

SUMMARY
The regulation and coordination of mitochondrial metabolism with hematopoietic stem cell (HSC) self-renewal and differentiation is not fully understood. Here we report that depletion of PTPMT1, a PTEN-like mitochondrial phosphatase, in inducible or hematopoietic-cell-specific knockout mice resulted in hematopoietic failure due to changes in the cell cycle and a block in the differentiation of HSCs. Surprisingly, the HSC pool was increased by ~40-fold in PTPMT1 knockout mice. Reintroduction of wild-type PTPMT1, but not catalytically deficient PTPMT1 or truncated PTPMT1 lacking mitochondrial localization, restored differentiation capabilities of PTPMT1 knockout HSCs. Further analyses demonstrated that PTPMT1 deficiency altered mitochondrial metabolism and that phosphatidylinositol phosphate substrates of PTPMT1 directly enhanced fatty-acid-induced activation of mitochondrial uncoupling protein 2. Intriguingly, depletion of PTPMT1 from myeloid, T lymphoid, or B lymphoid progenitors did not cause any defects in lineage-specific knockout mice. This study establishes a crucial role of PTPMT1 in the metabolic regulation of HSC function.
INTRODUCTION
Mitochondria are highly dynamic organelles that play multiple important roles in cells (Balaban et al., 2005). As the site of respiration/oxidative phosphorylation, these double-membrane organelles provide a highly efficient route for eukaryotic cells to generate ATP from energy-rich molecules. The role of the mitochondria in ATP generation is dependent on ambient oxygen tension. Under normal oxygen tensions, cells catabolize glucose to pyruvate. Pyruvate is then imported into the mitochondria for further catabolism through the Krebs cycle, which transfers electrons to the respiratory chain for ATP synthesis. In low oxygen tensions, or hypoxic conditions where there is a lack of oxygen as an electron acceptor, cells undergo anaerobic glycolysis as a default mode. Pyruvate is then used for low-efficiency energy production in the cytosol by glycolysis. Mitochondrial energy metabolism is regulated by multiple mechanisms. Uncoupling proteins (UCPs), especially UCP1, in the mitochondrial inner membrane attenuate mitochondrial energy production by conducting proton leak from the intermembrane space to the matrix and dissipating the proton gradient necessary for ATP generation (Brand and Esteves, 2005; Krauss et al., 2005). The functions of other UCPs, however, are still controversial. Emerging evidence suggests that UCP2 may regulate cellular bioenergetics by controlling mitochondrial substrate utilization. Specifically, increased UCP2 activity inhibits pyruvate entering into the mitochondria for Krebs cycle utilization while facilitating the oxidation of alternative carbon sources, such as fatty acids and glutamine (Bouillaud, 2009; Diano and Horvath, 2012; Samudio et al., 2009).
The function of mitochondria in the coordination of hematopoietic stem cell (HSC) maintenance and lineage differentiation is not well defined (Mantel et al., 2011; Suda et al., 2011). The ambient oxygen tension in the microenvironment (niches) of HSCs is believed to be low. To produce ATP in this environment, HSCs utilize glycolysis instead of mitochondrial oxidative phosphorylation for energy production (Simsek et al., 2010). Limited mitochondrial aerobic metabolism in HSCs helps to preserve this essential cell reservoir from oxidative damage by attenuating the production of reactive oxygen species (ROS), a byproduct generated during mitochondrial oxidative phosphorylation. A recent study has demonstrated that the mitochondrial content in HSCs is even slightly higher than that in myeloid progenitors, although lower than that in late erythroid progenitors (Norddahl et al., 2011), indicating that mitochondria in HSCs are relatively inactive. However, when HSCs undergo differentiation, a robust energy demand is expected for this rapid and energy-consuming cellular process. Indeed, ATP and ROS levels in lineage-committed progenitors are much higher than those in HSCs (Inoue et al., 2010; Simsek et al., 2010). Nonetheless, how mitochondrial energy metabolism is regulated and how mitochondrial bioenergetics and substrate utilization cooperatively coordinate both HSC differentiation and self-renewal in concert with other mechanisms is poorly understood.
PTPMT1, a PTEN-like phosphatase encoded by nuclear DNA, is relatively widely expressed in various tissues and is exclusively localized to the inner membrane of mitochondria via N-terminal amino acids 1–37 (Pagliarini et al., 2005). Little is known about the role of PTPMT1 in physiology and disease; however, recent studies have shown that global disruption of PTPMT1 results in developmental arrest and postimplantation lethality (Shen et al., 2011; Zhang et al., 2011a) due to the block of embryonic stem cell (ESC) differentiation (Shen et al., 2011). To further dissect the physiological function of PTPMT1, we have created a PTPMT1 conditional allele (PTPMT1fl/+) and determined the effects of PTPMT1 depletion on hematopoietic cell development.
RESULTS
PTPMT1 Depletion Results in Hematopoietic Failure While the Stem Cell Pool Is Drastically Expanded in Adult PTPMT1 Conditional Knockout Mice
PTPMT1 is highly expressed in HSCs (Lineage−Sca-1+c-Kit+ CD150+CD48−Flk2−), LSK (Lineage−Sca-1+c-Kit+) cells, and LK (Lineage−Sca-1−c-Kit+) cells relative to Lineage+ and mature cells (Figure 1A). To dissect the role of PTPMT1 in hematopoiesis, we created a PTPMT1 conditional allele (PTPMT1fl/+) (Figures S1A–S1C available online) and generated PTPMT1fl/fl/Mx1-Cre+ mice based on PTPMT1fl/+ mice and Mx1-Cre transgenic mice that express Cre in panhematopoietic cells (including HSCs) in response to polyinosinic-polycytidylic acid (pI-pC) treatment (Kühn et al., 1995). Four-week-old PTPMT1fl/fl/Mx1-Cre+ mice were treated with pI-pC to induce Cre expression and PTPMT1 deletion. As a result, nearly 80% of the mice died within 2–3 weeks of treatment (Figure 1B) due to pancytopenia and severe anemia (Figure S1D). Moreover, bone marrow (BM) myeloid (Figure 1C) and lymphoid (Figure 1D) progenitors detected by colony-forming unit (CFU) assays were drastically decreased as compared with those in control mice. Twenty percent of pI-pC-treated PTPMT1fl/fl/Mx1-Cre+ mice survived owing to the incomplete deletion of PTPMT1 in HSCs and the reconstitution of hematopoiesis from PTPMT1-undeleted HSCs. As shown in Figure 1E, 7 days after pI-pC treatment, a majority of peripheral blood cells showed PTPMT1 deletion (representing direct deletion from these cells). However, 3 weeks later, PTPMT1 deletion in peripheral blood cells was greatly decreased in surviving PTPMT1fl/fl/Mx1-Cre+ mice. Quantitative reverse transcription PCR (RT-PCR) analyses revealed that in these surviving animals PTPMT1 was deleted in 95%–98% of HSCs and LSK cells, whereas PTPMT1 deletion in common myeloid progenitors (CMPs; Lineage−Sca-1−c-Kit+CD34+ CD16/32low) and Lineage+ cells was only 20% (Figure 1F). Surprisingly, the percentage of HSCs in the BM of these mice was increased by ~40-fold (Figure 1G) (absolute numbers are shown in Table S1) despite PTPMT1 deletion in ~95%–98% of these cells, indicative of a robust expansion/accumulation of knockout stem cells. Phenotypic multipotent progenitor (MPP) cell populations (Lineage−Sca-1+c-Kit+CD150−CD48−Flk2−, Lineage−Sca-1+c-Kit+CD150+CD48+Flk2−, Lineage−Sca-1+c-Kit+CD150−CD48+Flk2−, and Lineage−Sca-1+c-Kit+ CD150−CD48+Flk2+) (Kiel et al., 2005; Wilson et al., 2008) were increased by 2.8- to 11-fold (Figure 1G). Remarkably, even 12 months after PTPMT1 ablation, the number of HSCs (~90% of which exhibited PTPMT1 depletion) in the knockout mice were still ~27 times greater than that in control mice (Figure S1E). The drastic expansion of the stem cell pool implies that hematopoietic failure caused by PTPMT1 ablation from the mitochondria was not due to an induction of apoptosis. In fact, apoptotic cells in HSCs were not appreciably changed in PTPMT1 knockout mice (Figure 1H).
Figure 1. PTPMT1 Depletion Results in Hematopoietic Failure While the Stem Cell Pool Is Drastically Expanded in Adult PTPMT1 Conditional Knockout Mice.

(A) HSC (Lineage−Sca-1+c-Kit+CD150+CD48−Flk2−), LSK (Lineage−Sca-1+c-Kit+), LK (Lineage−Sca-1−c-Kit+), and Lineage+ cells were sorted from mouse BM cells (n = 3/group). Macrophages were generated from BM cells as described in the Supplemental Information. Total RNA was extracted and PTPMT1 mRNA levels were determined by quantitative reverse transcription PCR (RT-PCR).
(B) Four-week-old PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ littermates (n = 30/group) were treated by intraperitoneal injection of pI-pC. Animal survival rates were determined.
(C and D) BM cells were harvested from PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ mice (n = 5/group) 2 weeks after pI-pC treatment and assessed for myeloid (C) and B lymphoid (D) progenitors by CFU assays.
(E) Peripheral blood was collected from PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ mice 1 and 4 weeks after pI-pC treatment. Genomic DNA was extracted from white blood cells and subjected to PCR genotyping.
(F) BM cells were harvested from PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ mice (n = 3/group) 4 weeks following pI-pC treatment. HSCs, LSK cells, CMPs (Lineage−Sca-1−c-Kit+CD34+CD16/32low), and Lineage+ cells were isolated. Total RNA was extracted and PTPMT1 mRNA levels were determined by quantitative RT-PCR.
(G and H) BM cells freshly harvested from PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ littermates 4 weeks following pI-pC treatment were assayed by multiparameter FACS analyses to determine frequencies of HSCs and MPPs (Lineage−Sca-1+c-Kit+CD150−CD48−Flk2−, Lineage−Sca-1+c-Kit+CD150+ CD48+Flk2−, Lineage−Sca-1+c-Kit+CD150−CD48+Flk2−, and Lineage−Sca-1+c-Kit+CD150−CD48+Flk2+) (n = 9/group) (G), and frequencies of early (Annexin V+/7-AAD−) and late (Annexin V+/7-AAD+) apoptotic cells in HSCs (n = 5/group) (H).
Data shown in (A), (C), (D), and (F)–(H) are presented as mean ± SD. Also see Figure S1.
Hematopoietic-Cell-Specific PTPMT1 Knockout Mice Fail to Establish Postnatal Hematopoiesis
To further verify the crucial role of PTPMT1 in hematopoiesis observed in inducible PTPMT1 knockout mice, we generated hematopoietic-cell-specific knockout (PTPMT1fl/fl/Vav1-Cre+) mice based on PTPMT1fl/+ mice and Vav1-Cre transgenic mice, in which Cre expression, and thus PTPMT1 deletion, was restricted to hematopoietic cells including stem cells. All PTPMT1fl/fl/Vav1-Cre+ mice died of pancytopenia and severe anemia within 5–9 days of birth (Figure 2A). These animals could be completely rescued by receiving transplants of wild-type (WT) BM cells (Figure 2A), verifying the cell-autonomous effect of PTPMT1 deficiency on neonatal hematopoiesis. Autopsies of PTPMT1fl/fl/Vav1-Cre+ pups revealed that the sizes of the spleen and the thymus were drastically decreased compared with those of WT littermates (Figure 2B). Histopathological examination of BM cells confirmed hematopoietic failure (Figure 2C). Total cell numbers of the BM from PTPMT1fl/fl/Vav1-Cre+ neonates were decreased by ~75-fold at postnatal day 5 (P5) (Figure S2A). Hematopoietic progenitors in the BM of knockout mice were undetectable (Figure 2D). The percentage of HSCs [Lineage−Sca-1+CD150+CD48−Flk2− cells were quantified because of the loss of c-Kit expression associated with severe hematopoietic failure (Simonnet et al., 2009; Yuan et al., 2004) in PTPMT1fl/fl/Vav1-Cre+ mice] in the BM of PTPMT1fl/fl/Vav1-Cre+ mice, however, was increased by ~30-fold (Figure 2E; PTPMT1 was completely deleted in these cells; data not shown), fully consistent with the stem cell effects in inducible adult PTPMT1 knockouts (Figure 1G). The expansion of HSCs in PTPMT1fl/fl/Vav1-Cre+ mice appeared to be developed gradually (Figure S2B). Notably, apoptosis in the BM cells (Figure S2C) and levels of ROS (Figure S2D) that are often elevated and cause cell death when mitochondria are dysfunctional were not altered.
Figure 2. Hematopoietic-Cell-Specific PTPMT1 Knockout Mice Fail to Establish Postnatal Hematopoiesis.

(A) Survival rates of PTPMT1fl/fl/Vav1-Cre+ (n = 50), PTPMT1+/+/Vav1-Cre+ (n = 50), and PTPMT1fl/fl/Vav1-Cre+ (n = 12) mice transplanted with WT BM cells were determined. Newborn PTPMT1fl/fl/Vav1-Cre+ pups were transplanted with BM cells (1 × 105) from WT pups by intravenous (superficial temporal vein) injection.
(B) Postnatal day 5 (P5) PTPMT1fl/fl/Vav1-Cre+ and PTPMT1+/+/Vav1-Cre+ neonates were dissected and photographed. Spleens dissected from P1 and P5 pups are shown on the right panel.
(C) Femurs isolated from P5 PTPMT1fl/fl/Vav1-Cre+ and PTPMT1+/+/Vav1-Cre+ pups were processed for histopathological examination (hematoxylin and eosin staining).
(D) BM cells harvested from P3 PTPMT1fl/fl/Vav1-Cre+ and PTPMT1+/+/Vav1-Cre+ littermates (n = 5/group) were assessed by CFU assays to determine myeloid progenitors.
(E) BM cells harvested from P3 to P5 PTPMT1fl/fl/Vav1-Cre+ and PTPMT1+/+/Vav1-Cre+ littermates (n = 8/group) were assayed by multiparameter FACS analyses to determine the frequency of Lineage−Sca-1+CD150+CD48−Flk2− cells.
Data shown in (D) and (E) are presented as mean ± SD. Also see Figure S2.
Differentiation and Repopulating Capabilities of HSCs Are Blocked in the Absence of PTPMT1
Since HSCs were drastically expanded and MPPs were increased to a much lesser extent, hematopoietic failure in PTPMT1 knockout mice appeared to be caused mainly by a block in the differentiation of stem cells. To test this hypothesis, HSCs were purified from PTPMT1-depleted mice and assessed for various functional activities. In response to cytokine stimulation, PTPMT1 knockout stem cells failed to differentiate to produce colonies in CFU assays (Figure 3A). The capability of PTPMT1-depleted HSCs to give rise to Lineage+ progeny when cocultured with OP9 stromal cells was essentially lost (Figure S3A). We also isolated HSCs and performed single-cell culture. In contrast to WT stem cells that vigorously differentiated, PTPMT1 knockout stem cells either did not divide or, at most, underwent one round of cell division under the in vitro culture conditions (Figure 3B). To further validate the impact of PTPMT1 deficiency on stem cell function, competitive repopulation assays were performed. BM cells (test cells) isolated from PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ mice (CD45.2+) were mixed with BM cells (competitor cells) harvested from BoyJ mice (CD45.1+) at the ratio of 1:1 and transplanted into lethally irradiated BoyJ mice. Six weeks after transplantation when donor cells were fully engrafted, recipient mice were treated with pI-pC. In this competitive setting, reconstitution from PTPMT1-depleted test cells was greatly decreased in primary transplants (Figures 3C and 3D) and barely detectable in secondary transplants (Figure 3E). The same results were obtained when BM cells from pI-pC-treated PTPMT1fl/fl/Mx1-Cre+ or PTPMT1+/+/Mx1-Cre+ mice were used for competitive transplantations. Contributions of PTPMT1-depleted donor cells to the whole hematopoietic cell population or each lineage in primary transplants (Figures S3B and S3C) and secondary transplants (Figure S3D) were blocked. We sorted test-cell-derived LSK (CD45.2+LSK), myeloid (CD45.2+Mac-1+Gr-1+), B lymphoid (CD45.2+B220+), and T lymphoid (CD45.2+CD3+) cells from primary recipients 20 weeks after pI-pC administration and examined PTPMT1 deletion in these cells. As shown in Figure 3F, whereas PTPMT1 was deleted in approximately 95% of CD45.2+LSK cells, PTPMT1 deletion in CD45.2+ lineage cells was barely detected, suggesting that these lineage cells were derived from PTPMT1-undeleted test cells. Remarkably, in both competitive repopulation assay setups, the percentages of CD45.2+LSK cells derived from PTPMT1-depleted test cells were still four to six times greater than those in control test-cell-derived populations in primary recipients (Figures 3G and S3E), reaffirming the profound cell-autonomous effects of PTPMT1 deficiency on stem/early progenitor cell homeostasis.
Figure 3. Repopulating Capabilities of PTPMT1-Depleted HSCs Are Blocked.

(A) HSCs were sorted from PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ mice (n = 3/group) 4 weeks after pI-pC treatment and assessed by the CFU assay. Colonies were counted 12 days later.
(B) Sorted HSCs were individually deposited into 96-well plates and cultured in Thrombopoietin (TPO) (50 ng/ml), Flt3 ligand (50 ng/ml), stem cell factor (SCF) (50 ng/ml), IL-3 (25 ng/ml), and IL-6 (20 ng/ml) containing medium for 7 days. The numbers of the wells containing 1, 2, or confluent cells were documented. Representative data from three independent experiments are shown.
(C and D) BM cells (1 × 106) (test cells) harvested from PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ mice (CD45.2+) (without pI-pC treatment) were mixed with the same number of BoyJ (CD45.1+) BM cells and transplanted into lethally irradiated BoyJ recipients (n = 9/group). Six weeks following transplantation, recipient mice were treated with pI-pC. Test cell reconstitution (CD45.2+ cells) in peripheral blood and BM cells was determined by FACS analyses 4, 8, 12, 16, and 20 weeks after pI-pC treatment. Representative FACS data are shown in (C) with summarized data shown in (D).
(E) BM cells harvested from primary recipients 20 weeks after pI-pC administration were transplanted into lethally irradiated secondary BoyJ recipient mice (2 × 106 cells/mouse) (n = 7/group). Test cell reconstitution in peripheral blood was determined 4, 8, 12, and 16 weeks after transplantation.
(F) CD45.2+LSK, CD45.2+Mac-1+Gr-1+, CD45.2+B220+, and CD45.2+CD3+ cells were sorted from the BM of primary transplants (n = 3/group). Total RNA was extracted and PTPMT1 mRNA levels were determined by quantitative RT-PCR.
(G) Frequencies of LSK cells in the test-cell-derived (CD45.2+) cell populations in primary recipient mice (n = 5/group) were quantified by multiparameter FACS analyses 20 weeks following pI-pC treatment.
Data shown in (A) and (C)–(G) are presented as mean ± SD. Also see Figure S3.
Catalytic Activity and Mitochondrial Localization Are Required for the Function of PTPMT1 in HSCs
PTPMT1 is a phosphatase that is exclusively localized to the mitochondria (Pagliarini et al., 2005). To elucidate the structural bases for the indispensable role of PTPMT1 in HSCs, WT PTPMT1, catalytically deficient PTPMT1 C132S, and truncated PTPMT1 lacking the mitochondrial localization signal (amino acids 1-37) (PTPMT1 Δ37) were transduced into PTPMT1fl/fl/Mx1-Cre+ BM cells through retroviral-mediated gene transfer. Transduced cells were sorted according to green fluorescent protein (GFP) expressed separately by the retroviral vector, and then assessed for repopulating/differentiation capabilities by competitive repopulation assays, as described in Figure 3D. Following deletion of endogenous PTPMT1 by pI-pC treatment, control-vector-transduced PTPMT1fl/fl/Mx1-Cre+ cells failed to reconstitute hematopoiesis (Figure 4A), consistent with the results shown in Figures 3C and 3D. Reintroduction of WT PTPMT1 in PTPMT1 knockout cells effectively rescued stem cell repopulating capabilities. In contrast, neither PTPMT1 C132S nor PTPMT1 Δ37 exhibited any rescue capacity (Figure 4A). In addition, embryonic day 15.5 (E15.5) fetal liver hematopoietic cells isolated from PTPMT1fl/fl/Vav1-Cre+ embryos were transduced with either WT PTPMT1, PTPMT1 C132S, PTPMT1 Δ37, or control vector, and then assessed for radioprotective function in lethally irradiated recipient mice. As shown in Figure 4B, only WT PTPMT1-transduced knockout cells rescued recipient animals, whereas all transplants receiving either PTPMT1 C132S, PTPMT1 Δ37, or control-vector-transduced knockout cells invariably died within 2–3 weeks of irradiation. Furthermore, the percentage of HSCs in WT PTPMT1-rescued recipients was restored to the normal range (data not shown). These add-back experiments, while confirming the crucial cell-intrinsic role of PTPMT1 in HSCs, suggest that this phosphatase functions in a catalytically dependent manner and that the mitochondrial localization is required for its function.
Figure 4. Catalytic Activity and Mitochondrial Localization Are Required for PTPMT1 Function in HSCs.
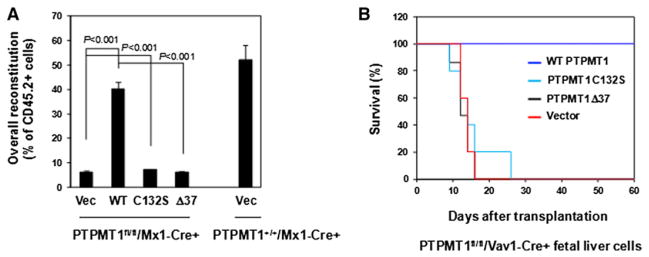
(A) BM cells freshly harvested from PTPMT1fl/fl/Mx1-Cre+ mice (without pI-pC treatment) were transduced with WT PTPMT1, PTPMT1 C132S, PTPMT1 Δ37, and control vector through retro-viral-mediated gene transfer. Transduced cells were sorted by FACS based on the expression of the GFP marker contained in the retroviral vector (MSCV-IRES-GFP). Sorted cells (test cells) were transplanted with the same number of the BM cells (competitor cells) isolated from BoyJ mice that had undergone the same culture procedures into lethally irradiated BoyJ recipients (n = 10/group). Six weeks after transplantation, recipient animals were treated with pI-pC. Test cell reconstitution was determined 16 weeks after the last dose of pI-pC.
(B) Embryonic day 15.5 (E.15.5) fetal liver cells freshly harvested from PTPMT1fl/fl/Vav1-Cre+ embryos were transduced with WT PTPMT1, PTPMT1 C132S, PTPMT1 Δ37, and vector control. Transduced cells (1 × 106 cells with around 20% transduction efficiency) were transplanted into lethally irradiated BoyJ recipients. Survival curves of recipient animals were determined (n = 10/group).
Data shown in (A) are presented as mean ± SD.
Depletion of PTPMT1 from Lineage Progenitors Does Not Cause Any Defects in Lineage-Specific Knockout Mice
To test whether PTPMT1 is generally important for all hematopoietic cells, we generated lineage-specific knockout mice PTPMT1fl/fl/LysM-Cre+ (in granulocyte-macrophage progenitors, GMPs), PTPMT1fl/fl/LCK-Cre+ (in CD4−/CD8− stage T lymphoid progenitors), and PTPMT1fl/fl/CD19-Cre+ (in pro-B stage B lymphoid progenitors) mice by crossing PTPMT1fl/+ mice with LysM-Cre, LCK-Cre, and CD19-Cre transgenic mice, respectively. Intriguingly, ablation of PTPMT1 from GMPs, T lymphoid, or B lymphoid progenitors in these mice did not disrupt normal cell development. In sharp contrast to panhematopoietic cell (including stem cell) knockouts (PTPMT1fl/fl/Vav1-Cre+) that died at the postnatal stage, PTPMT1fl/fl/LysM-Cre+, PTPMT1fl/fl/LCK-Cre+, and PTPMT1fl/fl/CD19-Cre+ mice showed no defects in myeloid, T lymphoid, or B lymphoid lineages (Figures S4A–S4C), nor was the cell cycle of lineage progenitors altered (Figure S4D). The lineage-specific knockout mice were active and fertile without any discernible abnormalities during the 1.5 years of follow-up. These in vivo data strongly suggest that while PTPMT1 plays a vital role in stem cells, it is dispensable for late lineage progenitors.
PTPMT1-Depleted HSCs Are Accumulated at the G1 Phase in the Cell Cycle
To further define the underlying mechanisms for the differentiation block of PTPMT1-depleted HSCs, we first tested whether knockout HSCs were replicatively senescent by examining senescence-associated beta-galactosidase (β-gal) activity using 5-dodecanoylaminofluorescein di-β-D-galactopyranoside (C12FDG), a fluorogenic substrate for β-gal. No significant differences in β-gal activities were detected in the HSCs of PTPMT1 knockout mice (Figure S4E). We also determined the expression of p16, a molecular hallmark of cellular senescence. Again, there were no significant changes in p16 levels between WT and knockout LSK cells (Figure 5F) or HSCs (Figure S4F). We then analyzed the cell cycle status of LSK cells and HSCs. These analyses revealed a significant increase in the percentages of knockout LSK cells and HSCs at the G1 phase while the percent-ages of S/G2/M cells were not changed (Figures 5A and 5B). Moreover, the percentages of LSK cells and HSCs at the G0 phase (quiescence) were decreased in PTPMT1-depleted mice. In vivo BrdU incorporation assays showed that after 16 hr of in vivo BrdU labeling, the percentages of BrdU+ cells in LSK and HSC populations in PTPMT1 knockout mice were not significantly altered (Figures 5C and 5D), verifying the S phase data obtained from the dye-based cell cycle analyses (Figures 5A and 5B). Furthermore, BrdU long-term label-retaining assays (Wilson et al., 2008) demonstrated that BrdU label-retaining cells in the HSC population in PTPMT1 knockout mice 100 days after the BrdU labeling were markedly decreased (Figure 5E), reaffirming the loss of quiescence in PTPMT1 deficient HSCs. Collectively, these cell cycle data suggest an enhanced entry of quiescent stem cells (G0 phase) into the cell cycle in PTPMT1 knockout mice, and that these cells were subsequently delayed at the G1 phase (the ratio of S/G2/M cells to G1 cells was decreased). In agreement with the cell cycle changes, p21 and p57, cyclin-dependent kinase inhibitors critical for controlling HSC cycling (Cheng et al., 2000; Matsumoto et al., 2011; Zou et al., 2011), were significantly upregulated in PTPMT1-depleted stem cells and early progenitors (Figure 5F).
Figure 5. PTPMT1-Depleted HSCs Are Delayed at the G1 Phase of the Cell Cycle.

(A and B) BM cells freshly harvested from PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ mice 4 weeks following pI-pC treatment were assayed to determine the cell cycle status of LSK cells (A, n = 9/group) and HSCs (B, n = 8/group). Percentages of LSK cells (A) and HSCs (B) at G0, G1, and S/G2/M phases were quantified. (C and D) PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ mice (n = 5/group) were injected i.p. with BrdU (1.0 mg/6.0 g body weight) 4 weeks following pI-pC treatment. Mice were sacrificed 16 hr later. BM cells were isolated and stained with antibodies against the indicated cell surface markers. BrdU staining was performed using the FTIC BrdU Flow Kit (BD Biosciences). Cells were then stained with the DNA dye 7-amino-actinomycin D (7-AAD) and subjected to FACS analyses so that percentages of BrdU+ cells in the LSK (C) and HSC (D) populations could be determined.
(E) PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ mice (n = 5/group) were injected i.p. with BrdU (1.0 mg/6.0 g body weight) 4 weeks following pI-pC treatment. The mice were fed with BrdU (1.0 mg/ml) containing water for 10 days and sacrificed 100 days later. BM cells were isolated and assessed for percentages of BrdU+ cells in the HSC population as above.
(F) LSK cells, HSCs, and various subsets of phenotypic MPPs were sorted from PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ mice (n = 3/group) 4 weeks following pI-pC treatment. mRNA levels of the indicated cell cycle regulatory genes in these cells were determined by quantitative RT-PCR.
Data shown in all panels are presented as mean ± SD. Also see Figure S4.
PTPMT1 Depletion Results in Defective Mitochondrial Aerobic Metabolism
We next sought to determine the fundamental impact of PTPMT1 ablation on the mitochondria. Ultra structures of mitochondria in PTPMT1-depleted stem cells were not altered (data not shown) and the overall mitochondrial DNA content in these cells was comparable to that in control cells (Figure 6A). Also, steady state total cellular ATP levels (Figure 6B) and ROS levels in PTPMT1-depleted HSCs (Figure 6C) were not significantly changed. We next analyzed mitochondrial aerobic metabolism in intact, viable LSK cells using real-time measurement of oxygen consumption. PTPMT1 knockout cells had much lower basal oxygen consumption and maximal oxidative capacity as compared with WT cells (Figure 6D). Interestingly, measurement of extracellular proton flux revealed that PTPMT1-depleted cells had significantly increased extracellular acidification rates (Figure 6E), suggesting that these cells exhibited enhanced glycolysis. In agreement with these data on the mitochondrial function, AMP-activated kinase (AMPK), an intracellular energy sensor (Hardie, 2011), was highly activated (determined by phosphorylation of Thr172) in PTPMT1 knockout LSK cells (Figure 6F) and HSCs (Figure 6G). Acetyl-CoA carboxylase (ACC), one of the targets of AMPK and a negative regulator of fatty acid oxidation (Viollet et al., 2009), was inhibited, as evidenced by the increase in the inhibitory phosphorylation of this enzyme (Figure 6F), implying that fatty acid metabolism might be enhanced in PTPMT1 knockout stem cells. Intriguingly, PTPMT1-depleted macrophages derived from myeloid-specific knockout (PTPMT1+/+/LysM-Cre+) mice also showed decreased mitochondrial aerobic metabolism at basal levels and maximal reserve capacities (Figure S5A). However, no defects in the growth (Figure S5B) and the cell cycle (Figure S5C) were detected. Also, there was no upregulation of p53, p21, or p57 in these knockout macrophages (Figure S5D), and AMPK phosphorylation was only marginally increased in these cells (Figure S5E).
Figure 6. Mitochondrial Aerobic Metabolism Is Defective in PTPMT1-Depleted LSK Cells.

BM cells were freshly harvested from PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ mice (n = 4 or 5/group) 4 weeks following pI-pC treatment. HSCs were sorted.
(A) Total DNA was extracted. Mitochondrial number was estimated by comparing mtDNA (Cytochrome B) levels to genomic DNA levels by quantitative PCR.
(B) Total cellular ATP levels were determined using an ATP assay kit.
(C) BM cells were immunostained with the antibodies recognizing HSC markers. Cells were then loaded with 2′-7′-dichlorofluorescein diacetate (DCF-DA) (5 μM) for 15 min. Mean fluorescence intensity in the gated HSC population was quantified by FACS to determine ROS (H2O2) levels.
(D and E) LSK cells were sorted from PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ mice (n = 3 or 4/group) 4 weeks following pI-pC treatment. Oxygen consumption rates (OCR) (D) and extracellular acidification rates (ECAR) (E) of live LSK cells were measured in the presence of the mitochondrial inhibitor (oligomycin, 350 nM), the uncoupling agent (FCCP, 5 μM), and the respiratory chain inhibitor (rotenone, 1 μM). **p < 0.01.
(F) Whole-cell lysates of sorted LSK cells were prepared and examined by immunoblotting with anti-phospho-AMPK, anti-phospho-ACC, and anti-UCP2 antibodies. Each lane represents an individual cell pool. Representative results from three independent experiments are shown.
(G) BM cells freshly harvested from PTPMT1fl/fl/Mx1-Cre+ and PTPMT1+/+/Mx1-Cre+ mice (n = 5/group) 4 weeks following pI-pC treatment were immunostained with antibodies recognizing HSC markers. Cells were then fixed, permeablized using the BD Cytofix/Cytoperm kit (BD Biosciences), and immunostained with anti-phospho-AMPK (Thr172) antibody (Cell Signaling Technology). Mean fluorescence intensity in the gated HSC population was quantified and normalized by FACS to determine phospho-AMPK levels. Light and dark gray lines indicate isotype controls.
Data shown in (A)–(E) and (G) are presented as mean ± SD. Also see Figure S5.
Excessive PIP Substrates of PTPMT1 Decrease Mitochondrial Aerobic by Enhancing UCP2 Activity
PTPMT1 favors phosphatidylinositol phosphates (PIPs), especially PI(3,5)P2, PI5P, and PI(3,4)P2, as substrates (Shen et al., 2011), similar to PTEN phosphatase. PIPs are a class of membrane phospholipids that regulate many important cellular processes, including membrane trafficking and ion channel/transporter functions (Di Paolo and De Camilli, 2006; Gamper and Shapiro, 2007). PTPMT1 is localized to the mitochondrial inner membrane (Pagliarini et al., 2005) where many ion channels/transporters, important for mitochondrial metabolism, reside. To determine the mechanisms of the altered mitochondrial function in PTPMT1-deficient cells, we assessed effects of overloading these PIP substrates on mitochondrial aerobic metabolism. Perfusion of PI(3,5)P2, PI(3,4)P2, or PI5P into WT LSK cells resulted in a significant reduction in oxygen consumption both at the basal level and at the maximal oxidative capacity (Figures 7A and 7B), recapitulating the metabolic changes of PTPMT1 knockout LSK cells (Figure 6D). These results indicate that PIP substrates accumulated due to the loss of dephosphorylation by PTPMT1 might account for the altered mitochondrial function in PTPMT1-depleted cells.
Figure 7. Excessive PIP Substrates of PTPMT1 Decrease Aerobic Metabolism by Enhancing UCP2 Activity.

(A and B) PI(3,5)P2, PI5P, and PI(3,4)P2 (di-C16) (20 μM) were delivered (shuttled) into purified WT LSK cells using Shuttle PIP kits (Echelon Biosciences, Inc.). Cells were assessed for OCR. A representative figure is shown in (A) and statistical data are shown in (B). **p < 0.01.
(C) Representative current-voltage characteristics of UCP2-containing membranes measured in the presence of PI(3,5)P2, arachidonic acid (AA), or both. Bilayer lipid membranes were made from E. coli polar lipid (1.8 mg/ml), AA (15 mol%), and hUCP2 (5 μg/mg lipid). Buffer solution contained 50 mM Na2S04, 10 mM TRIS, 10 mM MES, and 0.6 mM EGTA (pH = 7.35), T = 32°.
(D) Comparison of membrane conductance in the absence or presence of PI(3,5)P2, PI5P, or PI(3,4)P2. All PIPs were added at 4 μM. Statistical data from three independent experiments are shown.
(E) HSCs purified from PTPMT1fl/fl/Mx1-Cre+ mice 4 weeks after pI-pC treatment were cultured in SCF (50 ng/ml), IL-3 (25 ng/ml), and IL-6 (20 ng/ml) containing medium in the presence of PIKfyve inhibitor (YM201636) or DMSO (vehicle) for 7 days, immunostained for Mac-1 and B220, and then analyzed by FACS. Statistical data from three independent experiments are shown.
(F) Sorted HSCs were assessed by the CFU assay in the presence of PIKfyve inhibitor (1 μM) or DMSO. Twelve days later, colonies derived were counted and genotyped individually.
Data shown are mean ± SD of three independent experiments. Data shown in (A), (B), and (D)–(F) are presented as mean ± SD.
We next sought to identify direct effector proteins of these PIPs. A recent study has shown that ectopic expression of mitochondrial UCP2 blocks ESC differentiation with no impact on cell survival (Zhang et al., 2011b). Enhanced UCP2 activity impairs ESC differentiation by perturbing the metabolic transition from glycolysis to mitochondrial aerobic metabolism required for ESC differentiation (Zhang et al., 2011b). PTPMT1 depletion also blocks ESC differentiation without affecting cell survival (Shen et al., 2011). Similar effects caused by ectopic expression of UCP2 and PTPMT1 ablation prompted us to test for a potential link between PTPMT1 and UCP2. No differences in UCP2 mRNA levels were detected between WT and PTPMT1-depleted HSCs (data not shown). We then tested whether PTPMT1 PIP substrates might function as modulators of UCP2 activity using a UCP2-reconstituted planar lipid bilayer system. As shown in Figures 7C and 7D, the addition of PI(3,5)P2, PI5P, or PI(3,4)P2, but not their precursor PI, enhanced fatty acid (arachidonic acid)-induced activation of UCP2. Total membrane proton conductance carried out by UCP2 was markedly increased by the addition of PI(3,5)P2, PI5P, and PI(3,4)P2. These in vitro electrophysiological data provide direct evidence that PIP substrates, which accumulate in PTPMT1-depleted mitochondria, can alter mitochondrial function by enhancing UCP activities.
Finally, we reasoned that if excessive PIP substrates of PTPMT1, such as PI(3,5)P2, contributed to the mitochondrial dysfunction and thus the impaired differentiation of PTPMT1 knockout HSCs, then inhibition of PI(3,5)P2 production would partially correct this problem. We treated purified PTPMT1 knockout HSCs with YM201636, a selective inhibitor of type III PIP kinase PIKfyve (Jefferies et al., 2008) that is responsible for the production of PI(3,5)P2. Indeed, cytokine-induced differentiation of PTPMT1 knockout HSCs toward Mac-1+ cells was largely restored in liquid culture by the PIKfyve inhibitor (Figure 7E). Moreover, cytokine-induced colony formation of PTPMT1 knockout HSCs was also partially rescued (Figure 7F). Approximately 50% of the colonies formed in the YM201636-treated group were derived from PTPMT1-depleted HSCs while none of the colonies seen in the DMSO-treated group were generated from PTPMT1-deleted stem cells (they were derived from residual PTPMT1-undeleted cells) according to PCR genotyping results (data not shown). These rescue data provide further support to the notion that the differentiation block of PTPMT1 knockout stem cells is likely attributed to the accumulation of PTPMT1 PIP substrates.
DISCUSSION
By dissecting the role of mitochondrial phosphatase PTPMT1 in hematopoiesis, this study has revealed an essential mitochondrial metabolic regulation of HSC differentiation. HSCs contain similar numbers of mitochondria, as do myeloid progenitors (Norddahl et al., 2011). However, stem cells utilize glycolysis instead of aerobic metabolism for energy production (Simsek et al., 2010), suggesting that mitochondria are less active in these cells. In contrast, mitochondrial activities and ATP levels are much higher in lineage progenitors (Inoue et al., 2010; Simsek et al., 2010), indicating a robust upregulation of mitochondrial bioenergetics during stem cell differentiation. We now provide evidence that PTPMT1 plays a critical role in priming mitochondria for this rapid metabolic transition. PTPMT1 depletion caused hematopoietic failure in mice (Figures 1B, S1D, 2A, and 2B) and PTPMT1-depleted HSCs failed to differentiate in both in vitro (Figures 3A, 3B, and S3A) and in vivo (Figures 3C, 3D, S3B, and S3C) settings. This appears to be attributed to the alterations of mitochondrial metabolism in these cells (Figure 6D). Although steady state cellular ATP homeostasis was maintained in knockout stem cells (Figure 6B), this was likely due to a compensatory increase in alternative energy production pathways. Indeed, extracellular acidification rates, an indication of glycolysis, were elevated in PTPMT1-depleted cells (Figure 6E) and the activity of ACC, a negative regulator of fatty acid oxidation (Viollet et al., 2009), was inhibited (Figure 6F). Interestingly, PTPMT1 regulation of mitochondrial bioenergetics appears to be selectively important for differentiation versus self-renewal in stem cells. Differentiation of PTPMT1 knockout HSCs was completely blocked (Figure 3), but these cells were still cycling in vivo. Although knockout stem cells were delayed at the G1 phase, the percentage of S/G2/M cells was comparable to that of control cells. In addition, knockout stem cells lost quiescence (G0 phase) (Figure 5). Since the stem cell pool in knockout mice was drastically expanded (Figures 1G and 2E), these cell cycle changes caused by PTPMT1 ablation appear to mainly impact the differentiation-associated cycling while the cycling for stem cell self-renewal was less affected (PTPMT1-depleted HSCs failed to expand in vitro, likely because the culture conditions—cytokines, nutrients, oxidative stress, etc.—were not favorable). The underlying mechanism for this bias remains to be further determined.
Equally interesting is that PTPMT1 is required for the differentiation of stem cells and early progenitors but is not required for late lineage progenitor differentiation. While panhematopoietic cell (including stem cell) PTPMT1 knockout (PTPMT1fl/fl/Vav1-Cre+) mice died postnatally of the differentiation arrest of HSCs and early progenitors (Figures 2A and 2B), more restricted myeloid, T lymphoid, or B lymphoid lineage-specific PTPMT1 knockout mice did not show any abnormalities (Figures S4A–S4D). This suggests that PTPMT1 depletion has distinct effects in hematopoietic cells depending on the stage in which the cells are targeted. There appears to be a unique and specific requirement for mitochondrial energy metabolism for stem cell/early progenitor differentiation that does not exist in late lineage restricted progenitors or mature cells. Our previous studies showed that the loss of PTPMT1 also blocked ESC differentiation without impacting the function of mature embryonic fibroblasts (Shen et al., 2011). It seems that the bioenergetic stress-activated differentiation-controlling mechanism is well conserved between embryonic and somatic stem cells. Similar to HSC differentiation, ESC differentiation also requires a tremendous energy supply, which relies on a robust and rapid upregulation of mitochondrial activities. Unlike HSCs, however, ESCs also require substantial mitochondrial biogenesis for differentiation because the mitochondrial mass in ESCs is significantly lower than that in differentiated cells (Cho et al., 2006; Lonergan et al., 2007).
Another interesting finding in this report is that although mitochondrial metabolism in PTPMT1-depleted stem cells was only moderately perturbed, the cellular responses (cell cycle changes and differentiation block) were rather profound. This phenomenon is reminiscent of conditions involving nuclear DNA damage, or mitotic spindle assembly checkpoints, in which a single double-strand DNA break or misalignment of a single chromosome causes complete cell cycle arrest (Musacchio and Salmon, 2007; Reinhardt and Yaffe, 2009). The mitochondrial bioenergetic stress signal elicited by PTPMT1 ablation appears to be propagated and amplified through the p53-p21/p57 pathway, similar to the DNA-damage-induced cell cycle checkpoint. Cyclin-dependent kinase inhibitors, p21 and p57, were significantly upregulated, and p53 was moderately upregulated in PTPMT1 knockout stem cells and early progenitors (Figure 5F). As the energy stress sensor AMPK was highly activated in these cells (Figures 6F and 6G) and activation of AMPK has been demonstrated to induce p53-dependent cell cycle arrest (Imamura et al., 2001; Jones et al., 2005), it is likely that the altered mitochondrial metabolism caused by PTPMT1 deficiency is sensed by AMPK and then relayed to the p53-p21/p57 pathway.
PTPMT1 deficiency appears to exert its deleterious effects by disrupting PIP homeostasis in the mitochondria. PTPMT1 is localized to the inner membrane (Pagliarini et al., 2005) and breaks down PIPs (Pagliarini et al., 2004; Shen et al., 2011). Its catalytic activity and mitochondrial localization were required for HSC differentiation (Figure 4). Furthermore, overloading of PIP substrates, such as PI(3,5)P2, in WT LSK cells decreased mitochondrial oxygen consumption (Figure 7A), recapitulating the metabolic changes seen in PTPMT1-depleted LSK cells (Figure 6D). In contrast, inhibition of PI(3,5)P2 production by the PIK-fyve inhibitor partially restored differentiation capabilities of PTPMT1 knockout HSCs (Figures 7E and 7F). More importantly, PIP substrates of PTPMT1 directly enhanced fatty-acid-induced activation of mitochondrial UCP2 in in vitro proteoliposome assays (Figures 7C and 7D). Since UCP2 inhibits glucose-derived pyruvate oxidation in mitochondria (Bouillaud, 2009; Diano and Horvath, 2012; Samudio et al., 2009) and cellular differentiation may be intimately tied to the balance between glucose and fatty acid oxidation (Pearce et al., 2009; Zhang et al., 2011b), one intriguing hypothesis is that the build-up of PIP substrates in PTPMT1-depleted mitochondria enhances UCP2 function and thereby critically alters mitochondrial substrate utilization within the stem cell, leading to bioenergetic stress and differentiation block. Further studies will be essential to determine the structural bases of the interactions between PIPs and UCP2, how PTPMT1 might regulate other aspects of UCP-mediated physiology, and whether PTPMT1 plays a similar role in other tissue-specific stem cells.
EXPERIMENTAL PROCEDURES
Apoptosis and Cellular ROS Level Measurement
Fresh BM cells were stained with biotin-labeled antibodies against lineage markers (Gr-1, Mac-1, B220, Ter119, CD4, CD8, and CD3e) and then stained with Streptavidin eFluor® 450 and antibodies labeled with various fluorochromes (anti-c-Kit-APC-Cy7, anti-Sca-1-PE-Cy7, anti-CD150-APC, and anti-CD48-PE) purchased from BD Biosciences (San Jose, CA) or eBiosciences, Inc. (San Diego, CA). The cells were then stained with Annexin V-FITC and 7-amino-actinomycin D (7-AAD) using an Annexin V-FITC apoptosis Detection Kit I (BD Biosciences). Apoptotic cells in the gated HSC (Lineage−Sca-1+c-Kit+CD150+CD48−) population were quantified by FACS. To measure cellular ROS levels, cells were loaded with 2′-7′-dichlorofluorescein diacetate (DCF-DA) (5 μM) at 37°C for 15 min after being stained with the antibodies described as above. ROS (H2O2) levels in the gated HSC population were quantified using flow cytometry.
Cell Cycle Analysis
To analyze the cell cycle status of LSK cells, fresh BM cells were stained with Pyronin Y (1 μg/ml) and Hoechst 33342 (10 μg/ml). The cells were washed and then stained with antibodies labeled with various fluorochromes (anti-lineages-APC-Cy7 [Gr-1, Mac-1, B220, Ter119, CD4, CD8, and CD3e], anti-c-Kit-APC, and anti-Scal-1-PE-Cy7). Subsequent cell population gating and quantification of the cells at G0, G1, and S/G2/M phases by FACS were performed as previously reported (Xu et al., 2011; Xu et al., 2010). For HSC cycle analysis, HSCs (Lineage−Sca-1+c-Kit+CD150+CD48−) were sorted and deposited into 75% ethanol. Cells were fixed at −20°C for 24 hr and then stained with FITC-labeled anti-Ki67 antibody (BD Biosciences) followed by propidium iodide (PI) (50 μg/ml). Percentages of the cells at G0, G1, and S/G2/M phases were quantified by FACS.
Oxygen Consumption and ATP Measurement
Measurement of intact cellular respiration was performed using the Seahorse XF24 and XF96 analyzers as previously described (Liu et al., 2009). Respiration was measured under basal conditions, in the presence of mitochondrial inhibitor oligomycin (350 nM), mitochondrial uncoupling compound carbonyl-cyanide-4-trifluorometh-oxyphenylhydrazone (FCCP) (5 μM), and respiratory chain inhibitor rotenone (1 μM). To determine total cellular ATP levels, live HSCs (Lineage−Sca-1+c-Kit+CD150+CD48−) were sorted into PBS and assessed using a CellTiter-Glo® Luminescent Cell Viability Assay kit (Promega Corporation, Madison, WI), which is a homogeneous method to determine the number of viable cells in culture based on quantitation of the ATP present.
Reconstitution of UCP2 in Liposomes, Formation of UCP2-Containing Planar Bilayer, and Proton Conductance Measurement
Human recombinant UCP2 was purified from E. coli inclusion bodies and reconstituted into liposomes using a previously described protocol (Beck et al., 2007; Rupprecht et al., 2010). To introduce arachidonic acid (AA) and/or PIPs, proteoliposomes were mixed with AA- or PIP-containing liposomes in required proportions. Planar lipid bilayers were formed from proteoliposomes on the tip of plastic pipettes as previously described (Beck et al., 2006). Current-voltage (I-V) characteristics were measured by a patch-clamp amplifier (EPC 10, HEKA Elektronik Dr. Schulze GmbH, Germany). Total membrane conductance was determined from a linear fit of experimental data (I) at applied voltages ranging from −50 to 50 mV as described previously (Rupprecht et al., 2010).
Statistical Analysis
Statistical significance was determined using unpaired two-tailed Student’s t test. p < 0.05 was considered to be significant.
Supplementary Material
Acknowledgments
We are grateful to Dr. Thomas Graf at the Center for Genomic Regulation and ICREA, Spain, for Vav1-Cre transgenic mice; and Dr. Jaburek and Dr. Jezek at Academy of Sciences, Prague, Czech Republic, for providing us with E. coli inclusion bodies containing recombinant hUCP2. This work was supported by National Institutes of Health grants DK092722, HL068212, and HL095657 (to C.K.Q.) and the German Research Foundation grant Po 524/5-1 (to E.E.P).
Footnotes
Supplemental Information for this article includes five figures, one table, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.stem.2012.11.022.
References
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Beck V, Jabůrek M, Breen EP, Porter RK, Jezek P, Pohl EE. A new automated technique for the reconstitution of hydrophobic proteins into planar bilayer membranes. Studies of human recombinant uncoupling protein 1. Biochim Biophys Acta. 2006;1757:474–479. doi: 10.1016/j.bbabio.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Beck V, Jabůrek M, Demina T, Rupprecht A, Porter RK, Jezek P, Pohl EE. Polyunsaturated fatty acids activate human uncoupling proteins 1 and 2 in planar lipid bilayers. FASEB J. 2007;21:1137–1144. doi: 10.1096/fj.06-7489com. [DOI] [PubMed] [Google Scholar]
- Bouillaud F. UCP2, not a physiologically relevant uncoupler but a glucose sparing switch impacting ROS production and glucose sensing. Biochim Biophys Acta. 2009;1787:377–383. doi: 10.1016/j.bbabio.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005;2:85–93. doi: 10.1016/j.cmet.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- Cho YM, Kwon S, Pak YK, Seol HW, Choi YM, Park J, Park KS, Lee HK. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem Biophys Res Commun. 2006;348:1472–1478. doi: 10.1016/j.bbrc.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Diano S, Horvath TL. Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends Mol Med. 2012;18:52–58. doi: 10.1016/j.molmed.2011.08.003. [DOI] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS. Regulation of ion transport proteins by membrane phosphoinositides. Nat Rev Neurosci. 2007;8:921–934. doi: 10.1038/nrn2257. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun. 2001;287:562–567. doi: 10.1006/bbrc.2001.5627. [DOI] [PubMed] [Google Scholar]
- Inoue S, Noda S, Kashima K, Nakada K, Hayashi J, Miyoshi H. Mitochondrial respiration defects modulate differentiation but not proliferation of hematopoietic stem and progenitor cells. FEBS Lett. 2010;584:3402–3409. doi: 10.1016/j.febslet.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jefferies HB, Cooke FT, Jat P, Boucheron C, Koizumi T, Hayakawa M, Kaizawa H, Ohishi T, Workman P, Waterfield MD, Parker PJ. A selective PIKfyve inhibitor blocks PtdIns(3,5)P(2) production and disrupts endomembrane transport and retroviral budding. EMBO Rep. 2008;9:164–170. doi: 10.1038/sj.embor.7401155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Krauss S, Zhang CY, Lowell BB. The mitochondrial uncoupling-protein homologues. Nat Rev Mol Cell Biol. 2005;6:248–261. doi: 10.1038/nrm1592. [DOI] [PubMed] [Google Scholar]
- Kühn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, Liu H, Keyvanfar K, Chen H, Cao LY, et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–392. doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonergan T, Bavister B, Brenner C. Mitochondria in stem cells. Mitochondrion. 2007;7:289–296. doi: 10.1016/j.mito.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel C, Messina-Graham SV, Broxmeyer HE. Superoxide flashes, reactive oxygen species, and the mitochondrial permeability transition pore: potential implications for hematopoietic stem cell function. Curr Opin Hematol. 2011;18:208–213. doi: 10.1097/MOH.0b013e3283475ffe. [DOI] [PubMed] [Google Scholar]
- Matsumoto A, Takeishi S, Kanie T, Susaki E, Onoyama I, Tateishi Y, Nakayama K, Nakayama KI. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2011;9:262–271. doi: 10.1016/j.stem.2011.06.014. [DOI] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- Norddahl GL, Pronk CJ, Wahlestedt M, Sten G, Nygren JM, Ugale A, Sigvardsson M, Bryder D. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell. 2011;8:499–510. doi: 10.1016/j.stem.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Worby CA, Dixon JE. A PTEN-like phosphatase with a novel substrate specificity. J Biol Chem. 2004;279:38590–38596. doi: 10.1074/jbc.M404959200. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Wiley SE, Kimple ME, Dixon JR, Kelly P, Worby CA, Casey PJ, Dixon JE. Involvement of a mitochondrial phosphatase in the regulation of ATP production and insulin secretion in pancreatic beta cells. Mol Cell. 2005;19:197–207. doi: 10.1016/j.molcel.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol. 2009;21:245–255. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht A, Sokolenko EA, Beck V, Ninnemann O, Jaburek M, Trimbuch T, Klishin SS, Jezek P, Skulachev VP, Pohl EE. Role of the transmembrane potential in the membrane proton leak. Biophys J. 2010;98:1503–1511. doi: 10.1016/j.bpj.2009.12.4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samudio I, Fiegl M, Andreeff M. Mitochondrial uncoupling and the Warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009;69:2163–2166. doi: 10.1158/0008-5472.CAN-08-3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Liu X, Yu WM, Liu J, Nibbelink MG, Guo C, Finkel T, Qu CK. A critical role of mitochondrial phosphatase Ptpmt1 in embryogenesis reveals a mitochondrial metabolic stress-induced differentiation checkpoint in embryonic stem cells. Mol Cell Biol. 2011;31:4902–4916. doi: 10.1128/MCB.05629-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonnet AJ, Nehmé J, Vaigot P, Barroca V, Leboulch P, Tronik-Le Roux D. Phenotypic and functional changes induced in hematopoietic stem/progenitor cells after gamma-ray radiation exposure. Stem Cells. 2009;27:1400–1409. doi: 10.1002/stem.66. [DOI] [PubMed] [Google Scholar]
- Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, Schneider JW, Zhang CC, Sadek HA. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–390. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9:298–310. doi: 10.1016/j.stem.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Viollet B, Guigas B, Leclerc J, Hébrard S, Lantier L, Mounier R, Andreelli F, Foretz M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol (Oxf) 2009;196:81–98. doi: 10.1111/j.1748-1716.2009.01970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- Xu D, Wang S, Yu WM, Chan G, Araki T, Bunting KD, Neel BG, Qu CK. A germline gain-of-function mutation in Ptpn11 (Shp-2) phosphatase induces myeloproliferative disease by aberrant activation of hematopoietic stem cells. Blood. 2010;116:3611–3621. doi: 10.1182/blood-2010-01-265652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Liu X, Yu WM, Meyerson HJ, Guo C, Gerson SL, Qu CK. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J Exp Med. 2011;208:1977–1988. doi: 10.1084/jem.20110450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat Cell Biol. 2004;6:436–442. doi: 10.1038/ncb1126. [DOI] [PubMed] [Google Scholar]
- Zhang J, Guan Z, Murphy AN, Wiley SE, Perkins GA, Worby CA, Engel JL, Heacock P, Nguyen OK, Wang JH, et al. Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 2011a;13:690–700. doi: 10.1016/j.cmet.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, Wahjudi PN, Setoguchi K, Wang G, Do A, et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011b;30:4860–4873. doi: 10.1038/emboj.2011.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, Yoshihara H, Hosokawa K, Tai I, Shinmyozu K, Tsukahara F, Maru Y, Nakayama K, Nakayama KI, Suda T. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell. 2011;9:247–261. doi: 10.1016/j.stem.2011.07.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.