

Type 2 diabetes has rapidly emerged as a global health crisis. Because population-level genetic changes take many generations to occur, this epidemic is almost certainly primarily a consequence of recent environmental changes; nonetheless, diabetes does appear to occur preferentially in genetically predisposed populations, which suggests that the effects of pre-existing susceptibility genes have been triggered by recent shifts in nongenetic factors.
Predisposition is influenced by the level of certain environmental exposures, personal factors, access to good-quality primary care, and by genotype. Interactions between genetic and nongenetic risk factors are hypothesized to raise diabetes risk in a synergistic manner; reciprocally, health-enhancing changes in behavior, body composition, or medication may reduce the risk of disease conveyed by genetic factors. Defining the nature of these interactions and identifying ways through which reliable observations of gene-environment interactions (GEIs) can be translated into the public health setting might help 1) optimize targeting of health interventions to persons most likely to respond well to them, 2) improve cost- and health-effectiveness of existing preventive and treatment paradigms; 3) reduce unnecessary adverse consequences of interventions; 4) increase patient adherence to health practitioners’ recommendations; and 5) identify novel interventions that are beneficial only in a defined genetic subgroup of the population. In this Perspective, we describe the rationale and evidence relating to the existence of gene-environment and gene-treatment interactions in type 2 diabetes. We discuss the tried, tested, and often-failed approaches to investigating gene-lifestyle interactions in type 2 diabetes; we discuss some recent developments in gene-treatment interactions (pharmacogenetics); and we look forward to the strategies that are likely to dominate these fields of research in the future. We conclude with a discussion of the requirements for translating findings from these future studies into a form where they can be used to help predict, prevent, or treat diabetes. Here we describe the rationale and evidence concerning GEIs and gene-treatment interactions in type 2 diabetes, provide an interpretation of current findings and strategies, and offer a view for their future translation.
What is GEI?
The definition of GEI varies somewhat depending on the field of diabetes research. In this review, we adopt epidemiological definitions of interaction, also known as effect modification or effect modulation. For binary outcomes, an interaction would be present if the combined risk attributable to genetic and environmental exposures is significantly greater or less than expected if their effects were additive. For quantitative traits, an interaction would be present if the magnitude of the genetic effect estimate differs across the range of an environmental exposure or treatment. Although the word environment when used in the context of GEI can relate to any nongenetic factor to which a person is exposed, extending from the macro (e.g., urban planning) to the micro (e.g., circulating proteins) environment, in the fields of complex disease research, the word environment has most often referred to lifestyle behaviors (e.g., diet or physical activity), although this view is evolving (Fig. 1). The word interaction is sometimes used to describe the joint effects of a genetic exposure and a second factor that is positioned on the causal pathway between the genetic exposure and a disease phenotype; in epidemiology, this process is termed mediation, which differs in meaning from interaction. The term epistasis refers to the interaction between two or more genetic loci.
Figure 1.

The future of research on stratified diabetes medicine: a systems epidemiology approach to the discovery of interactions between the exposome (all nongenetic elements to which we are exposed) and the quantifiable elements of the human physiome.
Why do we think GEIs cause type 2 diabetes?
The evidence supporting the existence of gene-lifestyle interactions in type 2 diabetes comes primarily from 1) the pattern and distribution of diabetes across environmental settings and ethnic groups, 2) family-based intervention studies, in which response to interventions varies less between biologically related individuals than between unrelated individuals; and 3) animal studies in which genetic and environmental factors are experimentally manipulated to cause changes in the expression of metabolic phenotypes. A brief overview of pertinent literature from human studies is given below.
There is considerable global variation in the prevalence and incidence of type 2 diabetes (1). In societies of European origin, the prevalence of type 2 diabetes is generally 10% or less, with the disease confined primarily to overweight and obese older adults. However, in some nonwhite populations, including Native Americans (especially Pima Indians), Alaskan Natives, Micro-Indonesia islanders (especially Nauruans), and some Middle Eastern (esecially Saudis and Emiratis) and Canadian First Nation populations (2,3) (www.phac-aspc.gc.ca/cd-mc/publications/diabetes-diabete/facts-figures-faits-chiffres-2011/index-eng.php), the prevalence of type 2 diabetes is substantially higher than in the rest of the world.
The Pima Indians of Arizona have the highest recorded prevalence of type 2 diabetes, with more than half of the adult population affected by the disease (3), and diagnoses are often made in adolescence and occasionally in childhood (4). The damming of the Salt and Gila rivers around 1911 brought an abrupt end to the Pima’s traditional subsistence farming lifestyle and with it a sharp reduction in occupational physical activity and the consumption of fresh produce. A second group of Pima Indians live in the Sierra Madre Mountains of northern Mexico. Although Arizona and Mexican Pima are phylogenetically similar (5), their lifestyles stand in stark contrast, with the Mexican Pima still living a traditional way of life left behind by Arizona Pima almost a century ago. Probably because of this, the prevalence of type 2 diabetes in Mexican Pima is roughly five-times lower than that seen in their Arizonan cousins, with diabetes prevalence in the Mexican Pima comparable to that of other non-Pima populations of northern Mexico (5). This observation emphasizes how environmental changes can awaken an underlying, possibly genetic, susceptibility to obesity and type 2 diabetes.
A popular yet contentious explanation for why indigenous groups (whose evolution has involved long periods of migration, hunter-gatherer lifestyles, and frequent famine) are so susceptible to the adverse consequences of industrialized environments is termed the “thrifty genotype hypothesis,” first proposed by Neel in the 1960s (6). Whereas the original description of the thrifty genotype hypothesis focused on the over-production of insulin after meals and a corresponding period of hypoglycemia that induces appetite, the idea that efficient storage and utilization of energy in adipose tissue is a selected trait has also been widely discussed and attributed to thrifty genes (7). The hypothesis hinges on the notion that frequent exposure to famine and other physiologically stressful events, such as migration and cold temperatures, over thousands of years of evolution may have enriched certain populations with gene variants that promote metabolic thriftiness, which in turn conveyed a survival advantage during famine or other periods of energetic stress. In the modern world, however, where excessive automation and almost effortless access to energy-dense foods are rife, calorie accumulation and storage may become metabolically deleterious. Of note, however, there is little evidence of positive selection genetic signatures around established type 2 diabetes loci (8), suggesting these diabetes loci at least are not thrifty genes.
Caveats of the literature on gene-lifestyle interactions
A recent simulation study on the role gene-gene and GEIs are likely to play in risk prediction and targeted medicine reached a rather sobering conclusion (9). The authors estimated that the average improvement in predictive accuracy, as defined by the area under the receiver operating characteristic curve, for type 2 diabetes was ∼5% when between 4 and 20 interactions were added to a prediction model. To conduct their simulations, Aschard et al. (9) made a series of assumptions about the magnitude and frequency of interaction effects, based on published epidemiological studies that had focused on common diseases, common exposures, and common variants; however, it is possible that as geneticists begin to study lower frequency variants, fairly large magnitude interaction effects may be discovered, albeit affecting relatively few individuals, which would likely increase the value of data on interactions for disease prediction.
Epidemiological studies have been the predominant source of literature on gene-lifestyle interactions in cardiovascular and metabolic disease. Dozens of case-control and cohort studies have been published since the late 1990s purporting to have identified gene-lifestyle interactions in type 2 diabetes or related quantitative metabolic traits. Until recently, however, most of these studies were small and often relied on imprecise estimates of environmental exposures and outcomes. These are prone to error and bias, and exposures may not be assessed at the time when they conveyed their effects; for example, the causative exposures may have occurred very early in life, perhaps even in utero. Moreover, the complexities of modeling interaction effects have forced geneticists to focus primarily on very simple models of interaction, whereas clinically relevant interaction effects likely involve multiple genetic and nongenetic biomarkers. In addition, barely a handful of studies have examined incident type 2 diabetes as an outcome, with most focusing on cross-sectional measures of glucose and others relying on analyses that include prevalent cases of diabetes; this may introduce labeling bias, where the recall of well-known diabetes-associated behaviors is less likely to be accurate in individuals recently diagnosed with disease than in those who have not been diagnosed with disease.
In a systematic review published in 2006 (10), we found that almost all studies published at that time included fewer than 1,000 participants and none included more than 3,000 participants. Although all studies lacked rigorous replication, we identified four classes of genes harboring loci that showed most consistent evidence of gene-lifestyle interactions in diabetogenic traits: the β2 adrenergic receptor (ADRB2), uncoupling proteins (UCP) 1-3, lipid-related loci (LIPC, LPL, FABP2, APOC3, and APOE), and the peroxisome proliferator-activated receptor-γ (PPARG). Of these, PPARG (at its common missense polymorphism Pro12Ala) was perhaps the most promising candidate, with a number of studies reporting interactions with dietary fatty acids or exercise in relation to insulin concentrations (11–13), adiposity (12,14,15), and type 2 diabetes (16). Studies examining interactions between dietary fats and the Pro12Ala genotype have continued to accrue, and attempts have been made to formally summarize this literature through meta-analysis. These attempts have been unsuccessful, however, owing to the challenges of pooling unstandardized data and the inadequate descriptions of the methods and results in many published studies (17).
Other key caveats to small gene-lifestyle interaction studies include their likelihood to be underpowered and that they are prone to reporting biases (18). The problems with measurement imprecision in studies of gene-lifestyle interaction are eloquently outlined and discussed by Wong et al. (19), where the authors provided estimates of sample-size requirements to detect gene-lifestyle interactions in the presence of varying degrees of environmental exposure assessment and phenotyping measurement error. The authors show that for the detection of fairly large magnitude gene-lifestyle interactions (βGE = 2), a study of ∼2,000 individuals in which precise measures of environmental exposure and outcome had been made would be adequately powered (95% power, critical α P = 1 × 10−4); however, studies that imprecisely estimated environmental exposures and phenotypes, as is often the case in epidemiological studies, would require a sample collection ∼50-fold larger to afford comparable power.
Although the expected range of effects that are realistic for gene-lifestyle interactions in type 2 diabetes remains unclear, a doubling of the genetic risk estimate in the group exposed to adverse lifestyle factors compared with those who are unexposed (βGE = 2) is at the upper end of the interaction effect estimate ranges reported for common variants and common exposures (10). It is reasonable to conclude, therefore, that most of the interaction studies published to date report “lucky” true-positive results or false-positive results that may be underpinned by analytical and reporting biases. The replication of few examples of gene-lifestyle interactions in type 2 diabetes suggests that the literature is composed largely of the latter. Despite this, recent developments in the ways genetic association studies are performed, such as adoption of hypothesis-free approaches, the availability of comprehensive genotype arrays in large sample collections, global collaborations, and more rigorous analysis and reporting of data, have led to the emergence of many reproducible genetic association signals for type 2 diabetes and related glycemic traits, which has spurred a number of large-scale studies of gene-lifestyle interactions.
Using genome-wide association studies to inform the selection of loci for studies of gene-lifestyle interactions
The identification of more than 50 genetic loci that are reproducibly associated with type 2 diabetes (20) (Fig. 2) and 53 additional loci for glucose and insulin concentrations (21) has fueled multiple studies in which these loci have been tested for interactions with lifestyle risk factors for type 2 diabetes. One of the first publications of this kind focused on the interaction between the FTO rs9939609 variant and physical activity. Two cross-sectional cohort studies (22,23) and one clinical trial analysis (24), published at approximately the same time, provided nominal evidence that physical activity modifies the effects of the FTO variant on BMI or adipose tissue accumulation. Replication studies achieved mixed results; thus, we sought a definitive answer by conducting a prospective meta-analysis including 45 adult (n = 218,166) and 9 pediatric (n = 19,268) cohorts (25). Although the study yielded a statistically significant interaction summary statistic (Pinteraction= 0.005), which was directionally consistent with the original studies’ findings (22), the effect estimate was heterogeneous (I2 = 36%), suggesting the presence of one or more latent effect-modifiers (i.e., unidentified factors that change the magnitude of FTO’s effect on BMI). Further data exploration determined that the source of this heterogeneity was the geographic origin of the cohorts, with the interaction effect being driven almost entirely by the North American cohorts. Although this geographic difference remains unexplained, the observation strongly suggests that physical activity is not the causal effect-modifier; instead, factors that correlate with physical activity in North American but not in European cohorts, such as specific dietary factors, are likely to be the causal modifiers of FTO’s obesogenic effects. It is also important to bear in mind that almost all studies reporting significant FTO-lifestyle interactions are cross-sectional observational studies, from which causal effects and causal direction are almost impossible to ascertain. Thus, even if a causal relationship underlies the results reported above, it is possible that the direction of effect is reversed (i.e., a direct effect of FTO variation on lifestyle behaviors, which is stronger in fatter compared with leaner people). These alterative explanations are important to consider when discussing, as many do, the potential translational implications of studies of gene-lifestyle interactions.
Figure 2.
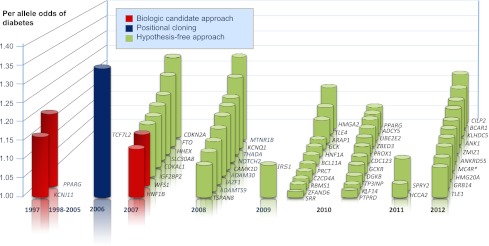
The evolving landscape of established type 2 diabetes-associated genetic loci. Effect estimates and list of loci are adapted from reference 21. Variants in CCND2 and GIPR (not shown in Fig.) have sex-heterogeneous effects, with the former having a larger effect in males and the latter in females (21). *This locus was initially associated with type 2 diabetes by Renström et al. (67).
The first large-scale study examining the interaction of established type 2 diabetes loci and physical activity included 16,000 men and women from southern Sweden, of whom 2,200 went on to develop diabetes during the ensuing 25 years of follow-up (27). Of the 17 established diabetes loci examined, the study identified a single locus, the noncoding polymorphism rs4430796 at the diabetes gene HNF1B, that interacted with baseline physical activity levels as estimated by questionnaire (Bonferroni corrected Pinteraction= 0.015). In homozygotes for the nonrisk allele (A), baseline physical activity apparently protected against the development of type 2 diabetes, as one would predict from previous studies of physical activity and diabetes; however, in carriers of the rs4430796 risk allele, the protective effect of physical activity appeared to be diminished in a dose-dependent fashion.
In a separate cohort from southern Sweden of ∼25,000, two gene-diet interactions have been reported in relation to type 2 diabetes. Sonestedt et al. (28) reported that dietary fat and dietary carbohydrate intake obtained by questionnaire modified the effect of the rs10423928 variant at GIPR on incident diabetes, such that the odds of diabetes associated with the established diabetes risk allele (A) were highest in individuals who consumed low levels of dietary fat or high levels of dietary carbohydrate. In the second study, Hindy et al. (29) reported that the diabetogenic effect of the TCF7L2 variant rs7903146 was augmented in individuals consuming high levels of dietary fiber; although the test of interaction was nominally statistically significant (Pinteraction= 0.049), it was no longer significant after correction for multiple testing. The findings of Hindy et al. contrast with those reported earlier in the Nurses Health Study, where Cornelis et al. (30) reported that for a correlated variant (rs12255372; r2 = 0.73 and D' = 0.93 with the rs7903146 TCF7L2 variant in white Europeans: www.hapmap.org), the effect of the diabetogenic genotype was greatest in women who reported consuming diets with a high glycemic load or index (uncorrected Pinteraction = 0.03). A further study of 46,000 individuals (31) reported no evidence of interaction between the rs4506565 variant (r2 = 0.92 and D' = 1.00 with the rs7903146 TCF7L2 variant in Europeans: www.hapmap.org) and dietary whole-grain intake in relation to insulin or glucose concentrations (nominal Pinteraction = 0.88).
Qi et al. (32) studied the interaction of a Western dietary pattern assessed by questionnaire and a genetic risk score (GRS) consisting of 10 genetic loci that had previously been reproducibly associated with type 2 diabetes (20). Cross-sectional analyses were conducted in a cohort of 1,196 prevalent and incident case participants with diabetes and 1,337 matched control participants from the Health Professionals Follow-up Study (32). The Western diet score was more strongly associated with diabetes risk in the health professionals with a higher GRS and less so in those with a lower GRS (Pinteraction = 0.02). Interaction analyses focusing on specific components of the Western diet score indicated that consumption of red and processed meat underlies the interactions described above and that heme iron intake, in particular, may be the central component of the diet score driving the interaction with the GRS. However, an analysis of ∼50,000 nondiabetic individuals by the Cohorts for Heart and Aging Research in Genomic Epidemiology consortium failed to find any evidence that established glucose- or insulin-associated loci modify the effects of Western dietary pattern on fasting insulin or glucose levels (33).
Individual groups and large consortia have embarked on genome-wide association study(GWAS) analyses stratified by potential effect modifiers such as sex and BMI. These studies are often much larger than single-cohort analyses but are restricted to cross-sectional data, which may hinder the interpretation of results for the reasons discussed above. This is so, particularly when focusing on BMI, because the onset of diabetes can correspond with weight loss as a consequence of lifestyle changes in the immediate aftermath of a diagnosis, treatment, or of the disease process itself. Nevertheless, in an analysis of 2,112 lean type 2 diabetes case subjects, 4,123 obese type 2 diabetes case subjects, and 54,412 unstratified nondiabetic control subjects, Perry et al. (34) identified a LAMA1 variant that conveyed a significantly higher odds of diabetes in lean compared with obese diabetic case subjects. Similar BMI stratum-specific genetic effects were observed for 29 of the 36 type 2 diabetes loci (binomial P = 0.0002) that had been identified previously in unstratified GWAS meta-analyses performed by the DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) consortium (20). These results indicate that when diabetes develops in a person who is lean, genetic risk factors are more likely to be present than in someone who is obese and develops the disease or that weight loss enhances the genetic risk of diabetes.
Genetic analyses performed in clinical trials involving intensive lifestyle modification provide an important adjunct to the epidemiological literature on gene-lifestyle interactions in type 2 diabetes. On one hand, a major advantage of randomized controlled trials is that interaction effects observed in trials are likely to reflect causal processes, whereas those observed in epidemiological investigations are more prone to confounding and reverse causality. Other advantages of well-designed clinical trials include relatively precise estimates of the environmental exposures (treatments) and of the phenotypes, careful ascertainment of participants, randomization of exposures, close predetermined follow-up, and hypothesis-driven design. On the other hand, trials are often smaller than epidemiologic cohorts, control poorly for changes in behavior outside the intervention sessions, and typically consist of individuals at high risk of diabetes; hence, results may not be generalizable to other population subgroups.
Only two randomized controlled trials, the Diabetes Prevention Program (DPP) from the U.S. (35) and the Finnish Diabetes Prevention Study (DPS) (36), have reported results for gene-lifestyle interactions in relation to diabetes incidence. Both studies focused on people at high risk of developing type 2 diabetes and implemented almost identical lifestyle intervention protocols. The DPP randomized 1,079 participants to an intensive lifestyle intervention, 1,082 to a placebo control arm, and 1,073 to metformin treatment. In the Finnish DPS, 522 participants were randomized to a lifestyle or control intervention. The DPP is well powered (∼80%) to detect genetic effects, with a hazard ratio of 1.2, but has appreciably lower power to detect gene-treatment interactions (37,38); statistical power to detect interactions in the Finnish DPS is less than in the DPP owing to its smaller sample size.
Notwithstanding the sample size constraints of the DPP, a number of interesting findings relating to gene-treatment interactions have emerged from the trial. For example, lifestyle intervention offsets the risk conveyed by the diabetogenic alleles at the TCF7L2 rs7903146 (39) and ENPP1 K121Q (40) loci, or by a genetic risk score consisting of 34 type 2 diabetes-associated variants (41). Elsewhere, the DPP investigators reported that the CDK2NA/B rs10811661 variant diminishes the effects of lifestyle intervention on diabetes risk and on estimated insulin secretion (Pinteraction ≤ 0.05) (37); interestingly a subsequent cohort study of 8,600 nondiabetic Swedish adults reported directionally consistent interactions between the same genotype and physical activity levels on the odds of impaired glucose regulation and on continuous 2-h glucose concentrations (Pinteraction ≤ 0.015) (27).
Hypothesis-free discovery of gene-lifestyle interaction effects
The decision to carry forward findings from conventional GWAS experiments to detect gene-lifestyle interactions is a simple, pragmatic, and relatively cost-efficient strategy. However, of the many loci associated with cardiometabolic traits, few have been reproducibly shown to interact with environmental factors; FTO (physical activity interactions in obesity) (25), chromosome 9q21 variants (prudent diet interactions in cardiovascular disease and myocardial infarction) (42), and an obesity GRS (sugar-sweetened beverages interaction in obesity) (43) are rare examples of gene-lifestyle interactions in cardiometabolic traits that have been robustly replicated.
The paucity of replicated examples of GWAS-derived loci that interact with lifestyle factors may be due to the low prioritization of follow-up studies by investigators and journal editors or that not all GWAS-derived loci have been examined for interactions in well-designed studies. Alternatively, it is possible that the statistical approaches used in conventional GWAS experiments bias against the detection of variants that interact with environmental factors that are reasonably prevalent within the populations in which the GWAS are performed. Indeed, the GWAS ranking system is typically based on the P value derived from the main effect regression model for each single nucleotide polymorphism (SNP). Of note, common, disease-associated variants have relatively small effect sizes (typically odds ratios <1.4 per risk allele); thus, for a genetic association signal to exceed the conservative genome-wide probability threshold used in most GWAS (P = 5 × 10−8), the estimates of the genetic effect are relatively consistent in magnitude within and between the populations included in GWAS meta-analyses, as reflected in the narrow CIs for the odds ratios of the top-ranked loci and low heterogeneity estimates. Broadly speaking, one would expect that the larger the magnitude of a GEI or gene-gene effect, the greater the variance associated with the main effects for the genetic and/or environmental components (44). Hence, with some exceptions (25,27), it is likely that the gene variants that are most relevant for GEI are those that rank poorly in most GWAS meta-analyses.
Genome-wide interaction studies have potential to identify gene variants that influence diabetes risk that might not be detected using hypothesis-driven approaches. However, the statistical power limitations of such studies when applying conventional tests of interaction, combined with the challenges of identifying large cohort collections with appropriately characterized environmental, genetic, and phenotypic data, pose challenges that conventional genetic association studies do not face. Several methods have been developed to mitigate these challenges; among the most promising is the joint meta-analysis approach, which is derived from the model with two degrees of freedom popularized by Kraft et al. (45) and developed further by Manning et al. (46). Manning et al. (47) went on to apply the joint meta-analysis approach in a genome-wide study of 52 cohorts in which they tested for SNP main effects and interactions (with BMI) on fasting glucose and insulin levels. The analysis yielded novel experiment-wide association signals for main effects, but none was discovered for interactions.
Recognizing that heterogeneous effect estimates are a signature of loci involved in interactions, Paré et al. (48) and Visscher and Posthuma (49) developed methods that model genetic associations with genotypic variance estimates rather than with phenotypic means, as is the case in conventional GWAS experiments. Both approaches involve two key steps: in step 1, a phenotypic variance estimate is obtained for each of the genotypes at a SNP locus. A statistical comparison of these variance estimates is then made and a P value obtained. These P values are ranked from lowest to highest, and those that exceed an experiment-wide threshold are carried forward to step 2, where conventional, pairwise tests of GEI are performed for an array of environmental exposures, with the intent of identifying one or more that underlie the SNP’s heterogeneous effect estimate revealed in step 1. In a recent application of method described by Visscher and Posthuma, Yang et al. (26) performed a meta-analysis for height and BMI in 170,000 samples and identified a single locus for BMI that met the genome-wide significance threshold; intriguingly, this locus was FTO, the most plausible candidate for GEI in obesity currently known. There was no genome-wide significant discovery for height, which is perhaps unsurprising given that this trait is under much tighter genetic control than weight and varies much less than weight across the adult life span.
Pharmacogenetics
Pharmacogenetics is a specialized example of GEI. Here, the environmental exposure is drug treatment; by studying interactions between gene variants and treatment, investigators seek to identify variants that are associated with adequate or inadequate response to diabetes therapies. The pharmacogenetics of diabetes therapies have been extensively reviewed elsewhere (50–52). However, three examples illustrate successful approaches and the potential clinical utility of pharmacogenetics in diabetes:
Firstly, studies of HNF1A mutations that cause a form of maturity-onset diabetes of the young showed that carriers of these mutations, who are often misdiagnosed as having type 1 or type 2 diabetes, respond better to sulfonylureas than metformin, thus facilitating their transition off insulin or metformin (53,54).
Secondly, common genetic variation in the gene that encodes a transporter responsible for disposing of metformin (MATE1, encoded by SLC47A1) has been associated with metformin response in a retrospective patient cohort (55), a preliminary finding corroborated in the DPP clinical trial (56).
Finally, in a discovery GWAS of 1,000 metformin-treated patients from the Genetics of Diabetes Audit and Research Tayside (GoDARTS) study, a locus including the ATM gene was associated with metformin response. This discovery was initially replicated in independent GoDARTS and UK Prospective Diabetes Study cohorts (57) as well as subsequent cohorts that were similarly ascertained (58); however, it was not reproduced in the DPP clinical trial, which differs from the earlier studies by its experimental, prospective design and its enrollment of nondiabetic participants in whom metformin was used for diabetes prevention (56). If ATM is eventually established as a causal regulator of metformin response, this will provide a novel unexpected role for this established oncogene in diabetes treatment. The challenge for pharmacogenetics is to establish clinical utility, which in adult diabetes is currently limited to the HNF1A paradigm. An elegant example of how pharmacogenetics influences therapy in neonatal diabetes has been reviewed by Greeley et al. (59).
Future directions
We have emphasized GEI in this Perspective because this is where most of the published research has been focused to date. A complementary set of disease predictors is being generated with the emergence of comprehensive metabolomic approaches, in which circulating small molecules present in human fluids are assayed in a high-throughput manner through liquid chromatography and mass spectroscopy. These molecules represent metabolic readouts of cellular states at a systems level and reflect the output of gene products and also their interactions with the environment. Using these platforms, independent groups have established a metabolomic signature of branched chain and aromatic amino acids as associated with obesity and insulin resistance (60) as well as future diabetes (61). How genes regulate circulating levels of these molecules, what they tell us about gene function, how much they reflect environmental factors, and to what extent they provide orthogonal information for diabetes prediction and treatment response is the subject of intense investigation. The participant-level integrated assessment of variation in the genome, metabolome, and other aspects of the physiome (e.g., microbiome, transcriptome, and proteome) in large cohorts has not previously been possible, but with recent advances in technology and analytical methods, and cost reductions, this is now feasible and is evolving into a new field called systems epidemiology (Fig. 1). This topic is eloquently reviewed elsewhere (62,63).
Although recent genetic discoveries in metabolic traits have typically illustrated novel pathways, pointed toward fundamental biology, confirmed prior epidemiological observations, highlighted the role of β-cell dysfunction in type 2 diabetes, and provided possible targets for pharmacotherapy, their role in genetic prediction is less clear. This is partly so because even in aggregate, they only explain a relatively small fraction of the disease’s heritability (41,64–66). The latter is likely due to insufficient sample sizes to detect small effects, a nearly exclusive focus on populations of European descent, an imperfect capture of infrequent genetic variants, an incomplete ascertainment of alternate (non-SNP) forms of genetic variation, and the limited exploration of additional genetic models, including those involving GEI. As the community embraces complementary approaches that include systematic fine-mapping, custom-made replication, denser genotyping arrays, platforms that focus on functional variation, next-generation sequencing techniques, expansion to non-European populations, and integration of other global biological measurements with genetic data, the coming years will continue to elucidate the genetic architecture of metabolic phenotypes and its interaction with the environment. A more refined characterization of the molecular basis of type 2 diabetes can then be translated into more detailed disease nosology, appropriate targeting of more effective and better tolerated therapeutic or preventive strategies, more rational and efficiently designed clinical trials, and stratification of risk groups so that costly public health interventions can be deployed intelligently.
We speculate that the future of diabetes medicine may involve genetic and molecular biomarker screening in patients to inform the prescription of lifestyle or drug therapy for diabetes prevention or management. However, the translation of this vision into clinical practice will require structured research programs that combine observational epidemiology to generate relevant hypotheses and experimental studies that test these hypotheses and demonstrate cause and effect. When reliable and causal interactions are discovered, it will be necessary to conduct studies proving that the inclusion of this information into conventional risk prediction algorithms improves predictive accuracy and/or reclassification, or that stratified medicine informed by biomarker data improves treatment outcomes; in addition, it will also be necessary to show that these strategies are cost-effective compared with conventional approaches.
The concept of stratified medicine (otherwise known as personalized or precision medicine) is an area of considerable research interest both within the pharmaceutical industry and academia. The European Union-Innovative Medicines Initiative (IMI)–funded DIabetes REsearCh on patient sTratification (DIRECT) study (www.direct-diabetes.org) is an academic-industry program that addresses most of these questions, spanning a discovery phase of comprehensive phenotyping, and large-scale omic analysis through to a validation and clinical trial phase to establish utility of a biomarker-stratified approach to diabetes medicine. The study focuses on two phenotypes where stratification approaches can be applied (glycemic deterioration of prediabetes and diabetes) and therapeutic response (to sulfonylureas, metformin, GLP-1R agonists, and obesity surgery). For each phenotype, the plan is to integrate physiological parameters with lifestyle measures, genetic (GWAS and sequencing), transcriptomic, metabolomic, proteomic, and metagenomic data to enable a comprehensive analysis for discovery of stratification and surrogate biomarkers. Biomarker stratified clinical trials will then be done to establish the utility of biomarker led therapeutics over traditional nonbiomarker led studies.
Similar technologies can be applied in the framework of comparative medicine. The National Institutes of Health-sponsored Glycemia Reduction Approaches for Diabetes: A Comparative Effectiveness Study (GRADE) trial will for the first time perform head-to-head comparisons of representative agents from four major drug classes for type 2 diabetes treatment—the sulfonylurea glimepiride, the DPP-4 inhibitor sitagliptin, the GLP-1 agonist liraglutide, and the basal insulin glargine—as adjuncts to metformin in achieving glycemic control. This large randomized clinical trial, planning to enroll 6,000 participants in 40+ centers throughout the United States, will be launched in the spring of 2013 and will collect phenotypes, covariates, end points, and biomaterials on all participants to enable the deployment of omics techniques to examine prediction and response to pharmacological manipulation.
Conclusions
We are witnessing a time in biomedical research when systems can be queried globally to establish the metabolic state of the organism in a single experiment. Such technologies can also be deployed across populations, tissues, and environmental conditions. The integration of all this information and its interpretation into a cogent vision presents enormous challenges, not least of which is the scientific imperative of reproducibility. As rigorous analytical standards are implemented and international collaborations enable the pursuit of these fundamental questions at an adequate scale, we stand on the verge of a true transformation of medicine as applied to the individual patient. While discovering and replicating evidence of GEIs is proceeding, the process of discussing and planning how such data can be translated into the clinical arena should already be underway. Current discoveries should also prompt us to consider the benefits and challenges (e.g., ethical, economic, logistic) that using genetic information in diabetes medicine is likely to present, which may enable the rapid translation of human genetics research into clinical practice.
Acknowledgments
P.W.F. is funded by Excellence in Diabetes Research in Sweden (EXODIAB), Lund University, Umeå University, Region Skåne Health Authority, the Swedish Diabetes Association, the Swedish Heart-Lung Foundation, the Swedish Research Council, the European Union, Novo Nordisk, and the National Institutes of Health (National Institute of Child Health and Human Development). J.C.F. is supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01 DK-072041 and R01 DK-088214.A1.
P.W.F. has received speaking honoraria from Novo Nordisk and other academic and not-for-profit organizations. J.C.F. has received consulting honoraria from Lilly and Pfizer.
P.W.F. and J.C.F. are DPP investigators. P.W.F. and E.P. are IMI-DIRECT Study investigators. This article is entirely the work of the authors, unless indicated otherwise.
No potential conflicts of interest relevant to this article were reported.
P.W.F. planned and wrote the initial draft of the manuscript, revised and edited its content, and approved its submission. E.P. and J.C.F. wrote key sections of the manuscript, revised and edited its content, and approved its submission.
The authors’ opinions are the result of many past interactions with mentors, colleagues, collaborators, students, and fellows, for which the authors express their thanks. The authors also thank Shafqat Ahmad (Genetic and Molecular Epidemiology Unit, Lund University, Sweden) for assistance with reference formatting.
References
- 1.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 2004;27:1047–1053 [DOI] [PubMed] [Google Scholar]
- 2.Ayach BB, Korda H. Type 2 diabetes epidemic in First Nations people of Canada. Ethn Dis 2010;20:300–303 [PubMed] [Google Scholar]
- 3.King H, Rewers M, WHO Ad Hoc Diabetes Reporting Group Global estimates for prevalence of diabetes mellitus and impaired glucose tolerance in adults. Diabetes Care 1993;16:157–177 [DOI] [PubMed] [Google Scholar]
- 4.Franks PW, Looker HC, Kobes S, et al. Gestational glucose tolerance and risk of type 2 diabetes in young Pima Indian offspring. Diabetes 2006;55:460–465 [DOI] [PubMed] [Google Scholar]
- 5.Schulz LO, Bennett PH, Ravussin E, et al. Effects of traditional and western environments on prevalence of type 2 diabetes in Pima Indians in Mexico and the U.S. Diabetes Care 2006;29:15866–1871 [DOI] [PubMed] [Google Scholar]
- 6.Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet 1962;14:353–362 [PMC free article] [PubMed] [Google Scholar]
- 7.Bouchard C. Genes and obesity. Preface. Prog Mol Biol Transl Sci 2010;94:xiii. [DOI] [PubMed] [Google Scholar]
- 8.Southam L, Soranzo N, Montgomery SB, et al. Is the thrifty genotype hypothesis supported by evidence based on confirmed type 2 diabetes- and obesity-susceptibility variants? Diabetologia 2009;52:1846–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aschard H, Chen J, Cornelis MC, Chibnik LB, Karlson EW, Kraft P. Inclusion of gene-gene and gene-environment interactions unlikely to dramatically improve risk prediction for complex diseases. Am J Hum Genet 2012;90:962–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franks PW, Mesa JL, Harding AH, Wareham NJ. Gene-lifestyle interaction on risk of type 2 diabetes. Nutr Metab Cardiovasc Dis 2007;17:104–124 [DOI] [PubMed] [Google Scholar]
- 11.Luan J, Browne PO, Harding AH, et al. Evidence for gene-nutrient interaction at the PPARgamma locus. Diabetes 2001;50:686–689 [DOI] [PubMed] [Google Scholar]
- 12.Kahara T, Takamura T, Hayakawa T, et al. PPARgamma gene polymorphism is associated with exercise-mediated changes of insulin resistance in healthy men. Metabolism 2003;52:209–212 [DOI] [PubMed] [Google Scholar]
- 13.Franks PW, Luan J, Browne PO, et al. Does peroxisome proliferator-activated receptor gamma genotype (Pro12ala) modify the association of physical activity and dietary fat with fasting insulin level? Metabolism 2004;53:11–16 [DOI] [PubMed] [Google Scholar]
- 14.Memisoglu A, Hu FB, Hankinson SE, et al. Interaction between a peroxisome proliferator-activated receptor gamma gene polymorphism and dietary fat intake in relation to body mass. Hum Mol Genet 2003;12:2923–2929 [DOI] [PubMed] [Google Scholar]
- 15.Robitaille J, Després JP, Pérusse L, Vohl MC. The PPAR-gamma P12A polymorphism modulates the relationship between dietary fat intake and components of the metabolic syndrome: results from the Québec Family Study. Clin Genet 2003;63:109–116 [DOI] [PubMed] [Google Scholar]
- 16.Lindi VI, Uusitupa MI, Lindström J, et al. Finnish Diabetes Prevention Study Association of the Pro12Ala polymorphism in the PPAR-gamma2 gene with 3-year incidence of type 2 diabetes and body weight change in the Finnish Diabetes Prevention Study. Diabetes 2002;51:2581–2586 [DOI] [PubMed] [Google Scholar]
- 17.Palla L, Higgins JP, Wareham NJ, Sharp SJ. Challenges in the use of literature-based meta-analysis to examine gene-environment interactions. Am J Epidemiol 2010;171:1225–1232 [DOI] [PubMed] [Google Scholar]
- 18.Franks PW, Nettleton JA. Invited commentary: Gene X lifestyle interactions and complex disease traits—inferring cause and effect from observational data, sine qua non. Am J Epidemiol 2010;172:992–997; discussion 998–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong MY, Day NE, Luan JA, Chan KP, Wareham NJ. The detection of gene-environment interaction for continuous traits: should we deal with measurement error by bigger studies or better measurement? Int J Epidemiol 2003;32:51–57 [DOI] [PubMed] [Google Scholar]
- 20.Morris AP, Voight BF, Teslovich TM, et al.; DIAGRAM Consortium. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012;44:981–990 [DOI] [PMC free article] [PubMed]
- 21.Scott RA, Lagou V, Welch RP, et al. DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet 2012;44:991–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andreasen CH, Stender-Petersen KL, Mogensen MS, et al. Low physical activity accentuates the effect of the FTO rs9939609 polymorphism on body fat accumulation. Diabetes 2008;57:95–101 [DOI] [PubMed] [Google Scholar]
- 23.Rampersaud E, Mitchell BD, Pollin TI, et al. Physical activity and the association of common FTO gene variants with body mass index and obesity. Arch Intern Med 2008;168:1791–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franks PW, Jablonski KA, Delahanty LM, et al. Diabetes Prevention Program Research Group Assessing gene-treatment interactions at the FTO and INSIG2 loci on obesity-related traits in the Diabetes Prevention Program. Diabetologia 2008;51:2214–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kilpeläinen TO, Qi L, Brage S, et al. Physical activity attenuates the influence of FTO variants on obesity risk: a meta-analysis of 218,166 adults and 19,268 children. PLoS Med 2011;8:e1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang J, Loos RJ, Powell JE, et al. FTO genotype is associated with phenotypic variability of body mass index. Nature 2012;490:267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brito EC, Lyssenko V, Renström F, et al. Previously associated type 2 diabetes variants may interact with physical activity to modify the risk of impaired glucose regulation and type 2 diabetes: a study of 16,003 Swedish adults. Diabetes 2009;58:1411–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonestedt E, Lyssenko V, Ericson U, et al. Genetic variation in the glucose-dependent insulinotropic polypeptide receptor modifies the association between carbohydrate and fat intake and risk of type 2 Diabetes in the Malmö Diet and Cancer Cohort. J J Clin Endocrinol Metab 2012;97:E810–E818 [DOI] [PubMed] [Google Scholar]
- 29.Hindy G, Sonestedt E, Ericson U, et al. Role of TCF7L2 risk variant and dietary fibre intake on incident type 2 diabetes. Diabetologia 2012;55:2646–2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cornelis MC, Qi L, Kraft P, Hu FB. TCF7L2, dietary carbohydrate, and risk of type 2 diabetes in US women. Am J Clin Nutr 2009;89:1256–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nettleton JA, McKeown NM, Kanoni S, et al. MAGIC Investigators Interactions of dietary whole-grain intake with fasting glucose- and insulin-related genetic loci in individuals of European descent: a meta-analysis of 14 cohort studies. Diabetes Care 2010;33:2684–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qi L, Cornelis MC, Zhang C, van Dam RM, Hu FB. Genetic predisposition, Western dietary pattern, and the risk of type 2 diabetes in men. Am J Clin Nutr 2009;89:1453–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nettleton JA, Hivert MF, Lemaitre RN, et al. Meta-analysis investigating associations between healthy diet and fasting glucose and insulin levels and modification by loci associated with glucose homeostasis in data from 15 cohorts. Am J Epidemiol 2013;177:103–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perry JR, Voight BF, Yengo L, et al. MAGIC. DIAGRAM Consortium. GIANT Consortium Stratifying type 2 diabetes cases by BMI identifies genetic risk variants in LAMA1 and enrichment for risk variants in lean compared to obese cases. PLoS Genet 2012;8:e1002741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knowler WC, Barrett-Connor E, Fowler SE, et al. Diabetes Prevention Program Research Group Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002;346:393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tuomilehto J, Lindström J, Eriksson JG, et al. Finnish Diabetes Prevention Study Group Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 2001;344:1343–1350 [DOI] [PubMed] [Google Scholar]
- 37.Moore AF, Jablonski KA, McAteer JB, et al. Diabetes Prevention Program Research Group Extension of type 2 diabetes genome-wide association scan results in the diabetes prevention program. Diabetes 2008;57:2503–2510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jablonski KA, McAteer JB, de Bakker PI, et al. Diabetes Prevention Program Research Group Common variants in 40 genes assessed for diabetes incidence and response to metformin and lifestyle intervention in the diabetes prevention program. Diabetes 2010;59:2672–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Florez JC, Jablonski KA, Bayley N, et al. Diabetes Prevention Program Research Group TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N Engl J Med 2006;355:241–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore AF, Jablonski KA, Mason CC, et al. Diabetes Prevention Program Research Group The association of ENPP1 K121Q with diabetes incidence is abolished by lifestyle modification in the diabetes prevention program. J Clin Endocrinol Metab 2009;94:449–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hivert MF, Jablonski KA, Perreault L, et al. DIAGRAM Consortium. Diabetes Prevention Program Research Group Updated genetic score based on 34 confirmed type 2 diabetes Loci is associated with diabetes incidence and regression to normoglycemia in the diabetes prevention program. Diabetes 2011;60:1340–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Do R, Xie C, Zhang X, et al. INTERHEART investigators The effect of chromosome 9p21 variants on cardiovascular disease may be modified by dietary intake: evidence from a case/control and a prospective study. PLoS Med 2011;8:e1001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qi Q, Chu AY, Kang JH, et al. Sugar-sweetened beverages and genetic risk of obesity. N Engl J Med 2012;367:1387–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Franks PW. Gene × environment interactions in type 2 diabetes. Curr Diab Rep 2011;11:552–561 [DOI] [PubMed] [Google Scholar]
- 45.Kraft P, Yen YC, Stram DO, Morrison J, Gauderman WJ. Exploiting gene-environment interaction to detect genetic associations. Hum Hered 2007;63:111–119 [DOI] [PubMed] [Google Scholar]
- 46.Manning AK, LaValley M, Liu CT, et al. Meta-analysis of gene-environment interaction: joint estimation of SNP and SNP × environment regression coefficients. Genet Epidemiol 2011;35:11–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manning AK, Hivert MF, Scott RA, et al. DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium. Multiple Tissue Human Expression Resource (MUTHER) Consortium A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 2012;44:659–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paré G, Cook NR, Ridker PM, Chasman DI. On the use of variance per genotype as a tool to identify quantitative trait interaction effects: a report from the Women’s Genome Health Study. PLoS Genet 2010;6:e1000981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Visscher PM, Posthuma D. Statistical power to detect genetic Loci affecting environmental sensitivity. Behav Genet 2010;40:728–733 [DOI] [PubMed] [Google Scholar]
- 50.Pearson ER. Pharmacogenetics in diabetes. Curr Diab Rep 2009;9:172–181 [DOI] [PubMed] [Google Scholar]
- 51.Manolopoulos VG, Ragia G, Tavridou A. Pharmacogenomics of oral antidiabetic medications: current data and pharmacoepigenomic perspective. Pharmacogenomics 2011;12:1161–1191 [DOI] [PubMed] [Google Scholar]
- 52.Huang C, Florez JC. Pharmacogenetics in type 2 diabetes: potential implications for clinical practice. Genome Med 2011;3:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 2003;362:1275–1281 [DOI] [PubMed] [Google Scholar]
- 54.Shepherd M, Pearson ER, Houghton J, Salt G, Ellard S, Hattersley AT. No deterioration in glycemic control in HNF-1alpha maturity-onset diabetes of the young following transfer from long-term insulin to sulphonylureas. Diabetes Care 2003;26:3191–3192 [DOI] [PubMed] [Google Scholar]
- 55.Becker ML, Visser LE, van Schaik RHN, Hofman A, Uitterlinden AG, Stricker BHC. Genetic variation in the multidrug and toxin extrusion 1 transporter protein influences the glucose-lowering effect of metformin in patients with diabetes: a preliminary study. Diabetes 2009;58:745–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Florez JC, Jablonski KA, Taylor A, et al. Diabetes Prevention Program Research Group The C allele of ATM rs11212617 does not associate with metformin response in the Diabetes Prevention Program. Diabetes Care 2012;35:1864–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou K, Bellenguez C, Spencer CC, et al. GoDARTS and UKPDS Diabetes Pharmacogenetics Study Group. Wellcome Trust Case Control Consortium 2. MAGIC investigators Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet 2011;43:117–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Leeuwen N, Nijpels G, Becker ML, et al. A gene variant near ATM is significantly associated with metformin treatment response in type 2 diabetes: a replication and meta-analysis of five cohorts. Diabetologia 2012;55:1971–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Greeley SA, Naylor RN, Philipson LH, Bell GI. Neonatal diabetes: an expanding list of genes allows for improved diagnosis and treatment. Curr Diab Rep 2011;11:519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Newgard CB, An J, Bain JR, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 2009;9:311–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang TJ, Larson MG, Vasan RS, et al. Metabolite profiles and the risk of developing diabetes. Nat Med 2011;17:448–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Voy BH. Systems genetics: a powerful approach for gene-environment interactions. J Nutr 2011;141:515–519 [DOI] [PubMed] [Google Scholar]
- 63.Hu FB. Metabolic profiling of diabetes: from black-box epidemiology to systems epidemiology. Clin Chem 2011;57:1224–1226 [DOI] [PubMed] [Google Scholar]
- 64.Meigs JB, Shrader P, Sullivan LM, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med 2008;359:2208–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lyssenko V, Jonsson A, Almgren P, et al. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med 2008;359:2220–2232 [DOI] [PubMed] [Google Scholar]
- 66.de Miguel-Yanes JM, Shrader P, Pencina MJ, et al. MAGIC Investigators. DIAGRAM+ Investigators Genetic risk reclassification for type 2 diabetes by age below or above 50 years using 40 type 2 diabetes risk single nucleotide polymorphisms. Diabetes Care 2011;34:121–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Renström F, Payne F, Nordström A, et al. GIANT Consortium Replication and extension of genome-wide association study results for obesity in 4923 adults from northern Sweden. Hum Mol Genet 2009;18:1489–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]