortality among men than the U.S. population at large (73). Reductions in cancer mortality among women were lesser, in part because of the burden of breast and other reproductive cancers that may not respond to changes in diet and smoking.
To summarize the preventability of cancer, we return to total cancer mortality due to the four cancers that cause 50% of all deaths. As noted in Table 2, since 1997, total cancer mortality has been declining significantly at 1.4–1.6% of mortality per year, reduction being greater in men than in women. These trends continue the decline that began in 1991.
Lung cancer is largely preventable through avoidance of smoking. Mortality is now declining in the United States overall and more clearly at younger ages. Lung cancer mortality in states such as Utah with a low prevalence of current cigarette smoking, 9.8% overall by 2009, can be compared with rates of up to 25% in states such as Kentucky and Oklahoma (14a). On the basis of the National Program of Cancer Registries, the Centers for Disease Control and Prevention (CDC) estimate that the corresponding lung cancer mortality varied from 26.4 per 100,000 in Utah to 97.7 deaths per 100,000 in Kentucky, a rate 73% lower when almost 10% of the population continues to smoke cigarettes (79). Thus the estimate that 90% or more of lung cancer cases can be prevented through elimination of cigarette smoking is achievable in the United States.
Colorectal cancer morality is declining largely owing to improved adherence to screening recommendations and improved lifestyle patterns (43). This result mixes medical interventions for prevention (screening) (3) and interventions (treatment after diagnosis to cancer reduce mortality).
Breast cancer incidence has recently declined owing to changes in patterns of postmenopausal hormone therapy. Mortality has a longer-term decline reflecting earlier diagnosis through screening and broader access to effective therapy. The potential for weight loss, increase in physical activity, and chemoprevention remains largely underutilized for prevention.
Prostate cancer mortality has largely been distorted by the advent of screening and changing surgical practices. Evidence for social and biologic strategies for prevention remains inconclusive.
LIMITATIONS AND FUTURE OPPORTUNITIES
A major gap in understanding the potential for prevention has been the absence of focus on time course of exposure to disease. Recent evidence from genomic analysis of pancreatic cancer suggests that risk or DNA damage accumulates over an average of 20+ years (70). Thus individual-level data from epidemiologic studies and many of the prevention trials conducted to date are likely to have data on a time frame that is not directly relevant to prevention (though it may approximate inhibition of very-late-stage promoters as seen for breast cancer). Most epidemiological data and almost all randomized trial data do not account for the multilevel nature of interactions among biologic pathways, social and physical environmental stressors, institutional and social context, and conditions that together have substantial impact on chronic disease risk (100). Future research to refine the understanding of the causes of cancer across these multiple levels will aid in refining prevention strategies.
SUMMARY POINTS.
Scientific evidene continues to mount indicating that cancer is preventable
Estimates of preventability often underestimate contributions of biologic, social, and environmental determinants to the cancer burden
The interplay of causes across biologic, social, and environemental levels is poorly understood and lack of knowledge hampers prevention strategies
Refined understnasing of causes across multiple levels can help inform strategies to prevent cancer
Acknowledgments
Supported by Sissy Hornig Clinical Resarch Professorship from the American Cacner Society and the Barnes-Jewish Hopsital Foundation, St Louis, MO. The authors acknowledge the technical assistance of Hank Dart, SM.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Contributor Information
Graham A. Colditz, Email: colditzg@wustl.edu.
Esther K. Wei, Email: weie@cpmcri.org.
LITERATURE CITED
- 1.Deleted in proof
- 2.Armitage P, Doll R. The age distribution of cancer and a multistage theory of carcinogenesis. Br J Cancer. 1954;8:1–12. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atkin WS, Edwards R, Kralj-Hans I, Wooldrage K, Hart AR, et al. Once-only flexible sigmoidoscopy screening in prevention of colorectal cancer: a multicentre randomised controlled trial. Lancet. 2010;375:1624–33. doi: 10.1016/S0140-6736(10)60551-X. [DOI] [PubMed] [Google Scholar]
- 4.Atwood K, Colditz GA, Kawachi I. From public health science to prevention policy: placing science in its social and political contexts. Am J Public Health. 1997;87:1603–6. doi: 10.2105/ajph.87.10.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baer HJ, Tworoger SS, Hankinson SE, Willett WC. Body fatness at young ages and risk of breast cancer throughout life. Am J Epidemiol. 2010;171:1183–94. doi: 10.1093/aje/kwq045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailar JC, Gornick HL. Cancer undefeated. N Engl J Med. 1997;336:1569–74. doi: 10.1056/NEJM199705293362206. [DOI] [PubMed] [Google Scholar]
- 7.Baron JA, Beach M, Mandel JS, van Stolk RU, Haile RW, et al. Calcium supplements for the prevention of colorectal adenomas. Calcium Polyp Prevention Study Group. N Engl J Med. 1999;340:101–7. doi: 10.1056/NEJM199901143400204. [DOI] [PubMed] [Google Scholar]
- 8.Beral V, Reeves G, Bull D, Green J. Breast cancer risk in relation to the interval between menopause and starting hormone therapy. J Natl Cancer Inst. 2011;103:296–305. doi: 10.1093/jnci/djq527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boffetta P. Epidemiology of environmental and occupational cancer. Oncogene. 2004;23:6392–403. doi: 10.1038/sj.onc.1207715. [DOI] [PubMed] [Google Scholar]
- 10.Boffetta P, McLaughlin JK, La Vecchia C. Reply: ‘environment’ in cancer causation and etiological fraction: limitations and ambiguities (by Boffetta, P. et al. 2007 Carcinogenesis 28:913–15) Carcinogenesis. 2008;29:1850. doi: 10.1093/carcin/bgm034. [DOI] [PubMed] [Google Scholar]
- 11.Boffetta P, McLaughlin JK, La Vecchia C, Autier P, Boyle P. ‘Environment’ in cancer causation and etiological fraction: limitations and ambiguities. Carcinogenesis. 2007;28:913–15. doi: 10.1093/carcin/bgm034. [DOI] [PubMed] [Google Scholar]
- 12.Brown CC, Chu KC. Use of multistage models to infer stage affected by carcinogenic exposure: example of lung cancer and cigarette smoking. J Chronic Dis. 1987;40(Suppl 2):S171–79. doi: 10.1016/s0021-9681(87)80020-6. [DOI] [PubMed] [Google Scholar]
- 13.Byers T, Mouchawar J, Marks J, Cady B, Lins N, et al. The American Cancer Society challenge goals. How far can cancer rates decline in the U.S by the year 2015? Cancer. 1999;86:715–27. [PubMed] [Google Scholar]
- 14.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–38. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 14a.Cent. Dis. Control Prev. (CDC) State-specific prevalence of cigarette smoking and smokeless tobacco use among adults—United States, 2009. MMWR. 2010;59:1400–6. [PubMed] [Google Scholar]
- 15.Chan AT, Giovannucci EL, Meyerhardt JA, Schernhammer ES, Curhan GC, Fuchs CS. Long-term use of aspirin and nonsteroidal anti-inflammatory drugs and risk of colorectal cancer. JAMA. 2005;294:914–23. doi: 10.1001/jama.294.8.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang MH, Chen CJ, Lai MS, Hsu HM, Wu TC, et al. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N Engl J Med. 1997;336:1855–59. doi: 10.1056/NEJM199706263362602. [DOI] [PubMed] [Google Scholar]
- 17.Chang MH, Chen TH, Hsu HM, Wu TC, Kong MS, et al. Prevention of hepatocellular carcinoma by universal vaccination against hepatitis B virus: the effect and problems. Clin Cancer Res. 2005;11:7953–57. doi: 10.1158/1078-0432.CCR-05-1095. [DOI] [PubMed] [Google Scholar]
- 18.Chang MH, You SL, Chen CJ, Liu CJ, Lee CM, et al. Decreased incidence of hepatocellular carcinoma in hepatitis B vaccinees: a 20-year follow-up study. J Natl Cancer Inst. 2009;101:1348–55. doi: 10.1093/jnci/djp288. [DOI] [PubMed] [Google Scholar]
- 19.Chen WY, Rosner B, Colditz GA. Moving forward with breast cancer prevention. Cancer. 2007;109:2387–91. doi: 10.1002/cncr.22711. [DOI] [PubMed] [Google Scholar]
- 20.Chlebowski RT, Kuller LH, Prentice RL, Stefanick ML, Manson JE, et al. Breast cancer after use of estrogen plus progestin in postmenopausal women. N Engl J Med. 2009;360:573–87. doi: 10.1056/NEJMoa0807684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho E, Rosner BA, Feskanich D, Colditz GA. Risk factors and individual probabilities of melanoma for whites. J Clin Oncol. 2005;23:2669–75. doi: 10.1200/JCO.2005.11.108. [DOI] [PubMed] [Google Scholar]
- 22.Cho E, Smith-Warner SA, Spiegelman D, Beeson WL, van den Brandt PA, et al. Dairy foods, calcium, and colorectal cancer: a pooled analysis of 10 cohort studies. J Natl Cancer Inst. 2004;96:1015–22. doi: 10.1093/jnci/djh185. [DOI] [PubMed] [Google Scholar]
- 23.Clarke CA, Glaser SL, Uratsu CS, Selby JV, Kushi LH, Herrinton LJ. Recent declines in hormone therapy utilization and breast cancer incidence: clinical and population-based evidence. J Clin Oncol. 2006;24:e49–50. doi: 10.1200/JCO.2006.08.6504. [DOI] [PubMed] [Google Scholar]
- 24.Colditz GA. Cohort studies of etiology and survival after cancer: the unique needs for uninterrupted funding. Cancer Causes Control. 2007;18:235–41. doi: 10.1007/s10552-007-0114-2. [DOI] [PubMed] [Google Scholar]
- 25.Colditz GA. Decline in breast cancer incidence due to removal of promoter: combination estrogen plus progestin. Breast Cancer Res. 2007;9:108. doi: 10.1186/bcr1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colditz GA. Ensuring long-term sustainability of existing cohorts remains the highest priority to inform cancer prevention and control. Cancer Causes Control. 2010;21:649–56. doi: 10.1007/s10552-009-9498-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colditz GA, DeJong D, Hunter DJ, Trichopoulos D, Willett WC, editors. Harvard Report on Cancer Prevention. Volume 1: Causes of Human Cancer Cancer Causes Control. 1996;7(Suppl):1–59. [Google Scholar]
- 28.Colditz GA, Rosner B. Cumulative risk of breast cancer to age 70 years according to risk factor status: data from the Nurses’ Health Study. Am J Epidemiol. 2000;152:950–64. doi: 10.1093/aje/152.10.950. [DOI] [PubMed] [Google Scholar]
- 29.
- 30.Colditz GA, Rosner BA, Chen WY, Holmes MD, Hankinson SE. Risk factors for breast cancer according to estrogen and progesterone receptor status. J Natl Cancer Inst. 2004;96:218–28. doi: 10.1093/jnci/djh025. [DOI] [PubMed] [Google Scholar]
- 31.Colditz GA, Rosner BA, Speizer FE. Risk factors for breast cancer according to family history of breast cancer. J Natl Cancer Inst. 1996;88:365–71. doi: 10.1093/jnci/88.6.365. [DOI] [PubMed] [Google Scholar]
- 32.Colditz GA, Sellers TA, Trapido E. Epidemiology—identifying the causes and preventability of cancer? Nat Rev Cancer. 2006;6:75–83. doi: 10.1038/nrc1784. [DOI] [PubMed] [Google Scholar]
- 33.Colditz GA, Taylor PR. Prevention trials: their place in how we understand the value of prevention strategies. Annu Rev Public Health. 2010;31:105–20. doi: 10.1146/annurev.publhealth.121208.131051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beral V, Doll R, Hermon C, Peto R, Reeves G Collab. Group Epidemiol. Stud. Ovarian Cancer. Ovarian cancer and oral contraceptives: collaborative reanalysis of data from 45 epidemiological studies including 23,257 women with ovarian cancer and 87,303 controls. Lancet. 2008;371:303–14. doi: 10.1016/S0140-6736(08)60167-1. [DOI] [PubMed] [Google Scholar]
- 35.Collab. Group Horm. Factors Breast Cancer. Breast cancer and hormone replacement therapy. Collaborative reanalysis of data from 51 epidemiological studies involving 52,705 women with breast cancer and 108,411 women without breast cancer. Lancet. 1997;350:1047–59. [PubMed] [Google Scholar]
- 36.Cuppone F, Bria E, Giannarelli D, Vaccaro V, Milella M, et al. Impact of hormonal treatment duration in combination with radiotherapy for locally advanced prostate cancer: meta-analysis of randomized trials. BMC Cancer. 2010;10:675. doi: 10.1186/1471-2407-10-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Danaei G, Vander Hoorn S, Lopez AD, Murray CJ, Ezzati M. Causes of cancer in the world: comparative risk assessment of nine behavioural and environmental risk factors. Lancet. 2005;366:1784–93. doi: 10.1016/S0140-6736(05)67725-2. [DOI] [PubMed] [Google Scholar]
- 38.Day NE. The Armitage-Doll multistage model of carcinogenesis. Stat Med. 1990;9:677–79. doi: 10.1002/sim.4780090614. [DOI] [PubMed] [Google Scholar]
- 39.Doll R, Peto R. Cigarette smoking and bronchial carcinoma: dose and time relationships among regular smokers and lifelong non-smokers. J Epidemiol Community Health. 1978;32:303–13. doi: 10.1136/jech.32.4.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.
- 41.Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Inst. 1981;66:1191–308. [PubMed] [Google Scholar]
- 42.Durand CP, Andalib M, Dunton GF, Wolch J, Pentz MA. A systematic review of built environment factors related to physical activity and obesity risk: implications for smart growth urban planning. Obes Rev Off J Int Assoc Study Obes. 2011;12:e173–82. doi: 10.1111/j.1467-789X.2010.00826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edwards BK, Ward E, Kohler BA, Eheman C, Zauber AG, et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116:544–73. doi: 10.1002/cncr.24760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Epstein S, Swartz J. Fallacies of lifestyle cancer theories. Nature. 1981;289:127–30. doi: 10.1038/289127a0. [DOI] [PubMed] [Google Scholar]
- 45.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90:1371–88. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 46.Fisher JC, Hollomon JH. A hypothesis for the origin of cancer foci. Cancer. 1951;4:916–18. doi: 10.1002/1097-0142(195109)4:5<916::aid-cncr2820040504>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 47.Flossmann E, Rothwell PM. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet. 2007;369:1603–13. doi: 10.1016/S0140-6736(07)60747-8. [DOI] [PubMed] [Google Scholar]
- 48.Frazier AL, Colditz GA, Fuchs CS, Kuntz KM. Cost-effectiveness of screening for colorectal cancer in the general population. JAMA. 2000;284:1954–61. doi: 10.1001/jama.284.15.1954. [DOI] [PubMed] [Google Scholar]
- 49.Freedman AN, Seminara D, Gail MH, Hartge P, Colditz GA, et al. Cancer risk prediction models: a workshop on development, evaluation, and application. J Natl Cancer Inst. 2005;97:715–23. doi: 10.1093/jnci/dji128. [DOI] [PubMed] [Google Scholar]
- 50.Gehlert S, Sohmer D, Sacks T, Mininger C, McClintock M, Olopade O. Targeting health disparities: a model linking upstream determinants to downstream interventions. Health Aff. 2008;27:339–49. doi: 10.1377/hlthaff.27.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glasziou PP, Irwig LM. An evidence based approach to individualising treatment. BMJ. 1995;311:1356–59. doi: 10.1136/bmj.311.7016.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gralow J, Ozols RF, Bajorin DF, Cheson BD, Sandler HM, et al. Clinical cancer advances 2007: major research advances in cancer treatment, prevention, and screening—a report from the American Society of Clinical Oncology. J Clin Oncol. 2008;26:313–25. doi: 10.1200/JCO.2007.15.4088. [DOI] [PubMed] [Google Scholar]
- 53.Haber DA, Gray NS, Baselga J. The evolving war on cancer. Cell. 2011;145:19–24. doi: 10.1016/j.cell.2011.03.026. [DOI] [PubMed] [Google Scholar]
- 54.Hazelton WD, Clements MS, Moolgavkar SH. Multistage carcinogenesis and lung cancer mortality in three cohorts. Cancer Epidemiol Biomark Prev. 2005;14:1171–81. doi: 10.1158/1055-9965.EPI-04-0756. [DOI] [PubMed] [Google Scholar]
- 55.Int. Agency Res. Cancer (IARC) Weight Control and Physical Activity. Lyon: IARC; 2002. p. 315. [Google Scholar]
- 56.Int. Agency Res. Cancer (IARC) Combined estrogen-progestogen contraceptives and combined estrogen-progestogen menopausal therapy. IARC Monogr Eval Carcinog Risks Hum. 2007;91:1–528. [PMC free article] [PubMed] [Google Scholar]
- 57.Int. Agency Res. Cancer (IARC) World Cancer Report 2008. Lyon: IARC; 2008. [Google Scholar]
- 58.Kaldor J, Day N. Mathematical models in cancer epidemiology. In: Schottenfeld D, Fraumeni J, editors. Cancer Epidemiology. New York: Oxford Univ. Press; 1996. pp. 127–37. [Google Scholar]
- 59.Kawachi I, Colditz GA, Hankinson S. Long-term benefits and risks of alternative methods of fertility control in the United States. Contraception. 1994;50:1–16. doi: 10.1016/0010-7824(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 60.Kenfield SA, Stampfer MJ, Rosner BA, Colditz GA. Smoking and smoking cessation in relation to mortality in women. JAMA. 2008;299:2037–47. doi: 10.1001/jama.299.17.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kerlikowske K, Miglioretti DL, Buist DS, Walker R, Carney PA. Declines in invasive breast cancer and use of postmenopausal hormone therapy in a screening mammography population. J Natl Cancer Inst. 2007;99:1335–39. doi: 10.1093/jnci/djm111. [DOI] [PubMed] [Google Scholar]
- 62.Kim D, Masyn KE, Kawachi I, Laden F, Colditz GA. Neighborhood socioeconomic status and behavioral pathways to risks of colon and rectal cancer in women. Cancer. 2010;116:4187–96. doi: 10.1002/cncr.25195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–23. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koutsky LA, Ault KA, Wheeler CM, Brown DR, Barr E, et al. A controlled trial of a human papillomavirus type 16 vaccine. N Engl J Med. 2002;347:1645–51. doi: 10.1056/NEJMoa020586. [DOI] [PubMed] [Google Scholar]
- 65.Lee JE, Willett WC, Fuchs CS, Smith-Warner SA, Wu K, et al. Folate intake and risk of colorectal cancer and adenoma: modification by time. Am J Clin Nutr. 2011;93:817–25. doi: 10.3945/ajcn.110.007781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leung GM, Thach TQ, Lam TH, Hedley AJ, Foo W, et al. Trends in breast cancer incidence in Hong Kong between 1973 and 1999: an age-period-cohort analysis. Br J Cancer. 2002;87:982–88. doi: 10.1038/sj.bjc.6600583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Linos E, Spanos D, Rosner BA, Linos K, Hesketh T, et al. Effects of reproductive and demographic changes on breast cancer incidence in China: a modeling analysis. J Natl Cancer Inst. 2008;100:1352–60. doi: 10.1093/jnci/djn305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Linos E, Spanos D, Rosner BA, Linos K, Hesketh T, et al. Effects of reproductive and demographic changes on breast cancer incidence in China: a modeling analysis. J Natl Cancer Inst. 2008;100:1339–41. doi: 10.1093/jnci/djn305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Little M, Hawkins M, Charles M, Hildreth N. Fitting the Armitage-Doll model to radiation-exposed cohorts and implications for population cancer risks. Radiat Res. 1992;132:207–21. [PubMed] [Google Scholar]
- 70.Luebeck EG. Cancer: genomic evolution of metastasis. Nature. 2010;467:1053–55. doi: 10.1038/4671053a. [DOI] [PubMed] [Google Scholar]
- 71.Martino S, Cauley JA, Barrett-Connor E, Powles TJ, Mershon J, et al. Continuing outcomes relevant to Evista: breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J Natl Cancer Inst. 2004;96:1751–61. doi: 10.1093/jnci/djh319. [DOI] [PubMed] [Google Scholar]
- 72.Meza R, Jeon J, Moolgavkar SH, Luebeck EG. Age-specific incidence of cancer: phases, transitions, and biological implications. Proc Natl Acad Sci USA. 2008;105:16284–89. doi: 10.1073/pnas.0801151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72a.Miettinen OS. Proportion of disease caused and prevented by a given exposure, trait, or intervention. Am J Epidemiol. 1974;99:325–33. doi: 10.1093/oxfordjournals.aje.a121617. [DOI] [PubMed] [Google Scholar]
- 73.Mills PK, Beeson WL, Phillips RL, Fraser GE. Cancer incidence among California Seventh-Day Adventists, 1976–1982. Am J Clin Nutr. 1994;59:S1136–42. doi: 10.1093/ajcn/59.5.1136S. [DOI] [PubMed] [Google Scholar]
- 74.Monson RR, Christiani DC. Summary of the evidence: occupation and environment and cancer. Cancer Causes Control. 1997;8:529–31. doi: 10.1023/a:1018429926593. [DOI] [PubMed] [Google Scholar]
- 75.Montague M, Borland R, Sinclair C. Slip! Slop! Slap! and SunSmart, 1980–2000: skin cancer control and 20 years of population-based campaigning. Health Educ Behav. 2001;28:290–305. doi: 10.1177/109019810102800304. [DOI] [PubMed] [Google Scholar]
- 76.Moolgavkar SH. Cancer models. Epidemiology. 1990;1:419–20. doi: 10.1097/00001648-199011000-00002. [DOI] [PubMed] [Google Scholar]
- 77.Moolgavkar SH, Day NE, Stevens RG. Two-stage model for carcinogenesis: epidemiology of breast cancer in females. J Natl Cancer Inst. 1980;65:559–69. [PubMed] [Google Scholar]
- 78.Moore LV, Diez Roux AV. Associations of neighborhood characteristics with the location and type of food stores. Am J Public Health. 2006;96:325–31. doi: 10.2105/AJPH.2004.058040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Natl. Program Cancer Regist. United States Cancer Statistics. 2011 http://apps.nccd.cdc.gov/uscs/statevsnational.aspx.
- 80.Nimptsch K, Giovannucci EL, Willett WC, Fuchs C, Wei EK, Wu K. Body fatness during childhood and adolescence, adult height and risk of colorectal adenoma in women. Cancer Prev Res. 2011;4:1710–8. doi: 10.1158/1940-6207.CAPR-11-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nordling CO. A new theory on cancer-inducing mechanism. Br J Cancer. 1953;7:68–72. doi: 10.1038/bjc.1953.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–44. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 83.Peto R, Chen ZM, Boreham J. Tobacco—the growing epidemic. Nat Med. 1999;5:15–17. doi: 10.1038/4691. [DOI] [PubMed] [Google Scholar]
- 84.Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ. 2000;321:323–29. doi: 10.1136/bmj.321.7257.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Peto R, Lopez AD, Boreham J, Thun M, Heath C, Jr, Doll R. Mortality from smoking worldwide. Br Med Bull. 1996;52:12–21. doi: 10.1093/oxfordjournals.bmb.a011519. [DOI] [PubMed] [Google Scholar]
- 86.Pike MC, Krailo MD, Henderson BE, Casagrande JT, Hoel DG. “Hormonal” risk factors, “breast tissue age” and the age-incidence of breast cancer. Nature. 1983;303:767–70. doi: 10.1038/303767a0. [DOI] [PubMed] [Google Scholar]
- 87.Prev. Work. Group. Cancer control objectives for the nation: 1985–2000. NCI Monogr. 1986;2:3–11. [PubMed] [Google Scholar]
- 88.Ravdin PM, Cronin KA, Howlader N, Berg CD, Chlebowski RT, et al. The decrease in breast-cancer incidence in 2003 in the United States. N Engl J Med. 2007;356:1670–74. doi: 10.1056/NEJMsr070105. [DOI] [PubMed] [Google Scholar]
- 89.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–78. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 90.Rosner B, Colditz GA. Nurses’ health study: log-incidence mathematical model of breast cancer incidence. J Natl Cancer Inst. 1996;88:359–64. doi: 10.1093/jnci/88.6.359. [DOI] [PubMed] [Google Scholar]
- 91.Rosner B, Colditz GA, Willett WC. Reproductive risk factors in a prospective study of breast cancer: the Nurses’ Health Study. Am J Epidemiol. 1994;139:819–35. doi: 10.1093/oxfordjournals.aje.a117079. [DOI] [PubMed] [Google Scholar]
- 92.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321–33. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 93.Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011;377:31–41. doi: 10.1016/S0140-6736(10)62110-1. [DOI] [PubMed] [Google Scholar]
- 94.Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376:1741–50. doi: 10.1016/S0140-6736(10)61543-7. [DOI] [PubMed] [Google Scholar]
- 95.Salois MJ. Obesity and diabetes, the built environment, and the ‘local’ food economy in the United States, 2007. Econ Hum Biol. 2011 doi: 10.1016/j.ehb.2011.04.001. E pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 96.Tarlov AR, St Peter RF. Introduction. In: Tarlov AR, St Peter RF, editors. The Society and Population Health Reader: Volume II. A State and Community Perspective. New York: New Press; 2000. pp. x–xi. [Google Scholar]
- 97.U.S. Dep. Health Hum. Serv. The Health Consequences of Smoking: A Report of the Surgeon General. Atlanta: Cent. Dis. Control Prev; 2004. [PubMed] [Google Scholar]
- 98.Viswanath K, Herbst RS, Land SR, Leischow SJ, Shields PG. Tobacco and cancer: an American Association for Cancer Research policy statement. Cancer Res. 2010;70:3419–30. doi: 10.1158/0008-5472.CAN-10-1087. [DOI] [PubMed] [Google Scholar]
- 99.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–32. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 100.Warnecke RB, Oh A, Breen N, Gehlert S, Paskett E, et al. Approaching health disparities from a population perspective: the National Institutes of Health Centers for Population Health and Health Disparities. Am J Public Health. 2008;98:1608–15. doi: 10.2105/AJPH.2006.102525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Waters EA, Weinstein ND, Colditz GA, Emmons K. Explanations for side effect aversion in preventive medical treatment decisions. Health Psychol. 2009;28:201–9. doi: 10.1037/a0013608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wei E, Giovannucci E, Liu J, Fuchs C, Willett W, et al. A prospective study of birthweight, height and body mass index at age 18 and the risk of colon cancer in women. Cancer Epidemiol Biomark Prev. 2005;14:S2758–59. [Google Scholar]
- 103.Wei EK, Colditz GA, Giovannucci EL, Fuchs CS, Rosner BA. Cumulative risk of colon cancer up to age 70 years by risk factor status using data from the Nurses’ Health Study. Am J Epidemiol. 2009;170:863–72. doi: 10.1093/aje/kwp210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wei EK, Wolin KY, Colditz GA. Time course of risk factors in cancer etiology and progression. J Clin Oncol Off J Am Soc Clin Oncol. 2010;28:4052–57. doi: 10.1200/JCO.2009.26.9324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weil D. OSHA: Beyond the politics. Frontline (online) 2003 http://www.pbs.org/wgbh/pages/frontline/shows/workplace/osha/weil.html.
- 106.Willett WC, Colditz GA, Mueller NE. Strategies for minimizing cancer risk. Sci Am. 1996;275:88–95. doi: 10.1038/scientificamerican0996-88. [DOI] [PubMed] [Google Scholar]
- 107.Wolin KY, Carson K, Colditz GA. Obesity and cancer. Oncologist. 2010;15:556–65. doi: 10.1634/theoncologist.2009-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wolin KY, Colditz GA. Implementing chronic disease prevention amongst cancer survivors. J Intern Med. 2011;269:85–87. doi: 10.1111/j.1365-2796.2010.02295.x. [DOI] [PubMed] [Google Scholar]
- 109.World Cancer Res. Fund. Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective. Washington, DC: Am. Inst. Cancer Res; 2007. [Google Scholar]
- 110.World Health Organ. Elimination of Asbestos-related Disease. Geneva: WHO; 2006. [Google Scholar]
- 111.World Health Organ. Cancer Control: Knowledge into Action (Prevention Module) Geneva: WHO; 2007. [Google Scholar]
- 112.Xu Z, Brown LM, Pan GW, Liu TF, Gao GS, et al. Cancer risks among iron and steel workers in Anshan, China, Part II: case-control studies of lung and stomach cancer. Am J Ind Med. 1996;30:7–15. doi: 10.1002/(SICI)1097-0274(199607)30:1<7::AID-AJIM2>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 113.Yachida S, Jones S, Bozic I, Antal T, Leary R, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–17. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zelen M. Are primary cancer prevention trials feasible? J Natl Cancer Inst. 1988;80:1442–44. doi: 10.1093/jnci/80.18.1442. [DOI] [PubMed] [Google Scholar]