

Abstract
The cystic fibrosis conductance regulator (CFTR) is a cAMP-regulated Cl− channel expressed predominantly at the apical membrane of secreting epithelial cells. Mutations in the CFTR gene lead to cystic fibrosis, the most frequent genetic disease in the Caucasian population. The most common mutation, a deletion of phenylalanine at position 508 (F508del), impairs CFTR folding and chloride channel function. Although an intense effort is under way to identify compounds that target the F508del CFTR structural defect and promote its expression and stability at the plasma membrane, so far their clinical efficacy has proven to be poor, highlighting the necessity to better understand the molecular mechanism of CFTR regulation and of the pathogenesis of the disease. Accumulating evidence suggests that the inclusion of the CFTR in macromolecular complexes and its interaction with the cortical cytoskeleton may play a key role in fine-tuning the regulation of channel function. Here we review some recent findings that support a critical role for protein–protein interactions involving CFTR and for the cytoskeleton in promoting local control of channel activity. These findings indicate that compounds that rescue and stabilize CFTR at the apical membrane may not be sufficient to restore its function unless the appropriate intracellular milieu is also reconstituted.
Keywords: CFTR, cAMP, PKA, cytoskeleton, cystic fibrosis, compartmentalization
Introduction
Cystic fibrosis (CF) is a lethal inherited autosomal disorder caused by mutations in the gene that encodes for the cystic fibrosis transmembrane conductance regulator (CFTR). CFTR is a 1480-amino-acid glycosylated protein that functions as a chloride channel regulated by cAMP and PKA-mediated phosphorylation (Riordan et al., 1989). It is expressed at high levels at the apical membrane of polarized epithelial secretory cells, where the chloride efflux also affects electrolyte and water transport; but it is also found at lower levels on the membrane of non-epithelial cells including cardiomyocytes (Gadsby et al., 1998), lymphocytes (Krauss et al., 1992) and endothelial cells (Tousson et al., 1996).
Nearly 2000 mutations of the cftr gene have been identified, affecting the folding, the localization or the activity of the channel. These mutated forms of CFTR fall into five functional groups: truncation mutations, processing mutations, activation mutations, channel mutations and splice mutations. Among these, the most frequent is a deletion of phenylalanine 508 (F508del), a mutation that prevents delivery of the channel to the apical membrane and impairs channel gating.
CF patients show an altered function of exocrine glands and exhibit gastrointestinal complications, with vitamin malabsorption and associated steatorrhoea, poor growth, increased risk of gallstones and hepatobiliary disease (Flume and Van Devanter, 2012). The most affected target is however the respiratory system, in which the reduced CFTR activity and a secondary increase in epithelial ENaC-mediated Na+ and fluid absorption results in volume depletion of the lung apical surface liquid (ASL). This leads to increased adhesiveness and cohesiveness of airway mucus with the consequent obstruction of small airways, air trapping and bronchial wall thickening (Matsui et al., 1998; Joo et al., 2006; Boucher, 2007). This event is accompanied by an associated neutrophilic inflammation, since bacterial opportunists enter the respiratory tract, from where it becomes very difficult to clear them; therefore, their growth and expansion leads to local inflammation. These processes result in a lifelong degradation of lung anatomy and function, so that respiratory failure is responsible for 80% of mortality in CF (Flume and Van Devanter, 2012).
There is presently no cure available for CF, and current treatment is symptomatic and relies on the administration of mucolytics to clear the mucus, antibiotics to fight lung infections, anti-inflammatory agents to prevent damage to lung tissue and pancreatic enzymes to correct pancreatic insufficiency. In recent years, growing efforts have been made to find therapeutic strategies aimed at targeting directly the cause of the disease. Since the discovery of CFTR, many expectations arose that gene therapy could provide a treatment for CF. A number of clinical trials using different gene transfer agents have been conducted; and, although some targets have been achieved, such as correction of nasal potential difference (PD) (Cuthbert, 2011), no real clinical benefits for patients has been shown so far. The finding that lentiviral carriers may be able to evade the immune system has recently revived interest in gene therapy strategies (Griesenbach and Alton, 2011). Nevertheless, the technical difficulties encountered with gene delivery have turned the focus towards pharmacological strategies, hoping for more immediate results. Initial clinical trials using small molecules to restore the function of mutant CFTR have been encouraging (Becq et al., 2011), but so far the efficacy of these compounds has been poor (Lukacs and Verkman, 2012). As major gaps still remain in our understanding of the molecular basis of CFTR regulation and of the pathogenesis of the disease, the general consensus is that a more detailed knowledge of these mechanisms may help to achieve the full translational potential of these compounds.
Pharmacological approaches to CF therapy
After its synthesis, wt CFTR is assembled in the ER; and, once properly folded, the immature form goes to the Golgi complex where it is fully glycosylated and subsequently transported and inserted into the apical membrane of polarized cells, where it is subjected to endocytosis (Lukacs et al., 1997). The majority of internalized wt CFTR is recycled back to the plasma membrane, and only a small amount is transferred to the late endosomes and lysosomes for degradation (Gentzsch et al., 2004; Sharma et al., 2004).
Seventy percent of CF patients carry one or two copies of a mutant allele encoding for a protein missing phenylalanine 508. F508del CFTR is a protein that does not reach complete maturation, being retained in internal membranes and ultimately degraded by the ubiquitin–proteasome complex. The incorrect functioning of F508del CFTR leads to an altered transepithelial ionic transport and, therefore, to the improper function of several organs.
F508del CFTR can be partially rescued to the cell surface either by lowering the temperature (Denning et al., 1992) or by using small molecules that act to release the mutant channel from protein degradation (Brown et al., 1996; Zeitlin, 2000). However, even when rescued to the cell surface, the F508del mutant is unstable, and its half-life in the membrane is reduced compared with that of the wild-type protein (Sharma et al., 2001; Swiatecka-Urban et al., 2005).
In addition, F508del CFTR exhibits abnormal cAMP regulation (Bebok et al., 2005) and defective channel gating (Wang et al., 2000). Interestingly, studies using isolated protein in cell-free systems showed that F508del CFTR retains normal cAMP-dependent PKA-dependent regulation and activity relative to wt CFTR (Li et al., 1993), suggesting that the intracellular milieu, rather than the mutation itself, is what determines the ability of F508del CFTR to respond to cAMP regulation.
The finding that F508del CFTR can be rescued to the cell surface, together with the evidence that restoration of small amounts of functional CFTR protein (5–10% of normal levels) (Davis et al., 1996) may greatly reduce disease severity, has stimulated much research to identify small molecule compounds that could either rescue the biosynthetic defect of F508del CFTR, thus restoring its folding and function (correctors) or enhance its regulated function once rescued to the surface (potentiators). Several efficacious F508del CFTR potentiators have been found to activate CFTR conductance (Yang et al., 2003; Pedemonte et al., 2005). VX 770, a Vertex Pharmaceutical compound, has been found to be efficacious in potentiating cAMP-mediated gating of F508del CFTR (Van Goor et al., 2006); and it was shown to have a significant efficacy in a phase II clinical trial in CF patients carrying the Gly551Asp mutation (for complete reviews see: Proesmans et al., 2008; Cai et al., 2011; Lukacs and Verkman, 2012). This field of research is expanding rapidly, primarily due to the automated methods of identifying and analysing potentially active compounds by high-throughput screening (Galietta et al., 2001).
In contrast to the potentiators, relatively few correctors have been found. Correctors may either stabilize F508del CFTR native state by directly binding to the mutant protein or enhance the protein folding efficiency, thus promoting its trafficking and rescuing F508del-CFTR activity. VX-809 (Van Goor et al., 2011) and the methylbithiazole analog, Corr-4a (Grove et al., 2009) are currently the most promising compounds. However, these drugs are not very efficacious, possibly due to the gating defect displayed by F508del CFTR rescued to the membrane, and it has been suggested that a combined administration of correctors and potentiators may be required to achieve clinical efficacy.
The drug profiling for many corrector compounds remains unclear. Such compounds could act either directly by promoting F508del CFTR escape from the ER and/or indirectly by altering cellular protein homeostasis and promoting F508del CFTR targeting and stability on the plasma membrane. However, some corrector compounds, such as VRT-325 and Corr 4a, while being able to rescue the apical expression of F508del CFTR to the apical membrane of airway cells are not able to restore normal stability of the mutant protein at the cell surface (Cholon et al., 2010). This suggests the need of combined pharmacological approaches that may increase the stability of F508del CFTR on the cell surface.
The finding that the effect of the corrector on the rescued CFTR-dependent chloride transport is strongly influenced by the cell background suggests that the corrector could regulate a plethora of proteins involved in CFTR maturation and/or degradation (e.g. proteostasis regulators) (Powers et al., 2009). In this regard, Pedemonte et al. (Pedemonte et al., 2010) suggest that any putative corrector effect needs to be tested in different cell systems, including primary airway cells. This means that other factors related to the intracellular environment are involved in rescuing the functional expression of F508del-CFTR. In support of this notion, it has been shown that cAMP-increasing agents, through receptor stimulation, direct activation of AC or inhibition of the cAMP-degrading enzymes PDEs, partially restore the defective chloride conductance of many CFTR mutants, including F508del CFTR from CF mice and patients (Liu et al., 2005).
Interaction of CFTR with other proteins and formation of macromolecular complexes
The regulation of CFTR has long been recognized to be highly localized (Huang et al., 2004). The interaction of the channel with different molecules and the consequent formation of a variety of macromolecular complexes within which the composition of the individual elements may change offer the possibility of a highly compartmentalized regulation of CFTR function whereby local control of channel activity at specific sites mediates specific functional outcomes. This is clearly exemplified by the ability of CFTR to form a complex with either NHERF1 or NHERF2. In vitro and in vivo studies demonstrated that, despite their structural similarities, NHERF1 and NHERF2 appear to differently tune CFTR activity as well as CFTR interactions with other transporters and receptors. As reported by Singh et al. (2009), in murine duodenum NHERF2 mediates CFTR inhibition by coupling LPA receptor to CFTR while NHERF1 stimulates CFTR activity by linking to β2-adrenergic receptors (β2AR).
CFTR is composed of two motifs, each of which consists of a hydrophobic membrane-spanning domain (MSD) and a cytosolic hydrophilic region (nucleotide binding domain, NBD) for binding ATP. These two motifs are linked by a cytoplasmic regulatory domain that contains several charged residues and multiple consensus sites for PKA phosphorylation, responsible for increasing the open probability of the channel and hence Cl− efflux. Both the amino- and carboxyl-terminal tails are located inside the cytoplasm and are involved in the binding between CFTR and a multitude of interacting partners including transporters, ion channels, receptors, kinases, phosphatases, signalling molecules and cytoskeletal elements. Such interactions have been shown to play a key role in the regulation of CFTR-mediated Cl− efflux both in vitro and in vivo (Li and Naren, 2010). In addition, these interactions have been suggested to mediate the ability of the CFTR to coordinate the activity of many other transmembrane ion fluxes via regulation of proteins such as the sodium channels amiloride-sensitive ENaC, responsible for sodium reabsorption, the potassium channels ROMK, responsible for K+ efflux, the chloride channels ORCC e CaCC, the Na+/H+ exchanger, responsible for the modulation of intracellular pH, the Cl−/HCO3− exchanger and aquaporin 3 (Schreiber et al., 1999).
The carboxyl terminus of CFTR contains the PDZ interacting domain, Asp-Thr-Arg-Leu, which is responsible for binding to several PDZ domain containing proteins including NHERF1 (Na+/H+ exchanger regulatory factor isoform 1), NHERF2, CAP-70 (CFTR-associated protein, 70 kDa) and CAL (CFTR associated ligand). The physiological significance of these adaptor proteins not only in the regulation of CFTR activity has been verified in several studies (Guggino and Stanton, 2006). NHERF1 interaction was demonstrated to affect both polarized expression of CFTR on the apical membrane of airway cells and the vectorial transport of chloride (Moyer et al., 2000). In addition, it was found that overexpression of NHERF1 stimulates CFTR-dependent chloride efflux by increasing apical expression of the channel in 16HBE14o- cells (expressing wt CFTR). Importantly, overexpression of NHERF1 was also shown to promote apical expression of the mutant channel in human CFBE41o- cell, a cell homozygous for F508del CFTR, resulting in a significant redistribution of F508del CFTR from the cytoplasm to the apical membrane and in rescued CFTR-dependent chloride secretion (Guerra et al., 2005). The finding that overexpression of NHERF1 is able to stimulate chloride secretion in CF cells is consistent with the hypothesis that some F508del CFTR is able to escape the degradative pathway and that NHERF1 overexpression may contribute to stabilize F508del CFTR that has been rescued on the plasma membrane, making it less susceptible to degradation (Kwon et al., 2007).
Role of the CFTR
That the cytoskeleton plays an important role in the regulation of CFTR activity was first suggested by early studies where cytoskeletal disruption with cytochalasin D completely blunted the cAMP-mediated activation of the channel (Prat et al., 1995; 1999). The integration of the CFTR into a macromolecular complex that is anchored to the subcortical cytoskeleton may underpin such regulation. For example, NHERF1 finely regulates the activity of the channel activity not only by directly interacting with CFTR but also by organizing the association of multiple scaffolding and signal transduction components in proximity of the channel (Guggino and Stanton, 2006). One of such interactions involves the carboxyl terminal of NHERF1 and the structural protein ezrin. Ezrin belongs to the Ezrin, Radixin and Moesin (ERM) protein family, and has the ability to interact with both the plasma membrane and filamentous actin thus providing a membrane–cytoskeletal linkage that is critical for the stability of the cell cortex. The ERM proteins are structured such that intramolecular interaction between the amino-terminal and carboxyl-terminal domains masks protein–protein interaction sites and maintains the protein in an inactive state in the cytoplasm. Disruption of this intra-molecular interaction activates ezrin, resulting in its recruitment to the plasma membrane via its N-terminal domain and binding to F-actin through its C-terminal domain. The activation of ezrin occurs essentially through conformational changes, resulting from binding to phosphatidylinositol the 4,5-bisphosphate (PIP2), a lipid that is selectively concentrated to the apical surface of polarized epithelia, and from phosphorylation of a conserved threonine in the actin binding domain (T567) (Yonemura et al., 2002; Fievet et al., 2004). In its activated state, the FERM domain of ezrin binds to target membrane proteins either directly or indirectly through the PDZ protein NHERF (Weinman et al., 2000; Fehon et al., 2010); and, once activated, ezrin has been demonstrated to play a fundamental role in control of cytoskeletal organization (Bretscher et al., 2002). Ezrin has been found to be essential in the functional expression of CFTR. On one side, it interacts with NHERF1, promoting CFTR stabilization on the apical membrane (Favia et al., 2010). On the other side, it can act as an A kinase anchoring protein (AKAP) as in its active conformation a central domain is exposed that binds to the regulatory subunit of PKA (Dransfield et al., 1997; Swiatecka-Urban et al., 2004). This allows the compartmentalization of PKA in proximity of CFTR and promotes its phosphorylation.
As documented by a number of studies, another mechanism by which the cytoskeleton may affect CFTR function involves recycling of the CFTR to the plasma membrane. The cytoskeleton network is strictly correlated with apical CFTR expression (Swiatecka-Urban et al., 2004), a process that has been shown to require the interaction with a complex of actin-binding proteins including myosin VI. Ganeshan et al., 2007 demonstrated that actin disassembly induced by Wiskostatin, dramatically decreased CFTR surface expression with a consequent increase in its internalization. Moreover, in HT29 colonic cell monolayers incubated with latrunculin B, an actin monomer sequestering agent, the same authors found a significant decrease of CFTR-dependent Cl− current. All together, these findings suggest that the observed inhibition of CFTR-dependent chloride secretion can be ascribed to an alteration of the balance between the internalization and the recycling at the plasma membrane that is dependent on a properly organized cytoskeleton. Moreover, it has been found that the interaction of wt CFTR with scaffolding proteins and the actin cytoskeleton is responsible for confined lateral diffusion of the channel within the plane of the membrane, since CFTR diffusion is highly increased after C-terminal CFTR truncations or overexpression of NHERF1 in which the carboxyl-terminus that interacts with ezrin is deleted (NHERF1-ΔERM), or after cytoskeletal disruption (Bates et al., 2006; Haggie et al., 2006). In line with these observations, we have found that NHERF1 overexpression in CF airway cell monolayers promotes the phosphorylation of ezrin via the activation of a RhoA/ROCK pathway, which results in increased F-actin organization and assembly. The activation of ezrin provides a regulated link between F508del CFTR, the actin filaments and the formation of a multi-protein complex that stabilizes the mutant CFTR in the apical membrane by delaying its internalization (Favia et al., 2010).
PKA exerts a local control on CFTR activity
The inclusion of the CFTR into macromolecular complexes that are anchored to the cortical cytoskeleton suggests an important spatial component to the regulation of the channel and provides a structural basis for local control of its activity (Huang et al., 2004).
Indeed, compartmentalization appears to be a key feature of PKA-mediated regulation of CFTR function, where all the elements required for the cAMP signal to be generated, affected, modulated and terminated are gathered in close proximity to the target CFTR. In airway epithelial cells, for example, Gs-protein coupled receptors that are involved in the regulation of the channel (e.g. Adenosine 2b receptors or β2AR) have been shown to be compartmentalized with NHERF1 and ezrin in close proximity to CFTR together with the cAMP-generating enzyme adenylate cyclase (AC) (Huang et al., 2001; Naren et al., 2003; Taouil et al., 2003). This complex promotes the rapid cAMP-dependent phosphorylation and activation of CFTR. The cAMP degrading enzyme PDE4 was also shown to be localized at the apical membrane of airway epithelial cells in proximity to CFTR. Degradation by PDE4 of cAMP newly synthesized by apical AC was shown to limit the lateral diffusion of the second messenger restricting its action to spatially confined microdomains within the apical membrane (Barnes et al., 2005). Furthermore, in airway and intestinal epithelial cells, the serine/threonine phosphatase PP2A was shown to physically and functionally interact with CFTR and to contribute to the regulation of channel activity (Thelin et al., 2005). Evidence also indicates that CFTR can be part of different macromolecular complexes that are functionally distinct. In one study, both β2AR and CFTR were shown to bind to NHERF1 through their PDZ domains at the apical membrane of epithelial cells (Naren et al., 2003). Deletion of the PDZ motif from CFTR resulted in uncoupling of the channel from the β2AR, both physically and functionally. Such uncoupling was shown to be receptor specific as deletion of the PDZ domain did not affect CFTR coupling to adenosine receptors (Naren et al., 2003). The specificity achieved by local regulation of CFTR activity is further illustrated by studies where Calu-3 cells treated with low doses of adenosine achieved maximal stimulation of CFTR-mediated Cl− efflux although no measurable change in global levels of cAMP could be detected (Huang et al., 2001). In another study, lysophosphatidic acid, a naturally occurring phospholipid that lowers cAMP levels via activation of a Gi-coupled receptor, was found to inhibit CFTR function in response to adenosine stimulation without causing a decrease in overall cAMP levels (Li et al., 2005). One interesting feature that emerges from these studies is that the regulation of CFTR appears to rely on a pool of cAMP that is confined to the sub-plasma membrane compartments and that stimuli that affect Cl− secretion do not substantially perturb overall levels of the second messenger. In line with this notion, inhibition of the multidrug resistance protein 4 (MRP4), a cAMP transporter that functionally and physically associates with CFTR, was shown to significantly potentiate CFTR function in response to adenosine without significantly increasing intracellular cAMP levels (Li et al., 2007).
Actin cytoskeleton, compartmentalization of cAMP and regulation of CFTR
Although cAMP is a small and highly hydrophilic molecule that in the aqueous intracellular environment is expected to diffuse very rapidly, in recent years, an increasing body of evidence clearly shows that cAMP is not free to diffuse inside the cells, but rather the propagation of cAMP signals is spatially regulated, resulting in the generation of restricted pools of second messenger within confined subcellular compartments (reviewed in Zaccolo, 2009). The local nature of cAMP signals leads to the activation of limited subsets of the effector PKA (Zaccolo and Pozzan, 2002) and, consequently, to the phosphorylation of a limited number of downstream targets (Di Benedetto et al., 2008). A key role in the spatial control of signal propagation is played by AKAPs, a large and diverse family of functionally related proteins that anchor PKA in proximity of its targets, thereby limiting the phosphorylation events to a restricted and specific subset of substrates (Wong and Scott 2004). The functional relevance of PKA anchoring to AKAPs has been demonstrated by several studies. For example, treatment of epithelial cells with Ht31, a competing peptide that displaces PKA from AKAPs, was shown to inhibit PKA-mediated phosphorylation of CFTR as well as to reduce cAMP-stimulated CFTR Cl− current (Sun et al., 2000). Some of the AKAPs have also been shown to bind PDEs, the enzymes that degrade cAMP and phosphatases, thus allowing for highly selective and local termination of the signal (Beene et al., 2007).
The compartmentalization of cAMP signalling allows for a specific extracellular stimulus to be translated into the required cellular response while avoiding inappropriate activation of the multiplicity of other cAMP-dependent pathways that are present in the cell but the activation of which is not required for that specific response.
The mechanism that limits the spread of cAMP signals generated at the plasma membrane to intracellular sites is the object of intense investigation as it is recognized that understanding how cAMP signalling operates at the subcellular level may provide novel avenues for the development of drugs with increased efficacy and reduced side effects (Zaccolo, 2011). PDEs have been shown to play a key role in differentially regulating the concentration of cAMP at defined intracellular sites (Mongillo et al., 2004; 2006) as well as specifically in regulating diffusion of cAMP away from the plasma membrane (Rich et al., 2001; Terrin et al., 2006; Oliveira et al., 2010). Other mechanisms, including the presence of a physical barrier due to the densely packed subcortical cytoskeleton or the close proximity of internal membranes to the plasma membrane, have been suggested (Rich et al., 2000), although direct evidence was so far lacking.
In a recent study, we used targeted FRET reporters to monitor in real time and in intact living cells the intracellular levels of cAMP and PKA activity in the sub-plasma membrane compartment and in the bulk cytosol of human pulmonary epithelial cells and found that cAMP is indeed compartmentalized. cAMP raising stimuli generate an increase in cAMP levels that is higher in the sub-plasma membrane compartment than in the bulk of the cytosol (Figure 1A) (Monterisi et al., 2012). We also found that the confinement of cAMP to the sub-plasma membrane domain requires an intact sub-cortical cytoskeleton as treatment of cells expressing wt CFTR with latrunculin B resulted in depletion of the sub-cortical pool of cAMP and accumulation of the second messenger in the cytosol and, consequently, in a redistribution of PKA phosphorylation activity from the subcortical space to the bulk cytosol (Monterisi et al., 2012). Notably, we found that in airway cells from CF patients homozygous for the delF508 mutation, the subcortical compartmentalization of cAMP is disrupted and cAMP levels, as well as PKA phosphorylation activity, are significantly increased in the bulk cytosol at the expenses of the sub-plasma membrane compartment (Figure 1B). The structural basis for cAMP compartmentalization appears to require the integrity of the CFTR/NHERF1 complex. Indeed, ablation of wild-type CFTR by small RNA interference was sufficient to disrupt cAMP compartmentalization resulting in loss of sub-cortical PKA activity, whereas overexpression of wild type CFTR in cells expressing F508del CFTR restored cAMP compartmentalization at the plasma membrane and local PKA activity.
Figure 1.
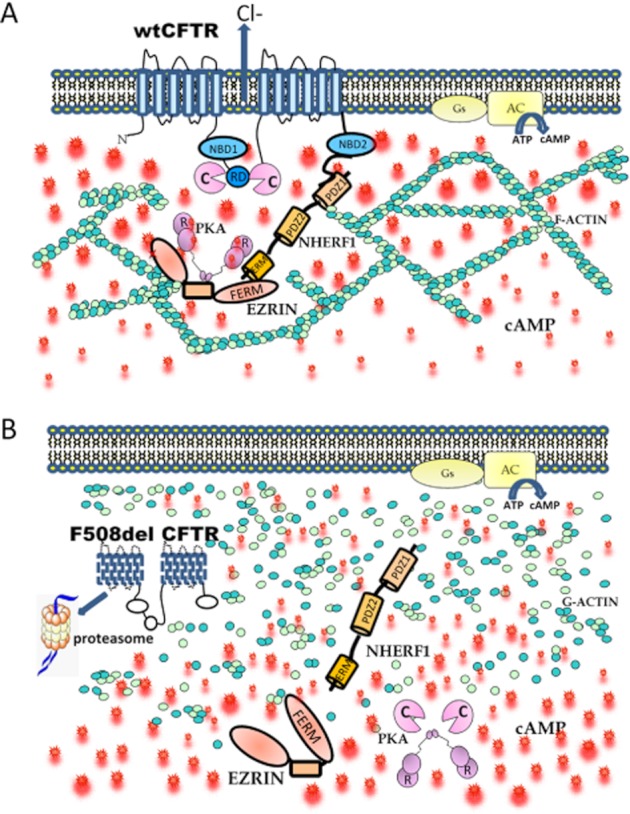
(A) Cells expressing wtCFTR show a well-organized subcortical cytoskeleton that limits cAMP diffusion away from the plasma membrane. As a result, in response to AC activation, the concentration of cAMP is higher in the sub-membrane compartment compared with the bulk cytosol. The local increase in cAMP activates ezrin-bound PKA, resulting in efficient phosphorylation of the CFTR and increased Cl− efflux. (B) Cells expressing F508del CFTR show a disorganized cytoskeleton and, consequently, diffusion of cAMP away from the membrane. Loss of an organized cortical cytoskeleton releases the PKA-anchoring protein ezrin with consequent relocalization of PKA to the cytosol. RD = regulatory domain; R = PKA regulatory subunit.
In the same system, disruption of the PKA–AKAP interaction after treatment with the anchoring inhibitor Ht31 not only dramatically reduced, in cells expressing wt CFTR, the ability of the channel to respond to cAMP modulation but it was also sufficient to completely ablate the Cl− efflux recovery mediated by NHERF1 overexpression in CFBE/sNHERF1 cells, indicating that PKA anchoring is required for the rescuing effect exerted by overexpression of NHERF1 (Monterisi et al., 2012). Overexpression of NHERF1 in cells expressing F508del CFTR, a manoeuvre previously shown to stabilize the mutant CFTR at the plasma membrane and to promote cytoskeleton organization (Favia et al., 2010), allowed for the barrier to cAMP diffusion to be reconstituted (Monterisi et al., 2012). The presence of a high level of cAMP in the sub-cortical compartment appears to be required for PKA-mediated regulation of CFTR activity as treatment with latrunculin B was sufficient to decrease the Cl− efflux in cells expressing wild type CFTR.
Potential implications of compartmentalized cAMP signalling
In the future, it will be important to establish whether altered compartmentalization of cAMP and PKA activity at the plasma membrane is a generalized defect of CF cells. However, the findings described above confirm the importance of a correctly organized intracellular milieu for the appropriate functioning of CFTR. Specifically, they point to the requirement for a sufficiently high concentration of cAMP in the sub-plasma membrane space to achieve effective activation of local PKA and consequent efficient regulation of CFTR activity. This may be relevant for the selection of pharmacological compounds aiming at re-establishing CFTR function. A number of these molecules appear to be unable to restore cAMP-mediated activation of CFTR despite their ability to stabilize F508del at the apical membrane (Pedemonte et al., 2005; Rowe et al., 2010). From a translational point of view, a number of compounds identified in primary screenings using cell lines expressing F508del CFTR gave disappointing results in clinical trials (Lukacs and Verkman, 2012). It may therefore be important to determine whether a local cAMP increase in the sub-cortical domain is a prerequisite for proficient rescue of CFTR activity. The ability of selected compounds to re-establish cAMP compartmentalization could potentially be an indicator of their effectiveness in restoring control of Cl− efflux in vivo.
Acknowledgments
The authors are supported by the Foundation Leducq (grant number O6 CVD 02), the British Heart Foundation (grant number PG/07/091/23698) and the NSF National Institutes of Health CRCNS program (grant number IH R01 AA18060) to M.Z and by the Italian Cystic Fibrosis Research Foundation (grant number FFC4/2011) with the contribution of the ParkinGO Oasi srl and the Delegazione FFC di Avellino, Loifur srl, Amici per la Ricerca Bassano; SM has been a PostDoc fellow of the Italian Cystic Fibrosis Foundation.
Glossary
- AKAP
A kinase anchoring protein
- CaCC
calcium-activated chloride channel
- CAL
CFTR-associated ligand
- CAP
CFTR-associated protein
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- ENaC
epithelial sodium channels
- ER
endoplasmic reticulum
- ERM
ezrin/radixin/moesin
- F508del
phenylalanine 508 deletion
- FERM
four point one, ezrin, radixin, moesin
- MRP4
multidrug resistance protein 4
- MSD
membrane-spanning domain
- NBD
nucleotide binding domain
- NHERF1
Na+/H+ exchanger regulatory factor isoform 1
- NHERF2
Na+/H+ exchanger regulatory factor isoform 2
- ORCC
outwardly rectifying chloride channel
- PD
nasal potential difference
- PDZ
postsynaptic density 95/disc-large/zona occludens
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PKA
cAMP-dependent protein kinase
- PP2A
protein phosphatase 2A
- ROMK
renal outer medullary potassium channel
- wt
wild type
Conflict of interest
The authors have no conflict of interest to declare.
References
- Barnes AP, Livera G, Huang P, Sun C, O'Neal WK, Conti M, et al. Phosphodiesterase 4D forms a cAMP diffusion barrier at the apical membrane of the airway epithelium. J Biol Chem. 2005;280:7997–8003. doi: 10.1074/jbc.M407521200. [DOI] [PubMed] [Google Scholar]
- Bates IR, Hebert B, Luo Y, Liao J, Bachir AI, Kolin DL, et al. Membrane lateral diffusion and capture of CFTR within transient confinement zones. Biophys J. 2006;91:1046–1058. doi: 10.1529/biophysj.106.084830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebok Z, Collawn JF, Wakefield J, Parker W, Li Y, Varga K, et al. Failure of cAMP agonists to activate rescued deltaF508 CFTR in CFBE41o- airway epithelial monolayers. J Physiol. 2005;569:601–615. doi: 10.1113/jphysiol.2005.096669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becq F, Mall MA, Sheppard DN, Conese M, Zegarra-Moran O. Pharmacological therapy for cystic fibrosis: from bench to bedside. J Cyst Fibros. 2011;10(Suppl. 2):S129–S145. doi: 10.1016/S1569-1993(11)60018-0. [DOI] [PubMed] [Google Scholar]
- Beene DL, Scott JD. A-kinase anchoring proteins take shape. Curr Opin Cell Biol. 2007;19:192–198. doi: 10.1016/j.ceb.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher RC. Cystic fibrosis: a disease of vulnerability to airway surface dehydration. Trends Mol Med. 2007;13:231–240. doi: 10.1016/j.molmed.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 2002;3:586–599. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- Brown CR, Hong-Brown LQ, Biwersi J, Verkman AS, Welch WJ. Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones. 1996;1:117–125. doi: 10.1379/1466-1268(1996)001<0117:ccctmp>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai ZW, Liu J, Li HY, Sheppard DN. Targeting F508del-CFTR to develop rational new therapies for cystic fibrosis. Acta Pharmacol Sin. 2011;32:693–701. doi: 10.1038/aps.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholon DM, O'Neal WK, Randell SH, Riordan JR, Gentzsch M. Modulation of endocytic trafficking and apical stability of CFTR in primary human airway epithelial cultures. Am J Physiol Lung Cell Mol Physiol. 2010;298:L304–L314. doi: 10.1152/ajplung.00016.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert AW. New horizons in the treatment of cystic fibrosis. Br J Pharmacol. 2011;163:173–183. doi: 10.1111/j.1476-5381.2010.01137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med. 1996;154:1229–1256. doi: 10.1164/ajrccm.154.5.8912731. [DOI] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Di Benedetto G, Zoccarato A, Lissandron V, Terrin A, Li X, Houslay MD, et al. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ Res. 2008;103:836–844. doi: 10.1161/CIRCRESAHA.108.174813. [DOI] [PubMed] [Google Scholar]
- Dransfield DT, Bradford AJ, Smith J, Martin M, Roy C, Mangeat PH, et al. Ezrin is a cyclic AMP-dependent protein kinase anchoring protein. EMBO J. 1997;16:35–43. doi: 10.1093/emboj/16.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favia M, Guerra L, Fanelli T, Cardone RA, Monterisi S, Di Sole F, et al. Na+/H+ exchanger regulatory factor 1 overexpression-dependent increase of cytoskeleton organization is fundamental in the rescue of F508del cystic fibrosis transmembrane conductance regulator in human airway CFBE41o- cells. Mol Biol Cell. 2010;21:73–86. doi: 10.1091/mbc.E09-03-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010;11:276–287. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fievet BT, Gautreau A, Roy C, Del Maestro L, Mangeat P, Louvard D, et al. Phosphoinositide binding and phosphorylation act sequentially in the activation mechanism of ezrin. J Cell Biol. 2004;164:653–659. doi: 10.1083/jcb.200307032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flume PA, Van Devanter DR. State of progress in treating cystic fibrosis respiratory disease. BMC Med. 2012;10:88. doi: 10.1186/1741-7015-10-88. doi: 10.1186/1741-7015-10-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby DC, Dousmanis AG, Nairn AC. ATP hydrolysis cycles and the gating of CFTR Cl- channels. Acta Physiol Scand Suppl. 1998;643:247–256. [PubMed] [Google Scholar]
- Galietta LJ, Springsteel MF, Eda M, Niedzinski EJ, By K, Haddadin MJ, et al. Novel CFTR chloride channel activators identified by screening of combinatorial libraries based on flavone and benzoquinolizinium lead compounds. J Biol Chem. 2001;276:19723–19728. doi: 10.1074/jbc.M101892200. [DOI] [PubMed] [Google Scholar]
- Ganeshan R, Nowotarski K, Di A, Nelson DJ, Kirk KL. CFTR surface expression and chloride currents are decreased by inhibitors of N-WASP and actin polymerization. Biochim Biophys Acta. 2007;1773:192–200. doi: 10.1016/j.bbamcr.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzsch M, Chang XB, Cui L, Wu Y, Ozols VV, Choudhury A, et al. Endocytic trafficking routes of wild type and DeltaF508 cystic fibrosis transmembrane conductance regulator. Mol Biol Cell. 2004;15:2684–2696. doi: 10.1091/mbc.E04-03-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesenbach U, Alton EW. Current status and future directions of gene and cell therapy for cystic fibrosis. BioDrugs. 2011;25:77–88. doi: 10.2165/11586960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Grove DE, Rosser MF, Ren HY, Naren AP, Cyr DM. Mechanisms for rescue of correctable folding defects in CFTRDelta F508. Mol Biol Cell. 2009;20:4059–4069. doi: 10.1091/mbc.E08-09-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra L, Fanelli T, Favia M, Riccardi SM, Busco G, Cardone RA, et al. Na+/H+ exchanger regulatory factor isoform 1 overexpression modulates cystic fibrosis transmembrane conductance regulator (CFTR) expression and activity in human airway 16HBE14o- cells and rescues DeltaF508 CFTR functional expression in cystic fibrosis cells. J Biol Chem. 2005;280:40925–40933. doi: 10.1074/jbc.M505103200. [DOI] [PubMed] [Google Scholar]
- Guggino WB, Stanton BA. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol. 2006;7:426–436. doi: 10.1038/nrm1949. [DOI] [PubMed] [Google Scholar]
- Haggie PM, Kim JK, Lukacs GL, Verkman AS. Tracking of quantum dot-labeled CFTR shows near immobilization by C-terminal PDZ interactions. Mol Biol Cell. 2006;17:4937–4945. doi: 10.1091/mbc.E06-08-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Lazarowski ER, Tarran R, Milgram SL, Boucher RC, Stutts MJ. Compartmentalized autocrine signaling to cystic fibrosis transmembrane conductance regulator at the apical membrane of airway epithelial cells. Proc Natl Acad Sci U S A. 2001;98:14120–14125. doi: 10.1073/pnas.241318498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Gilmore E, Kultgen P, Barnes P, Milgram S, Stutts MJ. Local regulation of cystic fibrosis transmembrane regulator and epithelial sodium channel in airway epithelium. Proc Am Thorac Soc. 2004;1:33–37. doi: 10.1513/pats.2306012. [DOI] [PubMed] [Google Scholar]
- Joo NS, Irokawa T, Robbins RC, Wine JJ. Hyposecretion, not hyperabsorption, is the basic defect of cystic fibrosis airway glands. J Biol Chem. 2006;281:7392–7398. doi: 10.1074/jbc.M512766200. [DOI] [PubMed] [Google Scholar]
- Krauss RD, Bubien JK, Drumm ML, Zheng T, Peiper SC, Collins FS, et al. Transfection of wild-type CFTR into cystic fibrosis lymphocytes restores chloride conductance at G1 of the cell cycle. EMBO J. 1992;11:875–883. doi: 10.1002/j.1460-2075.1992.tb05125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SH, Pollard H, Guggino WB. Knockdown of NHERF1 enhances degradation of temperature rescued DeltaF508 CFTR from the cell surface of human airway cells. Cell Physiol Biochem. 2007;20:763–772. doi: 10.1159/000110436. [DOI] [PubMed] [Google Scholar]
- Li C, Dandridge KS, Di A, Marrs KL, Harris EL, Roy K, et al. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J Exp Med. 2005;202:975–986. doi: 10.1084/jem.20050421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Krishnamurthy PC, Penmatsa H, Marrs KL, Wang XQ, Zaccolo M, et al. Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell. 2007;131:940–951. doi: 10.1016/j.cell.2007.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Naren AP. CFTR chloride channel in the apical compartments: spatiotemporal coupling to its interacting partners. Integr Biol (Camb) 2010;2:161–177. doi: 10.1039/b924455g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Ramjeesingh M, Reyes E, Jensen T, Chang X, Rommens JM, et al. The cystic fibrosis mutation (delta F508) does not influence the chloride channel activity of CFTR. Nat Genet. 1993;3:311–316. doi: 10.1038/ng0493-311. [DOI] [PubMed] [Google Scholar]
- Liu S, Veilleux A, Zhang L, Young A, Kwok E, Laliberte F, et al. Dynamic activation of cystic fibrosis transmembrane conductance regulator by type 3 and type 4D phosphodiesterase inhibitors. J Pharmacol Exp Ther. 2005;314:846–854. doi: 10.1124/jpet.105.083519. [DOI] [PubMed] [Google Scholar]
- Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol Med. 2012;18:81–91. doi: 10.1016/j.molmed.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs GL, Segal G, Kartner N, Grinstein S, Zhang F. Constitutive internalization of cystic fibrosis transmembrane conductance regulator occurs via clathrin-dependent endocytosis and is regulated by protein phosphorylation. Biochem J. 1997;328(Pt 2):353–361. doi: 10.1042/bj3280353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95:1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, et al. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004;95:67–75. doi: 10.1161/01.RES.0000134629.84732.11. [DOI] [PubMed] [Google Scholar]
- Mongillo M, Tocchetti CG, Terrin A, Lissandron V, Cheung YF, Dostmann WR, et al. Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ Res. 2006;98:226–234. doi: 10.1161/01.RES.0000200178.34179.93. [DOI] [PubMed] [Google Scholar]
- Monterisi S, Favia M, Guerra L, Cardone RA, Marzulli D, Reshkin SJ, et al. CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J Cell Sci. 2012;125:1106–1117. doi: 10.1242/jcs.089086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer BD, Duhaime M, Shaw C, Denton J, Reynolds D, Karlson KH, et al. The PDZ-interacting domain of cystic fibrosis transmembrane conductance regulator is required for functional expression in the apical plasma membrane. J Biol Chem. 2000;275:27069–27074. doi: 10.1074/jbc.M004951200. [DOI] [PubMed] [Google Scholar]
- Naren AP, Cobb B, Li C, Roy K, Nelson D, Heda GD, et al. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc Natl Acad Sci U S A. 2003;100:342–346. doi: 10.1073/pnas.0135434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira RF, Terrin A, Di Benedetto G, Cannon RC, Koh W, Kim M, et al. The role of type 4 phosphodiesterases in generating microdomains of cAMP: large scale stochastic simulations. Plos ONE. 2010;5:e11725. doi: 10.1371/journal.pone.0011725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N, Sonawane ND, Taddei A, Hu J, Zegarra-Moran O, Suen YF, et al. Phenylglycine and sulfonamide correctors of defective delta F508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol Pharmacol. 2005;67:1797–1807. doi: 10.1124/mol.105.010959. [DOI] [PubMed] [Google Scholar]
- Pedemonte N, Tomati V, Sondo E, Galietta LJ. Influence of cell background on pharmacological rescue of mutant CFTR. Am J Physiol Cell Physiol. 2010;298:C866–C874. doi: 10.1152/ajpcell.00404.2009. [DOI] [PubMed] [Google Scholar]
- Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- Prat AG, Xiao YF, Ausiello DA, Cantiello HF. cAMP-independent regulation of CFTR by the actin cytoskeleton. Am J Physiol. 1995;268:C1552–C1561. doi: 10.1152/ajpcell.1995.268.6.C1552. [DOI] [PubMed] [Google Scholar]
- Prat AG, Cunningham CC, Jackson GR, JR, Borkan SC, Wang Y, Ausiello DA, et al. Actin filament organization is required for proper cAMP-dependent activation of CFTR. Am J Physiol. 1999;277:C1160–C1169. doi: 10.1152/ajpcell.1999.277.6.C1160. [DOI] [PubMed] [Google Scholar]
- Proesmans M, Vermeulen F, De Boeck K. What's new in cystic fibrosis? From treating symptoms to correction of the basic defect. Eur J Pediatr. 2008;167:839–849. doi: 10.1007/s00431-008-0693-2. [DOI] [PubMed] [Google Scholar]
- Rich TC, Fagan KA, Nakata H, Schaack J, Cooper DM, Karpen JW. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J Gen Physiol. 2000;116:147–161. doi: 10.1085/jgp.116.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich TC, Fagan KA, Tse TE, Schaack J, Cooper DM, Karpen JW. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc Natl Acad Sci U S A. 2001;98:13049–13054. doi: 10.1073/pnas.221381398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [Erratum, Science 1989;245:1437.] [DOI] [PubMed] [Google Scholar]
- Rowe SM, Pyle LC, Jurkevante A, Varga K, Collawn J, Sloane PA, et al. DeltaF508 CFTR processing correction and activity in polarized airway and non-airway cell monolayers. Pulm Pharmacol Ther. 2010;23:268–278. doi: 10.1016/j.pupt.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R, Nitschke R, Greger R, Kunzelmann K. The cystic fibrosis transmembrane conductance regulator activates aquaporin 3 in airway epithelial cells. J Biol Chem. 1999;274:11811–11816. doi: 10.1074/jbc.274.17.11811. [DOI] [PubMed] [Google Scholar]
- Sharma M, Benharouga M, Hu W, Lukacs GL. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J Biol Chem. 2001;276:8942–8950. doi: 10.1074/jbc.M009172200. [DOI] [PubMed] [Google Scholar]
- Sharma M, Pampinella F, Nemes C, Benharouga M, So J, Du K, et al. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J Cell Biol. 2004;164:923–933. doi: 10.1083/jcb.200312018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AK, Riederer B, Krabbenhoft A, Rausch B, Bonhagen J, Lehmann U, et al. Differential roles of NHERF1, NHERF2, and PDZK1 in regulating CFTR-mediated intestinal anion secretion in mice. J Clin Invest. 2009;119:540–550. doi: 10.1172/JCI35541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Hug MJ, Bradbury NA, Frizzell RA. Protein kinase A associates with cystic fibrosis transmembrane conductance regulator via an interaction with ezrin. J Biol Chem. 2000;275:14360–14366. doi: 10.1074/jbc.275.19.14360. [DOI] [PubMed] [Google Scholar]
- Swiatecka-Urban A, Boyd C, Coutermarsh B, Karlson KH, Barnaby R, Aschenbrenner L, et al. Myosin VI regulates endocytosis of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2004;279:38025–38031. doi: 10.1074/jbc.M403141200. [DOI] [PubMed] [Google Scholar]
- Swiatecka-Urban A, Brown A, Moreau-Marquis S, Renuka J, Coutermarsh B, Barnaby R, et al. The short apical membrane half-life of rescued {Delta}F508-cystic fibrosis transmembrane conductance regulator (CFTR) results from accelerated endocytosis of {Delta}F508-CFTR in polarized human airway epithelial cells. J Biol Chem. 2005;280:36762–36772. doi: 10.1074/jbc.M508944200. [DOI] [PubMed] [Google Scholar]
- Taouil K, Hinnrasky J, Hologne C, Corlieu P, Klossek JM, Puchelle E. Stimulation of beta 2-adrenergic receptor increases cystic fibrosis transmembrane conductance regulator expression in human airway epithelial cells through a cAMP/protein kinase A-independent pathway. J Biol Chem. 2003;278:17320–17327. doi: 10.1074/jbc.M212227200. [DOI] [PubMed] [Google Scholar]
- Terrin A, Di Benedetto G, Pertegato V, Cheung YF, Baillie G, Lynch MJ, et al. PGE(1) stimulation of HEK293 cells generates multiple contiguous domains with different [cAMP]: role of compartmentalized phosphodiesterases. J Cell Biol. 2006;175:441–451. doi: 10.1083/jcb.200605050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelin WR, Kesimer M, Tarran R, Kreda SM, Grubb BR, Sheehan JK, et al. The cystic fibrosis transmembrane conductance regulator is regulated by a direct interaction with the protein phosphatase 2A. J Biol Chem. 2005;280:41512–41520. doi: 10.1074/jbc.M507308200. [DOI] [PubMed] [Google Scholar]
- Tousson A, Fuller CM, Benos DJ. Apical recruitment of CFTR in T-84 cells is dependent on cAMP and microtubules but not Ca2+ or microfilaments. J Cell Sci. 1996;109(Pt 6):1325–1334. doi: 10.1242/jcs.109.6.1325. [DOI] [PubMed] [Google Scholar]
- Van Goor F, Straley KS, Cao D, Gonzalez J, Hadida S, Hazlewood A, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1117–L1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zeltwanger S, Hu S, Hwang TC. Deletion of phenylalanine 508 causes attenuated phosphorylation-dependent activation of CFTR chloride channels. J Physiol. 2000;524:637–648. doi: 10.1111/j.1469-7793.2000.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinman EJ, Minkoff C, Shenolikar S. Signal complex regulation of renal transport proteins: NHERF and regulation of NHE3 by PKA. Am J Physiol Renal Physiol. 2000;279:F393–F399. doi: 10.1152/ajprenal.2000.279.3.F393. [DOI] [PubMed] [Google Scholar]
- Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- Yang H, Shelat AA, Guy RK, Gopinath VS, Ma T, Du K, et al. Nanomolar affinity small molecule correctors of defective Delta F508-CFTR chloride channel gating. J Biol Chem. 2003;278:35079–35085. doi: 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- Yonemura S, Matsui T, Tsukita S. Rho-dependent and -independent activation mechanisms of ezrin/radixin/moesin proteins: an essential role for polyphosphoinositides in vivo. J Cell Sci. 2002;115:2569–2580. doi: 10.1242/jcs.115.12.2569. [DOI] [PubMed] [Google Scholar]
- Zaccolo M. cAMP signal transduction in the heart: understanding spatial control for the development of novel therapeutic strategies. Br J Pharmacol. 2009;158:50–60. doi: 10.1111/j.1476-5381.2009.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo M. Spatial control of cAMP signalling in health and disease. Curr Opin Pharmacol. 2011;11:649–655. doi: 10.1016/j.coph.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo M, Pozzan T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 2002;295:1711–1715. doi: 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- Zeitlin PL. Pharmacologic restoration of delta F508 CFTR-mediated chloride current. Kidney Int. 2000;57:832–837. doi: 10.1046/j.1523-1755.2000.00922.x. [DOI] [PubMed] [Google Scholar]