

Abstract
Phospholipids (PLs), well known for their fundamental role in cellular structure, play critical signaling roles via their derivatives and cleavage products acting as second messengers in signaling cascades. Recent work has shown that intact PLs act as signaling molecules in their own right by modulating the activity of nuclear hormone transcription factors responsible for tuning genes involved in metabolism, lipid flux, steroid synthesis and inflammation. As such, PLs have been classified as novel hormones. This review highlights recent work in PL-driven gene regulation with a focus on the unique structural features of phospholipid-sensing transcription factors and what sets them apart from well known soluble phospholipid transporters.
Keywords: Phospholipid, Nuclear receptor, Signaling, Transcriptional regulation
1. Introduction
1.1. Phospholipids
PLs are ubiquitous to all forms of life serving as the major constituent of the membranes that isolate and protect cells from their external environment, and segregate organelles from the greater cellular milieu. PLs are composed of two hydrophobic tails, donated by a diacylglycerol (DAG), and a hydrophilic head group containing a phosphate, which is frequently conjugated to an additional hydrophilic metabolite (Fig. 1). This amphipathic, bipartite structure drives their spontaneous assembly into bilayers, which compartmentalize the cell and harbor an assortment of proteins, glycans, and other lipids that play critical roles in cell structure, function, metabolism, and signaling.
Fig. 1.
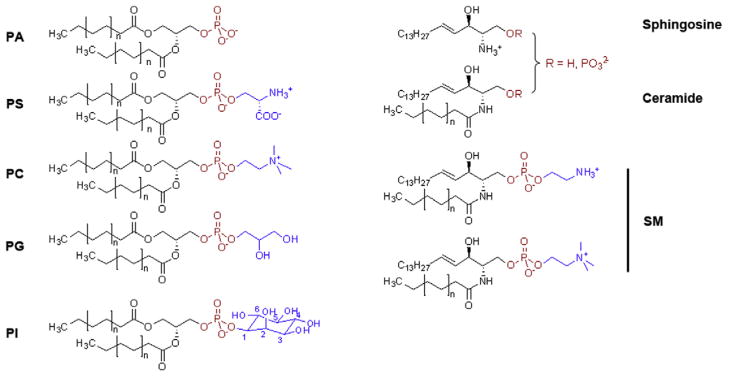
Structures of major phospholipid species. PLs consist of a hydrophobic diacyl tail (black), a phosphate (red), and a polar head group (blue). PA: phosphatidic acid; PS: phosphatidylserine; PC: phosphatidylcholine; PG: phosphatidyl glycerol; PI: phosphatidylinositol; SM: sphingomyelin.
1.1.1. PLs as signaling molecules
Though best known for their role in membrane construction, PLs play integral roles in a number of cellular signaling cascades at and within the membrane bilayer [1]. Arguably the most familiar of these are the IP3/DAG and Akt cascades. In the former, membrane-bound PI-bisphosphate (PIP2) is cleaved by PLC to yield inositol trisphosphate (IP3) and DAG; IP3 is released into the cytoplasm and triggers the release of Ca2+ from the endoplasmic reticulum, while DAG remains in the plasma membrane and activates PKC [2]. PI-trisphosphate (PIP3) is instrumental in recruiting Akt to the plasma membrane, where it is activated by PDK-1 [3]. In more recent years, additional PL derivatives have been implicated in cell signaling. Lysophospholipids, single-chain PLs that include sphingosine-1-phosphate (S1P) and lysophosphatidic acid (LPA), were found to bind and activate G protein coupled receptors (GPCRs) upstream of Ras homolog gene family, member A (RhoA) activation, affecting numerous signaling responses [4]. Furthermore, a family of tail-oxidized PLs are now known to play central roles in the regulation of the plasma membrane and the innate immune system [5]. PLs have therefore emerged as key players in the signal cascades that control many vital biological processes.
1.1.2. PLs outside the membrane
A significant fraction of the cellular PL pool resides outside of the membrane, particularly inside the nucleus. While some of this subpopulation may have structural roles as part of chromatin or the nuclear lamin [6], it is now evident that there is a PL signaling system distinct from that which occurs within the membrane bilayer [7]. PIs again are at the core of the known nuclear lipid signaling pathways [8], and while the nature of nuclear PLs remains enigmatic, it is now understood that PI and PIPs have important functions in the regulation of protein–chromatin interactions [9]. The close association of PLs with DNA [10] suggests that, in addition to their roles in cell structure and signal transduction, PLs play a role in driving gene expression and regulation.
1.1.3. PLs are a new class of hormone
Ernest Starling coined the term “hormone” in 1905, long before the isolation of the first nuclear receptor (NR) in 1958, to describe a substance that is able to travel throughout an organism serving as a chemical messenger to alter cell behavior. PLs have long been thought of as synthesis material for some hormones, but new evidence suggests they are transmitting their own unique signals to alter transcriptional patterns. The vast majority of evidence for direct PL-mediated transcription is among the NR family of transcription factors.
1.2. Nuclear receptors: lipid regulated transcription factors
1.2.1. Nuclear receptor structure and function
NRs are a family of ligand regulated transcription factors that are activated by a diverse group of lipophilic ligands including fatty acids, cholesterol derivatives, steroid hormones, vitamins, dietary components, and xenobiotics [11–14]. These ligands, primarily derived from lipids, act as messengers by transmitting chemical information that reflects the body’s nutritional and endocrine states [15]. This allows for the coordination of growth, reproduction, and homeostasis, and allows the body to appropriately respond to events, such as eating a meal, exercise, or stress.
NRs share a highly conserved multi-domain architecture including a variable N-terminal domain, often referred to as the activation function 1 (AF-1), a DNA binding domain (DBD), a flexible linker region, and a ligand binding domain (LBD) that contains a ligand sensitive transcriptional switch, the AF-2 [12,13]. Ligand dependent NR activation is centered on the LBD, a helical bundle containing a lipophilic cavity that can accommodate ligands. The hydrophobic pockets within NRs typically vary in size and shape to match their cognate hormone [13,14]. A mobile ligand sensing helix, termed the activation function helix (AF-H), responds to a bound ligand by rotating and packing against the LBD. This repositioning completes the AF-2 surface, enabling interaction with coactivator proteins contained in chromatin modifying complexes that promote gene transcription [12]. In the absence of ligand, NRs preferentially interact with corepressor complexes which displace the “active AF-H” from the body of the protein resulting in transcriptional repression [12]. Similarly, NR antagonists alter AF-H positioning to either prevent coactivator binding or promote binding of corepressor proteins to inhibit transcription.
NRs ligands are invariably hydrophobic and freely diffuse across membranes to allow for long-range signal transmission. In this way, hormones affect diverse groups of gene programs involved in pathophysiology ranging from diabetes to cancer making NRs ideal targets for pharmacological intervention. As such, NR-targeting drugs have a myriad of uses ranging from cancer treatments, and contraceptives, to treating allergic reactions and metabolic disorders and represent a major industrial and academic investment in basic research and drug development [14,16,17].
1.2.2. PL-driven NR activation
To date, four NRs have been identified as PL-binding proteins: liver receptor homolog 1 (LRH-1) and steroidogenic factor 1 (SF-1), members of the NR5a class of steroidogenic factor-like NRs; peroxisome proliferator-activated receptor alpha (PPARα), a member of the NR1 thyroid hormone receptor-like family of receptors; and ultraspiracle (USP), the insect homolog of the retinoid X receptor. This review will focus on the compelling evidence for PLs role in regulating these receptors, as well as a family of PL transporters that stimulate NR transactivation.
2. Case studies
2.1. LRH-1
LRH-1 is a member of the NR5, or Ftz-f1, subfamily of NR’s, and regulates the expression of genes involved in development, lipid and glucose homeostasis, steroidogenesis, and cell proliferation [18,19]. During the early stages of development, LRH-1 is responsible for maintaining levels of OCT-4, considered to be a master regulator of pluripotency [20]. Disruption of the LRH-1 gene in mice leads to the loss of Oct4 expression in the epiblast, causing lethality at embryonic day 6.5 [21]. Over expression of LRH-1 is sufficient to reprogram murine somatic cells to pluripotent cells without simultaneous overexpression of OCT-4. This makes LRH-1 the only known transcription factor that can replace OCT-4 in the cellular reprogramming identifying it as a new stem cell factor [22]. It is unknown what role LRH-1 plays in OCT4 regulation beyond development, however, the receptor was recently shown to regulate OCT4 expression in human cancer stem cells [23].
In adults, LRH-1 is expressed in liver, pancreas, intestine, brain and sex glands such as the ovaries and placenta [18,24]. In the liver, LRH-1 is a master regulator of lipid homeostasis [19] regulating bile acid and cholesterol flux through regulation of CYP7A1, which catalyzes the rate-limiting step in bile acid synthesis [18]. LRH-1 also regulates the transcription a number of other lipid, bile, and cholesterol synthesis enzymes and transporters required in the processes of lipid transport to the liver and elimination [25–32]. Recently, LRH-1 has been identified as a direct transcriptional regulator of glucokinase, responsible for glucose capture in the liver [33]. Disruption of the LRH-1 gene in healthy livers not only disrupted lipogenesis but resulted in reduced glycogen synthesis and glycolysis in response to acute and prolonged glucose exposure. Taken together, these studies demonstrate LRH-1’s influences on metabolic homeostasis by linking PL levels to glucose and lipid metabolism.
LRH-1 is also expressed in preadipocytes and adipocytes of estrogen receptor positive breast cancer cells [24]. Here, in conjunction with GATA and protein kinase A, LRH-1 drives the expression of CYP19 (aromatase), increasing the local estrogen concentration to fuel tumor growth [24,34]. Additionally, LRH-1 appears to take part in a positive feedback loop with active estrogen receptor further enhancing these effects [35].
In the colon, LRH-1 plays a markedly different role in cancer development and progression. Here, LRH-1 has been shown to synergize with the beta-catenin/TCF transcriptional complex to enhance the expression of cell proliferation, growth and survival genes such as cyclin’s D1 and E1 [21]. Additionally, LRH-1 has also been found to be overexpressed in gastric cancer [36].
2.1.1. Bound Escherichia coli PLs offer the first clue that LRH-1 may be PL regulated
In 2003, the crystal structure of mouse LRH-1 was reported, showing the receptor held in an active conformation in the absence of a ligand or co-regulatory peptide [37]. This structure suggested that LRH-1 may act in a ligand-independent manner, discouraging efforts to pursue LRH-1 as a drug target despite its therapeutic potential. In 2005, however, subsequent crystal structures of human LRH-1 all revealed a large >1400 Å3 ligand binding pocket (LBP) occupied by a diverse array of PLs including PG, PE, and a rare phosphatidylglycerol–phosphoglycerol [38–40]. Mutations designed to reduce PL binding showed decreased transcriptional activity in reporter gene assays and a decrease in the ability to recruit coregulators and coregulator fragments both in vitro and in cells [39,41]. These exciting new findings showed for the first time that LRH-1 may be regulated by PLs.
2.1.2. LRH-1–PIP interactions
To identify plausible mammalian PL ligands, Krylova et al. assessed binding of LRH-1 to immobilized PLs which revealed that LRH-1 bound to a range of PLs, but bound most strongly to PIP2 and PIP3 species [40]. Lipid binding was confirmed through non-denaturing mass spectrometry [40]. LBP mutations designed to prevent lipid binding decreased the ability of LRH-1 to bind these immobilized lipids [40]. Notably, this assay did not show PC binding for either LRH-1 or SF-1 [40], both of which were later shown to be activated by PC in cells and bind PC in vitro [41,42].
2.1.3. DLPC
Recently, Lee et al. showed that both human and mouse LRH-1 are specifically activated by the exogenous medium chain phosphatidylcholine isoforms – diundecanoyl (DUPC, PC 11:0/11:0) and dilauroyl (DLPC, PC 12:0/12:0) phosphatidylcholine [43]. These medium chain PC agonists selectively activate the receptor in luciferase assays, increase the ability of LRH-1 to interact with the coactivators and increase the production of LRH-1 target genes [43]. Moreover, DLPC lowers serum lipid levels and reduces blood glucose levels in diabetic mice in a LRH-1 dependent manner [43]. The X-ray crystal structure of the LRH-1–DLPC complex in combination with hydrogen–deuterium exchange assays confirmed that DLPC interacts directly with LRH-1 and revealed the mechanism dictating DLPC-driven transcriptional activation [41]. Unlike other NRs that rely on intra-protein interactions to coordinate activation, LRH-1 relies on intramolecular contacts between distal residues in the LBP and the PL to sense and transmit ligand status to the AF-H [41]. Additionally, generation and characterization of apo LRH-1, showed that ligand free LRH-1 LBD has a highly destabilized structure that is profoundly stabilized by lipids [41]. DLPC simultaneously enhanced co-activator peptide recruitment while disfavoring repressor peptide interaction [41]. These recent results show for the first time that LRH-1 is able to dynamically respond to a PL ligand.
2.2. SF-1
SF-1, another member of the Ftz-F1 NR5A subfamily, is a key regulator of steroidogenesis and the development of steroidogenic organs, such as the adrenal cortex and gonads [44]. It is expressed primarily in these tissues, and in tissues along the steroid hormone regulatory axes, including the hypothalamus and pituitary gland [45,46]. Genes involved in nearly all stages of steroid biosynthesis are regulated by SF-1, including those that encode HMG-CoA synthase [47], cholesterol transporters [48–50], 3β steroid dehydrogenase, and many of the cytochrome P450 enzymes that catalyze the conversion of cholesterol into steroid hormones [51].
Dysfunction of SF-1 has been linked to a number of human disorders [52,53]. Mutations in SF-1 have been detected in patients with disorders in sexual development [54–57], ovarian insufficiency [55], and adrenal failure [56], while SF-1 dysregulation has been linked to endometriosis [58] and adrenocortical carcinoma [59]. Like LRH-1, SF-1 makes an alluring drug target, yet a robust understanding of its ligand-binding properties is only now emerging.
However, some headway has been made in identifying synthetic compounds that act upon SF-1. In 2008, a number of inverse agonists for SF-1 were identified [60–62]. Not only could these compounds inhibit SF-1-dependent gene transcription in luciferase assays, they also inhibited StAR expression in human adrenocortical cells [60], suggesting a possible therapeutic value in the treatment of adrenocortical cancers. Isoquinolone-derived inverse agonists were subsequently shown to inhibit the expression of CYP21 and CYP17 mRNA in vitro, with a concurrent reduction in the secretion of aldosterone, cortisol, and DHEA-S, and inhibition of adrenocortical carcinoma cell proliferation [5,63]. These results indicate that pharmacological modulation of SF-1 may be a viable strategy in treating adrenocortical carcinomas, and possibly other human diseases. However, more research is needed to understand the intricacies of ligand-driven SF-1 activity, before its full potential as a drug target can be realized.
2.2.1. E. coli PL binding from early structural studies
The first crystal structures of SF-1 were reported in 2005, showing the LBD in complex with copurified E. coli medium chain PG and PE species [38,40,64]. The binding of SF-1 to immobilized eukaryotic PLs was tested along with LRH-1, and it was found that SF-1 could bind to an array of PL species, including PA, PI, PIP2, and PIP3, with a preference for PIPs phosphorylated at the 3- and 5-carbons [40]. Coactivator recruitment was enhanced by PEs [38,64] and PCs [64] identifying diverse PLs as activating ligands in vitro.
2.2.2. PA versus sphingosine
The discovery that SF-1 could bind exogenous PLs intensified the search for its endogenous ligands. By 2007, mass spectrometry experiments had identified sphingosine, lysoSM, PA, PE, and PI bound to SF-1 that had been immunoprecipitated from human adrenocarcinoma cells [65,66]. Further analysis showed that sphingosine acts as a SF-1 antagonist, blocking cAMP-stimulated CYP17 reporter gene activity and coactivator recruitment, which could be negated by inhibiting the acid ceramidases that produce sphingosine from ceramide, or by introducing mutations into the LBP that abrogated sphingosine binding [65]. Subsequently, it was found that PA activated SF-1-dependent CYP17 expression and transcriptional activity, SF-1 heterocomplex assembly, and steroidogenesis. These effects could be inhibited by sphingosine or by LBP mutations [66].
These data suggest a model, wherein SF-1 is maintained in an inactive conformation by sphingosine under basal conditions [65,67] and is activated by the binding of PA, which is generated subsequent to ACTH/cAMP signaling [66]. The two different lipid species have opposing effects on the activity of SF-1, suggesting a regulatory mechanism in which the levels of these two lipids control the expression of genes linked to SF-1.
2.2.3. PIP2 versus PIP3
While no structures of a SF-1–PI or SF-1–PIP complex have been reported, modeling studies showed that phosphorylated PIs may be stabilized by several histidine residues around the mouth of the SF-1 LBP [68]. Mutations to these residues greatly impaired exchange of bacterial PG with PIP2 and PIP3 and diminished SF-1 transcriptional activity, suggesting that the binding of PIPs to SF- 1 is a biologically relevant interaction [68]. Indeed, IPMK phosphorylates PIP2 only when bound to SF-1, increasing downstream gene transcription; likewise, PTEN cleaves PIP3 only when complexed with SF-1, attenuating downstream activity [69]. Thus, the PIP–SF-1 interaction appears to introduce a regulatory mechanism not previously seen in NRs, in which the phosphorylation status of a bound ligand dictates the activity of its receptor.
2.3. PPARs
The peroxisome proliferator-activated receptors (PPARs α, β/δ, and γ) are members of the NR1C subfamily of NRs and play integral roles in the regulation of lipid metabolism and inflammation [70– 72]. PPARs form heterodimers with the retinoid X receptor (RXR) [73], and recognize an array of ligands, including fatty acids, eicosinoids, and oxidized lipid products [72].
2.3.1. PPARα and PC 16:0/18:1
PPARα is expressed in the heart, liver, kidney, muscle, and brown adipose tissue [74]. As a fatty acid binding protein, PPARα regulates the expression of many proteins involved in cellular fatty acid homeostasis [75–77] and systemic lipid balance [78]. It has been implicated in atherosclerosis and dyslipidemia, and prolonged activation has been linked to oxidative damage and liver cancer [79]. As such, PPARα is an important pharmacological target. Fibrates, a class of drugs used to treat dyslipidemia, are pharmacological agonists of PPARα, and exert their therapeutic effects by lowering triglyceride levels [80].
PPARα is known to bind to many natural free fatty acids (FFAs) and while these are likely physiological ligands, proving that these are bona fide endogenous activators is technically challenging. Like PLs, FFAs are typically insoluble, partitioning into droplets, membranes and soluble lipid binding proteins making direct correlations between binding affinity and activation difficult. It is clear, however, that μM levels of exogenous FFAs (1–50 μM) activate PPARs in vivo and in animals [81]. This is on par with PL-dependent transactivation among NR5A receptors, which display EC50 values ranging from 30 to 100 μM for activating PC and PE isoforms [38,42]. This affinity for FFAs and PLs among nuclear receptors is likely a result of their “generous” lipid binding pockets (see Section 4) which allow binding to an array of lipid metabolites.
In 2009, mass spectrometry experiments identified PC 16:0/18:1 as one of several lipids bound to PPARα isolated from murine liver tissue, and the only one whose presence was dependent on fatty acid synthase (FAS) [81]. Binding of this PC species was selective for PPARα over PPARδ and PPARγ, and could be enhanced in vivo by FAS induction, and inhibited by treatment with a PPARα agonist [81]. Additionally, PC 16:0/18:1 treatment stimulated PPARα-dependent gene expression and decreased fatty liver symptoms in mice, lending further credence to its suggested role as an endogenous PPARα agonist [81].
2.3.2. PPARγ and tail-oxidized PLs
PPARγ, which regulates glucose and fatty acid metabolism, is an important target in the treatment of type II diabetes, and is the receptor upon which the thiazolidinedione class of drugs acts [82]. In addition to metabolic regulation, PPARγ is known to be an important player in anti-inflammatory pathways [83]. Recently, 15-KETE- and 15-HETE PE, two oxidized PE species, were shown to activate PPARγ in vitro. Reporter gene assays showed a dose dependent activation in HEK293 cells cotransfected with PPARγ and a PPRE-luciferase construct, and in macrophages harvested from PPRE-EGFP transgenic mice. Furthermore, these oxidized PEs induce the PPARγ-dependent expression of CD36 in human monocytes [84]. Unoxidized PE showed no PPARγ activation, suggesting that PPARγ may specifically recognize oxidized PLs. While the formation of oxidized PEs is not dependent on lipases, it remains possible that phospholipase A (PLA) isoforms may liberate oxidized fatty acids, which are also known PPAR activators. Earlier work showed that oxidized PLs bind directly to the LBP, and PPARγ protects these oxidized PLs from phospholipase A1 mediated cleavage; however, this same work showed that PLA1 treated oxidized PLs had a similar ability to stimulate PPARγ transactivation relative to untreated oxidized PLs [85]. For PPARα, however, PLA2 appears to be required for activation by oxidized PLs [86].
2.4. USP
Ultraspiracle protein (USP) was identified as the Drosophila homologue of mammalian RXR in 1990 [87,88]. Its major function is to serve as a binding partner for the ecdysone receptor (EcR); this heterodimer is a vital regulator of molting and metamorphosis, which is triggered by the binding of 20-hydroxyecdysone (20E) to the EcR subunit [89]. However, USP itself can bind to several farnesoid insect juvenile hormones [90], and it is hypothesized to be a ligand-activated NR in its own right [91].
2.4.1. E. coli PLs
Crystal structures of USP consistently show bacterially-derived PL bound in the LBP [92–95], stabilizing the receptor in an antagonist conformation [93]. While most data implicate farnesoid derivatives as the endogenous USP ligand, it is conceivable that insect PLs may play a role in USP-mediated gene regulation, given the emerging role of PLs in other NR pathways. Insects have coopted PLs in the regulation of SREBP processing and nuclear translocation and may have independently evolved PL sensitive NRs. A comparison of the USP-PL crystal structures reveals a nearly identical mode of PL binding versus LRH-1 and SF-1.
3. PL transport and PL dependent coactivation
3.1. PPAR and PC-TP
In addition to direct NR-mediated gene expression, PLs have been shown to indirectly affect gene regulation through lipid shuttling proteins such as phosphatidylcholine transfer protein (PC-TP). PC-TP is a member of the steroidogenic acute regulatory protein (StAR)-related lipid transfer (START) domain superfamily that shares a common fold for lipid binding [96,97]. PC-TP is exquisitely selective for PC’s [98], and was originally shown to catalyze both one-for-one PC exchange, and net PC transfer between membranes [99–101]. PC-TP has since been identified as an important metabolic regulator, participating in hepatobiliary cholesterol, lipoprotein, glucose and fatty acid metabolism as well as brown fatmediated thermogenesis [102].
Consistent with PC-TP’s participation in metabolic processes, it has been identified as a binding partner for multiple metabolic proteins [103]. Arguably, the most interesting of these interactions is with PPAR-α [104]. In addition to PPAR-α regulating the expression of PC-TP, PC-TP was shown to upregulate the transcriptional activity of both PPAR-α and HNF-4α [104]. The mechanism of this effect on the transcriptional activity of NRs is not currently understood. Additionally, the context in which NRs bind to PL transporters is also unclear. There is a possibility that in addition to its role in the distribution of lipids in membranes, PC-TP may also deliver PL ligands to PL-sensitive receptors.
4. Structural analysis of PL binding proteins
4.1. What does it take to bind to PLs as a ligand?
With a large aliphatic surface and significant conformational freedom for the bulk of the molecular structure, PLs certainly do not look like a traditional NR ligands (Fig. 1). Interaction with the hydrophobic tails, while energetically favorable, does not permit specificity by the usual suspects (e.g. H-bonds, salt bridges, cation–π interactions). Below, we discuss the distinction between soluble PL transporters and proteins that utilize the information contained in the PL headgroup to drive intermolecular signaling.
4.2. Shuttlers versus transcription factors
Structurally characterized soluble PL transport proteins such as PC-TP and PITPα, fully engulf PLs, interacting substantially with both the lipid tails and the headgroup (Fig. 2E and F) [98,105]. Headgroup specificity is generated via H-bonds, ionic interactions and cation–π interactions via residues located at the core of the protein. The lipid tails extend toward the protein surface but remain protected from bulk solvent. This binding mode is in stark contrast to PL-binding NRs which bury PL tails and present the headgroup at the protein surface (Fig. 2A). The average LBP volume in PC-TP and PITPα is 2297 and 3000 Å3, respectively; this is nearly twice as large as the LRH-1, SF-1 and USP LBPs. The molecular volume of their bound lipids, however, are 874 and 552 Å3, for PCTP and PITPα, respectively. It is tempting to speculate the excess cavity volume and “tails out” PL conformation may be due to the requirement that transporters deliver their PL cargo to a target membrane or PL binding receptor prohibiting tight molecular interactions. Consistent with these observations, holo structures of PC-TP and PITPα show that atomic disorder increases distally from the headgroup suggesting less than optimal contacts are made with the PL tails which have vastly more potential energy to contribute to the protein–ligand interaction.
Fig. 2.

Crystal structures of soluble PL signaling proteins. Proteins are depicted as ribbons with bound phospholipids represented as sticks (O, red; P, magenta; N, blue). Molecular surfaces are shown to highlight the ligand binding pockets. (A) LRH-1 (slate) bound to DLPC (magenta) [41], (B) SFH-1 (tan) bound to PI (cyan) [110], (C) CD-1 (yellow) bound to PC (magenta) [106], (D) CD-1 (pink) bound to PI (cyan) [107] showing the bound ligands with lipid head-groups exposed to solvent. In contrast, the lipid shuttling proteins, (E) PC-TP (light green) bound to PC (magenta) [98] and (F) PITP (almond) bound to PI (cyan) [114] completely engulf their lipid ligands.
4.3. Parallels in the immune system
Both exogenous and endogenous PLs have been implicated as lipid antigens capable of activating natural killer T cells when presented by CD1 proteins localized on human antigen presenting cells [106,107]. CD1 proteins play a critical role in presenting both pathogen derived lipids and glycoproteins to initiate cell-mediated immunity [108]. Like NRs, CD1 glycocproteins bind PLs in a “tails-first” orientation with the PL headgroup exposed to the protein surface. The binding and presentation of both PC and PI by CD1b and CD1d, respectively, is remarkably similar to the presentation of PLs by NRs (Fig. 2A and C–D), whereby the lipid tails are buried and the headgroup is exposed to solvent. Thus, PL headgroup presentation may be a hallmark of PL dependent signaling.
4.4. Comparison to the PL PI/PC transporter Sec14
Sec14, originally defined by its ability to promote the movement of PC and PI between membranes, is now known as an integrator of PL signaling at the membrane [109]. To accomplish this, Sec14 senses both PC and PI levels to stimulate PI4-K mediated PI phosphorylation – a process critical for vesicle biogenesis. Interestingly, Sec14 requires both PC binding and PI binding for activity [110], however, a PC/PI exchange model has been proposed whereby PC binding facilities PI loading. While a direct interaction between Sec14 and PI4-K has not been observed, presentation of PI for decoration requires that the inositol moiety is accessible to protein surface (Fig. 2B). Indeed, while Sec14 completely buries the PC headgroup, the inositol ring of PI requires only the movement of few side chains to access the solvent. These observations parallel what we know for LRH-1/SF-1; they both are capable of binding PC and PI and presentation of the phosphorylated inositol headgroup is required for signaling (SF-1). Furthermore, since DLPC binding has not yet been tested in vivo, it is possible that the PC binding ability of LRH-1 and SF-1 may facilitate the loading of PI in a similar exchange reaction.
4.5. PL presentation as a model for PL dependent signaling
Unlike widely prevalent PL binding domains such as PHD fingers that recognize PLs in the context of a membrane [111], NRs engulf PLs “tails first” making extensive hydrophobic contact with more than 15 residues and up to three hydrogen bonds near the surface of the receptor [112]. It is clear that most of the binding energy is derived from interaction with the aliphatic tails, which in all known structures, intertwine to fill large 1300–1750 Å3 binding pocket that starts at the core of the protein and terminates at the protein surface. Lipid tails occupy the very core of the receptor greatly enhancing protein stability [41]. In this way, PLs act as folding nuclei much like the hormones in other NR family members [113]. However, the vast diversity among PLs and the potential for lipid modifications suggests that PL dependent transcription factors may serve to integrate varying and complex signals to tune gene expression. This represents an added layer of complexity on the already complicated cistrome in which coregulators, DNA, chromatin modifying enzymes and accessory proteins orchestrate coordinated gene expression.
5. Closing remarks
Evolution has generated a highly complex system to control energy homeostasis, including allosteric mechanisms within key metabolic enzymes, and the nutritional control of gene expression via transcription factors. Lipids are a major source of energy for the cell, and it is well known that the composition and availability of these lipids plays a central role in regulating glycolysis. NR mediated gene program alteration, whether by responding to cellular PL content, PL delivery by transporters, or in place PL modification, connects PL levels not only to glucose and lipid homeostasis but to steroid synthesis, reproduction, inflammation, development and cell differentiation (Fig. 3).
Fig. 3.

Phospholipid mediated transcription control. (A) In the absence of a phospholipid agonist NRs are bound to corepressor proteins and block transcription. (B) Activating PLs from exogenous, membrane bound or cytoplasmic sources bind to NRs or are potentially delivered by PL transporter proteins. Once an activating PL is bound to the NR coactivator complexes along with other general transcription factors (GTFs) and RNA polymerase initiate the transcription of genes. (C) NRs can also be bound to non-activating lipids with lipid modifying enzymes alter the lipid in place to become an activating lipid.
Given the molecular properties of PLs, it is no surprise that PL-driven transcription factors have been largely recalcitrant to drug design. Proteins with large hydrophobic pockets typically require large ligands and the potential for specific interactions within core of the LBP are slim. While there have been a few successes in designing specific compounds targeting these receptors, improving these compounds and predicting their binding modes remain challenging. Clearly, modulating PL-driven transcriptional pathways remains an untapped therapeutic opportunity and advances in this area of research are desperately needed.
References
- 1.Michell RH. Inositol phospholipids and cell surface receptor function. Biochim Biophys Acta. 1975;415:81–147. doi: 10.1016/0304-4157(75)90017-9. 0304-4157(75)90017-9. [DOI] [PubMed] [Google Scholar]
- 2.Berridge MJ, Irvine RF. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature. 1984;312:315–321. doi: 10.1038/312315a0. [DOI] [PubMed] [Google Scholar]
- 3.Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. 009.001.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiang SY, Dusaban SS, Brown JH. Lysophospholipid receptor activation of RhoA and lipid signaling pathways. Biochim Biophys Acta. 2013;1831:213–222. doi: 10.1016/j.bbalip.2012.09.004. http://dx.doi.org/10.1016/j.bbalip.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Donnell VB, Murphy RC. New families of bioactive oxidized phospholipids generated by immune cells: identification and signaling actions. Blood. 2012;120:1985–1992. doi: 10.1182/blood-2012-04-402826. http://dx.doi.org/10.1182/blood-2012-04-402826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irvine RF, Divecha N. Phospholipids in the nucleus–metabolism and possible functions. Semin Cell Biol. 1992;3:225–235. doi: 10.1016/1043-4682(92)90024-p. [DOI] [PubMed] [Google Scholar]
- 7.Irvine RF. Nuclear lipid signalling. Nat Rev Mol Cell Biol. 2003;4:349–360. doi: 10.1038/nrm1100. http://dx.doi.org/10.1038/nrm1100nrm1100. [DOI] [PubMed] [Google Scholar]
- 8.Irvine RF. Nuclear lipid signaling. Sci STKE. 2002:re13. doi: 10.1126/stke.2002.150.re13. http://dx.doi.org/10.1126/stke.2002.150.re13. [DOI] [PubMed]
- 9.Viiri K, Maki M, Lohi O. Phosphoinositides as regulators of protein-chromatin interactions. Sci Signal. 2012;5:pe19. doi: 10.1126/scisignal.2002917. http://dx.doi.org/10.1126/scisignal.2002917. [DOI] [PubMed] [Google Scholar]
- 10.Fraschini A, Albi E, Gahan PB, Viola-Magni MP. TEM cytochemical study of the localization of phospholipids in interphase chromatin in rat hepatocytes. Histochemistry. 1992;97:225–235. doi: 10.1007/BF00267632. [DOI] [PubMed] [Google Scholar]
- 11.McEwan IJ. Nuclear receptors: one big family. Methods Mol Biol. 2009;505:3–18. doi: 10.1007/978-1-60327-575-0_1. http://dx.doi.org/10.1007/978-1-60327-575-0_1. [DOI] [PubMed] [Google Scholar]
- 12.Nagy L, Schwabe JW. Mechanism of the nuclear receptor molecular switch. Trends Biochem Sci. 2004;29:317–324. doi: 10.1016/j.tibs.2004.04.006. http://dx.doi.org/10.1016/j.tibs.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 13.Huang P, Chandra V, Rastinejad F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu Rev Physiol. 2010;72:247–272. doi: 10.1146/annurev-physiol-021909-135917. http://dx.doi.org/10.1146/annurev-physiol-021909-135917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sladek FM. What are nuclear receptor ligands? Mol Cell Endocrinol. 2011;334:3–13. doi: 10.1016/j.mce.2010.06.018. http://dx.doi.org/10.1016/j.mce.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kliewer SA, Lehmann JM, Willson TM. Orphan nuclear receptors: shifting endocrinology into reverse. Science. 1999;284:757–760. doi: 10.1126/science.284.5415.757. [DOI] [PubMed] [Google Scholar]
- 16.Schulman IG, Heyman RA. The flip side: identifying small molecule regulators of nuclear receptors. Chem Biol. 2004;11:639–646. doi: 10.1016/j.chembiol.2003.12.021. http://dx.doi.org/10.1016/j.chembiol.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 17.Moore JT, Collins JL, Pearce KH. The nuclear receptor superfamily and drug discovery. ChemMedChem. 2006;1:504–523. doi: 10.1002/cmdc.200600006. http://dx.doi.org/10.1002/cmdc.200600006. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez-Marcos PJ, Auwerx J, Schoonjans K. Emerging actions of the nuclear receptor LRH-1 in the gut. Biochim Biophys Acta. 2011;1812:947–955. doi: 10.1016/j.bbadis.2010.12.010. http://dx.doi.org/10.1016/j.bbadis.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee Y-k, Moore DD. Liver receptor homolog-1, an emerging metabolic modulator. Front Biosci. 2008;13:5950–5958. doi: 10.2741/3128. [DOI] [PubMed] [Google Scholar]
- 20.Kellner S, Kikyo N. Transcriptional regulation of the Oct4 gene, a master gene for pluripotency. Histol Histopathol. 2010;25:405–412. doi: 10.14670/hh-25.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Botrugno Oa, Fayard E, Annicotte JS, Haby C, Brennan T, Wendling O, Tanaka T, Kodama T, Thomas W, Auwerx J, et al. Synergy between LRH-1 and beta-catenin induces G1 cyclin-mediated cell proliferation. Mol Cell. 2004;15:499–509. doi: 10.1016/j.molcel.2004.07.009. http://dx.doi.org/10.1016/j.molcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 22.Heng JC, Feng B, Han J, Jiang J, Kraus P, Ng JH, Orlov YL, Huss M, Yang L, Lufkin T, et al. The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell. 2010;6:167–174. doi: 10.1016/j.stem.2009.12.009. http://dx.doi.org/10.1016/j.stem.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 23.Sung B, Do HJ, Park SW, Huh SH, Oh JH, Chung HJ, Kang MJ, Kim JH, Kim NH. Regulation of OCT4 gene expression by liver receptor homolog-1 in human embryonic carcinoma cells. Biochem Biophys Res Commun. 2012 doi: 10.1016/j.bbrc.2012.09.049. http://dx.doi.org/10.1016/j.bbrc.2012.09.049. [DOI] [PubMed]
- 24.Clyne CD, Speed CJ, Zhou J, Simpson ER. Liver receptor homologue-1 (LRH-1) regulates expression of aromatase in preadipocytes. J Biol Chem. 2002;277:20591–20597. doi: 10.1074/jbc.M201117200. http://dx.doi.org/10.1074/jbc.M201117200. [DOI] [PubMed] [Google Scholar]
- 25.Goodwin B, Jones Sa, Price RR, Watson Ma, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 26.Chen F, Ma L, Dawson PA, Sinal CJ, Sehayek E, Gonzalez FJ, Breslow J, Ananthanarayanan M, Shneider BL. Liver receptor homologue-1 mediates species- and cell line-specific bile acid-dependent negative feedback regulation of the apical sodium-dependent bile acid transporter. J Biol Chem. 2003;278:19909–19916. doi: 10.1074/jbc.M207903200. [DOI] [PubMed] [Google Scholar]
- 27.del Castillo-Olivares A, Gil G. Role of FXR and FTF in bile acid-mediated suppression of cholesterol 7alpha-hydroxylase transcription. Nucleic Acids Res. 2000;28:3587–3593. doi: 10.1093/nar/28.18.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delerive P, Galardi CM, Bisi JE, Nicodeme E, Goodwin B. Identification of liver receptor homolog-1 as a novel regulator of apolipoprotein AI gene transcription. Mol Endocrinol. 2004;18:2378–2387. doi: 10.1210/me.2004-0132. [DOI] [PubMed] [Google Scholar]
- 29.Freeman LA, Kennedy A, Wu J, Bark S, Remaley AT, Santamarina-Fojo S, Brewer HB., Jr The orphan nuclear receptor LRH-1 activates the ABCG5/ABCG8 intergenic promoter. J Lipid Res. 2004;45:1197–1206. doi: 10.1194/jlr.C400002-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Inokuchi A, Hinoshita E, Iwamoto Y, Kohno K, Kuwano M, Uchiumi T. Enhanced expression of the human multidrug resistance protein 3 by bile salt in human enterocytes. A transcriptional control of a plausible bile acid transporter. J Biol Chem. 2001;276:46822–46829. doi: 10.1074/jbc.M104612200. http://dx.doi.org/10.1074/jbc.M104612200. [DOI] [PubMed] [Google Scholar]
- 31.Lee YK, Schmidt DR, Cummins CL, Choi M, Peng L, Zhang Y, Goodwin B, Hammer RE, Mangelsdorf DJ, Kliewer SA. Liver receptor homolog-1 regulates bile acid homeostasis but is not essential for feedback regulation of bile acid synthesis. Mol Endocrinol. 2008;22:1345–1356. doi: 10.1210/me.2007-0565. http://dx.doi.org/10.1210/me.2007-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo Y, Liang CP, Tall AR. The orphan nuclear receptor LRH-1 potentiates the sterol-mediated induction of the human CETP gene by liver X receptor. J Biol Chem. 2001;276:24767–24773. doi: 10.1074/jbc.M100912200. [DOI] [PubMed] [Google Scholar]
- 33.Oosterveer MH, Mataki C, Yamamoto H, Harach T, Moullan N, van Dijk TH, Ayuso E, Bosch F, Postic C, Groen AK, et al. LRH-1-dependent glucose sensing determines intermediary metabolism in liver. J Clin Invest. 2012;122:2817–2826. doi: 10.1172/JCI62368. http://dx.doi.org/10.1172/JCI6236862368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouchard MF, Taniguchi H, Viger RS. Protein kinase A-dependent synergism between GATA factors and the nuclear receptor, liver receptor homolog-1, regulates human aromatase (CYP19) PII promoter activity in breast cancer cells. Endocrinology. 2005;146:4905–4916. doi: 10.1210/en.2005-0187. http://dx.doi.org/10.1210/en.2005-0187. [DOI] [PubMed] [Google Scholar]
- 35.Annicotte JS, Chavey C, Servant N, Teyssier J, Bardin A, Licznar A, Badia E, Pujol P, Vignon F, Maudelonde T, et al. The nuclear receptor liver receptor homolog-1 is an estrogen receptor target gene. Oncogene. 2005;24:8167–8175. doi: 10.1038/sj.onc.1208950. http://dx.doi.org/10.1038/sj.onc.1208950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang SL, Zheng DZ, Lan FH, Deng XJ, Zeng J, Li CJ, Wang R, Zhu ZY. Increased expression of hLRH-1 in human gastric cancer and its implication in tumorigenesis. Mol Cell Biochem. 2008;308:93–100. doi: 10.1007/s11010-007-9616-1. http://dx.doi.org/10.1007/s11010-007-9616-1. [DOI] [PubMed] [Google Scholar]
- 37.Sablin EP, Krylova IN, Fletterick RJ, Ingraham HA. Structural basis for ligand-independent activation of the orphan nuclear receptor LRH-1. Mol Cell. 2003;11:1575–1585. doi: 10.1016/s1097-2765(03)00236-3. [DOI] [PubMed] [Google Scholar]
- 38.Wang W, Zhang C, Marimuthu A, Krupka HI, Tabrizizad M, Shelloe R, Mehra U, Eng K, Nguyen H, Settachatgul C, et al. The crystal structures of human steroidogenic factor-1 and liver receptor homologue-1. Proc Natl Acad Sci USA. 2005;102:7505–7510. doi: 10.1073/pnas.0409482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ortlund EA, Lee Y, Solomon IH, Hager JM, Safi R, Choi Y, Guan Z, Tripathy A, Raetz CRH, McDonnell DP, et al. Modulation of human nuclear receptor LRH-1 activity by phospholipids and SHP. Nat Struct Mol Biol. 2005;12:357–363. doi: 10.1038/nsmb910. http://dx.doi.org/10.1038/nsmb910. [DOI] [PubMed] [Google Scholar]
- 40.Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, Juzumiene D, Bynum JM, Madauss K, Montana V, et al. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell. 2005;120:343–355. doi: 10.1016/j.cell.2005.01.024. http://dx.doi.org/10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 41.Musille PM, Pathak MC, Lauer JL, Hudson WH, Griffin PR, Ortlund EA. Antidiabetic phospholipid-nuclear receptor complex reveals the mechanism for phospholipid-driven gene regulation. Nat Struct Mol Biol. 2012;19 (532–537):S531–S532. doi: 10.1038/nsmb.2279. http://dx.doi.org/10.1038/nsmb.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee JM, Lee YK, Mamrosh JL, Busby Sa Griffin PR, Pathak MC, Ortlund Ea, Moore DD. A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature. 2011 doi: 10.1038/nature10111. http://dx.doi.org/10.1038/nature10111. [DOI] [PMC free article] [PubMed]
- 43.Moore D. Targeting nuclear receptors to treat type 2 diabetes. 14th International Congress of Endocrinology; Kyoto, Japan. 2010. [Google Scholar]
- 44.Hoivik EA, Lewis AE, Aumo L, Bakke M. Molecular aspects of steroidogenic factor 1 (SF-1) Mol Cell Endocrinol. 2010;315:27–39. doi: 10.1016/j.mce.2009.07.003. http://dx.doi.org/10.1016/j.mce.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 45.Shinoda K, Lei H, Yoshii H, Nomura M, Nagano M, Shiba H, Sasaki H, Osawa Y, Ninomiya Y, Niwa O, et al. Developmental defects of the ventromedial hypothalamic nucleus and pituitary gonadotroph in the Ftz-F1 disrupted mice. Dev Dyn. 1995;204:22–29. doi: 10.1002/aja.1002040104. http://dx.doi.org/10.1002/aja.1002040104. [DOI] [PubMed] [Google Scholar]
- 46.Ingraham HA, Lala DS, Ikeda Y, Luo X, Shen WH, Nachtigal MW, Abbud R, Nilson JH, Parker KL. The nuclear receptor steroidogenic factor 1 acts at multiple levels of the reproductive axis. Genes Dev. 1994;8:2302–2312. doi: 10.1101/gad.8.19.2302. [DOI] [PubMed] [Google Scholar]
- 47.Mascaro C, Nadal A, Hegardt FG, Marrero PF, Haro D. Contribution of steroidogenic factor 1 to the regulation of cholesterol synthesis. Biochem J. 2000;350 (Pt 3):785–790. [PMC free article] [PubMed] [Google Scholar]
- 48.Sugawara T, Holt JA, Kiriakidou M, Strauss JF., 3rd Steroidogenic factor 1-dependent promoter activity of the human steroidogenic acute regulatory protein (StAR) gene. Biochemistry. 1996;35:9052–9059. doi: 10.1021/bi960057r. http://dx.doi.org/10.1021/bi960057r. [DOI] [PubMed] [Google Scholar]
- 49.Cao G, Garcia CK, Wyne KL, Schultz RA, Parker KL, Hobbs HH. Structure and localization of the human gene encoding SR-BI/CLA-1. Evidence for transcriptional control by steroidogenic factor 1. J Biol Chem. 1997;272:33068–33076. doi: 10.1074/jbc.272.52.33068. [DOI] [PubMed] [Google Scholar]
- 50.Lopez D, Shea-Eaton W, McLean MP. Characterization of a steroidogenic factor-1-binding site found in promoter of sterol carrier protein-2 gene. Endocrine. 2001;14:253–261. doi: 10.1385/ENDO:14:2:253. http://dx.doi.org/10.1385/ENDO:14:2:253. [DOI] [PubMed] [Google Scholar]
- 51.Parker KL, Schimmer BP. Steroidogenic factor 1: a key determinant of endocrine development and function. Endocr Rev. 1997;18:361–377. doi: 10.1210/edrv.18.3.0301. [DOI] [PubMed] [Google Scholar]
- 52.Schimmer BP, White PC. Minireview: steroidogenic factor 1: its roles in differentiation, development, and disease. Mol Endocrinol. 2010;24:1322–1337. doi: 10.1210/me.2009-0519. http://dx.doi.org/10.1210/me.2009-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferraz-de-Souza B, Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, NR5A1) and human disease. Mol Cell Endocrinol. 2011;336:198–205. doi: 10.1016/j.mce.2010.11.006. http://dx.doi.org/10.1016/j.mce.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Correa RV, Domenice S, Bingham NC, Billerbeck AE, Rainey WE, Parker KL, Mendonca BB. A microdeletion in the ligand binding domain of human steroidogenic factor 1 causes XY sex reversal without adrenal insufficiency. J Clin Endocrinol Metab. 2004;89:1767–1772. doi: 10.1210/jc.2003-031240. [DOI] [PubMed] [Google Scholar]
- 55.Camats N, Pandey AV, Fernandez-Cancio M, Andaluz P, Janner M, Toran N, Moreno F, Bereket A, Akcay T, Garcia-Garcia E, et al. Ten novel mutations in the NR5A1 gene cause disordered sex development in 46, XY and ovarian insufficiency in 46, XX individuals. J Clin Endocrinol Metab. 2012;97:E1294–E1306. doi: 10.1210/jc.2011-3169. http://dx.doi.org/10.1210/jc.2011-3169. [DOI] [PubMed] [Google Scholar]
- 56.Achermann JC, Ito M, Hindmarsh PC, Jameson JL. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet. 1999;22:125–126. doi: 10.1038/9629. http://dx.doi.org/10.1038/9629. [DOI] [PubMed] [Google Scholar]
- 57.Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sex Dev. 2008;2:200–209. doi: 10.1159/000152036. http://dx.doi.org/10.1159/000152036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bulun SE, Utsunomiya H, Lin Z, Yin P, Cheng YH, Pavone ME, Tokunaga H, Trukhacheva E, Attar E, Gurates B, et al. Steroidogenic factor-1 and endometriosis. Mol Cell Endocrinol. 2009;300:104–108. doi: 10.1016/j.mce.2008.12.012. http://dx.doi.org/10.1016/j.mce.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 59.Pianovski MA, Cavalli LR, Figueiredo BC, Santos SC, Doghman M, Ribeiro RC, Oliveira AG, Michalkiewicz E, Rodrigues GA, Zambetti G, et al. SF-1 overexpression in childhood adrenocortical tumours. Eur J Cancer. 2006;42:1040–1043. doi: 10.1016/j.ejca.2006.01.022. http://dx.doi.org/10.1016/j.ejca.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 60.Del Tredici AL, Andersen CB, Currier EA, Ohrmund SR, Fairbain LC, Lund BW, Nash N, Olsson R, Piu F. Identification of the first synthetic steroidogenic factor 1 inverse agonists: pharmacological modulation of steroidogenic enzymes. Mol Pharmacol. 2008;73:900–908. doi: 10.1124/mol.107.040089. http://dx.doi.org/10.1124/mol.107.040089. [DOI] [PubMed] [Google Scholar]
- 61.Madoux F, Li X, Chase P, Zastrow G, Cameron MD, Conkright JJ, Griffin PR, Thacher S, Hodder P. Potent, selective and cell penetrant inhibitors of SF-1 by functional ultra-high-throughput screening. Mol Pharmacol. 2008;73:1776–1784. doi: 10.1124/mol.108.045963. http://dx.doi.org/10.1124/mol.108.045963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roth J, Madoux F, Hodder P, Roush WR. Synthesis of small molecule inhibitors of the orphan nuclear receptor steroidogenic factor-1 (NR5A1) based on isoquinolinone scaffolds. Bioorg Med Chem Lett. 2008;18:2628–2632. doi: 10.1016/j.bmcl.2008.03.027. http://dx.doi.org/10.1016/j.bmcl.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doghman M, Cazareth J, Douguet D, Madoux F, Hodder P, Lalli E. Inhibition of adrenocortical carcinoma cell proliferation by steroidogenic factor-1 inverse agonists. J Clin Endocrinol Metab. 2009;94:2178–2183. doi: 10.1210/jc.2008-2163. http://dx.doi.org/10.1210/jc.2008-2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Y, Choi M, Cavey G, Daugherty J, Suino K, Kovach A, Bingham NC, Kliewer SA, Xu HE. Crystallographic identification and functional characterization of phospholipids as ligands for the orphan nuclear receptor steroidogenic factor-1. Mol Cell. 2005;17:491–502. doi: 10.1016/j.molcel.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 65.Urs AN, Dammer E, Sewer MB. Sphingosine regulates the transcription of CYP17 by binding to steroidogenic factor-1. Endocrinology. 2006;147:5249–5258. doi: 10.1210/en.2006-0355. http://dx.doi.org/10.1210/en.2006-0355. [DOI] [PubMed] [Google Scholar]
- 66.Li D, Urs AN, Allegood J, Leon A, Merrill AH, Jr, Sewer MB. Cyclic AMP-stimulated interaction between steroidogenic factor 1 and diacylglycerol kinase theta facilitates induction of CYP17. Mol Cell Biol. 2007;27:6669–6685. doi: 10.1128/MCB.00355-07. http://dx.doi.org/10.1128/MCB.00355-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Urs AN, Dammer E, Kelly S, Wang E, Merrill AH, Jr, Sewer MB. Steroidogenic factor-1 is a sphingolipid binding protein. Mol Cell Endocrinol. 2007;265–266:174–178. doi: 10.1016/j.mce.2006.12.016. http://dx.doi.org/10.1016/j.mce.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sablin EP, Blind RD, Krylova IN, Ingraham JG, Cai F, Williams JD, Fletterick RJ, Ingraham HA. Structure of SF-1 bound by different phospholipids: evidence for regulatory ligands. Mol Endocrinol. 2009;23:25–34. doi: 10.1210/me.2007-0508. http://dx.doi.org/10.1210/me.2007-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blind RD, Suzawa M, Ingraham HA. Direct modification and activation of a nuclear receptor–PIP(2) complex by the inositol lipid kinase IPMK. Sci Signal. 2012;5:ra44. doi: 10.1126/scisignal.2003111. http://dx.doi.org/10.1126/scisignal.2003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. doi: 10.1038/nature07202. http://dx.doi.org/10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- 71.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 72.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. http://dx.doi.org/10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 73.Miyata KS, McCaw SE, Marcus SL, Rachubinski RA, Capone JP. The peroxisome proliferator-activated receptor interacts with the retinoid X receptor in vivo. Gene. 1994;148:327–330. doi: 10.1016/0378-1119(94)90707-2. [DOI] [PubMed] [Google Scholar]
- 74.Auboeuf D, Rieusset J, Fajas L, Vallier P, Frering V, Riou JP, Staels B, Auwerx J, Laville M, Vidal H. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-alpha in humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes. 1997;46:1319–1327. doi: 10.2337/diab.46.8.1319. [DOI] [PubMed] [Google Scholar]
- 75.Martin G, Schoonjans K, Lefebvre AM, Staels B, Auwerx J. Coordinate regulation of the expression of the fatty acid transport protein and acyl-CoA synthetase genes by PPARalpha and PPARgamma activators. J Biol Chem. 1997;272:28210–28217. doi: 10.1074/jbc.272.45.28210. [DOI] [PubMed] [Google Scholar]
- 76.Motojima K, Passilly P, Peters JM, Gonzalez FJ, Latruffe N. Expression of putative fatty acid transporter genes are regulated by peroxisome proliferator-activated receptor alpha and gamma activators in a tissue- and inducer-specific manner. J Biol Chem. 1998;273:16710–16714. doi: 10.1074/jbc.273.27.16710. [DOI] [PubMed] [Google Scholar]
- 77.Dreyer C, Keller H, Mahfoudi A, Laudet V, Krey G, Wahli W. Positive regulation of the peroxisomal beta-oxidation pathway by fatty acids through activation of peroxisome proliferator-activated receptors (PPAR) Biol Cell. 1993;77:67–76. doi: 10.1016/s0248-4900(05)80176-5. [DOI] [PubMed] [Google Scholar]
- 78.Schoonjans K, Peinado-Onsurbe J, Lefebvre AM, Heyman RA, Briggs M, Deeb S, Staels B, Auwerx J. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 1996;15:5336–5348. [PMC free article] [PubMed] [Google Scholar]
- 79.Pyper SR, Viswakarma N, Yu S, Reddy JK. PPARalpha: energy combustion, hypolipidemia, inflammation and cancer. Nucl Recept Signal. 2010;8:e002. doi: 10.1621/nrs.08002. http://dx.doi.org/10.1621/nrs.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci USA. 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chakravarthy MV, Lodhi IJ, Yin L, Malapaka RR, Xu HE, Turk J, Semenkovich CF. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell. 2009;138:476–488. doi: 10.1016/j.cell.2009.05.036. http://dx.doi.org/10.1016/j.cell.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang JV, Greyson CR, Schwartz GG. PPAR-gamma as a therapeutic target in cardiovascular disease: evidence and uncertainty. J Lipid Res. 2012;53:1738–1754. doi: 10.1194/jlr.R024505. http://dx.doi.org/10.1194/jlr.R024505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Martin H. Role of PPAR-gamma in inflammation. Prospects for therapeutic intervention by food components. Mutat Res. 2009;669:1–7. doi: 10.1016/j.mrfmmm.2009.06.009. http://dx.doi.org/10.1016/j.mrfmmm.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 84.Hammond VJ, Morgan AH, Lauder S, Thomas CP, Brown S, Freeman BA, Lloyd CM, Davies J, Bush A, Levonen AL, et al. Novel keto-phospholipids are generated by monocytes and macrophages, detected in cystic fibrosis, and activate peroxisome proliferator-activated receptor-gamma. J Biol Chem. 2012;287:41651–41666. doi: 10.1074/jbc.M112.405407. http://dx.doi.org/10.1074/jbc.M112.405407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Davies SS, Pontsler AV, Marathe GK, Harrison KA, Murphy RC, Hinshaw JC, Prestwich GD, Hilaire AS, Prescott SM, Zimmerman GA, et al. Oxidized alkyl phospholipids are specific, high affinity peroxisome proliferator-activated receptor gamma ligands and agonists. J Biol Chem. 2001;276:16015–16023. doi: 10.1074/jbc.M100878200. http://dx.doi.org/10.1074/jbc.M100878200. [DOI] [PubMed] [Google Scholar]
- 86.Delerive P, Furman C, Teissier E, Fruchart J, Duriez P, Staels B. Oxidized phospholipids activate PPARalpha in a phospholipase A2-dependent manner. FEBS Lett. 2000;471:34–38. doi: 10.1016/s0014-5793(00)01364-8. [DOI] [PubMed] [Google Scholar]
- 87.Oro AE, McKeown M, Evans RM. Relationship between the product of the Drosophila ultraspiracle locus and the vertebrate retinoid X receptor. Nature. 1990;347:298–301. doi: 10.1038/347298a0. http://dx.doi.org/10.1038/347298a0. [DOI] [PubMed] [Google Scholar]
- 88.Henrich VC, Sliter TJ, Lubahn DB, MacIntyre A, Gilbert LI. A steroid/thyroid hormone receptor superfamily member in Drosophila melanogaster that shares extensive sequence similarity with a mammalian homologue. Nucleic Acids Res. 1990;18:4143–4148. doi: 10.1093/nar/18.14.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schwedes CC, Carney GE. Ecdysone signaling in adult Drosophila melanogaster. J Insect Physiol. 2012;58:293–302. doi: 10.1016/j.jinsphys.2012.01.013. http://dx.doi.org/10.1016/j.jinsphys.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 90.Jones G, Sharp PA. Ultraspiracle: an invertebrate nuclear receptor for juvenile hormones. Proc Natl Acad Sci USA. 1997;94:13499–13503. doi: 10.1073/pnas.94.25.13499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jones D, Jones G. Farnesoid secretions of dipteran ring glands: what we do know and what we can know. Insect Biochem Mol Biol. 2007;37:771–798. doi: 10.1016/j.ibmb.2007.05.014. http://dx.doi.org/10.1016/j.ibmb.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 92.Clayton GM, Peak-Chew SY, Evans RM, Schwabe JW. The structure of the ultraspiracle ligand-binding domain reveals a nuclear receptor locked in an inactive conformation. Proc Natl Acad Sci USA. 2001;98:1549–1554. doi: 10.1073/pnas.041611298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Billas IM, Moulinier L, Rochel N, Moras D. Crystal structure of the ligand-binding domain of the ultraspiracle protein USP, the ortholog of retinoid X receptors in insects. J Biol Chem. 2001;276:7465–7474. doi: 10.1074/jbc.M008926200. [DOI] [PubMed] [Google Scholar]
- 94.Billas IM, Iwema T, Garnier JM, Mitschler A, Rochel N, Moras D. Structural adaptability in the ligand-binding pocket of the ecdysone hormone receptor. Nature. 2003;426:91–96. doi: 10.1038/nature02112. http://dx.doi.org/10.1038/nature02112. [DOI] [PubMed] [Google Scholar]
- 95.Browning C, Martin E, Loch C, Wurtz JM, Moras D, Stote RH, Dejaegere AP, Billas IM. Critical role of desolvation in the binding of 20-hydroxyecdysone to the ecdysone receptor. J Biol Chem. 2007;282:32924–32934. doi: 10.1074/jbc.M705559200. http://dx.doi.org/10.1074/jbc.M705559200. [DOI] [PubMed] [Google Scholar]
- 96.Iyer LM, Koonin EV, Aravind L. Adaptations of the helix-grip fold for ligand binding and catalysis in the START domain superfamily. Proteins. 2001;43:134–144. doi: 10.1002/1097-0134(20010501)43:2<134::aid-prot1025>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 97.Ponting CP, Aravind L. START: a lipid-binding domain in StAR, HD-ZIP and signalling proteins. Trends Biochem Sci. 1999;24:130–132. doi: 10.1016/s0968-0004(99)01362-6. [DOI] [PubMed] [Google Scholar]
- 98.Roderick SL, Chan WW, Agate DS, Olsen LR, Vetting MW, Rajashankar KR, Cohen DE. Structure of human phosphatidylcholine transfer protein in complex with its ligand. Nat Struct Biol. 2002;9:507–511. doi: 10.1038/nsb812. http://dx.doi.org/10.1038/nsb812. [DOI] [PubMed] [Google Scholar]
- 99.Wirtz KW, Devaux PF, Bienvenue A. Phosphatidylcholine exchange protein catalyzes the net transfer of phosphatidylcholine to model membranes. Biochemistry. 1980;19:3395–3399. doi: 10.1021/bi00555a046. [DOI] [PubMed] [Google Scholar]
- 100.Kamp HH, Wirtz WA, Baer PR, Slotboom AJ, Rosenthal AF, Paltauf F, van Deenem LL. Specificity of the phosphatidylcholine exchange protein from bovine liver. Biochemistry. 1977;16:1310–1316. doi: 10.1021/bi00626a011. [DOI] [PubMed] [Google Scholar]
- 101.Johnson LW, Zilversmit DB. Catalytic properties of phospholipid exchange protein from bovine heart. Biochim Biophys Acta. 1975;375:165–175. doi: 10.1016/0005-2736(75)90186-8. [DOI] [PubMed] [Google Scholar]
- 102.Kang HW, Wei J, Cohen DE. PC-TP/StARD2: Of membranes and metabolism. Trends Endocrinol Metab. 2010;21:449–456. doi: 10.1016/j.tem.2010.02.001. http://dx.doi.org/10.1016/j.tem.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kanno K, Wu MK, Agate DS, Fanelli BJ, Wagle N, Scapa EF, Ukomadu C, Cohen DE. Interacting proteins dictate function of the minimal START domain phosphatidylcholine transfer protein/StarD2. J Biol Chem. 2007;282:30728–30736. doi: 10.1074/jbc.M703745200. http://dx.doi.org/10.1074/jbc.M703745200. [DOI] [PubMed] [Google Scholar]
- 104.Kang HW, Kanno K, Scapa EF, Cohen DE. Regulatory role for phosphatidylcholine transfer protein/StarD2 in the metabolic response to peroxisome proliferator activated receptor alpha (PPARalpha) Biochim Biophys Acta. 2010;1801:496–502. doi: 10.1016/j.bbalip.2009.12.013. http://dx.doi.org/10.1016/j.bbalip.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zheng J, Singh VK, Jia Z. Identification of an ITPase/XTPase in Escherichia coli by structural and biochemical analysis. Structure. 2005;13:1511–1520. doi: 10.1016/j.str.2005.07.007. http://dx.doi.org/10.1016/j.str.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 106.Giabbai B, Sidobre S, Crispin MD, Sanchez-Ruiz Y, Bachi A, Kronenberg M, Wilson IA, Degano M. Crystal structure of mouse CD1d bound to the self ligand phosphatidylcholine: a molecular basis for NKT cell activation. J Immunol. 2005;175:977–984. doi: 10.4049/jimmunol.175.2.977. [DOI] [PubMed] [Google Scholar]
- 107.Gadola SD, Zaccai NR, Harlos K, Shepherd D, Castro-Palomino JC, Ritter G, Schmidt RR, Jones EY, Cerundolo V. Structure of human CD1b with bound ligands at 2.3 A, a maze for alkyl chains. Nat Immunol. 2002;3:721–726. doi: 10.1038/ni821. http://dx.doi.org/10.1038/ni821. [DOI] [PubMed] [Google Scholar]
- 108.Jullien D, Stenger S, Ernst WA, Modlin RL. CD1 presentation of microbial nonpeptide antigens to T cells. J Clin Invest. 1997;99:2071–2074. doi: 10.1172/JCI119378. http://dx.doi.org/10.1172/JCI119378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bankaitis VA, Mousley CJ, Schaaf G. The Sec14 superfamily and mechanisms for crosstalk between lipid metabolism and lipid signaling. Trends Biochem Sci. 2010;35:150–160. doi: 10.1016/j.tibs.2009.10.008. http://dx.doi.org/10.1016/j.tibs.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schaaf G, Ortlund EA, Tyeryar KR, Mousley CJ, Ile KE, Garrett TA, Ren J, Woolls MJ, Raetz CR, Redinbo MR, et al. Functional anatomy of phospholipid binding and regulation of phosphoinositide homeostasis by proteins of the sec14 superfamily. Mol Cell. 2008;29:191–206. doi: 10.1016/j.molcel.2007.11.026. http://dx.doi.org/10.1016/j.molcel.2007.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gozani O, Karuman P, Jones DR, Ivanov D, Cha J, Lugovskoy AA, Baird CL, Zhu H, Field SJ, Lessnick SL, et al. The PHD finger of the chromatin-associated protein ING2 functions as a nuclear phosphoinositide receptor. Cell. 2003;114:99–111. doi: 10.1016/s0092-8674(03)00480-x. [DOI] [PubMed] [Google Scholar]
- 112.Ingraham Ha, Redinbo MR. Orphan nuclear receptors adopted by crystallography. Curr Opin Struct Biol. 2005;15:708–715. doi: 10.1016/j.sbi.2005.10.009. http://dx.doi.org/10.1016/j.sbi.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 113.Gee AC, Katzenellenbogen JA. Probing conformational changes in the estrogen receptor: evidence for a partially unfolded intermediate facilitating ligand binding and release. Mol Endocrinol. 2001;15:421–428. doi: 10.1210/mend.15.3.0602. [DOI] [PubMed] [Google Scholar]
- 114.Tilley SJ, Skippen A, Murray-Rust J, Swigart PM, Stewart A, Morgan CP, Cockcroft S, McDonald NQ. Structure-function analysis of human [corrected] phosphatidylinositol transfer protein alpha bound to phosphatidylinositol. Structure. 2004;12:317–326. doi: 10.1016/j.str.2004.01.013. http://dx.doi.org/10.1016/j.str.2004.01.013. [DOI] [PubMed] [Google Scholar]