

Abstract
OBJECTIVE
Pharmacological agents for diabetic peripheral neuropathy (DN) target a number of mechanisms, including sodium channel function and γ-aminobutyric acid–minergic processes. At present, prescription is undertaken on a trial-and-error basis, leading to prolonged medication trials and greater healthcare costs. Nerve-excitability techniques are a novel method of assessing axonal ion channel function in the clinical setting. The aim of this study was to determine the effects of axonal ion channel dysfunction on neuropathy-specific quality-of-life (QoL) measures in DN.
RESEARCH DESIGN AND METHODS
Fifty-four patients with type 2 diabetes mellitus underwent comprehensive neurologic assessment, nerve-conduction studies, and nerve-excitability assessment. Neuropathy severity was assessed using the Total Neuropathy Score. Neuropathy-specific QoL was assessed using a DN-specific QoL questionnaire (Neuropathy-Specific Quality of Life Questionnaire [NeuroQoL]). Glycosylated hemoglobin and BMI were recorded in all patients.
RESULTS
NeuroQoL scores indicated significant QoL impairment (mean 9.08 ± 5.93). Strength-duration time constant (SDTC), an excitability parameter reflecting sodium channel function, was strongly correlated with QoL scores (r = 0.545; P < 0.005). SDTC was prolonged in 48.6% of patients who experienced neuropathic symptoms. A significant correlation was also noted between SDTC and neuropathy severity (r = 0.29; P < 0.05). This relationship was strengthened when looking specifically at patients with clinically graded neuropathy (r = 0.366; P < 0.05).
CONCLUSIONS
The current study has demonstrated an association between markers of sodium channel function and QoL in DN. The study demonstrates that excitability techniques may identify patients in whom altered sodium channel function may be the dominant abnormality. The findings suggest that excitability techniques may have a role in clinical decision making regarding neuropathic treatment prescription.
Diabetic peripheral neuropathy (DN) is a frequent complication of diabetes mellitus (DM) affecting 30–50% of patients with type 2 DM (T2DM) (1). DN manifests as a length-dependent polyneuropathy, and as such, patients typically present with sensory symptoms such as pain, paraesthesia, and numbness in distal lower limb segments, which may progress to motor and upper limb involvement in more severe cases (2).
It is well established that quality of life (QoL) is significantly impaired in DN patients, affecting physical, social, and emotional well-being (3,4). Currently, treatment options for the neuropathic symptoms of DN include anticonvulsant medications that have effects on voltage-gated ion channels, γ-aminobutyric acid (GABA)–mimetic agents, and selective noradrenaline- and serotonin-reuptake inhibitors. These medications either block sodium (Na+) channels or alter the balance between inhibitory and excitatory neurotransmitters, reducing peripheral and central sensitization (5,6). At present, the selection of a specific drug for an individual patient is largely undertaken on a trial-and-error basis (7,8), frequently leading to prolonged trials of medication, development of significant side effects, and greater health care costs (8,9).
Previously, human studies have demonstrated that ectopic sensory symptoms such as paraesthesia and pain may be related to changes in axonal ion channel properties (10). Specifically, these studies have provided evidence that axonal persistent Na+ channels may play a major role in the generation of ectopic symptoms, both sensory and motor (11). These insights have been established through the use of nerve-excitability testing, a novel technique that provides information on axonal ion channel function in the clinical setting (12). Studies of nerve excitability have been applied to a number of neuropathic processes, including metabolic, toxic, and demyelinating neuropathies (13). Previous studies using nerve-excitability techniques in patients with DM have demonstrated prominent alterations in axonal ion channel properties, including changes in parameters reflecting Na+ channel function (14–16). Although these changes in ion channel properties have been postulated to underlie the development of neuropathic symptoms (17), the relationship between these changes and QoL in a patient cohort with DM has not been evaluated. The aim of the current study was to investigate the potential relationship between axonal ion channel dysfunction, ectopic sensory symptoms, and QoL measures in a cohort of patients with T2DM.
RESEARCH DESIGN AND METHODS
A screening clinical neurologic assessment was conducted in 87 patients with T2DM, consecutively recruited from the Diabetes Centre at Prince of Wales Hospital in Sydney. Exclusion criteria for this study were current treatment with neuropathic medications, clinical or electrophysiological features of carpal tunnel syndrome, and a history of neuropathic symptoms for <6 months. On the basis of these criteria, 33 patients were excluded from further testing. The 54 remaining patients (male/female 28:26) underwent a full suite of neurophysiological assessments including nerve-excitability testing and standard nerve-conduction studies. Patients were grouped according to neuropathy severity, and all neuropathic patients underwent QoL assessment.
All subjects gave written informed consent in accordance with the Declaration of Helsinki, and the study was approved by the South Eastern Sydney Area Health Service and the University of New South Wales Research Ethics committees.
Clinical assessment and neuropathy staging
A comprehensive neurologic assessment was conducted in all patients, and neuropathy severity was graded according to a revised version of the Total Neuropathy Score (TNS) (18) in which autonomic symptoms and quantitative assessment of vibration were excluded from assessment and tibial motor responses assessed in place of peroneal responses. This revised version has been used in a previous study of patients with T2DM (19).
The TNS is a composite score of eight categories. The categories included the extent and severity of sensory and motor symptoms, assessment of deep-tendon reflexes, muscle strength, vibration sensibility (128-Hz tuning fork), pinprick sensibility (Neurotip, Owen Mumford, U.K.), and tibial and sural nerve amplitudes (Medelec Synergy System; Oxford Instruments, Oxfordshire, U.K.). Each category was scored from 0 to 4 (0, no dysfunction; 4, severe dysfunction) and summed to give a total score from 0 to 32 (32 is maximum dysfunction). The TNS was further subdivided into total neuropathy grades (TNG) to indicate severity of neuropathy: TNG 0, TNS 0–1; TNG 1, TNS 2–8; TNG 2, TNS 9–16; TNG 3, TNS 17–24; and TNG 4, TNS 25–32 (18). For the purpose of this study, we refer to TNG 0 as no neuropathy, TNG 1 as mild neuropathy, TNG 2 as moderate neuropathy, TNG 3 as severe neuropathy, and TNG 4 as very severe neuropathy.
Assessment of axonal ion channel properties
Nerve-excitability studies were undertaken in all patients (20). These studies involved stimulating the median nerve at the wrist and measuring the compound muscle action potential (CMAP) of abductor pollicis brevis using surface electrodes (Unomedical, Bikerod, Denmark). Specifically, the TRONDNF protocol (12) through QTRAC automated software (Digitimer, London, U.K.) was used. This protocol and software allowed for the rapid acquisition of a number of excitability parameters derived from five distinct testing paradigms: 1) stimulus-response behavior, 2) strength-duration-time-constant (SDTC), 3) threshold electrotonus (TE), 4) current threshold relationship (I/V), and 5) recovery cycle (RC).
Stimulus-response curves were obtained by applying a 1-ms–duration current of increasing intensity until the maximal CMAP response was achieved. From this, a target for the remaining tests was calculated (∼40% of maximum CMAP). The stimulus required to reach this target was termed “threshold.” SDTC was obtained by plotting the relationship between stimulus strength and stimulus duration using at least two duration points. SDTC was then determined to be the ratio of threshold current increase to stimulus duration decrease. This time constant is partly determined by the activity of nodal persistent Na+ conductances (21), which are thought to underlie the generation of ectopic neuropathic symptoms (11,22,23). TE was determined by plotting percentage change of threshold when 1-ms test impulses were delivered during and after 100-ms subthreshold conditioning currents of +40% and −40% control threshold. This paradigm provides a reflection of internodal properties and overall axonal membrane potential (24). I/V was obtained by plotting threshold change with 1-ms test impulses postdelivery of 200-ms depolarizing and hyperpolarizing conditioning currents (+50 to −100 ms). I/V provides information on rectifying properties of the internode (12,23). The RC examines recovery of axonal conduction following supramaximal stimulus. Percentage threshold change in RC was measured by varying the delay between a test impulse at threshold and a supramaximal conditioning stimulus (12).
Assessment of neuropathy-specific QoL
Impairment in QoL was assessed using the Neuropathy-Specific Quality of Life Questionnaire (NeuroQoL). The NeuroQoL assess symptoms, emotional burden, and restrictions on activities of daily living specific to diabetic neuropathy (3). It has been used to measure the impact of DN on QoL (25) and for assessment of the effectiveness of potential neuropathic treatments (26). Importantly, NeuroQoL has been shown to discriminate more clearly between varying severities of DN compared with more generic QoL scales such as the 36-item short-form health survey (27) and 12-item short-form health survey (3). The questionnaire consists of 35 questions and rates the effect of neuropathy on QoL as a product of their presence and total level of bother. In the current study, the following domains were assessed: 1) symptomatic affliction, 2) psychosocial impairment, 3) DN-specific impact, and 4) overall QoL. The average rating for the questions in domains 1–3 were summed to give a total NeuroQoL impairment score of 3–35 (3 is nil QoL impairment from neuropathy, and 35 is maximal QoL impairment from neuropathy).
Statistical analysis
Statistical analysis was performed using SPSS statistics software v. 20 (IBM, Chicago, IL). Normality of the data was first assessed using the Shapiro-Wilk test. To investigate differences in nerve-excitability parameters between the patients with DM and normal control (NC) subjects (n = 50), independent t tests or Mann-Whitney U tests were conducted where appropriate. To compare between TNG groups, one-way ANOVA or Kruskal-Wallis tests were conducted depending on normality of data distribution. Following this, the Bonferroni post hoc test was applied to establish differences between groups. Finally, to determine relationships among clinical characteristics, nerve-excitability parameters, and NeuroQoL scores, Spearman ρ correlations (two-tailed) were conducted. Findings were considered statistically significant when P < 0.05 and trending when 0.05 < P < 0.1.
RESULTS
Clinical characteristics and neuropathy grading
Of the 87 patients with T2DM who were screened, 54 were enrolled in the study (Fig. 1). As a group, the enrolled patients had an average age of 62.19 ± 1.28 years, BMI of 31.26 ± 0.81 kg/m2, duration of diabetes of 147.32 ± 15.53 months, and glycosylated hemoglobin (GHb) of 7.98 ± 0.23%. Mean characteristics per TNG are expressed in Table 1.
Figure 1.

Recruitment flow chart. Total of 87 patients with T2DM underwent clinical neurologic assessment. Patients who were on neuropathic medications, those with carpal tunnel syndrome (CTS), and those with neuropathic symptoms that had lasted <6 months were excluded from further study (n = 33). The remaining 54 patients underwent further neurologic and neurophysiological assessment.
Table 1.
Patient clinical characteristics according to neuropathy severity
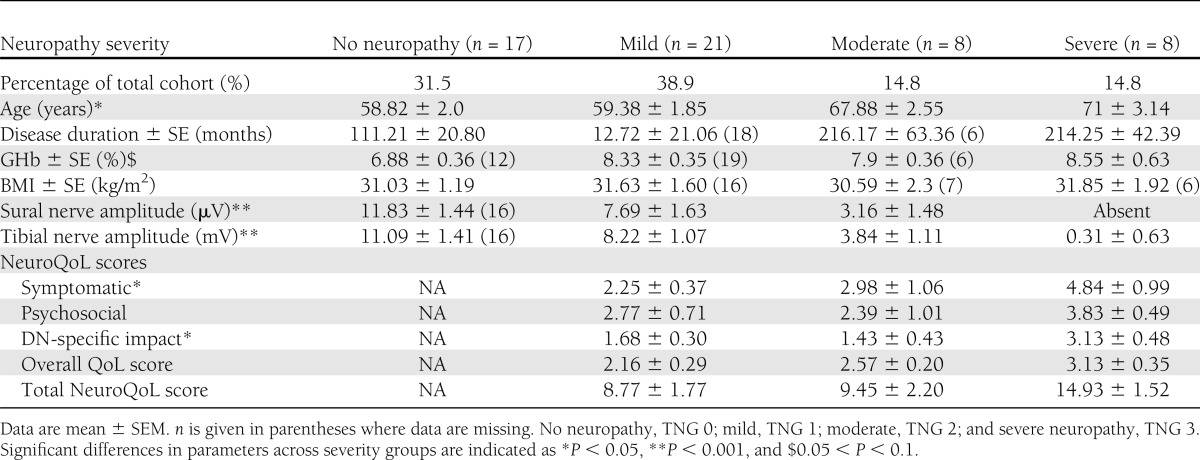
Clinical neurologic examination demonstrated a wide range of neurologic impairment, with TNS scores ranging from 0–23 and a mean TNS score of 7.00 ± 1.02 (18). Further grading of patients according to TNS revealed that 31.5% of the cohort had no neuropathy (TNG 0), 38.9% had mild neuropathy (TNG 1), and 29.6% had moderate to severe neuropathy (TNG 2 to 3). Ectopic sensory neurologic symptoms such as paresthesia were reported in 53.7% of patients. Duration of diabetes was correlated with increasing neuropathy severity (r = 0.427; P < 0.001). There were no significant correlations between BMI and GHb with neuropathy severity (TNS scores).
Neuropathy-specific QoL findings
The NeuroQoL (3) was administered to patients with evidence of clinical neuropathy (TNG 1–3, n = 37) (Table 1). Overall NeuroQoL scores in this group ranged from 4–26.25, with a mean of 10.33 ± 1. The NeuroQoL was further divided into symptomatic, psychosocial, and diabetic neuropathy–specific scores, all of which showed impairment (symptomatic score: 2.07 ± 0.14; psychosocial score: 2.28 ± 0.18; and diabetic neuropathy–specific score: 1.88 ± 0.22). Significant correlations were noted between TNS scores and overall QoL, as well as symptomatic and psychosocial scores (r = 0.61, P < 0.001; r = 0.44, P < 0.05; and r = 0.445, P < 0.05, respectively).
Significant differences were also observed in mean NeuroQoL domain scores across the different severity groups. Specifically, symptomatic NeuroQoL scores were significantly higher in patients with severe neuropathy compared with patients with mild neuropathy (mild: 2.25 ± 0.37; severe: 4.84 ± 0.99; P < 0.05). Psychosocial scores also demonstrated greater impairment in patients with severe neuropathy (0.383 ± 0.49) compared with those with mild (2.77 ± 0.71; P < 0.005) and moderate neuropathy (2.39 ± 1.01; P < 0.005). Similarly, DN-specific QoL impairment was higher in the severe group (3.13 ± 0.48) compared with those in the mild (1.68 ± 0.30; P < 0.005) and moderate neuropathy group (1.14 ± 0.43; P < 0.05).
Nerve-excitability abnormalities
Prominent changes in excitability parameters were noted in DN patients compared with NC subjects (Fig. 2A), and these changes were progressively greater with increasing neuropathy severity (Fig. 1B). With increasing neuropathy severity, there was a reduction in mean CMAP amplitude (mV), with significant differences noted between NC subjects and patients with severe neuropathy (NC, 6.56; severe group, 4.66 ± 1.18; P < 0.05). Distal motor latency also increased with greater neuropathy severity (TNG: r = 0.485, P < 0.005; TNS: r = 0.431, P < 0.01). SDTC, a neurophysiological parameter reflecting the behavior of persistent Na+ conductances (11,21), was increased in patients with moderate to severe neuropathy when compared with NC (TNG 2 to 3, 0.50 ± 0.02; NC, 0.44 ± 0.01; P < 0.05).
Figure 2.

A: Nerve-excitability plots for patients with DN are flatter. Mean curve for patients with DN (block line) for TE and RC paradigms are plotted against 95% confidence limits of NC (dotted lines). By convention, TE is expressed as percent change in threshold over time (ms), and RC is expressed as percent change in threshold against interstimulus interval (ms). TE and RC plots for DN patients showed a flattened morphology, sitting at or outside lower limits of NC data. Mean superexcitability, subexcitability, RRP, and S2 accommodation were found to be significantly different between patients with DM and NC subjects. *P < 0.05, **P < 0.005. B: Progressive changes were found in a number of nerve-excitability parameters with increasing neuropathy severity in patients with DM. Mean values ± SE expressed: black filled bar, patients with no neuropathy; diagonal line bar, mild neuropathy; dotted bar, moderate neuropathy; and horizontal line bar, severe neuropathy. Patients were compared with NC data (not shown), and statistical significance was determined using a Mann-Whitney U test: *P < 0.05, **P < 0.005, and #0.05 < P < 0.1. Superexcitability (i), subexcitability (ii), and S2 accommodation (iv) are expressed as percentage (%) change of threshold (NC mean: S2 accommodation, 22.84 ± 0.488; superexcitability, 23.811; and subexcitability, 14.75 ± 0.592). The RRP (iii) is expressed in ms (NC: 3.028 ms). Taken together, these parameters indicate a progressive shift toward a more depolarized axonal-membrane potential in the DM cohort.
The changes in SDTC were accompanied by changes in other excitability parameters that were qualitatively similar to those reported in previous studies of patients with DM (15,16,28) (Fig. 2B). Progressive alterations were noted in the RC parameters superexcitability (P < 0.05), late subexcitability (P < 0.005), and relative refractory period (RRP) in addition to the TE parameter S2 accommodation (P < 0.005) (Fig. 2). Taken together, these changes are consistent with axonal-membrane depolarization, a finding that has been demonstrated in previous excitability studies of DN patients (15,19).
Correlations between nerve-excitability abnormalities and neuropathy-specific QoL
Correlations were undertaken to explore the potential relationship between excitability parameters and QoL measures. A strong positive relationship was observed between SDTC, a marker of the activity of persistent Na+ conductances (11), and QoL impairment as a result of neuropathic symptoms (symptomatic NeuroQoL score: r = 0.545; P < 0.005) (Fig. 3). There was a significant correlation between SDTC and total NeuroQoL score (r = 0.515; P < 0.05). Of the patients who experienced neuropathic symptoms, SDTC was prolonged in 48.6% of this group (upper limit 95% CI 0.462 ms) (Fig. 3). In addition, a positive correlation was noted between SDTC and increasing neuropathy severity (n = 54; r = 0.29; P < 0.05). This relationship was strengthened when looking specifically at patients with clinically graded neuropathy (n = 37; r = 0.366; P < 0.05).
Figure 3.

Correlation between SDTC and symptomatic NeuroQoL score. r = 0.545; P < 0.05. Dotted line indicates the upper limit of normal range for SDTC of NC cohort (upper 95% confidence limit: 0.4622 ms). SDTC was prolonged in 48.6% of symptomatic DN patients compared with NC.
In addition, correlations were undertaken between SDTC and symptoms of nerve hyperexcitability (i.e., paraesthesia and burning). A strong correlation was noted between increasing SDTC and symptoms of hyperexcitability (r = 0.745; P < 0.05). Further analysis demonstrated a weaker correlation between the occurrence of negative sensory symptoms (i.e., numbness and loss of sensation) and SDTC (r = 0.539; P < 0.05). There was no significant correlation between SDTC and symptoms of unsteadiness or impaired balance. Similarly, psychosocial, DN-specific, and overall QoL scores were not significantly correlated with SDTC.
Analyses were also undertaken between NeuroQoL scores and other excitability measures that were not specifically related to Na+ channel function, namely TE parameters and superexcitability. There was no significant correlation between QoL and these other neurophysiological parameters. In total, the findings suggest that the correlation between SDTC and QoL scores was not due to a generalized disturbance in axonal function but more likely reflected a specific effect of DM on persistent Na+ conductances.
CONCLUSIONS
The current study has demonstrated an association between clinical markers of Na+ channel dysfunction and QoL measures in a cohort of patients with DN. Specifically, the study has shown correlations between SDTC, a parameter reflecting upregulation of nodal persistent Na+ conductances (21), and symptomatic neuropathy-specific QoL. These correlations were even stronger when analysis was limited to symptoms of nerve hyperexcitability. The importance of this association lies in the prominent causal role that has been attributed to nodal persistent Na+ conductances in generating ectopic impulse activity in the peripheral nervous system (10,22).
It may be argued that the underlying cause of the prolongations in SDTC may relate to axonal membrane depolarization (15,16), given the pattern of change noted in TE and RC parameters with increasing neuropathy severity. Critically, however, there was no correlation between other excitability parameters that reflect changes in membrane potential and neuropathy-specific QoL measures, suggesting that the correlation between SDTC and neuropathy-specific QoL was intrinsically related to upregulation of persistent Na+ conductances rather than a more generalized change in axonal function. Although the current study focused on upper limb motor recordings, the findings provide further evidence that excitability changes in diabetes may occur in a generalized distribution, despite the clinical features of DN being largely lower limb predominant (15,19).
Our findings suggest that persistent Na+ conductances may be upregulated in patients with DN. These findings are supported by studies in animal studies that have demonstrated that increased Na+ channel expression may play an important role in the generation of diabetic neuropathic symptoms (29). Specifically, these studies have shown that there is upregulation of Na+ channel isoforms in diabetic nerves and that these changes are associated with the development of neuropathic symptoms (29). The increase in Na+ currents has been attributed to Na+ channel phosphorylation, which modifies channel properties and may cause an increase in channel conductance (29). Studies specifically exploring the contributions of persistent Na+ currents to diabetic neuropathic pain have demonstrated increases in mRNA levels for Na+ channel isoforms (30,31). Such changes have been postulated to result from alterations in signal transduction cascades and the increased levels of protein kinase A and protein kinase C induced by diabetes, which further enhance Na+ channel expression and ectopic firing (30).
Persistent Na+ conductances play an important role in the generation of ectopic neuropathic symptoms, and a number of neuropathic pain treatments preferentially modulate persistent Na+ conductances (32,33). In the current study, 48.6% of patients with neuropathic symptoms had prolonged SDTC, consistent with an upregulation of persistent Na+ conductances, whereas the remainder had a normal or reduced SDTC. This finding suggests that the contribution of altered Na+ channel function toward the development of neuropathic symptoms may vary between individual patients. In patients with a normal SDTC, it is possible that other mechanisms may play a greater role in the development of neuropathic symptoms, including reduced descending inhibition to pain (5,34), altered levels of excitatory neurotransmitters (35), or increased sensitization of central pain-related areas (36,37). These mechanisms may not cause changes in voltage-gated ion channel function of peripheral nerve axons and therefore may not alter axonal excitability properties (38).
From a clinical perspective, the current study has demonstrated that excitability techniques have the potential to identify diabetic neuropathy patients in whom alterations in Na+ channel properties may be the dominant physiological abnormality. The study further demonstrates that these changes have a very clear clinical correlate, in that they are associated with greater impairments in QoL scores. At present, neuropathic pain treatments are administered largely on a trial-and-error basis, reflecting the contribution of multiple discrete mechanisms in the generation of diabetic neuropathic symptoms (8,38).
Current neuropathic treatments for DN include anticonvulsant medications, tricyclic antidepressants, and selective serotonin reuptake inhibitors, all of which block axonal Na+ channels (6). In contrast, GABA-mimetic agents, such as gabapentin and pregabalin, have clearly demonstrated benefits in the treatment of DN-related symptoms (39) and yet have no specific effects on Na+ channel properties (6). These medications are thought to improve DN symptoms through effects on the central nervous system, including enhancement of inhibitory input of GABA-mediated pathways and antagonism of excitatory N-methyl-D-aspartic acid receptors, thereby reducing central sensitization (36). Such medications may be less efficacious in patients in whom altered Na+ conductances are a primary mechanism for neuropathic symptom generation. Importantly, the current study suggests that nerve-excitability techniques, applied in the clinical setting, may have a role in determining the differential mechanisms underlying neuropathic symptom generation in DN. Further to this, the study provides a basis for future work investigating whether excitability measures may play a role in clinical decision making pertaining to the commencement and monitoring of neuropathic pain treatment in patients with type 2 diabetes.
Acknowledgments
N.C.G.K. and R.A. were supported by an Australian Postgraduate Award scholarship. A.V.K. was supported by a Career Development Award of the National Health and Medical Research Council of Australia (Grant 568680).
No potential conflicts of interest relevant to this article were reported.
N.C.G.K. was involved in study design, recruitment, data collection, data interpretation, and manuscript composition. R.A. contributed to recruitment, data collection, discussion, and manuscript composition. C.W. was involved in recruitment, data collection, and interpretation. C.S.-Y.L. and M.C.K. were involved in interpretation of data. A.M.P. was involved in recruitment and discussion. A.V.K. was involved in study design, data interpretation, and manuscript composition. A.V.K. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Preliminary findings from this report were presented as an abstract at the Annual Scientific Meeting of the Australian Diabetes Society and Australian Diabetes Educators Association [“Neurophysiological parameters and impaired quality of life in diabetic peripheral neuropathy (DPN)”], Perth, Australia, 31 August–2 September 2011 and the 14th Clinical Neurophysiology Workshop of the Australian and New Zealand Association of Neurologists (“Effects of altered axonal biophysical properties on quality of life in diabetic peripheral neuropathy”), Gold Coast, Australia, 2–5 October 2011.
References
- 1.National Association of Diabetes Centres Australian National Diabetes Information Audit and Benchmarking (ANDIAB) Project Report. Australian Capital Territory, National Association of Diabetes Centres, 2010, p. 174 [Google Scholar]
- 2.Sugimoto K, Murakawa Y, Sima AAF. Diabetic neuropathy—a continuing enigma. Diabetes Metab Res Rev 2000;16:408–433 [DOI] [PubMed] [Google Scholar]
- 3.Vileikyte L, Peyrot M, Bundy C, et al. The development and validation of a neuropathy- and foot ulcer-specific quality of life instrument. Diabetes Care 2003;26:2549–2555 [DOI] [PubMed] [Google Scholar]
- 4.Quattrini C, Tesfaye S. Understanding the impact of painful diabetic neuropathy. Diabetes Metab Res Rev 2003;19(Suppl. 1):S2–S8 [DOI] [PubMed] [Google Scholar]
- 5.Maneuf YP, Hughes J, McKnight AT. Gabapentin inhibits the substance P-facilitated K(+)-evoked release of [(3)H]glutamate from rat caudial trigeminal nucleus slices. Pain 2001;93:191–196 [DOI] [PubMed] [Google Scholar]
- 6.Lenkey N, Karoly R, Lukacs P, et al. Classification of drugs based on properties of sodium channel inhibition: a comparative automated patch-clamp study. PLoS One 2010. Available from http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0015568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Connor AB. Neuropathic pain: quality-of-life impact, costs and cost effectiveness of therapy. Pharmacoeconomics 2009;27:95–112 [DOI] [PubMed] [Google Scholar]
- 8.Yarnitsky D, Granot M, Nahman-Averbuch H, Khamaisi M, Granovsky Y. Conditioned pain modulation predicts duloxetine efficacy in painful diabetic neuropathy. Pain 2012;153:1193–1198 [DOI] [PubMed] [Google Scholar]
- 9.Vinik AI. Diabetic neuropathy: pathogenesis and therapy. Am J Med 1999;107:17S–26S [DOI] [PubMed] [Google Scholar]
- 10.Mogyoros I, Kiernan MC, Burke D, Bostock H. Excitability changes in human sensory and motor axons during hyperventilation and ischaemia. Brain 1997;120:317–325 [DOI] [PubMed] [Google Scholar]
- 11.Mogyoros I, Kiernan MC, Burke D. Strength-duration properties of human peripheral nerve. Brain 1996;119:439–447 [DOI] [PubMed] [Google Scholar]
- 12.Kiernan MC, Burke D, Andersen KV, Bostock H. Multiple measures of axonal excitability: a new approach in clinical testing. Muscle Nerve 2000;23:399–409 [DOI] [PubMed] [Google Scholar]
- 13.Nodera H, Kaji R. Nerve excitability testing and its clinical application to neuromuscular diseases. Clin Neurophysiol 2006;117:1902–1916 [DOI] [PubMed] [Google Scholar]
- 14.Misawa S, Kuwabara S, Ogawara K, Kitano Y, Yagui K, Hattori T. Hyperglycemia alters refractory periods in human diabetic neuropathy. Clin Neurophysiol 2004;115:2525–2529 [DOI] [PubMed] [Google Scholar]
- 15.Krishnan AV, Kiernan MC. Altered nerve excitability properties in established diabetic neuropathy. Brain 2005;128:1178–1187 [DOI] [PubMed] [Google Scholar]
- 16.Misawa S, Sakurai K, Shibuya K, et al. Neuropathic pain is associated with increased nodal persistent Na(+) currents in human diabetic neuropathy. J Peripher Nerv Syst 2009;14:279–284 [DOI] [PubMed] [Google Scholar]
- 17.Krishnan AV, Lin CS, Kiernan MC. Activity-dependent excitability changes suggest Na+/K+ pump dysfunction in diabetic neuropathy. Brain 2008;131:1209–1216 [DOI] [PubMed] [Google Scholar]
- 18.Cornblath DR, Chaudhry V, Carter K, et al. Total neuropathy score: validation and reliability study. Neurology 1999;53:1660–1664 [DOI] [PubMed] [Google Scholar]
- 19.Sung J-Y, Park SB, Liu Y-T, et al. Progressive axonal dysfunction precedes development of neuropathy in type 2 diabetes. Diabetes 2012;61:1592–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burke D, Kiernan MC, Bostock H. Excitability of human axons. Clin Neurophysiol 2001;112:1575–1585 [DOI] [PubMed] [Google Scholar]
- 21.Bostock H, Rothwell JC. Latent addition in motor and sensory fibres of human peripheral nerve. J Physiol 1997;498:277–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bostock H. The strength-duration relationship for excitation of myelinated nerve: computed dependence on membrane parameters. J Physiol 1983;341:59–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiernan MC, Bostock H. Effects of membrane polarization and ischaemia on the excitability properties of human motor axons. Brain 2000;123:2542–2551 [DOI] [PubMed] [Google Scholar]
- 24.Krishnan AV, Lin CS, Park SB, Kiernan MC. Axonal ion channels from bench to bedside: a translational neuroscience perspective. Prog Neurobiol 2009;89:288–313 [DOI] [PubMed] [Google Scholar]
- 25.Davies M, Brophy S, Williams R, Taylor A. The prevalence, severity, and impact of painful diabetic peripheral neuropathy in type 2 diabetes. Diabetes Care 2006;29:1518–1522 [DOI] [PubMed] [Google Scholar]
- 26.Lavery LA, Murdoch DP, Williams J, Lavery DC. Does anodyne light therapy improve peripheral neuropathy in diabetes? A double-blind, sham-controlled, randomized trial to evaluate monochromatic infrared photoenergy. Diabetes Care 2008;31:316–321 [DOI] [PubMed] [Google Scholar]
- 27.Vileikyte L, Boulton A. Assessing Quality Of Life In Diabetic Neuropathy: Condition-Specific Or Generic Approach? J Peripher Nerv Syst 2000;5:180–181 [Google Scholar]
- 28.Misawa S, Kuwabara S, Kanai K, et al. Axonal potassium conductance and glycemic control in human diabetic nerves. Clin Neurophysiol 2005;116:1181–1187 [DOI] [PubMed] [Google Scholar]
- 29.Hong S, Morrow TJ, Paulson PE, Isom LL, Wiley JW. Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J Biol Chem 2004;279:29341–29350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Craner MJ, Klein JP, Renganathan M, Black JA, Waxman SG. Changes of sodium channel expression in experimental painful diabetic neuropathy. Ann Neurol 2002;52:786–792 [DOI] [PubMed] [Google Scholar]
- 31.Hong S, Wiley JW. Altered expression and function of sodium channels in large DRG neurons and myelinated A-fibers in early diabetic neuropathy in the rat. Biochem Biophys Res Commun 2006;339:652–660 [DOI] [PubMed] [Google Scholar]
- 32.Spadoni F, Hainsworth AH, Mercuri NB, et al. Lamotrigine derivatives and riluzole inhibit INa,P in cortical neurons. Neuroreport 2002;13:1167–1170 [DOI] [PubMed] [Google Scholar]
- 33.Lenkey N, Karoly R, Epresi N, Vizi E, Mike A. Binding of sodium channel inhibitors to hyperpolarized and depolarized conformations of the channel. Neuropharmacology 2011;60:191–200 [DOI] [PubMed] [Google Scholar]
- 34.Maneuf YP, Blake R, Andrews NA, McKnight AT. Reduction by gabapentin of K+-evoked release of [3H]-glutamate from the caudal trigeminal nucleus of the streptozotocin-treated rat. Br J Pharmacol 2004;141:574–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cardoso S, Carvalho C, Santos R, et al. Impact of STZ-induced hyperglycemia and insulin-induced hypoglycemia in plasma amino acids and cortical synaptosomal neurotransmitters. Synapse 2011;65:457–466 [DOI] [PubMed] [Google Scholar]
- 36.Rose MA, Kam PC. Gabapentin: pharmacology and its use in pain management. Anaesthesia 2002;57:451–462 [DOI] [PubMed] [Google Scholar]
- 37.Fischer TZ, Tan AM, Waxman SG. Thalamic neuron hyperexcitability and enlarged receptive fields in the STZ model of diabetic pain. Brain Res 2009;1268:154–161 [DOI] [PubMed] [Google Scholar]
- 38.Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain 1999;83:389–400 [DOI] [PubMed] [Google Scholar]
- 39.Rosenstock J, Tuchman M, LaMoreaux L, Sharma U. Pregabalin for the treatment of painful diabetic peripheral neuropathy: a double-blind, placebo-controlled trial. Pain 2004;110:628–638 [DOI] [PubMed] [Google Scholar]