

Abstract
The Duchenne Muscular dystrophy (DMD) is the most frequent muscle disorder in childhood caused by mutations in the Xlinked dystrophin gene (about 65% deletions, about 7% duplications, about 26% point mutations and about 2% unknown mutations). The clinically milder Becker muscular dystrophy (BMD) is allelic to DMD. About 33% of all patients are due to de novo mutations and germ line mosaicism is frequently observed. While in earlier studies equal mutation rates in males and females had been reported, a breakdown by mutation types can better explain the sex ratio of mutations: Point mutations and duplications arise preferentially during spermatogenesis whereas deletions mostly arise in oogenesis.
With current analytical methods, the underlying mutation can be identified in the great majority of cases and be used for carrier detection. However, in families with no mutation carrier available, the genetic model to be used for counselling of relatives can be quite complex.
Key words: Duchenne muscular dystrophy, Becker muscular dystrophy, dystrophin gene, molecular genetic diagnosis, genetic model, germ line mosaicism
Duchenne muscular dystrophy (DMD, OMIM # 310200) is the most common muscle disorder in childhood and also the most frequent X-linked recessive disease. The incidence of DMD in male newborns has been assessed at 3 * 10-4 in several populations while the allelic more benign Becker muscular dystrophy (BMD) is rarer with an incidence of about 0.54 * 10-4 (1).
Mutations in the dystrophin gene (OMIM * 300377) are the genetic causes of DMD and BMD. The predominant mutation type are deletions encompassing one or more of the 79 exons (about 65%) (2). In a smaller group of patients, the molecular cause is due to duplications of one or more exons (about 7%) (3). Point mutations affecting only one or a few base pairs in the coding sequence or the splice consensus sites account for another 26% of all mutations. However, with routine diagnostic strategies focusing on the coding and splice sequences of the dystrophin gene, the causative mutation remains undetected in approximately 2% of all DMD / BMD patients (4).
Diagnostic strategy
In patients with clinically suspected DMD or BMD, the molecular genetic workup should be performed as a two-step procedure:
Step 1: MLPA (multiplex ligation-dependent probe amplification) has become the method-ofchoice for the detection of exon deletions and duplications in all 79 exons of the dystrophin gene (5) but alternative screening methods are available (6).
Step 2: When a deletion / duplication in the dystrophin gene has been excluded, sequencing of the coding regions and splice sites for the detection of point mutations should be performed.
The analytical sensitivity of MLPA alone is about 71.3% and for the full analysis (MLPA and sequencing) about 97.3%.
The detection of a deletion or duplication in the dystrophin gene confirms the diagnosis of DMD or BMD. If only one exon is missing or duplicated the mutation should be verified by a second independent method in order to exclude technical artifacts and rare sequence variants affecting proper probe binding.
The interpretation of point mutations is less straightforward. In DMD, the great majority of point mutations generates premature stop codons or affects splice consensus sequences. These mutations ablate proper protein expression and are compatible with the DMD phenotype. In a minority DMD cases, and more frequently in BMD, point mutations lead to the substitution of individual amino acids. In a large protein like dystrophin, the functional consequence of such mutations is more difficult to interpret even with modern prediction algorithms.
The identification of a causative mutation usually renders a muscle biopsy unnecessary for diagnosis. For very young patients whose clinical course (DMD or BMD) is not yet known, a prognostic interpretation of molecular results is often desired. The basis for the correlation of deletions in the dystrophin gene with the clinical course and severity is the "reading frame hypothesis" (7). Several retrospective studies (8) found that in more than 95% of DMD patients the deletion had induced a shift of the translational reading frame of the mRNA ("out-of-frame" or "frame shift" deletions). Conversely, in more than 95% of BMD patients, the original reading frame was not altered by the deletion ("in-frame" deletions). The most common exception from this rule is the deletion of exons 3-7 which is formally "out-of-frame" but leads to a milder phenotype due to alternative splicing of exon 8 (9). Very large "inframe" deletions removing important protein domains may also not follow the reading frame hypothesis. With duplications, predictions based on the reading frame hypothesis should be made with caution since the MLPA method does not allow to determine whether or not the gene regions are duplicated in a head-to-tail arrangement. The homepage of the Institute of Human Genetics of Leiden University provides software to determine the reading frame for every possible deletions and duplications in the dystrophin gene ("reading frame checker", www.dmd.nl).
Although the reading frame hypothesis is a useful tool for predicting disease course and severity, a muscle biopsy may be considered in special cases since a quantification of dystrophin protein shows an even better correlation with the severity of the disease.
Diagnosis of potentially heterozygous women (carrier testing)
The diagnosis of women who may be carriers of DMD or BMD has been a puzzling problem for decades. Heterozygous females are usually clinically normal. In rare cases (less than 5%) female carriers show clinical symptoms which can vary from very mild to clinically severe muscle disease. Therefore, in an adult female patient with mild muscle weakness a carrier status for DMD/ BMD should always be considered.
For mutation detection in potential carriers, the same methods can be applied as for index cases, i.e. MLPA for deletions and duplications and sequencing for point mutations. Wherever possible, it is sensible to identify the mutation in the index case or an obligatory carrier first. Otherwise, a negative genetic test result (i.e. no mutation found) is difficult to interpret. Considering that the analytical sensitivity of the molecular methods is not 100% (MLPA about 99%; sequencing approximately 93%) a significant residual risk will remain if the familial mutation cannot be identified (Table 1).
Table. 1.
Risk of the daughter being a carrier with negative test results.
| Familial mutation unknown | Risk of daughter being carrier |
|---|---|
| Mother obligatory carrier, deceased | 50% |
| Daughter: negative test result for MLPA | 26,5% |
| Daughter: negative test result for MLPA and sequencing | 3,0% |
The residual risk figures given in Table 1 are subject to change conditional on additional information as available (e.g., CK-levels, haplotypes). Bayesian logic can be used for risk assessment in order to incorporate all available information which is relevant for DMD carrier status.
The genetic model for risk assessment in DMD / BMD families with mutation unknown
Haldane was first to postulate a mutation-selection equilibrium in DMD (10). Since DMD boys do not pass on their defective allele to the following generation, in every generation one third of mutated alleles is 'lost' from the population. This should lead to a rapid drop in disease incidence. Neither at Haldane's times nor in the nearly three generations since has a decline in disease incidence been observed. Therefore, Haldane concluded that the selection of mutated alleles from the population must be compensated for by an equivalent rate (i.e. one third) of de novo mutations.
Assuming this mutation-selection equilibrium, Haldane elaborated a formula to express the proportion of female carriers and of male patients in the population as a multiple of the mutation rates (with "v" being the mutation rate in men and "u" in women; Table 2).
Table 2.
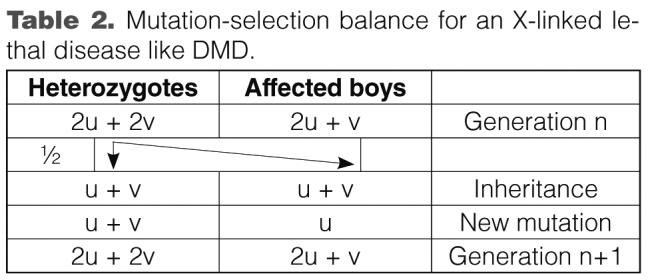
Mutation-selection balance for an X-linked lethal disease like DMD.
After cloning of the dystrophin gene in 1986 and with the advent of molecular mutation detection methods, it soon became clear that a peculiar type of de novo mutations is not infrequent in DMD / BMD: germinal mosaicism. This term was coined after several reports had been published of a second mutation carrier (male or female) who had been born to mothers not carrying the causative mutation of the index case in their somatic cells. Bakker et al. (11) estimated a recurrence risk of 14% for brothers of sporadic cases with the same haplotype when the mutation could not be detected in lymphocytes of the mother. Van Essen et al. (12) gave an estimate of 20% (95% CI 10% - 30%). Based on CK levels, Passos-Bueno et al. (13) concluded that mothers of isolated DMD cases have likelihoods of 62.3% to be carriers, of 6.7% to carry a germline mosaic and of 31% to bear two normal alleles. From the largest published study on this issue with some 2000 families, Barbujani et al. concluded that at least 10% of sporadic cases are due to mutations that arose in the early stages of germ cell development (14).
Thus, Haldane's model must be expanded to include the possibility of a germ cell mosaic (GLM) (15).
Taking into account a relative fitness (w) of 0.0 for DMD and 0.7 for BMD cases, an equilibrium mutation selection can be described as follows (Table 3):
Table 3.
Mutation-selection equilibrium for X-linked disease (eg DMD or BMD) in consideration of a germ cell mosaic (u = female mutation rate; v = male mutation rate; g = proportion of new mutations in mitosis; 1-g = proportion of new mutations in meiosis; f = segregation of the mutation in GLM; w = relative fitness).
| Genetic model for DMD (w = 0) and BMD (w = 0.7) | |||
| Heterozygotes | GLM in women | Affected males | GLM in males |
| [(2u + 2v + 2wu)(1-g+fg)] / [1-w] | 2 gu | [(2u + v)(1-g+fg)] / [1-w] | gv |
According to this genetic model, there are therefore three situations for the inheritance of a dystrophin mutation:
Women are female carriers because their mother is already a carrier (Fig. 1);
A de novo mutation has arisen in meiosis either in the grand-parental generation (in spermatogenesis of the grandfather or oogenesis of the grandmother; Fig. 2a), or in the mother (de novo mutation in oogenesis; Fig. 2b);
Mitotic de novo mutations with the consequence of germline mosaics can occur in the spermatogenesis of the grandfather (Fig. 3a), in the oogenesis of the grandmother (Fig. 3b) or in the oogenesis of the mother (Fig. 3c)
Figure 1.
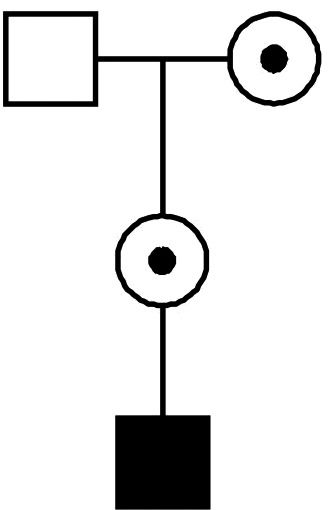
Mother of an affected boy is carrier because her mother is carrier.
Figure 2.
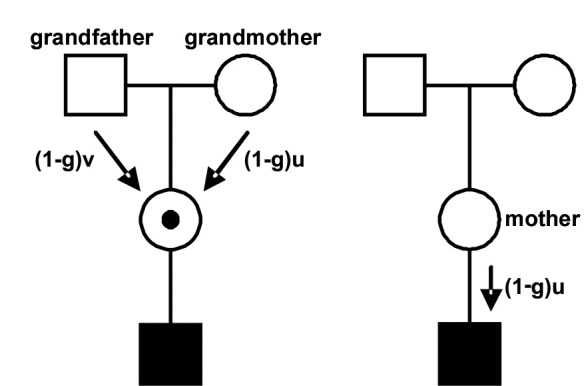
Left pedigree: mother of an affected boy is carrier as a result of a de novo mutation in one of her parents' gametes (1-g = proportion of de novo mutations occurring in the meiosis; u = female mutation rate; v = male mutation rate). Right pedigree: the affected boy is a de novo mutation.
Figure 3.

Left and middle pedigree: the mother of an affected boy is carrier because her father (a) of her mother (b) carried a germline mosaic (f = segregation in germline mosaics; g = probability of de novo mutations in the mitosis; u = female mutation rate; v = male mutation rate). Right pedigree: a mother of an affected boy carries a germline mosaic.
Best estimates for the parameters required for this genetic model are listed in Table 4.
Table 4.
Parameters for the DMD model.
| Incidence (DMD) | I = 0,000333 | (1) |
| Proportion of deletions | d = 0,650 | (2) |
| Proportion of duplikations | o = 0,070 | (3) |
| Proportion of point mutations | p = 0,280 | p=1-d-o |
| Fitness (DMD) | w = 0 | |
| Sex ratio of the mutation rates | k = 1,10 | (14) |
| Sex ratio of deletions | kd= 0,385 | modified from (16) |
| Sex ratio of duplications | ko= 5,000 | modified from (17) |
| Sex ratio of point mutations | kp= 5,000 | modified from (16) |
| Probability of de novo mutations in mitosis | g = 0,81 | (15) |
| Probability of de novo mutations in meiosis | 1-g = 0,19 | |
| Segregation in germline mosaic | f = 0,34 | (20) |
These parameters account for germinal mosaicisms and consider the relative probabilities of de novo mutation per mutation type: Large deletions arise predominantly in oogenesis, whereas point mutations and duplications result largely form errors in spermatogenesis (16, 17). This has direct impact on the risk assessment in DMD families with mutations unknown (18). Table 5 gives the probabilities of a mother of a sporadic DMD cases being carrier based on the various situations (assuming no healthy brothers or maternal uncles).
Table 5.
Probabilities (depending on the type of mutation) that the mother of a sporadic DMD patient is heterozygous, carries a germline mosaicism or the affected son has a meiotic de novo mutation.
| All mutations | |
| Mother is a carrier (heterozygote) | 0,677 |
| Mother is a germline mosaicism | 0,190 |
| Meiotic de novo mutation in the affected son | 0,133 |
| Deletions | |
| Mother is a carrier (heterozygote) | 0,581 |
| Mother is a germline mosaicism | 0,247 |
| Meiotic de novo mutation in the affected son | 0,173 |
| Point mutations or duplications | |
| Mother is a carrier (heterozygote) | 0,857 |
| Mother is a germline mosaicism | 0,084 |
| Meiotic de novo mutation in the affected son | 0,059 |
Examples of two programs for risk calculation which are freely available:
MLINK and LinkMap, part of the LINKAGE package (19), an MS-DOS program which does not allow the consideration of germline mosaicism and the distinction of deletions, duplications and point mutations.
RISCALW (21), a Windows program, which allows risk calculation in families with DMD, including all the important parameters with the exception of duplications.
Concluding remarks
Progress in nucleic acid analytical techniques has greatly facilitated and reduced in cost the identification of potential female carriers in DMD and BMD families. Thus, the vast majority of females at risk can now be diagnosed by direct assessment of the causative mutation. However, due to the reduced life expectancy of affected males and the reduction in kindred size in many societies, no mutation carrier may be available in a subset of families with a sporadic index case. Due to the complexity of mutation generation in the dystrophin gene, risk assessment for genetic counseling can be quite complex in these situations.
References
- 1.Emery AE. Population frequencies of inherited neuromuscular diseases--a world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 2.Dunnen JT, Grootscholten PM, Bakker E, et al. Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. Am J Hum Genet. 1989;45:835–847. [PMC free article] [PubMed] [Google Scholar]
- 3.White SJ, Aartsma-Rus A, Flanigan KM, et al. Duplications in the DMD gene. Hum Mutat. 2006;27:938–945. doi: 10.1002/humu.20367. [DOI] [PubMed] [Google Scholar]
- 4.Deburgrave N, Daoud F, Llense S, et al. Protein- and mRNA-based phenotype-genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with nonsense and frameshift mutations in the DMD gene. Hum Mutat. 2007;28:183–195. doi: 10.1002/humu.20422. [DOI] [PubMed] [Google Scholar]
- 5.Lalic T, Vossen RH, Coffa J, et al. Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet. 2005;13:1231–1234. doi: 10.1038/sj.ejhg.5201465. [DOI] [PubMed] [Google Scholar]
- 6.Abbs S, Tuffery-Giraud S, Bakker E, et al. Best Practice Guidelines on molecular diagnostics in Duchenne/Becker muscular dystrophies. Neuromuscul Disord. 2010;20:422–427. doi: 10.1016/j.nmd.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Monaco AP, Bertelson CJ, Liechti-Gallati S, et al. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2:90–95. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- 8.Koenig M, Beggs AH, Moyer M, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet. 1989;45:498–506. [PMC free article] [PubMed] [Google Scholar]
- 9.Malhotra SB, Hart KA, Klamut HJ, et al. Frameshift deletions in patients with Duchenne and Becker muscular dystrophy. Science. 1988;242:755–759. doi: 10.1126/science.3055295. [DOI] [PubMed] [Google Scholar]
- 10.Haldane JSB. The rate of spontaneous mutation of a human gene. J Genet. 1935;31:317–326. doi: 10.1007/BF02717892. [DOI] [PubMed] [Google Scholar]
- 11.Bakker E, Veenema H, Dunnen JT, et al. Germinal mosaicism increases the recurrence risk for 'new' Duchenne muscular dystrophy mutations. J Med Genet. 1989;26:553–559. doi: 10.1136/jmg.26.9.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Essen AJ, Abbs S, Baiget M, et al. Parental origin and germline mosaicism of deletions and duplications of the dystroph in gene: a European study. Hum Genet. 1992;88:249–257. doi: 10.1007/BF00197255. [DOI] [PubMed] [Google Scholar]
- 13.Passos-Bueno MR, Rapaport D, Love D, et al. Screening of deletions in the dystrophin gene with the cDNA probes Cf23a, Cf56a, and Cf115. J Med Genet. 1990;27:145–150. doi: 10.1136/jmg.27.3.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barbujani G, Russo A, Danieli GA, et al. Segregation analysis of 1885 DMD families: significant departure from the expected proportion of sporadic cases. Hum Genet. 1990;84:522–526. doi: 10.1007/BF00210802. [DOI] [PubMed] [Google Scholar]
- 15.Grimm T, Muller B, Muller CR, et al. Theoretical considerations on germline mosaicism in Duchenne muscular dystrophy. J Med Genet. 1990;27:683–687. doi: 10.1136/jmg.27.11.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimm T, Meng G, Liechti-Gallati S, et al. On the origin of deletions and point mutations in Duchenne muscular dystrophy: most deletions arise in oogenesis and most point mutations result from events in spermatogenesis. J Med Genet. 1994;31:183–186. doi: 10.1136/jmg.31.3.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawamura J, Kato S, Ishihara T, et al. Difference of new mutation rates in dystrophin gene between deletion and duplication mutation in Duchenne and Becker muscular dystrophy. Rinsho Shinkeigaku. 1997;37:212–217. [PubMed] [Google Scholar]
- 18.Fischer C, Gross W, Krüger J, et al. Modelling germline mosaicism and different new mutation rates simultaneously for appropriate risk calculations in families with Duchenne muscular dystrophy. Ann Hum Genet. 2006;70:237–248. doi: 10.1111/j.1469-1809.2005.00226.x. [DOI] [PubMed] [Google Scholar]
- 19.Lathrop GM, Lalouel JM, Julier C, et al. Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA. 1984;81:3443–3446. doi: 10.1073/pnas.81.11.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Müller B, Grimm T, Golla A. Estimating the proportion of affected germ-cells in cases of germinal mosaicism in Duchenne muscular dystrophy (DMD) Med Genetik. 1995;7:119–119. [Google Scholar]
- 21.Fischer C, Krüger J, Gross W. RISCALW: a Windows program for risk calculation in families with Duchenne muscular dystrophy. Ann Hum Genet. 2006;70:249–253. doi: 10.1111/j.1529-8817.2005.00227.x. [DOI] [PubMed] [Google Scholar]