

INCRETIN EFFECT IN TYPE 2 DIABETES
Whereas glucose-tolerant individuals are capable of adjusting their insulin secretion to their actual insulin sensitivity, people with type 2 diabetes are incapable of doing so (1). β-Cell failure is therefore the hallmark of this disease, although failure may be precipitated by the development of insulin resistance, typically as a consequence of obesity. In healthy subjects, a considerable part of the postprandial insulin response is due to the actions of the incretin hormones glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). Together, the two hormones are responsible for the so-called incretin effect, i.e., the amplification of insulin secretion that is observed when glucose is taken orally as opposed to infused intravenously to provide identical plasma glucose concentrations (2).
Although frequently ignored, the effect strongly depends on the dose of glucose (3). A convenient way of describing the effect is to calculate the gastrointestinally mediated glucose disposal (GIGD) (4). Here the amount of glucose required by intravenous infusion to copy the glucose excursions after the oral load is related to the oral load. Thus, if 25 g is required to copy a 75-g oral glucose load, the GIGD amounts to 100 × (75 – 25)/75 = 66%. In other words, mechanisms associated with and activated by the oral ingestion resulted in a disposal of 75 – 25 = 50 g of the ingested glucose. In healthy subjects, most of the GIGD is accounted for by the actions of the incretin hormones, but inhibition of hepatic glucose production by suppression of glucagon secretion, hepatic uptake of glucose from the portal vein, and gut-brain or liver-brain reflex activity may also play a role. GIGD is particularly useful in the study of oral glucose handling in C-peptide–negative patients with type 1 diabetes, where the classical incretin definitions have no meaning (4). In a study of oral administration of 25, 50, and 100 g glucose (3), the amounts of intravenous glucose required to match the excursions after oral administration amounted to ∼20 g uniformly. Calculated as indicated above, the GIGD varied from 20% to as much as 80%. Thus, the healthy human body has a remarkable capacity to handle the intake of increasing amounts of glucose and is therefore capable of maintaining almost unchanged postprandial glucose excursions, regardless of the oral load. There is no doubt that the incretin hormones play a major role in GIGD in healthy subjects, and it can be concluded that the incretin effect plays a major role for normal glucose tolerance. In people with type 2 diabetes, this ability is dramatically reduced (5), as illustrated by calculation of the GIGD, which may be close to zero. Thus, if a patient with type 2 diabetes is given an oral glucose load of 50 g glucose, it typically takes close to 50 g intravenous glucose to copy the oral excursions (6). In other words, in these individuals, there is no mechanism available to dispose of the glucose taken in orally, or put in another way, the oral and the intravenous glucose loads are handled equally. The almost complete loss of GIGD is typically accompanied by a greatly reduced difference between the insulin responses to the oral and the intravenous glucose load, i.e., the incretin effect (5,6). This effect is often expressed as the integrated incremental insulin response (area under the curve [AUC]) to the oral glucose load [iAUCoral] minus the integrated incremental insulin response to the isoglycemic intravenous glucose infusion [iAUCiv] divided by the iAUCoral. When expressed in percent, this amounts to 100% × (iAUCoral − iAUCiv)/iAUCoral. This value is typically around 70% (for 75 g glucose) in healthy subjects, whereas individuals with type 2 diabetes may have values around 30% (for 50 g glucose). As indicated by the almost complete loss of GIGD, the incretin effect (∼30%) remaining in the patients with type 2 diabetes has little effect on glucose disposal, probably as a result of the simultaneously occurring insulin resistance. The loss of incretin effect is therefore likely to contribute importantly to the postprandial hyperglycemia in type 2 diabetes.
In the present article, we review a number of central studies elucidating the mechanisms involved in the dramatic loss of ability to handle dietary carbohydrates in type 2 diabetes.
SECRETION OF INCRETIN HORMONES IN PATIENTS WITH TYPE 2 DIABETES
Research carried out by several groups during the last decades has indicated that the incretin effect is mediated mainly by GIP and GLP-1 (7,8). No other gut hormones fulfill all criteria to act as incretin hormones, i.e., being secreted during glucose ingestion and being capable of stimulating insulin secretion during similar glycemic levels and in those concentrations that are reached during glucose ingestion (9,10). The concentrations of GIP have been reported to be both elevated, decreased, and unchanged in patients with type 2 diabetes (11), but many of the early results were obtained with assays that cross-react with substances in plasma that are unrelated to GIP (12). Using COOH-terminal GIP assays without such cross-reaction, Toft-Nielsen et al. (13) found slightly decreased postprandial GIP concentrations in a large group of type 2 diabetic patients compared with a control group of carefully matched (for weight, age, and sex) nondiabetic subjects. The impairment was significant but small, and it is probable that results obtained in smaller cohorts would not show significance. Obesity may be associated with increased GIP secretion (14,15) and will therefore be a confounding factor unless accounted for in the matching process. Thus, a major secretory defect regarding GIP secretion does not seem to exist in type 2 diabetes. When GLP-1 was identified as the other important incretin hormone (16,17), it was relevant to evaluate GLP-1 secretion in type 2 diabetes also. In the already-mentioned rather large group of type 2 diabetic subjects, Toft-Nielsen et al. (13) found a pronounced impairment of the postprandial GLP-1 response, particularly during the later postprandial phase (after the first 60 min). Similar findings were made in subsequent studies (18), which also indicated that the plasma concentration profiles of the intact hormone (GLP-1 is rapidly degraded by the enzyme dipeptidyl peptidase-4 [DPP-4] and the concentrations of the active intact hormone are much lower than the concentrations of the metabolite thus formed, GLP-1 9–36amide) were similarly decreased in the late postprandial phase. The decreased secretion was thought to be secondary to the disease, since it was noted only in the diabetic twin of identical twins discordant for diabetes (19). In addition, first-degree relatives of type 2 diabetic patients exhibited normal 24-h GLP-1 profiles (20). In further studies of the secretion of GLP-1 in type 2 diabetes, it was possible to identify obesity particularly (13) but also insulin resistance (21) and glucose intolerance (22) as factors that would associate with decreased secretion. The impairment seems to mainly affect responses to mixed-meal ingestion (23). Some studies (24,25) could not confirm decreased GLP-1 responses, and we have therefore compared the various studies to look for explanations. From such comparisons, it appears that long duration and severity of type 2 diabetes (poor glycemic control, high HbA1c levels, poor insulin secretory reserve) are associated with poor GLP-1 responses (13,22). BMI is a powerful regulator of the GLP-1 response and comes out as a significant determinant in most larger studies (13,22,24,26,27). It has been suggested that it is particularly the GLP-1 response to carbohydrates that is impaired in obesity (26). It is also known that gastric emptying rate is important for meal-induced GLP-1 secretion (28). To the extent that there are differences in gastric emptying between control subjects and type 2 diabetic patients (impaired emptying rates in the patients, partly because of hyperglycemia [29]), this may result in differences in GLP-1 responses. It has also been suggested that decreased emptying of the gall bladder, sometimes seen in type 2 diabetic patients and thought to result in impaired GLP-1 secretion, could be involved (30). Finally, it is now known that antidiabetic treatment may influence GLP-1 secretion, most clearly demonstrated for metformin, which increases proglucagon expression in the L-cells (31–33) and enhances postprandial GLP-1 responses. Therefore, concurrent therapy of patients with type 2 diabetes represents an important confounding factor. Figure 1 shows the correlations between GLP-1 secretion (expressed as AUCs after oral glucose) and both BMI and the 120-min glucose values in a rather large study of subjects with varying degrees of BMI and glucose tolerance (22).
Figure 1.
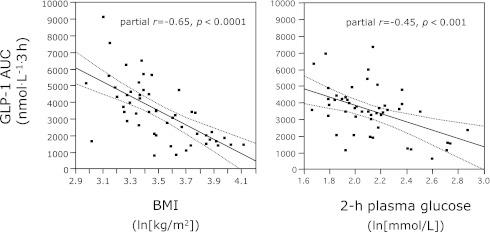
Correlation between GLP-1 secretion (calculated as total GLP-1 responses to oral glucose tolerance test) in subjects with varying degrees of glucose intolerance and BMI and 2-h glucose concentrations or BMI (after logarithmic transformation). From Muscelli et al. (22).
In conclusion, impaired secretion of the incretin hormones may not be a constant finding, but a decreased secretion of GLP-1 after mixed meals is observed in most studies. Given that the sensitivity of the pancreatic islets to the actions of the incretin hormones is decreased in type 2 diabetes (see below), it is evident that impaired secretion, when present, will aggravate the loss of incretin effect in these patients.
EFFECTS OF THE INCRETIN HORMONES ON THE PANCREATIC ISLETS
It was observed early that the insulinotropic effects of GIP are lost in type 2 diabetes (34). In a seminal study from 1993, Nauck et al. (35) demonstrated that whereas GIP, even in supra-physiological doses, had little effect on insulin secretion, GLP-1 infused to reach slightly supra-physiological concentrations had profound effects on insulin secretion and actually normalized the insulin response to a mildly hyperglycemic clamp in a group of patients with relatively mild type 2 diabetes. In a subsequent study, Kjems et al. (36) infused GLP-1 in increasing doses in combination with a ramp of ascending glucose infusions in relatively mild type 2 diabetic patients and matched control subjects and found that GLP-1 strongly increased β-cell sensitivity to glucose (the slope of the relationship between insulin secretion rate and plasma glucose concentrations), so that even with the lowest dose of GLP-1, it was possible to completely restore the β-cell sensitivity to glucose to normal values, i.e., to normalize insulin secretion in response to glucose (but of course in the continued presence of a certain level of GLP-1). Nevertheless, the dose-response relationship for this effect of GLP-1 was significantly impaired in the subjects with type 2 diabetes. From these studies, therefore, it was predicted that in type 2 diabetic patients, a lower postprandial secretion of GLP-1 might result in impaired β-cell secretion and therefore contribute to the loss of the incretin effect. In further studies, Højberg et al. (37) studied infusion of GLP-1 and GIP to reach normal postprandial levels during a 15 mmol/L hyperglycemic clamp, which in healthy subjects resulted in dramatic rises in insulin secretion, reaching levels >4,000 pmol/L. In patients with type 2 diabetes and rather severe disease, the same infusions had no significant effects on insulin secretion. These studies clearly identified a decreased sensitivity to the insulinotropic actions of both GIP and GLP-1 as important factors behind the loss of incretin effects in type 2 diabetes. In studies by Vilsbøll et al. (38), the effects of supra-physiological doses of GIP and GLP-1 were investigated. In these studies, which involved clamping of glucose at 15 mmol/L (to allow comparisons between patients and control subjects at identical plasma glucose levels), GLP-1 was infused at a rate of 1.2 pmol/kg/min (a dose that resulted in complete normalization of fasting glucose in patients with type 2 diabetes in studies by Nauck et al. in 1993 [39]), resulting in clearly supra-physiological plasma concentrations, and GIP was given at rates of 4 and 16 pmol/kg × min (resulting in extremely high plasma concentrations of GIP). But whereas GIP had virtually no effect on insulin secretion, GLP-1 generated insulin levels that were identical to those measured in the control subjects given the glucose clamp alone. In other words, this dose of GLP-1 completely normalized the β-cell responsiveness to glucose in the patients. Importantly, during the GIP infusion, there was no increase in the glucose turnover (as measured by the glucose infusion rates required to maintain the clamp), whereas the GLP-1 infusion was associated with major increases in glucose turnover. The molecular mechanism underlying this remarkable difference between GIP and GLP-1 is not known. The retained insulinotropic effect of GLP-1 is the basis for the use of GLP-1 receptor agonists for the treatment of type 2 diabetes. It was suggested that GIP receptors may be downregulated in type 2 diabetes (40), but in studies of bolus injections of GIP and GLP-1, which both cause short-lived insulin responses, the response to GIP was reduced to the same extent as the response to GLP-1 in patients with type 2 diabetes compared with control subjects (38), suggesting that it is not expression of the GIP receptor that is decreased, but postreceptor mechanisms associated with particularly the later phase of insulin secretion that are suffering in type 2 diabetes. Interestingly, Højberg et al. (37) demonstrated that improvement of glycemic control (with intensive insulin therapy for 4 weeks) significantly improved the β-cell sensitivity to both GIP and GLP-1, suggesting that the loss of the effect of GIP and of the potency of GLP-1 might be at least partly secondary to glycemic control. Aaboe et al. (41) investigated the possibility that a defective regulation of ATP-sensitive K+ (KATP) channels in the β-cells might be responsible for the defective response to GIP in patients with type 2 diabetes, as suggested from studies in mice with KATP channel subunit deletions (42), and were able to demonstrate enhanced responses to GIP in the acute presence of sulfonylureas (which cause KATP channel closure), but the responses were still not normal (when compared with historical controls).
In conclusion, the dramatic loss in patients with type 2 diabetes of the ability to dispose of orally ingested glucose (with GIGD values close to zero as opposed to up to 80% in healthy subjects) is clearly related to the inability of the incretin hormones to increase insulin secretion in the concentrations reached after meal or glucose ingestion.
IS THE LOSS OF INCRETIN EFFECT A PRIMARY EVENT IN THE PATHOGENESIS OF TYPE 2 DIABETES?
Several lines of evidence support that the loss of incretin effect is secondary to the development of diabetes. Thus, patients with diabetes secondary to destruction of insular tissues in patients with chronic pancreatitis exhibit an almost complete loss of incretin effect (6), whereas patients with a similar degree of chronic pancreatitis, as judged from pancreatic imaging and function tests but normal glucose tolerance, have a normal incretin effect. The studies also indicated that the incretin effect was lost at very low elevations of fasting or postprandial glucose concentrations. Initial studies by Meier et al. (43) suggested that the effect of GIP was impaired in first-degree relatives of patients with type 2 diabetes, but subsequent investigation showed that these individuals exhibited a similar reduction in glucose-induced insulin secretion, indicating that the reduced response was not specific for GIP (44). In further studies of first-degree relatives, the incretin effect and the secretory responses of GIP and GLP-1 were normal (45). Thus, a preexisting GIP deficiency was not identified. Women with previous gestational diabetes, who are at high risk for developing type 2 diabetes, also had normal insulinotropic responses to GIP (46). That the loss of GIP insulinotropic efficacy is secondary to the development of diabetes is consistent with the finding that similar losses of efficacy were observed in patients with diabetes secondary to chronic pancreatitis, monogenic maturity-onset diabetes of the young (MODY-3), and type 1 diabetes (with preserved β-cell function) and in patients with late-onset autoimmune diabetes of the adult (LADA) (47). By correlating insulin responses to GIP and to intravenous glucose in various groups of type 2 diabetic patients, Meier and Nauck (44) recently found a significant relationship (r2 = 0.58) suggesting that the loss of the GIP response may be related to the loss of glucose-induced insulin secretion.
WHEN DO PATIENTS WITH TYPE 2 DIABETES LOSE THE INCRETIN EFFECTS?
Given that the loss of incretin effect is secondary to the development of diabetes, it becomes of interest to determine whether insulin resistance, defective β-cell function, or glucose intolerance are associated with impaired incretin function. To investigate this, two studies were recently carried out. In the first study (48), insulin resistance was induced in completely healthy young individuals (a family history of diabetes was also excluded) by means of physical inactivity, increased energy intake, and a daily dose of 371/2 mg prednisolone for 12 days. The incretin effect was then determined using isoglycemic oral and intravenous glucose challenges. The incretin effect was reduced from 72 ± 5 to 43 ± 7% and GIGD decreased from 56 ± 4 to 19 ± 8%. The decrease in incretin effect was obviously the result of a smaller difference between insulin response to oral and intravenous glucose, but the major change was an increase in the insulin response to intravenous glucose. In agreement with this, the disposition index (insulin secretion related to the prevailing insulin sensitivity) during intravenous glucose was unchanged by the intervention. In other words, these healthy individuals were capable of compensating completely for the impaired insulin sensitivity with increased insulin secretion, when challenged with intravenous glucose, but they were unable to upregulate their ability to handle glucose delivered by the oral route, as also indicated by the dramatically decreased GIGD value. In the second study (49), insulin resistance was induced by 5 days of dexamethasone treatment in a group of completely glucose-tolerant first-degree relatives of type 2 diabetic patients. This protocol was chosen because previous studies had indicated that insulin resistance would be induced in all subjects, but about half of the subjects would in addition develop impaired glucose tolerance, whereas the rest would remain glucose tolerant. The group developing impaired glucose tolerance represents individuals who have a particularly high risk of developing subsequent diabetes. The results indicated that in the individuals developing insulin resistance only, there was a significant reduction in the incretin effect from 71 ± 3 to 58 ± 5%, whereas β-cell function was completely normal, as determined by calculations of disposition indices based on measurements of levels of insulin, C-peptide, or insulin secretion rates. In the group developing impaired glucose tolerance, the incretin effect decreased from 67 ± 5 to 32 ± 8% (similar to the effect in individuals with overt type 2 diabetes [6]), but this was accompanied by clearly impaired β-cell function. These studies, therefore, indicate that impairment of the incretin effect is a very early sign of impaired glucose metabolism that may be observed before other signs of β-cell dysfunction are apparent, but that is aggravated further when β-cell function is impaired. This result raises the question whether an impairment of the incretin effect might be observed at what perhaps represents the first step toward glucose intolerance—namely, obesity. This scenario was actually observed in a recent study of middle-aged obese insulin-resistant individuals with normal glucose tolerance and normal GIP and GLP-1 responses during oral glucose compared with lean insulin-sensitive but otherwise matched control subjects (50).
It has recently been suggested (44) that the loss of incretin effect observed in type 2 diabetes merely represents a diminished β-cell secretory capacity that will affect the stronger stimulus elicited by oral glucose more than the weaker stimulus provided by intravenous glucose. Thus, a situation might occur where the intravenous glucose elicits a response corresponding to the maximum secretory capacity of the β-cells, and in this situation, oral administration of glucose would not be able to elicit a greater insulin response. However, as indicated above, impairment of incretin function can be observed without measurable changes in β-cell function. The hypothesis that the intravenous glucose causes a stimulation that corresponds to the maximum secretory capacity of the β-cells has actually been tested in several experiments where β-cell function was stimulated by hyperglycemic clamps. In fact, the maximum β-cell response cannot be brought about by intravenous glucose. Several experiments have indicated that combinations of intravenous glucose and, for example, arginine or glucagon, independent of glucose tolerance, stimulate insulin secretion significantly more than glucose alone. This was demonstrated very elegantly by Ward et al. (51), introducing the term “maximal secretory capacity.” It is also well established that reduced sensing and response of the β-cell to intravenous glucose may be observed during developing type 2 diabetes, whereas the insulin response to nonglucose stimuli is much better preserved (52). Furthermore, as discussed above, in patients with type 2 diabetes, infusions of even very large supra-physiological doses of GIP were unable to enhance insulin secretion further, a finding that could be compatible with the assumption that the maximum secretory capacity was reached (38). However, in the same experiments, GLP-1 infused to pharmacological levels were associated with greatly increased insulin secretion, even exceeding the levels observed in healthy subjects given the same hyperglycemic clamp, proving that, in these individuals, the β-cell secretory capacity was not maximally stimulated by intravenous glucose. Importantly, similar findings were observed in patients with a clear reduction in insulin secretory capacity because of diabetes secondary to chronic pancreatitis. In these patients, GIP had no effect in addition to the effect of the clamp alone, whereas supra-physiological levels of GLP-1 were capable of generating a large insulin response (47). The loss of incretin effect in such patients, therefore, can only be explained by a specific loss of insulinotropic activity of the incretin hormones at physiological levels.
GENETIC VARIANTS AND INCRETIN EFFECT
Several risk genes for type 2 diabetes have been identified. All identified variants are common (minor allele frequency >5%) and have a weak impact on the phenotype, but the majority seem to potentially influence pancreatic β-cell function (53). The TCF7L2 polymorphisms are thought to make a greater contribution to the development of type 2 diabetes than other genetic markers (53). The TCF7L2 variants (rs7903146 and rs12255372) are associated with a reduced incretin effect as evaluated by both the isoglycemic oral and intravenous glucose technique and a hyperglycemic clamp despite normal secretion of GIP and GLP-1 (54–57). In in vitro studies of islets, knockdown of the TCF7L2 gene resulted in decreased β-cell proliferation and insulin secretion and increased β-cell apoptosis combined with a reduced expression of both GLP-1 and GIP receptors (58,59), whereas overexpression of TCF7L2 attenuated apoptosis and improved β-cell function. TCF7L2 encodes a transcription factor involved in Wnt signaling, important for the development of the pancreatic β-cells and for β-cell survival. Therefore, TCF7L2 variants may underlie the loss of incretin effect. However, the effect of the different variants is small, and analysis of the β-cell responsiveness to the incretin hormones is technically demanding. Thus, the insight into the importance of the TCF7L2 system has had little clinical impact.
A variant (rs10423928) in the GIP receptor was recently demonstrated to associate with slightly increased 2-h glucose concentrations and a reduced incretin effect (60). The GIP receptor variant also associated with type 2 diabetes (odds ratio 1.07), in contrast to an older study where no association between polymorphisms in the GIP receptor and type 2 diabetes could be demonstrated, presumably because of a lack of statistical power (61). Additional type 2 diabetes risk loci, including WFS1 and KCNQ1, may also be relevant because they influence insulin secretion. Wolfram syndrome 1 gene (WFS1) has been associated with type 2 diabetes and with impaired insulin secretion during a GLP-1 infusion (62,63), and the variant rs151290 in KCNQ1 has been associated with changes in insulin secretion and increased GIP and GLP-1 secretion (64). Variants in THADA have been associated with diminished insulin secretion during GLP-1 treatment (65), whereas variants in the MTNR1B gene were associated with reduced insulin responses to GLP-1 stimulation, but the attenuated responsiveness was also observed for other β-cell stimuli and also during intravenous glucose, indicating a global β-cell defect (53,55). Thus, genetic variants may indeed influence the incretin effect and possibly also the responsiveness to incretin-based therapy in some patients. Nevertheless, the studies also demonstrate that common variants have a low impact on these parameters, and a clear genetic explanation for the loss of incretin effect cannot be provided currently.
Therefore, our conclusion from the discussion presented above is that an impaired incretin effect, most likely due to impaired islet responses to the incretin hormones, is an early sign of impaired glucose metabolism, perhaps associated with insulin resistance, and that a further impairment is seen as glucose intolerance develops. In overt type 2 diabetes, the consequence of the impaired incretin effect is that the ability of the patients to efficiently dispose of orally as opposed to intravenously administered glucose is completely lost.
Acknowledgments
No potential conflicts of interest relevant to this article were reported.
Footnotes
This publication is based on the presentations at the 3rd World Congress on Controversies to Consensus in Diabetes, Obesity and Hypertension (CODHy). The Congress and the publication of this supplement were made possible in part by unrestricted educational grants from AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Daiichi Sankyo, Eli Lilly, Ethicon Endo-Surgery, Generex Biotechnology, F. Hoffmann-La Roche, Janssen-Cilag, Johnson & Johnson, Novo Nordisk, Medtronic, and Pfizer.
References
- 1.Kahn SE, Prigeon RL, McCulloch DK, et al. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 1993;42:1663–1672 [DOI] [PubMed] [Google Scholar]
- 2.Holst JJ, Gromada J. Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans. Am J Physiol Endocrinol Metab 2004;287:E199–E206 [DOI] [PubMed] [Google Scholar]
- 3.Nauck MA, Homberger E, Siegel EG, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab 1986;63:492–498 [DOI] [PubMed] [Google Scholar]
- 4.Hare KJ, Vilsbøll T, Holst JJ, Knop FK. Inappropriate glucagon response after oral compared with isoglycemic intravenous glucose administration in patients with type 1 diabetes. Am J Physiol Endocrinol Metab 2010;298:E832–E837 [DOI] [PubMed] [Google Scholar]
- 5.Nauck M, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986;29:46–52 [DOI] [PubMed] [Google Scholar]
- 6.Knop FK, Vilsbøll T, Højberg PV, et al. Reduced incretin effect in type 2 diabetes: cause or consequence of the diabetic state? Diabetes 2007;56:1951–1959 [DOI] [PubMed] [Google Scholar]
- 7.Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev 2007;87:1409–1439 [DOI] [PubMed] [Google Scholar]
- 8.Drucker DJ. The biology of incretin hormones. Cell Metab 2006;3:153–165 [DOI] [PubMed] [Google Scholar]
- 9.Creutzfeldt W, Ebert R. New developments in the incretin concept. Diabetologia 1985;28:565–573 [DOI] [PubMed] [Google Scholar]
- 10.Fahrenkrug J, Schaffalitzky de Muckadell OB, Kühl C. Effect of secretin on basal- and glucose-stimulated insulin secretion in man. Diabetologia 1978;14:229–234 [DOI] [PubMed] [Google Scholar]
- 11.Krarup T. Immunoreactive gastric inhibitory polypeptide. Endocr Rev 1988;9:122–134 [DOI] [PubMed]
- 12.Krarup T, Holst JJ. The heterogeneity of gastric inhibitory polypeptide in porcine and human gastrointestinal mucosa evaluated with five different antisera. Regul Pept 1984;9:35–46 [DOI] [PubMed] [Google Scholar]
- 13.Toft-Nielsen MB, Damholt MB, Madsbad S, et al. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab 2001;86:3717–3723 [DOI] [PubMed] [Google Scholar]
- 14.Yip RG, Wolfe MM. GIP biology and fat metabolism. Life Sci 2000;66:91–103 [DOI] [PubMed] [Google Scholar]
- 15.Vilsbøll T, Krarup T, Sonne J, et al. Incretin secretion in relation to meal size and body weight in healthy subjects and people with type 1 and type 2 diabetes mellitus. J Clin Endocrinol Metab 2003;88:2706–2713 [DOI] [PubMed] [Google Scholar]
- 16.Holst JJ, Orskov C, Nielsen OV, Schwartz TW. Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett 1987;211:169–174 [DOI] [PubMed] [Google Scholar]
- 17.Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet 1987;2:1300–1304 [DOI] [PubMed] [Google Scholar]
- 18.Vilsbøll T, Krarup T, Deacon CF, Madsbad S, Holst JJ. Reduced postprandial concentrations of intact biologically active glucagon-like peptide 1 in type 2 diabetic patients. Diabetes 2001;50:609–613 [DOI] [PubMed] [Google Scholar]
- 19.Vaag AA, Holst JJ, Vølund A, Beck-Nielsen HB. Gut incretin hormones in identical twins discordant for non-insulin-dependent diabetes mellitus (NIDDM): evidence for decreased glucagon-like peptide 1 secretion during oral glucose ingestion in NIDDM twins. Eur J Endocrinol 1996;135:425–432 [DOI] [PubMed] [Google Scholar]
- 20.Nyholm B, Walker M, Gravholt CH, et al. Twenty-four-hour insulin secretion rates, circulating concentrations of fuel substrates and gut incretin hormones in healthy offspring of type II (non-insulin-dependent) diabetic parents: evidence of several aberrations. Diabetologia 1999;42:1314–1323 [DOI] [PubMed] [Google Scholar]
- 21.Rask E, Olsson T, Söderberg S, et al. Impaired incretin response after a mixed meal is associated with insulin resistance in nondiabetic men. Diabetes Care 2001;24:1640–1645 [DOI] [PubMed] [Google Scholar]
- 22.Muscelli E, Mari A, Casolaro A, et al. Separate impact of obesity and glucose tolerance on the incretin effect in normal subjects and type 2 diabetic patients. Diabetes 2008;57:1340–1348 [DOI] [PubMed] [Google Scholar]
- 23.Deacon CF. What do we know about the secretion and degradation of incretin hormones? Regul Pept 2005;128:117–124 [DOI] [PubMed] [Google Scholar]
- 24.Vollmer K, Holst JJ, Baller B, et al. Predictors of incretin concentrations in subjects with normal, impaired, and diabetic glucose tolerance. Diabetes 2008;57:678–687 [DOI] [PubMed] [Google Scholar]
- 25.Ryskjaer J, Deacon CF, Carr RD, et al. Plasma dipeptidyl peptidase-IV activity in patients with type-2 diabetes mellitus correlates positively with HbA1c levels, but is not acutely affected by food intake. Eur J Endocrinol 2006;155:485–493 [DOI] [PubMed] [Google Scholar]
- 26.Ranganath LR, Beety JM, Morgan LM, Wright JW, Howland R, Marks V. Attenuated GLP-1 secretion in obesity: cause or consequence? Gut 1996;38:916–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verdich C, Toubro S, Buemann B, Lysgård Madsen J, Juul Holst J, Astrup A. The role of postprandial releases of insulin and incretin hormones in meal-induced satiety: effect of obesity and weight reduction. Int J Obes Relat Metab Disord 2001;25:1206–1214 [DOI] [PubMed] [Google Scholar]
- 28.Miholic J, Orskov C, Holst JJ, Kotzerke J, Meyer HJ. Emptying of the gastric substitute, glucagon-like peptide-1 (GLP-1), and reactive hypoglycemia after total gastrectomy. Dig Dis Sci 1991;36:1361–1370 [DOI] [PubMed] [Google Scholar]
- 29.Vollmer K, Gardiwal H, Menge BA, et al. Hyperglycemia acutely lowers the postprandial excursions of glucagon-like peptide-1 and gastric inhibitory polypeptide in humans. J Clin Endocrinol Metab 2009;94:1379–1385 [DOI] [PubMed] [Google Scholar]
- 30.Knop FK. Bile-induced secretion of glucagon-like peptide-1: pathophysiological implications in type 2 diabetes? Am J Physiol Endocrinol Metab 2010;299:E10–E13 [DOI] [PubMed] [Google Scholar]
- 31.Migoya EM, Bergeron R, Miller JL, et al. Dipeptidyl-peptidase-4 inhibitors administered in combination with metformin result in an additive increase in the plasma concentration of active GLP-1. Clin Pharmacol Ther 2010;88:801–808 [DOI] [PubMed] [Google Scholar]
- 32.Yasuda N, Inoue T, Nagakura T, et al. Enhanced secretion of glucagon-like peptide 1 by biguanide compounds. Biochem Biophys Res Commun 2002;298:779–784 [DOI] [PubMed] [Google Scholar]
- 33.Cuthbertson J, Patterson S, O’Harte FP, Bell PM. Addition of metformin to exogenous glucagon-like peptide-1 results in increased serum glucagon-like peptide-1 concentrations and greater glucose lowering in type 2 diabetes mellitus. Metabolism 2010;60:52–56 [DOI] [PubMed] [Google Scholar]
- 34.Krarup T, Saurbrey N, Moody AJ, Kühl C, Madsbad S. Effect of porcine gastric inhibitory polypeptide on beta-cell function in type I and type II diabetes mellitus. Metabolism 1987;36:677–682 [DOI] [PubMed] [Google Scholar]
- 35.Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 1993;91:301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kjems LL, Holst JJ, Vølund A, Madsbad S. The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes 2003;52:380–386 [DOI] [PubMed] [Google Scholar]
- 37.Højberg PV, Vilsbøll T, Rabøl R, et al. Four weeks of near-normalisation of blood glucose improves the insulin response to glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. Diabetologia 2009;52:199–207 [DOI] [PubMed] [Google Scholar]
- 38.Vilsbøll T, Krarup T, Madsbad S, Holst JJ. Defective amplification of the late phase insulin response to glucose by GIP in obese type II diabetic patients. Diabetologia 2002;45:1111–1119 [DOI] [PubMed] [Google Scholar]
- 39.Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7-36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993;36:741–744 [DOI] [PubMed] [Google Scholar]
- 40.Piteau S, Olver A, Kim SJ, et al. Reversal of islet GIP receptor down-regulation and resistance to GIP by reducing hyperglycemia in the Zucker rat. Biochem Biophys Res Commun 2007;362:1007–1012 [DOI] [PubMed] [Google Scholar]
- 41.Aaboe K, Knop FK, Vilsboll T, et al. KATP channel closure ameliorates the impaired insulinotropic effect of glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes. J Clin Endocrinol Metab 2009;94:603–608 [DOI] [PubMed] [Google Scholar]
- 42.Miki T, Minami K, Shinozaki H, et al. Distinct effects of glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 on insulin secretion and gut motility. Diabetes 2005;54:1056–1063 [DOI] [PubMed] [Google Scholar]
- 43.Meier JJ, Hücking K, Holst JJ, Deacon CF, Schmiegel WH, Nauck MA. Reduced insulinotropic effect of gastric inhibitory polypeptide in first-degree relatives of patients with type 2 diabetes. Diabetes 2001;50:2497–2504 [DOI] [PubMed] [Google Scholar]
- 44.Meier JJ, Nauck MA. Is the diminished incretin effect in type 2 diabetes just an epi-phenomenon of impaired beta-cell function? Diabetes 2010;59:1117–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nauck MA, El-Ouaghlidi A, Gabrys B, et al. Secretion of incretin hormones (GIP and GLP-1) and incretin effect after oral glucose in first-degree relatives of patients with type 2 diabetes. Regul Pept 2004;122:209–217 [DOI] [PubMed] [Google Scholar]
- 46.Meier JJ, Gallwitz B, Askenas M, et al. Secretion of incretin hormones and the insulinotropic effect of gastric inhibitory polypeptide in women with a history of gestational diabetes. Diabetologia 2005;48:1872–1881 [DOI] [PubMed] [Google Scholar]
- 47.Vilsbøll T, Knop FK, Krarup T, et al. The pathophysiology of diabetes involves a defective amplification of the late-phase insulin response to glucose by glucose-dependent insulinotropic polypeptide-regardless of etiology and phenotype. J Clin Endocrinol Metab 2003;88:4897–4903 [DOI] [PubMed] [Google Scholar]
- 48.Hansen KB, Vilsboll T, Bagger JI, Holst JJ, Knop FK. Reduced glucose tolerance and insulin resistance induced by steroid treatment, relative physical inactivity, and high-calorie diet impairs the incretin effect in healthy subjects. J Clin Endocrinol Metab 2010;95:3309–3317 [DOI] [PubMed]
- 49.Jensen DH, Aaboe K, Henriksen JE, et al. Insulin resistance and glucose intolerance independently reduce the incretin effect. Diabetologia; 2010;53(Suppl. 1):S262 [Google Scholar]
- 50.Knop FK, Aaboe K, Vilsboll T, Madsbad S, Holst JJ, Krarup T. Reduced incretin effect in obese subjects with normal glucose tolerance as compared to lean control subjects (Abstract). Diabetes 2008;57:A410 [Google Scholar]
- 51.Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D., Jr Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest 1984;74:1318–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Porte D., Jr Banting lecture 1990. Beta-cells in type II diabetes mellitus. Diabetes 1991;40:166–180 [DOI] [PubMed] [Google Scholar]
- 53.Sparsø T, Grarup N, Andreasen C, et al. Combined analysis of 19 common validated type 2 diabetes susceptibility gene variants shows moderate discriminative value and no evidence of gene-gene interaction. Diabetologia 2009;52:1308–1314 [DOI] [PubMed] [Google Scholar]
- 54.Schäfer SA, Tschritter O, Machicao F, et al. Impaired glucagon-like peptide-1-induced insulin secretion in carriers of transcription factor 7-like 2 (TCF7L2) gene polymorphisms. Diabetologia 2007;50:2443–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lyssenko V, Lupi R, Marchetti P, et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J Clin Invest 2007;117:2155–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Villareal DT, Robertson H, Bell GI, et al. TCF7L2 variant rs7903146 affects the risk of type 2 diabetes by modulating incretin action. Diabetes 2010;59:479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pilgaard K, Jensen CB, Schou JH, et al. The T allele of rs7903146 TCF7L2 is associated with impaired insulinotropic action of incretin hormones, reduced 24 h profiles of plasma insulin and glucagon, and increased hepatic glucose production in young healthy men. Diabetologia 2009;52:1298–1307 [DOI] [PubMed] [Google Scholar]
- 58.Shu L, Matveyenko AV, Kerr-Conte J, Cho JH, McIntosh CH, Maedler K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum Mol Genet 2009;18:2388–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shu L, Sauter NS, Schulthess FT, Matveyenko AV, Oberholzer J, Maedler K. Transcription factor 7-like 2 regulates beta-cell survival and function in human pancreatic islets. Diabetes 2008;57:645–653 [DOI] [PubMed] [Google Scholar]
- 60.Saxena R, Hivert MF, Langenberg C, et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet 2010;42:142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Almind K, Ambye L, Urhammer SA, et al. Discovery of amino acid variants in the human glucose-dependent insulinotropic polypeptide (GIP) receptor: the impact on the pancreatic beta cell responses and functional expression studies in Chinese hamster fibroblast cells. Diabetologia 1998;41:1194–1198 [DOI] [PubMed] [Google Scholar]
- 62.Sandhu MS, Weedon MN, Fawcett KA, et al. Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet 2007;39:951–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schäfer SA, Müssig K, Staiger H, et al. A common genetic variant in WFS1 determines impaired glucagon-like peptide-1-induced insulin secretion. Diabetologia 2009;52:1075–1082 [DOI] [PubMed] [Google Scholar]
- 64.Müssig K, Staiger H, Machicao F, et al. Association of type 2 diabetes candidate polymorphisms in KCNQ1 with incretin and insulin secretion. Diabetes 2009;58:1715–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simonis-Bik AM, Nijpels G, van Haeften TW, et al. Gene variants in the novel type 2 diabetes loci CDC123/CAMK1D, THADA, ADAMTS9, BCL11A, and MTNR1B affect different aspects of pancreatic beta-cell function. Diabetes 2010;59:293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]