

Abstract
Significance: Phagocytosis is required for the clearance of dying cells. The subsequent regulation of inflammatory responses by phagocytic cells is mediated by both innate and adaptive immune responses. Autophagy, an evolutionarily ancient process of lysosomal self-digestion of organelles, protein aggregates, apoptotic corpses, and cytosolic pathogens, has only recently become appreciated for its dynamic relationship with phagocytosis, including newly discovered autophagic-phagocytosis “hybrid” processes such as microtubule-associated protein 1 light chain 3-associated phagocytosis (LAP). Recent Advances: Signal transduction by reactive oxygen species (ROS) plays a critical role in the modulation of autophagy, phagocytosis, and LAP, and serves as both a link and an additional layer of regulation between these processes. Furthermore, specific targets for oxidation by ROS molecules have recently begun to become identified in each of these processes, as have “shared” proteins that facilitate the successful completion of both autophagy and phagocytosis. High mobility group box 1 is at the crossroads of autophagy, phagocytosis, and oxidative stress. Critical Issues: In this review, we discuss the most recent findings that link elements of autophagy and phagocytosis, specifically through redox-dependent signal transduction. These interconnected cellular processes are placed in the context of cell death and immunity in both health and disease. Future Directions: Given the broad roles that autophagy, phagocytosis, and ROS signaling play in human health, disease, and the maintenance of cellular and organismal homeostatic balance, it is important to delineate intersections between these pathways and uncover targets for potential therapeutic intervention in the setting of autoimmune and inflammatory diseases. Antioxid. Redox Signal. 18, 677–691.
Introduction
Most cell types have a limited life span that ends physiologically or pathophysiologically through the process of cell death. Cell death can be an important part of normal animal development and tissue homeostasis. Until recently, the mechanism of cell death and how dead cells are removed by healthy neighboring cells remained largely unknown (38). The two primary cell death modalities are apoptosis and necrosis. Apoptosis is a process that is indispensable for the maintenance of tissue development and homeostasis and in this regard, organismal survival. Likewise, mechanisms for both the facilitation and resolution of apoptosis are equally indispensible. Biochemical events leading to characteristic cell changes observed during apoptosis include blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, and chromosomal DNA fragmentation. Necrotic cell death resulting from cell or tissue injury requires the recruitment of immune cells to promote pathogen clearance and reparative wound healing. Clearance of dying cells is important in preventing autoimmunity (80), as animals that are deficient in mechanisms responsible for the phagocytosis of dying cells often suffer from autoimmune disorders (42, 103). Moreover, failure to properly clear dying cells has been linked to unresolved inflammation and developmental abnormalities (35). Phagocytosis is crucial for the clearance of the apoptotic bodies that are the remnants of completed programmed cell death, necrotic cell debris, and opsonized pathogens (1, 27, 86). Unlike the latter that are recognized by phagocytic cells such as macrophages by pattern recognition and Fc receptors (FcRs) for degradation and presentation to adaptive immune cells, phagocytosis of apoptotic cells is immunologically silent (36). Typically accompanied by the secretion of regulatory cytokines such as interleukin-10 (IL-10) and transforming growth factor beta (TGFβ), disruption of this silence has drastic consequences for the host, including the development of autoimmunity and pathological inflammatory responses (24). This distinction necessitates the utilization of unique signaling pathways once cargo has become engulfed to ensure proper intracellular processing for either presentation to immune cells, or the silent resolution of successful apoptosis.
The clearance of dying cells involves many surface molecules, adaptors, and chemotactic proteins. As such, it is regulated at multiple levels. Despite differences in outcomes (pro- versus anti-inflammatory), there is a certain degree of crosstalk between intracellular compartments once cargo has been internalized. Autophagy, literally “self-eating,” is the process by which cells clear protein aggregates, whole organelles, and other components for the concomitant purposes of nutrient derivation and the mitigation of cell stress that is mediated by potentially cytotoxic aggregates and organelles (e.g., mitochondria) which have persisted past their usefulness, and it is also an important source of antigen derived from degraded pathogens and abnormal self-proteins (19, 64, 128). As a constitutive process involving the formation of double-membrane vesicles that receive input from endosomal compartments and ultimately fuse with lysosomes (70, 119), this process has a dynamic relationship with both immuno-stimulatory and silent phagocytosis. This relationship is made further complex by the active role redox-dependent signal transduction plays in modulating both phagocytosis and autophagy (Fig. 1) (3, 49, 98).
FIG. 1.

Schematic representation of the recognition of target cells and pathogens by phagocytosis and autophagy. Reactive oxygen species (ROS) have been identified as signaling molecules in various pathways regulating both phagosome and autophagosome formation and maturation. Eventually, phagosomes and autophagosomes fuse with the lysosome, form the phagolysosome and autolysosome, respectively, and mediate the degradation of pathogens, dying cells, or cellular debris. NADPH, nicotinamide adenine dinucleotide phosphate reduced.
Given the commonalities between the phagocytic and autophagic pathways, including shared molecules and their sensitivity to redox signaling, here we review the most recent findings that link elements of both pathways, specifically through redox-dependent signal transduction. Furthermore, these interconnected cellular processes are placed in the context of cell death and immunity in both health and disease.
Eat-Me, Find-Me, and Damage-Associated Molecular Pattern Molecule Signals
In order for a dying cell to become phagocytosed, it should be rendered recognizable to phagocytes (tissue-resident macrophages, monocytes, and dendritic cells [DCs]). Generally speaking, the initiation of successful phagocytosis relies on four steps (85, 86): (i) the release of “find-me” signals that recruit phagocytes; (ii) the receptor ligation-dependent recognition of “eat-me” signals and the down-regulation of “don't eat me” signals such as CD47, and the complement regulatory proteins, CD200, which facilitate engulfment; (iii) the processing and degradation of corpses involving a series of phagosomal maturation steps; and (iv) the suppression or initiation of inflammatory responses depending on additional innate immune stimuli. Recently, potential find-me signals released by apoptotic cells were reported, namely lysophosphatidylcholine (63), CX3CL1/fractalkine (121), sphingosine 1-phosphate (S1P) (40), nucleotides adenosine triphosphate (ATP), and uridine 5 triphosphate (UTP) (23), which bind to G-protein-coupled receptors, including P2Y2, CX3CR1, S1P1–5, and G2A in macrophages and monocytes (Fig. 2). Unfortunately, current understanding of “find-me” signals is still limited. Interestingly, the iron-binding protein lactoferrin could serve as an anti-attraction (“keep-out”) signal by apoptotic cells to inhibit the migration of granulocytes (12). Although macrophages and DCs are highly motile under basal conditions, certain factors associated with cell death are known to possess chemotactic properties. Many damage-associated molecular pattern molecules (DAMPs) that are released actively during cell stress or passively during necrosis (Fig. 3) or late-stage apoptosis are associated with chemotaxis (72). High mobility group box-1 (HMGB1), the prototypic DAMP, when released into the extracellular milieu attracts both macrophages and DCs by itself, and also binds to chemokines such as CCL19 and CXCL12 (21), thereby promoting more efficient gradient-dependent migration toward them. Other DAMPs, including nucleotides, ATP, and UDP, have also been ascribed chemoattractant properties and are known to be passively released from necrotic cells when the integrity of the plasma membrane is compromised and actively released from apoptotic cells via pannexin 1 channels (14). Given the fluidity of the plasma membrane during both forms of cell death, it is possible that additional, unidentified intracellular proteins may also escape into the extracellular space and recruit phagocytes to sites of injury and apoptosis.
FIG. 2.

Eat-me signals, find-me signals, and phagocytic receptors for the engulfment of apoptotic cells. Apoptotic cells express or release various eat-me and find-me signals, which bind to phagocyte-cell-surface receptors either directly or indirectly through bridging molecules. Multiple eat-me signals and their receptors can form a cluster within the phagocytic cup at the engulfment synapse to facilitate the clearance of apoptotic cells. LPC, lysophosphatidylcholine; S1P, sphingosine 1-phosphate; ATP, adenosine triphosphate; UTP, uridine 5 triphosphate; MEG-E8, globule EGF factor 8 protein; Gas6, growth-arrest-specific 6; BAI1, brain angiogenesis inhibitor 1; PtdSer, phosphatidylserine; TIM4, T-cell immunoglobulin domain and mucin domain 4; Tulp1, tubby-like protein 1; β2-GPI, β2-glycoprotein-I; TREM2, Triggering receptor expressed on myeloid cells 2; MerTK, Mer tyrosine kinase; LRP, low density lipoprotein receptor-related protein. Some don't-eat-me signals such as CD24 have also been identified.
FIG. 3.

Damage-associated molecular pattern molecule (DAMP) signals, macropinocytosis, and phagocytosis for the engulfment of necrotic cells. DAMPs are molecules that can initiate and perpetuate immune response during the noninfectious inflammatory responses. Many DAMPs are nuclear or cytosolic proteins that are released after necrosis. Once released into the extracellular space, DAMPs bind DAMP receptors on macrophages or other phagocytes, and mediate inflammation and/or the removal of necrotic cells by macropinocytosis and phagocytosis. HMGB1, high mobility group box-1; ATP, adenosine triphosphate; HSP, heat shock protein; TSP1, thrombospondin-1; HRG, histidine-rich glycoprotein; C1q, complement 1q; C3/C4, complement 3 and 4; MBL, mannose-binding lectin; TREM1, Triggering receptor expressed on myeloid cells 1; RAGE, the receptor for advanced glycation end products; TLR, Toll-like receptor.
By contrast, “eat-me” signals that facilitate recognition by phagocytes are much better understood (85, 86). Eat-me signals or phagocytosis ligands can be classified into two major categories, membrane-anchored eat-me signals (e.g., phosphatidylserine [PtdSer], intercellular adhesion molecule-1 [ICAM-1], calreticulin) and soluble bridging molecules (e.g., growth-arrest-specific 6 [Gas6], protein S, globule EGF factor 8 protein [MFG-E8], β2-glycoprotein-I [β2-GPI], and annexin I). These signals can be trafficked to the cell surface by canonical and noncanonical secretory pathways, as well as “revealed” by both conformational and enzymatic modification of the plasma membrane (66). PtdSer (which localizes to the inner leaflet of the plasma membrane) becomes displayed on the cell surface when portions of the lipid bilayer flip during apoptosis (25). This allows interactions with receptors and bridging molecules such as T-cell immunoglobulin domain and mucin domain 4 (TIM4), MFG-E8, brain angiogenesis inhibitor 1 (BAI1), and Mer tyrosine kinase (MerTK) expressed on phagocytes, thereby promoting phagocytosis (42, 103). Proteins, such as calreticulin, can also be up-regulated on the surfaces of cells during apoptosis and interact with and activate distinct phagocytic receptors such as low-density lipoprotein receptor-related protein (LRP or CD91) (32).
Unlike apoptosis, cells that release intracellular contents during necrosis or are opsonized with antibodies due to the expression of abnormal self or pathogen-derived antigen are recognized by phagocytes such as macrophages through macropinocytosis mechanisms which are independent of apoptotic cells (Fig. 3) (62). Macropinocytosis is a form of bulk uptake of fluid and solid cargo into cytoplasmic vacuoles, called macropinosomes, and has been studied mostly in relation to antigen presentation. In contrast, necrotic cells can also be phagocytosed by DCs (92). In addition to the activation of complement cascades, an antibody-coated cell or pathogen becomes immediately recognizable to phagocytes which express cell surface receptors that bind to the Fc region of an antibody termed FcR (31). Activation of these receptors initiates the phagocytosis of the target cell, and in concert with pattern recognition receptor (PRR) ligation by pathogen-associated molecular pattern (PAMP) molecules or DAMPs, initiates maturational programs within the phagocyte that stimulate adaptive immune responses via presentation of antigenic peptides in class I and class II major histocompatability complexes (MHC-II) derived from the phagocytosed target cell (43).
Phagocytosis: A Process of Killing Cells or Microbes
Unlike autophagy, phagocytosis has been a recognized cellular process for more than a century (78). Macropinocytosis involves the constitutive “imbibing” of extracellular fluid in a nonspecific fashion, whereas phagocytosis is a highly regulated process that is dependent on precise receptor-ligand interactions. The most well-characterized form of phagocytosis is the internalization of antibody-coated (opsonized) particles by the FcR, and is, therefore, discussed as an example of how phagocytosis proceeds at the molecular level (37, 75). It should be noted, however, that although there are numerous forms of phagocytosis which operate under both parallel and distinct initial molecular regulation, the downstream mechanisms of vesicular formation and fusion are largely conserved.
The ligation of a singular FcR by an opsonized particle is not sufficient to initiate phagocytosis (105). In order to propagate a successful signal that initiates phagocytosis, engagement of multiple receptors is required. This is accomplished by the clustering of receptors that occurs immediately after initial engagement. In turn, this clustering facilitates the ligation of more unoccupied FcR sites and so forth. After this, a tyrosine phosphorylation-dependent signaling cascade (33, 37) is transduced within the phagocyte that causes the recruitment of specialized adaptor proteins such as Grb2-associated binder 2 (Gab2) (39) and CrkII (88), both of which mediate the recruitment of additional scaffolding proteins and initiate the formation of the cellular machinery required for the reorganization of both the actin cytoskeleton (77) and the plasma membrane (125) which are necessary for successful phagocytosis. Significantly, phagocytosis is significantly obstructed when these adaptor proteins are chromosomally deleted in mice (39). Rapid and efficient removal of dying cells and foreign microbes by phagocytes is important during development, tissue homeostasis, and immune responses (Fig. 4). Phagocytosis of apoptotic cells results in tolerance or anti-inflammatory responses. Necrotic cells release DAMPs that stimulate pro-inflammatory responses. Autophagy plays dual roles in promoting or discouraging inflammation. Initially, the plasma membrane serves as the source of lipid membrane for a newly forming phagosome (125), although there is evidence suggesting that additional organelles such as the endoplasmic reticulum contribute to the membrane as the phagosome matures (120). The early phagophore forms after scission from the plasma membrane and begins to mature as it traffics toward the lysosome in a GTPase-dependent fashion (13). The late phagosome acquires a much more acidic pH (59) and fuses with the lysosome via interactions with lysosome-associated membrane proteins (LAMP)-1 and LAMP-2 (51). This newly formed vesicle is termed the phagolysosome and is a degradative organelle in which lytic proteins process the phagocytic cargo and ultimately derive both nutrients and antigenic peptides for MHC-II presentation to immune effector cells (43). Toll-like receptor (TLR) signaling contributes to the regulation of phagosome maturation in macrophages and DCs (10, 11). Moreover, engaging the autophagic pathway via TLR signaling enhances phagosome maturation by microtubule-associated protein 1 light chain 3 (LC3)-associated phagocytosis (LAP) (94). In contrast, other independent studies demonstrate that TLR stimulation does not influence phagosome maturation assayed by defined particles and quantitative methodology; therefore, additional studies are needed to more clearly define the role of TLRs in both autophagy and phagocytosis (89, 129, 130).
FIG. 4.

Discriminating between necrosis, apoptosis, and autophagy. Cells can respond to stress in a variety of ways ranging from the activation of survival pathways such as autophagy to the initiation of apoptosis. When apoptotic cells are rapidly cleared by phagocytes, the immune system is not stimulated. In contrast, necrotic cells release DAMPs that stimulate pro-inflammatory responses. Autophagy plays a dual role in the regulation of inflammation depending on the context. TLR4, Toll-like receptor-4.
Autophagy: A Lysosomal Degradation Pathway
Much like phagocytosis, autophagy is a process that impinges on the formation, maturation, and fusion of vesicles encapsulated by the lipid membrane and facilitates the degradation of selective cargo derived from intracellular components, including whole organelles such as mitochondria and ribosomes, as well as cytotoxic protein aggregates. In addition, foreign pathogens acquired by cytoplasmic sequestration are also degraded by a specialized form of autophagy termed xenophagy (19, 64). There are at least three recognized types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (Fig. 5) (128). This review focuses on macroautophagy and will hereafter refer to macroautophagy as simply “autophagy.”
FIG. 5.

Types of autophagy. Microautophagy refers to the sequestration of cytosolic components directly by lysosomes through invaginations within their limiting membrane. Chaperone-mediated autophagy involves the direct translocation of unfolded substrate proteins (KFERQ-like motif ) across the lysosomal membrane through the action of a cytosolic and lysosomal chaperone heat shock cognate protein of 70 kDa (Hsc70), and the integral membrane receptor lysosome-associated membrane protein type 2A (LAMP-2A). In the case of macroautophagy, the cargo is sequestered within a unique double-membraned cytosolic vesicle, termed autophagosome. The autophagosome itself is formed by expansion of the phagophore. The autophagosome undergoes fusion with the lysosome to form an autolysosome, in which the sequestered material is degraded. Autophagy, an intrinsically nonselective process, can also target selective cargo for degradation such as mitophagy, lipophagy, ribophagy and xenophagy. NDP52, nuclear dot protein 52 kDa; NBR1, neighbor of BRCA1 gene 1.
The initial steps of autophagy involve the formation of a specialized, double-membranous vesicle termed the isolation membrane, namely phagophore. Recent studies have found that the sources of membrane for these nascent autophagic vesicles can be from the plasma membrane itself, the golgi complex, and even the mitochondrial membrane (119). In addition, under conditions of differing autophagy-initiating events, the primary membrane source may also be different. For example, during cellular starvation-induced autophagy, studies suggest that mitochondria are the primary source from which the membrane is derived (41). As the isolation membrane matures, the protein LC3-I (called Atg8 in yeast) becomes covalently lipidated into LC3-II and incorporated into the membrane as a crucial scaffolding protein (52). Given this role, the conversion of LC3-I to LC3-II serves as a marker for heightened autophagic flux and not surprisingly, cells deficient for LC3 are unable to successfully initiate autophagy (79). Before fusion of the isolation membrane and the formation of the closed vesicle termed the autophagosome, autophagic cargo is recruited via adaptor molecules such as p62 (9, 81), Nix (93, 102), nuclear dot protein 52 kDa (NDP52) (117), neighbor of BRCA1 gene 1 (NBR1) (60), optineurin (126), and galectin 8 (118) (Fig. 5). These molecules contain ubiquitin-binding domains that recognize poly-ubiquitinated protein aggregates, organelles, and bacteria. The autophagosome then traffics to and fuses with the lysosome, forming the autolysosome in which the cargo is proteolytically degraded. Significantly, autophagosomes continuously receive input from endosomes and have been demonstrated to fuse with MHC-II loading compartments, thereby making the autophagosome an essential source of antigen for presentation to CD4+ helper T cells (15). Furthermore, antigens specifically targeted to the autophagosome by fusion with an LC3 construct are much more effectively presented to adaptive immune cells and elicit functionally superior responses (100). Notably, autophagy contributes to dying cell clearance during apoptosis (83).
The molecular mechanisms governing the initiation of autophagy in response to various stimuli are complex and not fully delineated. In a general sense, autophagy can be classified as being either a mammalian target of rapamycin (mTOR) dependent or independent (95). mTOR is a nutrient sensor that is associated with the lysosome which, when inhibited, initiates signaling events that lead to heightened autophagy (87). This is due to mTOR's function of inhibiting Atg1, which is required during the initiation of autophagy (58). As such, many pharmacological agents that induce autophagy operate through this pathway (mTOR inhibitors) and include the drug rapamycin (87) and its analogues. The protein Beclin 1 (called Atg8 in yeast) appears to be central to preautophagic signaling (56). Usually, Beclin 1 is bound to the anti-apoptotic protein Bcl-2. This binding is abrogated during autophagic signaling (82), and promotes Beclin 1 interaction with class III phosphatidylinositol 3-kinase (PI3KC3) to further transduce the message.
Signal Transduction by Reactive Oxygen Species
Reactive oxygen species (ROS) are formed as a metabolic by-product of electron transfer to molecular oxygen, and predate the wholesale oxygenation of the atmosphere 2bya and acquisition of mitochondria 1bya. This generation can occur in a number of ways that are generally associated with mitochondrial function and respiration. Given the drastic physiological and pathological outcomes which are observed through the modulation of ROS, including tumorigenesis and various neurodegenerative and metabolic disorders, it can be concluded that ROS signal transduction plays a critical role in maintaining eukaryotic homeostasis. ROS is generated by both complexes within the electron transport chain (Complex I and III) as well as by nicotinamide adenine dinucleotide phosphate reduced (NADPH) oxidase (Fig. 6) and additional enzymes, which are tightly associated with the mitochondria, including the monoamine oxidases that are critical for the metabolism of monoamine signaling proteins such as dopamine and serotonin (16). In the presence of mitochondrial superoxide dismutase (SOD), O2•− can be converted to hydrogen peroxide (H2O2), which can then diffuse out of the mitochondria into the cytoplasm. In the presence of high iron concentrations, H2O2 can form the highly reactive hydroxyl radical (•OH) via the Fenton reaction. Catalase is responsible for converting H2O2 to water and oxygen. O2•− can also react with nitric oxide to form the highly reactive peroxynitrite (ONOO•). Other sources of ROS include the endoplasmic reticulum and peroxisome (44, 101). Interestingly, many signaling events result in dramatic elevations in ROS levels. Ligation of receptors by growth factors such as epidermal growth factor (EGF) induces this response (5), but given the reactivity of ROS molecules, it is difficult to imagine how specific ROS-mediated signaling can occur. However, evidence is beginning to emerge regarding the trafficking of ROS across the plasma membrane. ROS, specifically hydrogen peroxide, appears to cross the membrane in a specific manner via aquaporin channels, representing a method of potential regulation (8). Regardless of this, there are certainly many identifiable targets of ROS signaling. Studies have demonstrated that phosphotases become transiently inactivated after increases in ROS levels, and many cellular processes, including the regulation of responses to hypoxia, inflammatory responses (specifically formation of the NLRP3 or NOD-like receptor pyrin domain-containing 3 inflammasome), phagocytosis, and autophagy are all responsive to ROS signaling (3, 49, 122).
FIG. 6.

ROS and the regulation of autophagy. Mitochondria and NADPH oxidase are associated with phagocytosis and are major sources of ROS. ROS act as signaling molecules in the regulation of autophagy by targeting autophagy genes (e.g., ATG4), transcription factors (e.g., hypoxia-inducible factor [HIF]-1α), and signal transduction systems (e.g., mammalian target of rapamycin [mTOR] and mitogen-activated protein kinases [MAPKs]). In contrast, up-regulated autophagy inhibits ROS production by mitophagy-mediated impaired mitochondria removal, or increases ROS production by degrading catalases. LC3, microtubule-associated protein 1 light chain 3; ATM, ataxia telangiectasia mutated; TSC2, tuberous sclerosis protein 2; BNIP3, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3; PKB, protein kinase B.
Redox Signaling and Phagocytosis
ROS signal transduction plays a critical role in the cellular physiology of phagocytic cells (3). Oxidant bursts observed after growth receptor ligation also occur in macrophages, neutrophils, and so on, both during phagocytosis and in response to various stimulatory signals. Specific ROS-sensitive targets in these cells have also been identified. Nuclear factor (NF)-κB is a dimeric transcription factor that is involved in the regulation of a large number of genes which control various aspects of the immune and inflammatory response. NF-κB, which is usually localized to the cytosol, is translocated to the nucleus in its active form in response to oxidative stress (34, 65). In addition, mitogen-activated protein kinases (MAPK), including p38 and JNK which require phosphorylation for activation, are responsive to the kinase apoptosis-signal kinase-1 (ASK-1), which is maintained in an inactive state until ROS-mediated oxidation of its binding partner thioredoxin liberates it (47).
However, the most well-known role for ROS within phagocytes is for their bactericidal properties. NADPH oxidase is a multicomponent enzyme that is localized in the plasma membrane of phagocytic leukocytes. The generation of ROS in these cells is mediated by NADPH oxidase, which, when absent, results in an inhibited capacity to clear pathogens (4). It is possible that the ROS generated by NADPH oxidase contribute to host defenses not only through their microbicidal action but also through the modulation of redox-sensitive pathways in phagocytes. A recent study suggests that NADPH oxidase 2 (NOX2) activity decreases proteolytic efficiency of the phagosome through prolonged modification of the lumenal redox environment and oxidation of cysteine cathepsins (90). Anti-oxidant enzymes are required to neutralize phagocytosis and stimulatant-induced oxidant bursts and return ROS levels to basal concentrations in these cells. In addition, as previously mentioned, ROS signaling, particularly through NF-κB, may play an important role in the transcription and translation of NF-κB target genes, which include pro-inflammatory cytokines and chemokines that both recruit and stimulate phagocytes (28, 65).
Redox Signaling and Autophagy
Similar to phagocytosis, autophagy is also responsive to ROS signaling (49, 98). This is beginning to be extensively characterized given the often observed concurrent ROS generation and heightened autophagic flux (Fig. 6). Initially, given the consequences of ROS accumulation on organelles and genomic integrity, autophagy was thought to be enhanced purely as a means to mitigate oxidative stress (76). Although this remains true, and has drastic implications in many pathological states such as cancer and neurodegenerative diseases when the integrity of the autophagic pathway is disrupted, there is increasing evidence of direct regulation of this process by ROS molecules modulating specific autophagy proteins. When LC3 becomes lipidated via direct conjugation to phosphoethanolamine, a deconjugation event should occur in order for the molecule to be properly recycled. This deconjugation is mediated by the protease, Atg4. Studies have demonstrated that in the setting of heightened autophagy, particularly starvation-induced, Atg4 is a target for oxidation by hydrogen peroxide (99). This mechanism for regulation serves as a contributing factor toward the heightened autophagic state observed in a cell treated directly with exogenous hydrogen peroxide. Hypoxia-inducible factor 1 (HIF-1) plays a key role in the regulation of oxygen homeostasis. ROS induces HIF-1α-dependent expression of BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), which promotes the dissociation of Beclin 1 from its Bcl-2 inhibitors (132). In addition, ROS regulates autophagic signaling transduction pathways that induce autophagy such as mTOR and MAPK.
Negative regulation of ROS-mediated autophagy is facilitated by anti-oxidant enzymes that prevent ROS from being elevated for prolonged measures of time. Interestingly, we know that autophagy is induced in the setting of oxidative stress and that autophagy can serve as its own negative regulator by removing sources of ROS such as mitochondria (mitophagy) and clearing oxidized proteins from the cytosol (a process in which p62 may be required) (97). However, in some conditions, autophagy can contribute to abnormal ROS accumulation by selectively promoting the degradation of the major enzymatic ROS scavenger, catalase (131).
HMGB1 at the Crossroads of Autophagy, Phagocytosis, and Oxidative Stress
HMGB1, which was originally thought to function only as a DNA chaperone that enhances replication, repair, and recombination, was “re-discovered” to be a crucial DAMP that mediates the response to infection, injury, inflammation, tissue generation, and cell migration (2, 71, 113). To perform its role as a DAMP, HMGB1 should transit from the nucleus, through the cytoplasm, to the extracellular environment (7, 71). This process can occur during cell activation as well as necrosis, autophagy, and late-stage apoptosis (57, 96, 109, 116). HMGB1, composed of an A box, B box, and C tail domains, is a redox-sensitive protein. There are three cysteines at positions 23, 45, and 106 (C23, C45, and C106, respectively). C23 and C45 readily form an intra-molecular disulfide bridge, whereas the C106 remains in a reduced form (46). C23 and C45 are required for HMGB1 binding to Beclin 1 during autophagy (109), whereas C106 is required for HMGB1 binding to TLR4 in macrophages (127) and appears to be important for transit to the cytosol.
HMGB1 is a stress sensor, and it is up-regulated or released in response to several forms of stress, including oxidative stress (Fig. 7) (114). Oxidative stress such as H2O2 or knockdown of SOD1 induces HMGB1 release in many cell types (111, 115, 123), whereas anti-oxidants such as N-Acetyl Cysteine and quercetin inhibit HMGB1 release as does knockdown of the autophagy gene, Atg5 (109, 112, 123). The oxidation of C106 (and not C23 or C45) abrogates the ability of HMGB1 to function as an immuno-stimulatory protein when encountered by DCs (57). Exogenous HMGB1 stimulates neutrophil NADPH oxidase activation and ROS generation in a TLR4-dependent manner (26), suggesting the presence of a positive feedback loop for HMGB1 release during oxidative stress. Moreover, means to distinguish the three forms of HMGB1, the “all reduced”, dithiol, and oxidized forms are necessary to understand its disparate role in the nucleus, the cytosol, and outside the cell.
FIG. 7.

HMGB1 at the crossroads of autophagy, phagocytosis, and oxidative stress. HMGB1 is a DNA-binding nuclear protein that is released actively after cytokine stimulation as well as passively during necrosis. As a DNA chaperone and DAMPs, the redox properties of HMGB1 play a role in many processes, including DNA nuclear events, inflammation, and immunity. There is a complex relationship between autophagy, phagocytosis, and oxidative stress. Recent studies suggest that HMGB1 is a central player in the crosstalk between autophagy, phagocytosis, and oxidative stress (detail in text). HSPB1, heat shock protein β-1.
HMGB1 is also an important regulator of autophagy (108) (Fig. 7). Nuclear HMGB1 regulates small heat shock protein β-1 (HSPB1) expression (110). The phosphorylation of HSPB1 is necessary for the regulation of the actin cytoskeleton, which affects the cellular transport required for autophagy in response to mitochondrial injury. Thus, the HMGB1-HSPB1 pathway controls mitochondrial quality by autophagy/mitophagy. Cytosolic HMGB1 is a novel Beclin 1 binding protein that dissociates its inhibitory partner, Bcl-2 (109). The loss of p53 increases cytosolic HMGB1 binding to Beclin 1, thereby promoting autophagy and decreasing apoptosis (69). Extracellular reduced HMGB1 binds the receptor for advanced glycation end products (RAGE), but not TLR4, which inhibits mTOR and promotes the formation of the Beclin 1-PI3KC3 complexes (107). In contrast, oxidized HMGB1 induces the initiation of the intrinsic mitochondrial apoptotic pathway by unknown receptors (107). The induction of autophagy by both intracellular and extracellular HMGB1 is important for tumor development and a novel target for cancer therapy (50, 68, 113, 124). In addition, its receptor RAGE also regulates autophagy in pancreatic cancer cells (53–55). Targeted ablation of RAGE in mice delays pancreatic tumorigenesis and inhibits IL-6/STAT3-mediated autophagy (53).
Likewise, HMGB1 is also a regulator of phagocytosis (Fig. 7). Extracellular HMGB1 inhibits phagocytosis by binding PtdSer or ανβ3 in apoptotic neutrophils or phagocytic macrophages, respectively (30, 67). These findings provide another mechanism by which exogenous HMGB1 enhances inflammatory responses by targeting phagocytosis-mediated anti-inflammatory responses during apoptosis, although it is unknown whether redox status regulates HMGB1's function in this instance. Moreover, intracellular HMGB1 is a negative regulator of phagocytosis by associating with Src kinase and inhibiting the interaction between Src and focal adhesion kinase (FAK) in macrophages (6). RAGE enhances phagocytosis in macrophages by binding to both PtdSer (29, 45) and free DNA. Future studies are needed to determine why both HMGB1 and RAGE interact with PtdSer, but induce opposite outcomes in the context of phagocytosis.
Autophagy and Phagocytosis in Cell Death, Immunity, and Inflammation
Given the nature of both autophagy and phagocytosis and the respective roles they play in maintaining the homeostatic balance at the level of both the cell and the organism, it becomes easy to accept that a dynamic relationship between both exist, particularly within the setting of cell death. This is so much so the case, that molecules previously thought to be specific to one process, such as LC3 in the case of autophagy, are actually playing similar roles in the other. In macrophages phagocytosing pathogens and cellular debris from apoptotic, necrotic, or the newly described “necroptotic” cells that display PtdSer, LC3 is rapidly recruited to phagosomes which lack the typical double-membrane coat that classical autophagosomes exhibit despite the requirement of Beclin 1, PI3KC3, Atg5, and Atg7, but not unc-51-like kinase 1 (ULK1). (48, 74, 94). This process has been termed LC3-associated phagocytosis or LAP (Fig. 8) (74). As upstream signals, TLRs, TIM4, FcγR, and NADPH oxidase-mediated ROS signal are required for LAP (48, 74). LAP is required for the efficient clearance of cell corpses as well as for phagosomal maturation.
FIG. 8.

The process of LC3-associated phagocytosis (LAP). LAP is the degradation of phagocytosed cellular corpses (e.g., apoptotic, necrotic, and necroptotic cells) and pathogens in phagosomes that utilize autophagic machinery. This process is dependent on some members of the classical autophagy pathway, including Beclin1, PI3KC3, ATG5, and ATG7, but not unc-51-like kinase 1 (ULK1). PtdSer is an eat-me signal exhibited by dying cells, and the receptors, including TLR, TIM4, and FcγR in phagocytes (e.g., macrophages and neutrophils) are required for recognizing PtdSer and recruiting LC3 to the phagosome to form a single-membrane structure. NADPH oxidase (e.g., NOX2) is one of the major sources of ROS that regulates LAP. PI3KC3, class III phosphatidylinositol 3-kinase; RIPK3, receptor-interacting serine-threonine kinase 3; NOX2, NADPH oxidase 2.
Abnormalities in cell death resolution can contribute to a variety of diseases, including cancer, autoimmunity, and neurodegenerative disorders. There is now a substantial body of literature that explores the relationship between autophagy and apoptosis (73). In fact, autophagic proteins are often directly inhibited by apoptotic proteins. Furthermore, evidence that apoptotic caspases can serve as autophagic substrates suggests another layer of direct crosstalk between apoptosis and autophagy (20). This has compelled many in these fields to orient autophagy opposite to apoptosis and term the process as a form of “programmed cell survival” (55, 56). It has also been suggested that autophagy itself can serve as an alternative form of cell death, although this has not been described in a consistent manner (61). Similar to phagocytosis, autophagy has been demonstrated as helping clear apoptotic cells during embryogenesis (83). They are also linked to nutrient acquisition and energy generation (84, 104), which is important for cell survival during cytotoxic insults.
The primary function of most phagocytes is to destroy pathogens. Phagocytosis plays an essential role in innate immune sensing, and degrading products of internalized pathogens may traffic and be modified as nutrients (104). In addition, phagocytosis is essential in antigen presentation and adaptive immunity (104). Several studies reveal a crucial role for autophagy in adaptive and innate immunity, including pathogen elimination, limiting or promoting virus replication, T- and B-cell homeostasis, and antigen processing and presentation, with the term “immunophagy” (17, 18) referring to all such processes collectively (Fig. 9).
FIG. 9.

Function of phagocytosis, autophagy, and LAP in immune cells. ROS are emerging as regulators of the eat-me processes such as autophagy, phagocytosis, and LAP in various cellular contexts. These eat-me mechanisms have been linked to the removal of dying cells, immune regulation, and inflammation in immune cells such as macrophages, monocytes, dendritic cells, and neutrophils.
The apoptosis of inflammatory cells and their subsequent clearance by phagocytosis (also called efferocytosis) is key to orchestrating the successful resolution of inflammation and suppressing autoimmune responses. This is in part achieved through the release of anti-inflammatory cytokines IL-10, TGFβ, platelet activating factor (PAF), and prostaglandin E2 (PGE2) (24) and the inhibition of pro-inflammatory cytokines such as TNFα and IL-1β (106). In the absence of LAP, engulfment of dead cells results in increased production of pro-inflammatory cytokines (e.g., IL-1β) and decreased production of anti-inflammatory cytokines (74). A key mechanism of inflammation is the activation of the “inflammasome,” which leads to caspase-1 activation and the maturation and release of IL-1 family cytokines and other inflammatory mediators (Fig. 10). Interestingly, autophagy was previously considered as an anti-inflammatory mechanism when enhanced IL-1β release was observed during sepsis in Atg16L1-null mice (91). However, recent studies note that the induction of autophagy by starvation promotes inflammasome-dependent IL-1β secretion (22). These findings suggest that autophagy plays dual roles in the regulation of inflammation depending on the timing and type of autophagic activation.
FIG. 10.
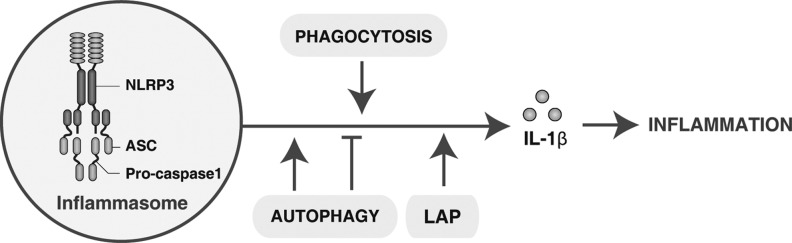
Phagocytosis, autophagy, and LAP regulate interleukin (IL)-1β production and release. The nucleotide-binding oligomerization domain-like receptors (NLRs) family, pyrin domain-containing 3 (NLRP3) inflammasome is a multiprotein complex, including NLRP3, ASC, and the effector cysteine protease caspase 1, which activates caspase 1, leading to the processing and secretion of the pro-inflammatory cytokine IL-1β. Phagocytosis and LAP have been demonstrated to contribute to this process. In contrast, autophagy plays a dual role in the regulation of IL-1β production and release. ASC, also known as PYCARD, the adaptor protein apoptosis-associated speck-like protein containing a CARD.
Conclusion
The crucial role for both phagocytosis and autophagy in maintaining homeostatic balance and both inciting and regulating host responses to cellular injury makes the identification of cross-talk between the two pathways paramount. Given the wide range of pathologies in which either heightened or inhibited autophagic flux has been implicated as a contributor, the potential uncovering of therapeutic targets continues to be an important task. Cancer biologists and clinicians, in particular, have recognized this and have begun to thoroughly characterize combinatorial approaches to therapy in which autophagy modulators (specifically inhibitors) have been incorporated with promising results. Considering the active role phagocytes play in modulating inflammatory responses in cancers and other diseases, it becomes important to understand how manipulation of the autophagic pathway may affect phagocytic cells.
Making the relationship between phagocytosis and autophagy more complex is the role that ROS signal transduction plays in both processes (Fig. 9). Although direct targets for ROS modulation of both pathways such as NF-κB and Atg4 are beginning to become identified and characterized at the molecular level, much remains unknown regarding the specificity and directionality of ROS signaling. However, it is becoming increasingly clear that signal transduction by ROS molecules occurs in a much more regulated and orchestrated manner than previously thought, and ROS messengers directly modulate both phagocytosis and autophagy.
Abbreviations Used
- ASC
the adaptor protein apoptosis associated speck-like protein containing a CARD
- ASK-1
apoptosis-signal kinase-1
- ATM
ataxia telangiectasia mutated
- ATP
adenosine triphosphate
- β2-GPI
β2-glycoprotein-I
- BAI1
brain angiogenesis inhibitor 1
- BNIP3
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3
- C1q
complement 1q
- C3/C4
complement 3 and 4
- DAMP
damage-associated molecular pattern molecule
- EGF
epidermal growth factor
- FAK
focal adhesion kinase
- FcR
Fc receptor
- Gab2
Grb2-associated binder 2
- Gas6
growth-arrest-specific 6
- H2O2
hydrogen peroxide
- HIF-1
hypoxia-inducible factor 1
- HMGB1
high-mobility group box-1
- HRG
histidine-rich glycoprotein
- Hsc70
heat shock cognate protein of 70 kDa
- HSP
heat shock protein
- HSPB1
heat shock protein β-1
- ICAM-1
intercellular adhesion molecule-1
- IL
interleukin
- LAMP
lysosome-associated membrane proteins
- LAP
LC3-associated phagocytosis
- LC3
microtubule-associated protein 1 light chain 3
- LPC
lysophosphatidylcholine
- LRP
low density lipoprotein receptor-related protein
- MAPK
mitogen-activated protein kinases
- MBL
mannose-binding lectin
- MerTK
Mer tyrosine kinase
- MFG-E8
globule EGF factor 8 protein
- MHC-II
class II major histocompatability complexes
- mTOR
mammalian target of rapamycin
- NADPH
nicotinamide adenine dinucleotide phosphate reduced
- NBR1
neighbor of BRCA1 gene 1
- NDP52
nuclear dot protein 52 kDa
- NF-κB
nuclear factor-κB
- NLRs
nucleotide-binding oligomerization domain-like receptors
- NLRP3
NOD-like receptor pyrin domain-containing 3
- NOX2
NADPH oxidase 2
- PAF
platelet activating factor
- PAMP
pathogen associated molecular pattern
- PGE2
prostaglandin E2
- PI3KC3
class III phosphatidylinositol 3-kinase
- PKB
protein kinase B
- PRR
pattern recognition receptor
- PtdSer
phosphatidylserine
- RAGE
the receptor for advanced glycation end products
- RIPK3
receptor-interacting serine-threonine kinase 3
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- S1P
sphingosine 1-phosphate
- TGF
transforming growth factor
- TIM4
T-cell immunoglobulin domain and mucin domain 4
- TLR
Toll-like receptor
- TREM1
triggering receptor expressed on myeloid cells 1
- TREM2
triggering receptor expressed on myeloid cells 2
- TSC2
tuberous sclerosis protein 2
- TSP1
thrombospondin-1
- Tulp1
tubby-like protein 1
- ULK1
unc-51-like kinase 1
- UTP
uridine 5 triphosphate
Acknowledgments
The authors apologize in advance to the researchers who were not referenced due to space limitations. D. T. is funded by the Department of Surgery and the University of Pittsburgh Cancer Institute. They appreciate careful review, fruitful discussions, and criticism by Dr. Michael T. Lotze from the Damage-Associated Molecular Pattern Laboratory.
References
- 1.Aderem A. Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 2.Andersson U. Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babior BM. Phagocytes and oxidative stress. Am J Med. 2000;109:33–44. doi: 10.1016/s0002-9343(00)00481-2. [DOI] [PubMed] [Google Scholar]
- 4.Babior BM. NADPH oxidase. Curr Opin Immunol. 2004;16:42–47. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Bae YS. Kang SW. Seo MS. Baines IC. Tekle E. Chock PB. Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 6.Banerjee S. de Freitas A. Friggeri A. Zmijewski JW. Liu G. Abraham E. Intracellular HMGB1 Negatively Regulates Efferocytosis. J Immunol. 2011;187:4686–4694. doi: 10.4049/jimmunol.1101500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 8.Bienert GP. Moller AL. Kristiansen KA. Schulz A. Moller IM. Schjoerring JK. Jahn TP. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem. 2007;282:1183–1192. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 9.Bjorkoy G. Lamark T. Brech A. Outzen H. Perander M. Overvatn A. Stenmark H. Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blander JM. Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 11.Blander JM. Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 12.Bournazou I. Pound JD. Duffin R. Bournazos S. Melville LA. Brown SB. Rossi AG. Gregory CD. Apoptotic human cells inhibit migration of granulocytes via release of lactoferrin. J Clin Invest. 2009;119:20–32. doi: 10.1172/JCI36226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caron E. Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–1721. doi: 10.1126/science.282.5394.1717. [DOI] [PubMed] [Google Scholar]
- 14.Chekeni FB. Elliott MR. Sandilos JK. Walk SF. Kinchen JM. Lazarowski ER. Armstrong AJ. Penuela S. Laird DW. Salvesen GS. Isakson BE. Bayliss DA. Ravichandran KS. Pannexin 1 channels mediate/’find-me/’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crotzer VL. Blum JS. Autophagy and intracellular surveillance: Modulating MHC class II antigen presentation with stress. Proc Natl Acad Sci U S A. 2005;102:7779–7780. doi: 10.1073/pnas.0503088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D'Autreaux B. Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 17.Deretic V. Autophagy as an immune defense mechanism. Curr Opin Immunol. 2006;18:375–382. doi: 10.1016/j.coi.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 18.Deretic V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev. 2011;240:92–104. doi: 10.1111/j.1600-065X.2010.00995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deretic V. Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Djavaheri-Mergny M. Maiuri MC. Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene. 2010;29:1717–1719. doi: 10.1038/onc.2009.519. [DOI] [PubMed] [Google Scholar]
- 21.Dumitriu IE. Bianchi ME. Bacci M. Manfredi AA. Rovere-Querini P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukoc Biol. 2007;81:84–91. doi: 10.1189/jlb.0306171. [DOI] [PubMed] [Google Scholar]
- 22.Dupont N. Jiang S. Pilli M. Ornatowski W. Bhattacharya D. Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011;30:4701–4711. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elliott MR. Chekeni FB. Trampont PC. Lazarowski ER. Kadl A. Walk SF. Park D. Woodson RI. Ostankovich M. Sharma P. Lysiak JJ. Harden TK. Leitinger N. Ravichandran KS. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fadok VA. Bratton DL. Konowal A. Freed PW. Westcott JY. Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fadok VA. Voelker DR. Campbell PA. Cohen JJ. Bratton DL. Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 26.Fan J. Li Y. Levy RM. Fan JJ. Hackam DJ. Vodovotz Y. Yang H. Tracey KJ. Billiar TR. Wilson MA. Hemorrhagic Shock Induces NAD(P)H Oxidase Activation in Neutrophils: Role of HMGB1-TLR4 Signaling. J Immunol. 2007;178:6573–6580. doi: 10.4049/jimmunol.178.10.6573. [DOI] [PubMed] [Google Scholar]
- 27.Flannagan RS. Jaumouille V. Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol. 2012;7:61–98. doi: 10.1146/annurev-pathol-011811-132445. [DOI] [PubMed] [Google Scholar]
- 28.Forman HJ. Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Respir Crit Care Med. 2002;166:S4–S8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- 29.Friggeri A. Banerjee S. Biswas S. de Freitas A. Liu G. Bierhaus A. Abraham E. Participation of the receptor for advanced glycation end products in efferocytosis. J Immunol. 2011;186:6191–6198. doi: 10.4049/jimmunol.1004134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friggeri A. Yang Y. Banerjee S. Park YJ. Liu G. Abraham E. HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3-integrin. Am J Physiol Cell Physiol. 2010;299:C1267–C1276. doi: 10.1152/ajpcell.00152.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia-Garcia E. Rosales C. Signal transduction during Fc receptor-mediated phagocytosis. J Leukoc Biol. 2002;72:1092–1108. [PubMed] [Google Scholar]
- 32.Gardai SJ. McPhillips KA. Frasch SC. Janssen WJ. Starefeldt A. Murphy-Ullrich JE. Bratton DL. Oldenborg PA. Michalak M. Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 33.Ghazizadeh S. Bolen JB. Fleit HB. Physical and functional association of Src-related protein tyrosine kinases with Fc gamma RII in monocytic THP-1 cells. J Biol Chem. 1994;269:8878–8884. [PubMed] [Google Scholar]
- 34.Gloire G. Legrand-Poels S. Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 35.Green DR. The end and after: how dying cells impact the living organism. Immunity. 2011;35:441–444. doi: 10.1016/j.immuni.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 36.Green DR. Ferguson T. Zitvogel L. Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353–363. doi: 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenberg S. Chang P. Silverstein SC. Tyrosine phosphorylation is required for Fc receptor-mediated phagocytosis in mouse macrophages. J Exp Med. 1993;177:529–534. doi: 10.1084/jem.177.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griffith TS. Ferguson TA. Cell death in the maintenance and abrogation of tolerance: the five ws of dying cells. Immunity. 2011;35:456–466. doi: 10.1016/j.immuni.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu H. Botelho RJ. Yu M. Grinstein S. Neel BG. Critical role for scaffolding adapter Gab2 in Fc gamma R-mediated phagocytosis. J Cell Biol. 2003;161:1151–1161. doi: 10.1083/jcb.200212158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gude DR. Alvarez SE. Paugh SW. Mitra P. Yu J. Griffiths R. Barbour SE. Milstien S. Spiegel S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008;22:2629–2638. doi: 10.1096/fj.08-107169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hailey DW. Rambold AS. Satpute-Krishnan P. Mitra K. Sougrat R. Kim PK. Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanayama R. Tanaka M. Miyasaka K. Aozasa K. Koike M. Uchiyama Y. Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 43.Harding CV. Geuze HJ. Class II MHC molecules are present in macrophage lysosomes and phagolysosomes that function in the phagocytic processing of Listeria monocytogenes for presentation to T cells. J Cell Biol. 1992;119:531–542. doi: 10.1083/jcb.119.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haynes CM. Titus EA. Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol Cell. 2004;15:767–776. doi: 10.1016/j.molcel.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 45.He M. Kubo H. Morimoto K. Fujino N. Suzuki T. Takahasi T. Yamada M. Yamaya M. Maekawa T. Yamamoto Y. Yamamoto H. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 2011;12:358–364. doi: 10.1038/embor.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoppe G. Talcott KE. Bhattacharya SK. Crabb JW. Sears JE. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp Cell Res. 2006;312:3526–3538. doi: 10.1016/j.yexcr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 47.Hsieh CC. Papaconstantinou J. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. FASEB J. 2006;20:259–268. doi: 10.1096/fj.05-4376com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang J. Canadien V. Lam GY. Steinberg BE. Dinauer MC. Magalhaes MA. Glogauer M. Grinstein S. Brumell JH. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A. 2009;106:6226–6231. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang J. Lam GY. Brumell JH. Autophagy signaling through reactive oxygen species. Antioxid Redox Signal. 2011;14:2215–2231. doi: 10.1089/ars.2010.3554. [DOI] [PubMed] [Google Scholar]
- 50.Huang J. Ni J. Liu K. Yu Y. Xie M. Kang R. Vernon P. Cao L. Tang D. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res. 2012;72:230–238. doi: 10.1158/0008-5472.CAN-11-2001. [DOI] [PubMed] [Google Scholar]
- 51.Huynh KK. Eskelinen EL. Scott CC. Malevanets A. Saftig P. Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007;26:313–324. doi: 10.1038/sj.emboj.7601511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kabeya Y. Mizushima N. Ueno T. Yamamoto A. Kirisako T. Noda T. Kominami E. Ohsumi Y. Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kang R. Loux T. Tang D. Schapiro NE. Vernon P. Livesey KM. Krasinskas A. Lotze MT. Zeh HJ., 3rd. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc Natl Acad Sci U S A. 2012;109:7031–7036. doi: 10.1073/pnas.1113865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kang R. Tang D. Livesey KM. Schapiro NE. Lotze MT. Zeh HJ., 3rd. The Receptor for Advanced Glycation End-products (RAGE) protects pancreatic tumor cells against oxidative injury. Antioxid Redox Signal. 2011;15:2175–2184. doi: 10.1089/ars.2010.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang R. Tang D. Schapiro NE. Livesey KM. Farkas A. Loughran P. Bierhaus A. Lotze MT. Zeh HJ. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010;17:666–676. doi: 10.1038/cdd.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kang R. Zeh HJ. Lotze MT. Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kazama H. Ricci JE. Herndon JM. Hoppe G. Green DR. Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32. doi: 10.1016/j.immuni.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim J. Kundu M. Viollet B. Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kinchen JM. Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008;9:781–795. doi: 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kirkin V. Lamark T. Sou YS. Bjorkoy G. Nunn JL. Bruun JA. Shvets E. McEwan DG. Clausen TH. Wild P. Bilusic I. Theurillat JP. Overvatn A. Ishii T. Elazar Z. Komatsu M. Dikic I. Johansen T. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 61.Kroemer G. Galluzzi L. Vandenabeele P. Abrams J. Alnemri ES. Baehrecke EH. Blagosklonny MV. El-Deiry WS. Golstein P. Green DR. Hengartner M. Knight RA. Kumar S. Lipton SA. Malorni W. Nunez G. Peter ME. Tschopp J. Yuan J. Piacentini M. Zhivotovsky B. Melino G. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krysko DV. Denecker G. Festjens N. Gabriels S. Parthoens E. D'Herde K. Vandenabeele P. Macrophages use different internalization mechanisms to clear apoptotic and necrotic cells. Cell Death Differ. 2006;13:2011–2022. doi: 10.1038/sj.cdd.4401900. [DOI] [PubMed] [Google Scholar]
- 63.Lauber K. Bohn E. Krober SM. Xiao YJ. Blumenthal SG. Lindemann RK. Marini P. Wiedig C. Zobywalski A. Baksh S. Xu Y. Autenrieth IB. Schulze-Osthoff K. Belka C. Stuhler G. Wesselborg S. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–730. doi: 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- 64.Levine B. Mizushima N. Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li N. Karin M. Is NF-kappaB the sensor of oxidative stress? FASEB J. 1999;13:1137–1143. [PubMed] [Google Scholar]
- 66.Li W. Eat-me signals: keys to molecular phagocyte biology and “appetite” control. J Cell Physiol. 2012;227:1291–1297. doi: 10.1002/jcp.22815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu G. Wang J. Park YJ. Tsuruta Y. Lorne EF. Zhao X. Abraham E. High mobility group protein-1 inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J Immunol. 2008;181:4240–4246. doi: 10.4049/jimmunol.181.6.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu L. Yang M. Kang R. Wang Z. Zhao Y. Yu Y. Xie M. Yin X. Livesey KM. Lotze MT. Tang D. Cao L. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia. 2011;25:23–31. doi: 10.1038/leu.2010.225. [DOI] [PubMed] [Google Scholar]
- 69.Livesey K. Kang R. Vernon P. Buchser W. Loughran P. Watkins SC. Zhang L. Manfredi JJ. Zeh HJ. Li L. Lotze M. Tang D. p53/HMGB1 Complexes Regulate Autophagy and Apoptosis. Cancer Res. 2012;72:1996–2005. doi: 10.1158/0008-5472.CAN-11-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Longatti A. Tooze SA. Vesicular trafficking and autophagosome formation. Cell Death Differ. 2009;16:956–965. doi: 10.1038/cdd.2009.39. [DOI] [PubMed] [Google Scholar]
- 71.Lotze MT. Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 72.Lotze MT. Zeh HJ. Rubartelli A. Sparvero LJ. Amoscato AA. Washburn NR. Devera ME. Liang X. Tor M. Billiar T. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 73.Maiuri MC. Zalckvar E. Kimchi A. Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 74.Martinez J. Almendinger J. Oberst A. Ness R. Dillon CP. Fitzgerald P. Hengartner MO. Green DR. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108:17396–17401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 75.Massol P. Montcourrier P. Guillemot JC. Chavrier P. Fc receptor-mediated phagocytosis requires CDC42 and Rac1. EMBO J. 1998;17:6219–6229. doi: 10.1093/emboj/17.21.6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mathew R. Karp CM. Beaudoin B. Vuong N. Chen G. Chen HY. Bray K. Reddy A. Bhanot G. Gelinas C. Dipaola RS. Karantza-Wadsworth V. White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.May RC. Machesky LM. Phagocytosis and the actin cytoskeleton. J Cell Sci. 2001;114:1061–1077. doi: 10.1242/jcs.114.6.1061. [DOI] [PubMed] [Google Scholar]
- 78.Metschnikoff E. Lecture on Phagocytosis and Immunity. Br Med J. 1891;1:213–217. doi: 10.1136/bmj.1.1570.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mizushima N. Yoshimori T. Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nagata S. Hanayama R. Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–630. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 81.Pankiv S. Clausen TH. Lamark T. Brech A. Bruun JA. Outzen H. Overvatn A. Bjorkoy G. Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 82.Pattingre S. Tassa A. Qu X. Garuti R. Liang XH. Mizushima N. Packer M. Schneider MD. Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 83.Qu X. Zou Z. Sun Q. Luby-Phelps K. Cheng P. Hogan RN. Gilpin C. Levine B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 84.Rabinowitz JD. White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. 2010;207:1807–1817. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. 2011;35:445–455. doi: 10.1016/j.immuni.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ravikumar B. Vacher C. Berger Z. Davies JE. Luo S. Oroz LG. Scaravilli F. Easton DF. Duden R. O'Kane CJ. Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 88.Reddien PW. Horvitz HR. CED-2/CrkII and CED-10/Rac control phagocytosis and cell migration in Caenorhabditis elegans. Nat Cell Biol. 2000;2:131–136. doi: 10.1038/35004000. [DOI] [PubMed] [Google Scholar]
- 89.Russell DG. Vanderven BC. Glennie S. Mwandumba H. Heyderman RS. The macrophage marches on its phagosome: dynamic assays of phagosome function. Nat Rev Immunol. 2009;9:594–600. doi: 10.1038/nri2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rybicka JM. Balce DR. Khan MF. Krohn RM. Yates RM. NADPH oxidase activity controls phagosomal proteolysis in macrophages through modulation of the lumenal redox environment of phagosomes. Proc Natl Acad Sci U S A. 2010;107:10496–10501. doi: 10.1073/pnas.0914867107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Saitoh T. Fujita N. Jang MH. Uematsu S. Yang BG. Satoh T. Omori H. Noda T. Yamamoto N. Komatsu M. Tanaka K. Kawai T. Tsujimura T. Takeuchi O. Yoshimori T. Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 92.Sancho D. Joffre OP. Keller AM. Rogers NC. Martinez D. Hernanz-Falcon P. Rosewell I. Reis e Sousa C. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature. 2009;458:899–903. doi: 10.1038/nature07750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sandoval H. Thiagarajan P. Dasgupta SK. Schumacher A. Prchal JT. Chen M. Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sanjuan MA. Dillon CP. Tait SW. Moshiach S. Dorsey F. Connell S. Komatsu M. Tanaka K. Cleveland JL. Withoff S. Green DR. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 95.Sarkar S. Ravikumar B. Floto RA. Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16:46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- 96.Scaffidi P. Misteli T. Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 97.Scherz-Shouval R. Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17:422–427. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 98.Scherz-Shouval R. Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 99.Scherz-Shouval R. Shvets E. Fass E. Shorer H. Gil L. Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schmid D. Pypaert M. Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schrader M. Fahimi HD. Mammalian peroxisomes and reactive oxygen species. Histochem Cell Biol. 2004;122:383–393. doi: 10.1007/s00418-004-0673-1. [DOI] [PubMed] [Google Scholar]
- 102.Schweers RL. Zhang J. Randall MS. Loyd MR. Li W. Dorsey FC. Kundu M. Opferman JT. Cleveland JL. Miller JL. Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Scott RS. McMahon EJ. Pop SM. Reap EA. Caricchio R. Cohen PL. Earp HS. Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–211. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- 104.Stuart LM. Ezekowitz RA. Phagocytosis and comparative innate immunity: learning on the fly. Nat Rev Immunol. 2008;8:131–141. doi: 10.1038/nri2240. [DOI] [PubMed] [Google Scholar]
- 105.Swanson JA. Hoppe AD. The coordination of signaling during Fc receptor-mediated phagocytosis. J Leukoc Biol. 2004;76:1093–1103. doi: 10.1189/jlb.0804439. [DOI] [PubMed] [Google Scholar]
- 106.Takahashi K. Rochford CD. Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tang D. Kang R. Cheh CW. Livesey KM. Liang X. Schapiro NE. Benschop R. Sparvero LJ. Amoscato AA. Tracey KJ. Zeh HJ. Lotze MT. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29:5299–5310. doi: 10.1038/onc.2010.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tang D. Kang R. Coyne CB. Zeh HJ., 3rd Lotze MT. PAMPs and DAMPs: Signal 0's that Spur autophagy and immunity. Immunol Rev. 2012;249:158–175. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tang D. Kang R. Livesey KM. Cheh CW. Farkas A. Loughran P. Hoppe G. Bianchi ME. Tracey KJ. Zeh HJ., 3rd Lotze MT. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tang D. Kang R. Livesey KM. Kroemer G. Billiar TR. Van Houten B. Zeh HJ., 3rd Lotze MT. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011;13:701–711. doi: 10.1016/j.cmet.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tang D. Kang R. Livesey KM. Zeh HJ., 3rd Lotze MT. High mobility group box 1 (HMGB1) activates an autophagic response to oxidative stress. Antioxid Redox Signal. 2011;15:2185–2195. doi: 10.1089/ars.2010.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tang D. Kang R. Xiao W. Zhang H. Lotze MT. Wang H. Xiao X. Quercetin prevents LPS-induced high-mobility group box 1 release and proinflammatory function. Am J Respir Cell Mol Biol. 2009;41:651–660. doi: 10.1165/rcmb.2008-0119OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tang D. Kang R. Zeh HJ., 3rd Lotze MT. High-mobility group box 1 and cancer. Biochim Biophys Acta. 2010;1799:131–140. doi: 10.1016/j.bbagrm.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tang D. Kang R. Zeh HJ., 3rd Lotze MT. High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal. 2011;14:1315–1335. doi: 10.1089/ars.2010.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tang D. Shi Y. Kang R. Li T. Xiao W. Wang H. Xiao X. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J Leukoc Biol. 2007;81:741–747. doi: 10.1189/jlb.0806540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Thorburn J. Horita H. Redzic J. Hansen K. Frankel AE. Thorburn A. Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 2009;16:175–183. doi: 10.1038/cdd.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Thurston TL. Ryzhakov G. Bloor S. von Muhlinen N. Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 118.Thurston TL. Wandel MP. von Muhlinen N. Foeglein A. Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482:414–418. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tooze SA. Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12:831–835. doi: 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- 120.Touret N. Paroutis P. Terebiznik M. Harrison RE. Trombetta S. Pypaert M. Chow A. Jiang A. Shaw J. Yip C. Moore HP. van der Wel N. Houben D. Peters PJ. de Chastellier C. Mellman I. Grinstein S. Quantitative and dynamic assessment of the contribution of the ER to phagosome formation. Cell. 2005;123:157–170. doi: 10.1016/j.cell.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 121.Truman LA. Ford CA. Pasikowska M. Pound JD. Wilkinson SJ. Dumitriu IE. Melville L. Melrose LA. Ogden CA. Nibbs R. Graham G. Combadiere C. Gregory CD. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 2008;112:5026–5036. doi: 10.1182/blood-2008-06-162404. [DOI] [PubMed] [Google Scholar]
- 122.Tschopp J. Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 123.Tsung A. Klune JR. Zhang X. Jeyabalan G. Cao Z. Peng X. Stolz DB. Geller DA. Rosengart MR. Billiar TR. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204:2913–2923. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Weiner LM. Lotze MT. Tumor-cell death, autophagy, and immunity. N Engl J Med. 2012;366:1156–1158. doi: 10.1056/NEJMcibr1114526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Werb Z. Cohn ZA. Plasma membrane synthesis in the macrophage following phagocytosis of polystyrene latex particles. J Biol Chem. 1972;247:2439–2446. [PubMed] [Google Scholar]
- 126.Wild P. Farhan H. McEwan DG. Wagner S. Rogov VV. Brady NR. Richter B. Korac J. Waidmann O. Choudhary C. Dotsch V. Bumann D. Dikic I. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yang H. Hreggvidsdottir HS. Palmblad K. Wang H. Ochani M. Li J. Lu B. Chavan S. Rosas-Ballina M. Al-Abed Y. Akira S. Bierhaus A. Erlandsson-Harris H. Andersson U. Tracey KJ. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107:11942–11947. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang Z. Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yates RM. Hermetter A. Taylor GA. Russell DG. Macrophage activation downregulates the degradative capacity of the phagosome. Traffic. 2007;8:241–250. doi: 10.1111/j.1600-0854.2006.00528.x. [DOI] [PubMed] [Google Scholar]
- 130.Yates RM. Russell DG. Phagosome maturation proceeds independently of stimulation of toll-like receptors 2 and 4. Immunity. 2005;23:409–417. doi: 10.1016/j.immuni.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 131.Yu L. Wan F. Dutta S. Welsh S. Liu Z. Freundt E. Baehrecke EH. Lenardo M. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang H. Bosch-Marce M. Shimoda LA. Tan YS. Baek JH. Wesley JB. Gonzalez FJ. Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]