

Abstract
Background and Purpose
Endoplasmic reticulum (ER) stress has been implicated in the pathogeneses of insulin resistance and type 2 diabetes, and extracellular signal-regulated kinase (ERK) antagonist is an insulin sensitizer that can restore muscle insulin responsiveness in both tunicamycin-treated muscle cells and type 2 diabetic mice. The present study was undertaken to determine whether the chemical or genetic inhibition ER stress pathway targeting by ERK results in metabolic benefits in muscle cells.
Experimental Approach
ER stress was induced in L6 myotubes using tunicamycin (5 μg·mL−1) or thapsigargin (300 nM) and cells were transfected with siRNA ERK or AMPKα2. Changes in ER stress and in the ERK and AMPK signalling pathways were explored by Western blotting. The phosphorylation levels of insulin receptor substrate 1 were analysed by immunoprecipitation and using glucose uptake assay.
Key Results
ER stress dampened insulin-stimulated signals and glucose uptake, whereas treatment with the specific ERK inhibitor U0126 (25 μM) rescued impaired insulin signalling via AMPK activation. In db/db mice, U0126 administration decreased markers of insulin resistance and increased the phosphorylations of Akt and AMPK in muscle tissues.
Conclusions and Implications
Inhibition of ERK signalling pathways by a chemical inhibitor and knockdown of ERK improved AMPK and Akt signallings and reversed ER stress-induced insulin resistance in L6 myotubes. These findings suggest that ERK signalling plays an important role in the regulation of insulin signals in muscle cells under ER stress.
Keywords: AMPK, endoplasmic reticulum, ERK, glucose uptake, insulin resistance
Introduction
Insulin resistance is defined as a failure of normal insulin levels to trigger downstream metabolic actions and is closely associated with the development of type 2 diabetes (Kahn et al., 2006). The precise mechanism responsible for the link between obesity and insulin resistance remains unknown but likely involves alterations in fatty acid metabolism and excess triglyceride accumulation in liver and muscle (Neschen et al., 2005; Savage et al., 2006; Deivanayagam et al., 2008; Korenblat et al., 2008).
Endoplasmic reticulum (ER) stress has been shown to contribute to insulin resistance associated with obesity in experimental models (Ozcan et al., 2004; Gregor and Hotamisligil, 2007). The ER is responsible for the synthesis, folding and trafficking of secretory and membrane proteins, and disruption of ER homeostasis results in an adaptive unfolded protein response (UPR), which aims to restore ER folding capacity and mitigate stress (Ron, 2002). Increased ER stress is associated with impaired insulin action in obese mice (Ozcan et al., 2004), and chemical or genetic amelioration of this stress has been shown to improve insulin sensitivity and glucose homeostasis (Ozcan et al., 2006).
Previous studies have indicated ERK as a positive regulator of adipogenesis and obesity (Bost et al., 2005; Kortum et al., 2005; Lee et al., 2010). In addition, the targeting of ERK could protect obese mice against insulin resistance and liver steatosis by decreasing adipose tissue inflammation and increasing muscle glucose uptake (Jager et al., 2011). Interestingly, the administration of U0126, a MEK inhibitor, has been reported to suppress ER stress-induced insulin resistance in HEK293 cells (Tang et al., 2011).
These observations suggest that a strategy based on targeting the ERK pathway offers therapeutic potential for the treatment of metabolic disorders. However, although data from studies conducted in animal models and cell systems demonstrate beneficial metabolic effects, the mechanism responsible for the effect of ERK on insulin action has not been studied under ER stress.
AMP-activated protein kinase (AMPK) acts on glucose utilization by increasing the expression of glut4 and its translocation to the plasma membrane, which increases glucose uptake in skeletal muscle, independently of insulin (Hayashi et al., 2000). Furthermore, AMPKα2 knockout mice display impaired insulin-stimulated whole-body glucose utilization and skeletal muscle glycogen synthesis, although the underlying mechanisms are unknown (Viollet et al., 2003). These studies indicate that AMPK can regulate glucose metabolism and insulin sensitivity.
The present study was undertaken to determine whether the chemical or genetic inhibition ER stress pathway targeting by ERK results in metabolic benefits in muscle cells. We found that treatment of L6 myotubes with an ER stressor (tunicamycin or thapsigargin) inhibited the ability of insulin to stimulate insulin receptor substrate-1 (IRS-1) tyrosine phosphorylation and glucose uptake, and thus, drastically reduced endogenous AMPK activity. In contrast, ER stress significantly increased ERK phosphorylation, and ER stress-induced ERK phosphorylation was diminished by the MEK inhibitor U0126 and by the siRNA silencing of ERK. Importantly, AMPK phosphorylation and glucose uptake were recovered by U0126 or ERK siRNA in ER-stressed L6 myotubes. Furthermore, siRNA-mediated knockdown of AMPKα2 markedly abolished repressed insulin-sensitizing Akt phosphorylation and glucose uptake via ERK inhibition under ER stress. Taken together, these findings demonstrate that AMPK might have protective effects against ER stress-induced insulin resistance. To confirm this possibility, we compared glucose homeostasis in U0126-injected db/db mice and rosiglitazone (an anti-diabetic)-injected db/db mice. U0126-injected and rosiglitazone-injected db/db mice were found to exhibit significantly lower blood glucose concentrations than control db/db mice, possibly due to the specific activity of AMPK in muscle tissue. We hypothesize that a deficiency of ERK activity with a subsequent increase in AMPK phosphorylation might be beneficial for attenuating insulin resistance.
Methods
Materials
Tunicamycin (an inhibitor of N-linked glycoprotein synthesis), thapsigargin (an irreversible inhibitor of ER Ca2+-ATPase), rosiglitazone and insulin were obtained from Sigma-Aldrich (St. Louis, MO, USA). U0126 (MEK inhibitor) was from Calbiochem-Novabiochem (La Jolla, CA, USA). The adenoviral expression vector of dominant-negative AMPKα1 (Ad-DN-AMPKα1) was produced as previously described (Mu et al., 2001) (generously donated by Dr I. K. Lee, Kyungpook National University School of Medicine). FBS, αMEM, trypsin/EDTA and penicillin/streptomycin were obtained from Gibco (Auckland, New Zealand). Reagents for polyacrylamide gel electrophoresis were from Bio-Rad (Hercules, CA, USA). 2-Deoxy-[3H]d-glucose was obtained from PerkinElmer Life Sciences (Boston, MA, USA). Antibodies against total and phospho-AMPK (Thr172), phospho-acetyl CoA carboxylase (ACC) (Ser79), phospho-Akt (Ser473), phospho-PERK (Tyr980), phospho-JNK (Thr183/Tyr185) and phospho-ERK1/2 (Thr202/Tyr204) were purchased from Cell Signaling Technology (Beverly, MA, USA). Phospho-IRE1 (Ser724) and β-tubulin were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). All other reagents were of the highest analytical grade available.
Cell culture
L6 myotube cells were obtained from the American Type Culture Collection (Manassas, VA, USA), and maintained in αMEM culture media supplemented with 10% v/v FBS, 100 units·mL−1 of penicillin, and 100 μg·mL−1 of streptomycin in a humidified 5% CO2 atmosphere at 37°C. After reaching confluence, cells were cultured in αMEM containing 2% FBS, and maintained for 5∼7 days with media replacement every 48 h prior to use in the experiments.
Glucose uptake assay
Radiolabelled 2-deoxyglucose uptake was measured for 10 min in transport buffer containing 20 mM HEPES (pH 7.4), 140 mM NaCl, 5 mM KCl, 2.5 mM MgSO4, 1 mM CaCl2, 10 μM unlabeled 2-deoxyglucose and 10 μM 2-deoxy-[3H]d-glucose (1 μCi·mL−1). The reaction was terminated by washing in ice-cold 0.9% NaCl (w/v). Non-specific uptake was determined in the presence of 10 μM cytochalasin B. Cells were collected in 0.05 N NaOH, and cell-associated radioactivity was determined by scintillation counting. Results are expressed as pmole of 2-deoxyglucose transported min−1 mg−1 of protein.
Immunoblotting and immunoprecipitation
Immunoblotting was performed as described previously (Jorgensen et al., 2004). In brief, L6 myotubes were stimulated with reagents or incubated under indicated conditions. After stimulation, cells were immediately lysed in lysis buffer [20 mM Tris-HCl, pH 8.0, 1% Nonidet P-40, 1 mM EDTA, 1 mM EGTA, 1 mM sodium orthovanadate, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 2 μg·mL−1 of aprotinin, 2 μg·mL−1 of leupeptin, and 1 μg·mL−1 of pepstatin]. Cell debris was then removed by centrifugation at 14 000× g for 15 min at 4°C, and resulting supernatants (cell lysate) were subjected to Western blotting. Protein concentrations were measured using a bicinchoninic acid assay (BCA Protein Assay kit; Pierce, Rockford, IL, USA). Samples were separated by 8% SDS-PAGE and then transferred to polyvinylidene difluoride membranes in transfer buffer containing 20 mM Tris-HCl, 154 mM glycine and 20% methanol. Membranes were blocked with 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween 20 and then incubated with specific antibodies, followed by horseradish peroxidase-conjugated secondary antibodies. Immunoreactive bands were visualized using an enhanced chemiluminescence detection system. For immunoprecipitation, 1 mg protein in total cell lysate was incubated with anti-IRS-1 or anti-ERK antibody for 2 h at 4°C. Immunocomplexes were precipitated with 20 μL protein A-Sepharose and extensively washed (three times) with ice-cold lysis buffer. These precipitates or total cell lysates were then subjected to SDS-PAGE and immunoblotted with indicated antibodies.
Transfection with small-interfering RNA (siRNA)
For experiments with RNAi, SMARTpool for rat ERK (ONTARGETplus SMARTpool targeting rat ERK, L-100592-00-0010), and AMPKα2 (L-100623-00-0020) were obtained from Dharmacon (Lafayette, CO, USA). Non-specific siRNA (ONTARGETplus siCONTROL Non-Targeting Pool, D-001810-10-20) was used as a control. For transient expression experiments, L6 myotubes were serum-starved for 16 h in serum-free media, and then transfected with DharmaFECT transfection reagent (Dharmacon) according to the manufacturer's protocol in 12-mm or 60-mm plates containing 100 nM AMPKα2 or ERK siRNA, or non-targeting control (mock) siRNA per plate. After 48 h, cells were treated with vehicle or tunicamycin (5 μg·mL−1) for 3 h before harvesting, or treated with vehicle for insulin signalling or glucose uptake experiments.
Animal care and experimental procedures
Male C57BL/KsJ-Leprdb/Leprdb mice and lean C57BL/6Jms mice [the wild type (WT)] were purchased from the Jackson Laboratories (Bar Harbor, ME, USA) and housed in a temperature-controlled facility (22 ± 2°C) under a 12 h/12 h light/dark cycle. Water and a normal standard pellet diet were available ad libitum throughout the experimental period. For the studies, 8-week-old male db/db mice received U0126 (10 mg·kg−1 day−1), rosiglitazone (5 mg·kg−1 day−1) or phosphate-buffered saline by intraperitoneal injection daily for 2 weeks. After administering U0126, the changes in body weight, food intake, blood glucose levels, serum adiponectin contents, insulin, high-density lipoprotein (HDL), low-density lipoprotein (LDL), triglycerides and total cholesterol levels were measured. These effects were compared to those of rosiglitazone, which improves insulin resistance, at 5 mg·kg−1 day−1 in db/db mice. For immunoblot analysis, muscle tissue was expeditiously isolated after the animal was killed and homogenized in ice-cold buffer (50 nmol·L−1 of Hepes (pH 7.4), 150 mmol·L−1 of NaCl, 10 mmol·L−1 of NaF, 1 mmol·L−1 of sodium pyrophosphate, 0.5 mmol·L−1 of EDTA, 250 mmol·L−1 of sucrose, 1 mmol·L−1 of dithiothreitol, 1% TritonX-100, 1 mmol·L−1 of Na3VO4 and one Roche protease inhibitor tablet per 50 mL of buffer). Lysates were prepared as previously described (Jorgensen et al., 2004) and stored at −80°C until required for analysis. Protein contents in the lysates were quantified using the bicinchoninic acid method (Pierce). Experiments using mice were approved beforehand by the Institutional Animal Care and Use Committee of Yeungnam University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Biochemical analysis
At sacrifice, about 1 mL of venous blood was collected from the vena cava under anaesthesia. All blood samples were centrifuged at 15 000 rpm for 10 min at room temperature in clotting-activated serum tubes. Serum triglycerides, total cholesterol, LDL and HDL levels were assayed using an automated blood analyser (AU400, Olympus, Japan). Serum insulin levels were measured using an insulin-enzyme immunosorbent assay test kit (Mercodia, Uppsala, Sweden).
Measurement of serum adiponectin
To measure of serum adiponectin levels, blood was collected at sacrifice from the vena cava after overnight fasting. Serum adiponectin levels were measured using a commercially available ELISA kit (Otsuka Pharmaceutical Co. Ltd., Tokyo, Japan).
Statistical analysis
Calculations and the statistical analysis were performed using GraphPad Prism 3–5.0 software. The statistical significances of differences between groups were determined using the Student's t-test and multiple comparisons were analysed using one-way anova using treatments and experiments as factors. All results are presented as means ± SEMs. Differences were considered statistically significant for P-values of <0.05.
Results
Induction of ER stress impairs insulin action
Tunicamycin is a potent activator of ER stress (Barbosa-Tessmann et al., 2000; Shen et al., 2001). First, in order to examine whether or not an ER stressor (tunicamycin or thapsigargin) would trigger insulin resistance, we evaluated the phosphorylation patterns of several molecular indicators of insulin resistance. The phosphorylation statuses of RNA-activated protein kinase-like ER resident kinase (PERK) and inositol-requiring kinase-1 (IRE-1) are key indicators of the presence of ER stress (Shi et al., 1998; 2003; Harding et al., 1999; Chawla et al., 2011). It was found that the phosphorylations of PERK and IRE-1 were significantly increased by tunicamycin or thapsigargin treatment in L6 myotubes (Figure 1A and B respectively). A previous study showed activation of the ERK pathway is strongly associated with whole-body insulin resistance, and indicated that ERK is probably a key modulator of the development of insulin resistance (Zheng et al., 2009). In the present study, we used tunicamycin and thapsigargin to identify the pathway involved in insulin resistance. Time- and dose-dependent experiments showed that tunicamycin or thapsigargin caused the phosphorylation of ERK in L6 myotubes (Figure 1A and B). Several studies have reported reduced AMPK activity in skeletal muscle in type 2 diabetic subjects (Kelley and Simoneau, 1994; Gaster et al., 2004). Accordingly, we tested the hypothesis that suppressed rates of glucose uptake are mediated by the ER stress-induced inhibition of AMPK signalling. ER stress was found to suppress the phosphorylations of AMPK and ACC time- and dose-dependently (Figure 1A and B). To determine whether ER stress alters insulin receptor signalling, L6 myotubes were exposed to tunicamycin (5 μg·mL−1) for 3 h to activate ER stress, and then treated with insulin (100 nM) for 10 min. Insulin treatment was found to increase IRS-1 tyrosine phosphorylation significantly, but this stimulatory effect was inhibited by tunicamycin (Figure 1C). Next, we analysed Akt serine phosphorylation and found that insulin-stimulated Akt serine phosphorylation was reduced to an undetectable level when the cells were subjected to ER stress (Figure 1C). In addition, we examined the effects of tunicamycin on the activations of AMPK and ERK, which are known to be insulin resistance. Interestingly, ER stress increased the phosphorylation of ERK but decreased the phosphorylation of AMPK (Figure 1C). We also examined the effect of tunicamycin on insulin-stimulated glucose uptake in L6 myotubes. As shown in Figure 1E, tunicamycin reduced endogenous glucose uptake levels and insulin-stimulated glucose uptake, suggesting that ER stress inhibits insulin signalling at all steps. These experiments were repeated with thapsigargin (another ER stress-inducing agent) to rule out the possibility that the tunicamycin-mediated blockade insulin signalling was due to a non-specific effect (Figure 1D and E). It was found that thapsigargin had the same effect on ER stress activation as tunicamycin. Overall, these results demonstrated that the activation of ER stress blocked AMPK activity and insulin signalling, and thus, inhibited glucose uptake.
Figure 1.
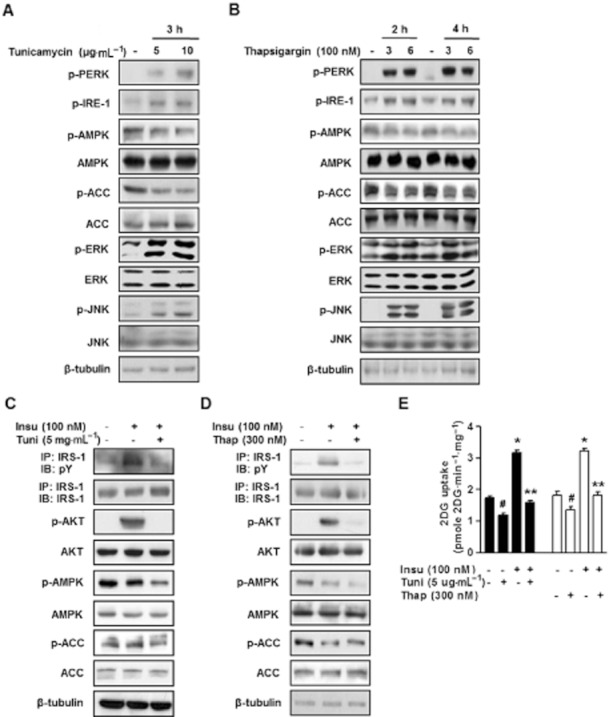
ER stress inhibits insulin signalling ER stress markers, including ERK phosphorylation (p-ERK), PERK (p-PERK), IRE-1 (p-IRE-1), and JNK (p-JNK) in L6 muscle cells. (A) ER stress was induced in L6 myotubes by treating them with 5 μg·mL−1 of tunicamycin for 3 h. (B) Thapsigargin (300 nM or 600 nM) induced ER stress after 2 h of treatment, and its effect was maintained for 4 h. Furthermore, thapsigargin time- and dose-dependently suppressed basal AMPK phosphorylation and ACC phosphorylation. Phosphorylated ERK, PERK, IRE-1, JNK, and AMPK levels and their total protein levels were examined by immunoblotting. ER stress was induced in L6 myotubes by 5 μg·mL−1 of tunicamycin for 3 h (C) or 300 nM thapsigargin for 4 h (D). Cells were subsequently stimulated with 100 nM insulin. Phosphorylated IRS-1 (at tyrosine; pY), Akt, AMPK, and ACC levels and their total protein levels were examined either by immunoprecipitation (IP) following by immunoblotting (IB) or by direct immunoblotting. (E) Cells were treated with 5 μg·mL−1 of tunicamycin for 3 h, or 5 μg·mL−1 of tunicamycin plus 100 nM insulin for 10 min, 300 nM thapsigargin for 4 h or with 300 nM thapsigargin plus 100 nM insulin for 10 min. Glucose uptake were then measured as described in ‘the Experimental section’. Results are expressed as means ± SEMs (n = 4). *P < 0.05, #P < 0.05 versus untreated controls (Student's t-test) and **P < 0.05 versus insulin-treated cells (Student's t-test or anova).
U0126 stimulates glucose uptake via the activation of AMPK in L6 myotubes
In muscle cells, there are two important signal pathways that regulate glucose uptake, namely, the insulin signalling pathway (Hayashi et al., 2000) and the AMPK pathway (Saltiel and Kahn, 2001). Thus, we considered that if U0126 and insulin stimulate glucose uptake via different mechanisms, the combination of U0126 plus insulin should be additive. To confirm this hypothesis, we examined whether U0126 and/or insulin regulate glucose uptake in L6 myotubes. Treatment of the cells with 100 nM insulin for 10 min increased glucose uptake 1.9-fold (2.8 ± 0.05 pmole·mg−1 min−1) (P < 0.01) as compared with untreated cells (1.5 ± 0.1 pmole·mg−1 min−1) (Figure 2A), whereas L6 myotubes exposed to 25 μM U0126 for 1 h showed a 2.0-fold increase in glucose uptake (3.0 ± 0.1 pmole·mg−1 min−1) (P < 0.01) (Figure 2A). Importantly, treatment of L6 myotubes with U0126 plus insulin significantly increased glucose uptake by 2.3-fold (3.5 ± 0.1 pmole·mg−1 min−1) (P < 0.01) (Figure 2A), indicating a partial additive effect. These results demonstrate that U0126 stimulates glucose uptake via an insulin-independent signalling pathway.
Figure 2.
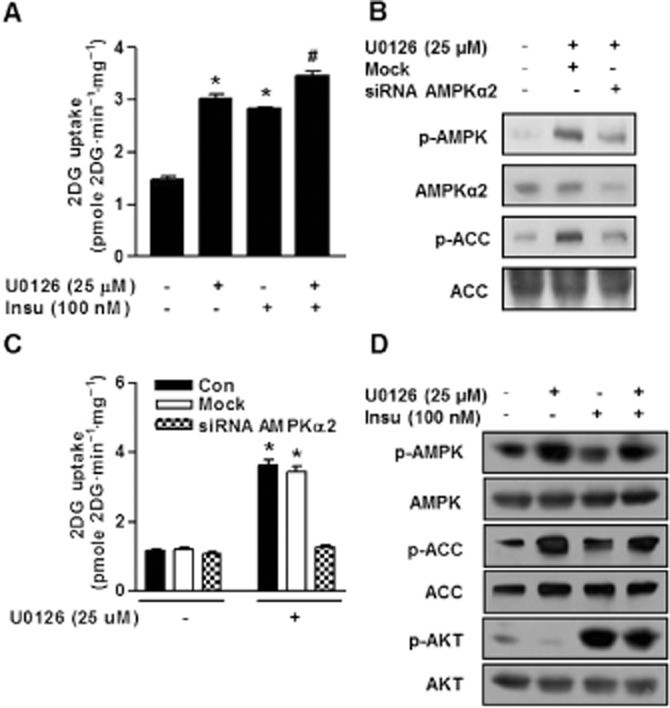
Additive effects of U0126 plus insulin on glucose uptake (A) Cells were serum-starved for 6 h, followed by treatment either for 30 min with 25 μM U0126 or for 10 min with 100 nM insulin, or U0126 plus insulin as indicated. After treatment, glucose uptake was measured as described in ‘the Experimental section’. Results are expressed as means ± SEMs (n = 4). *P < 0.05 compared with untreated control (Student's t-test) and #P < 0.05 versus untreated control and/or insulin-treated cells (Student's t-test or anova). (B) L6 myotubes were transfected with control siRNA (mock) or AMPKα2 siRNA for 48 h and then treated with U0126 for 30 min. Phosphorylated forms of AMPK and ACC were examined via immunoblotting using specific antibodies. (C) Cells were transfected with AMPKα2 siRNA and then exposed to U0126 for 30 min. Next, glucose uptake measurement was determined as described in ‘the Experimental section’ Results are expressed as means ± SEMs (n = 4). *P < 0.05 versus untreated control (Student's t-test). (D) Cells were serum-starved for 6 h, followed by treatment either for 30 min with 25 μM U0126 or for 10 min with 100 nM insulin, or U0126 plus insulin as indicated. Phosphorylated forms of AMPK and ACC were examined via immunoblotting using specific antibodies.
One of the major acute effects of AMPK is the induction of glucose uptake in muscle (30). To verify the specificity of U0126 with respect to AMPK activity, we examined the effect of the siRNA-mediated knockdown of AMPKα2. As expected, AMPK phosphorylation was abrogated by AMPKα2 siRNA (Figure 2B). Moreover, AMPKα2 knockdown significantly abolished U0126-stimulated glucose uptake (Figure 2C). In order to confirm that U0126 acts via the phosphorylation of AMPK, we infected L6 myotubes with a control adenovirus (Ad-GFP) and with a kinase defective Ad-DN-AMPKα1 that inhibits the activity of α1 AMPK (Mu et al., 2001). Interestingly, L6 myotubes expressing dominant negative Ad-DN-AMPKα1 displayed significantly attenuated effects of U0126 on AMPK phosphorylation and glucose uptake (Supporting Information Figure S1A and B). These findings suggest that U0126-mediated AMPK activation and glucose uptake are required either AMPKα1 or AMPKα2. To evaluate the effect of U0126 on insulin signalling, we treated L6 myotubes with insulin or U0126 or insulin plus U0126. We found that AMPK was activated when L6 myotubes were treated with U0126 (Figure 2D), but interestingly, U0126 did not cause Akt phosphorylation (Figure 2D), suggesting inhibition of ERK preferentially associates with AMPK activity, but it was not associate with insulin signalling cascade under normal conditions.
The inhibition of ERK may derepress glucose uptake derived from insulin resistance via AMPK activation
The ERK pathway mediates the insulin-induced down regulation of IRS-1 tyrosine phosphorylation. Indeed, ERK has been shown to play a major role in the development of obesity and insulin resistance (Carlson et al., 2003; Tanti and Jager, 2009). To examine the effects of acute ERK inhibition, we investigated the effect of U0126 (a pharmacological MEK inhibitor) under ER stress. The phosphorylations of AMPK and ACC were significantly inhibited and ERK phosphorylation was increased by tunicamycin, whereas these effects were abolished by U0126 (Figure 3A). For this reason, we attempted to clarify the potential role played by ERK in the AMPK signalling pathway under ER stress. We found that the ERK-AMPK interaction was significantly increased by ER stress and reversed by U0126 (Supporting Information Figure S2). These results suggest that ER stress increases the interaction between ERK and AMPK and that the inhibition ERK results in the retention of AMPK, and thereby, the activation of AMPK. Moreover, U0126 reduced tunicamycin-induced ERK phosphorylation (Figure 3A). However, in U0126-treated L6 myotubes, the phosphorylations of PERK and IRE-1 were not lower than in tunicamycin-treated cells (Figure 3A), suggesting that U0126 was not reduced under ER stress. As expected, treatment of U0126 significantly increased glucose uptake under ER stress (Figure 3B). These results demonstrate that U0126 prevented ER stress-repressed AMPK activity and insulin-mediated glucose uptake in muscle cells. We then examined the role played by ERK in ER stress-impaired insulin signalling. Inhibition of ERK activity by U0126 reversed the ER stress-induced reduction of IRS-1 tyrosine phosphorylation and insulin-stimulated glucose uptake (Figure 3A and B). Interestingly, U0126 preserved insulin-stimulated glucose uptake in ER-stressed L6 myotubes, and this uptake was blocked by wortmannin (a PI3K inhibitor) (Figure 3B). These results strongly support the notion that U0126 improves ER stress-induced reductions of insulin sensitivity in L6 myotubes.
Figure 3.
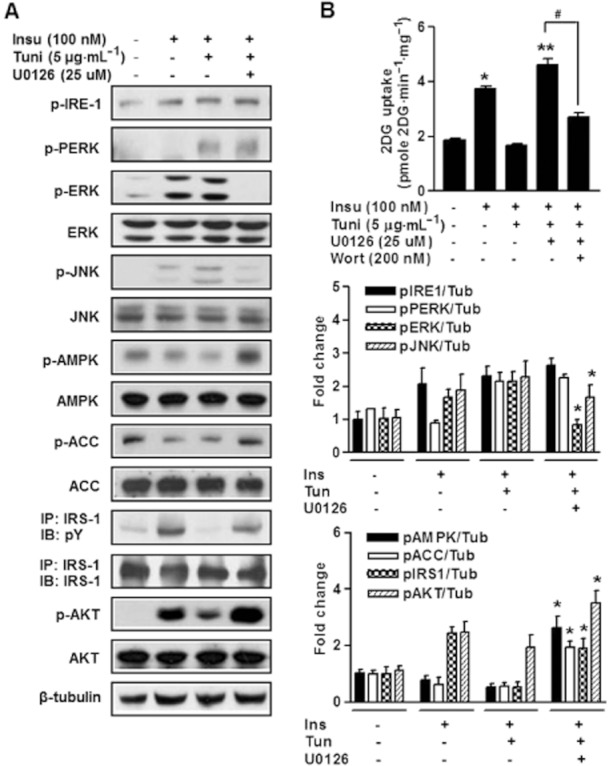
ERK inhibition increases phosphorylation of AMPK and glucose uptake (A) L6 myotubes were pretreated with U0126 (25 μM) for 30 min and then stimulated or not with tunicamycin (5 μg·mL−1) for 3 h and then stimulated for 10 min with 100 nM insulin. IRS-1 tyrosine phosphorylation (pY), Akt phosphorylation, PERK phosphorylation, IRE-1 phosphorylation and JNK phosphorylation and their total protein levels were examined either with immunoprecipitation (IP) following by immunoblotting (IB) or by direct immunoblotting in cell lysates of U0126-treated L6 myotubes. Anti-β-tubulin antibody was used as a loading control. All blots are representative of at least four to six blots from four to six independent experiments. *P < 0.05 compared with tunicamycin plus insulin-treated cells (Student's t-test). (B) Glucose uptake experiments were performed as described in ‘the Experimental section’. Representative glucose uptake and quantification of five independent experiments are shown. Results are expressed as means ± SEMs (n = 4). *P < 0.05 versus untreated control (Student's t-test) and **P < 0.01 versus untreated control and/or insulin-treated cells (Student's t-test or anova). #P < 0.05 for the indicated comparisons (Student's t-test).
Knockdown of ERK improves ER stress-induced insulin resistance in L6 myotubes
To understand the molecular mechanism whereby knockdown of ERK leads to enhanced AMPK activity and insulin sensitivity under ER stress, we depleted ERK in muscle cells by transducing them with ERK specific siRNA. Non-related siRNA (Mock) was used as a control. In skeletal muscle, insulin promote glucose uptake by activating the IRS-1-PI3K-Akt signalling cascade. Although the negative insulin signalling that lead to the inhibitory IRS-1-PI3K-Akt signalling cascade is still being investigated, it is apparent that several serine kinases, including ERK, JNK, IKK and mTOR-S6K (Taniguchi et al., 2006; Thirone et al., 2006). In the present study, ER stress was induced in ERK-depleted or control L6 myotubes, and levels of phospho-ERK were determined. Interestingly, AMPK phosphorylation was significantly increased in ERK-depleted cells versus treatment naïve controls (Figure 4A), which was consistent with our previous finding (Figure 3A). These results demonstrate ERK activation was induced by ER stress and that it seemed to antagonize AMPK activation, which is consistent with observations in U0126-treated cells. Thus, ERK depletion reversed ER stress-induced reductions of Akt phosphorylation and insulin-stimulated glucose uptake (Figure 4A and B). Importantly, ERK knockdown significantly increased glucose uptake to a similar magnitude (Figure 4B). However, this effect was not observed under the same conditions in the presence of wortmannin (Figure 4B), suggesting that U0126-mediated glucose uptake is dependent on PI3K activity under ER stress.
Figure 4.
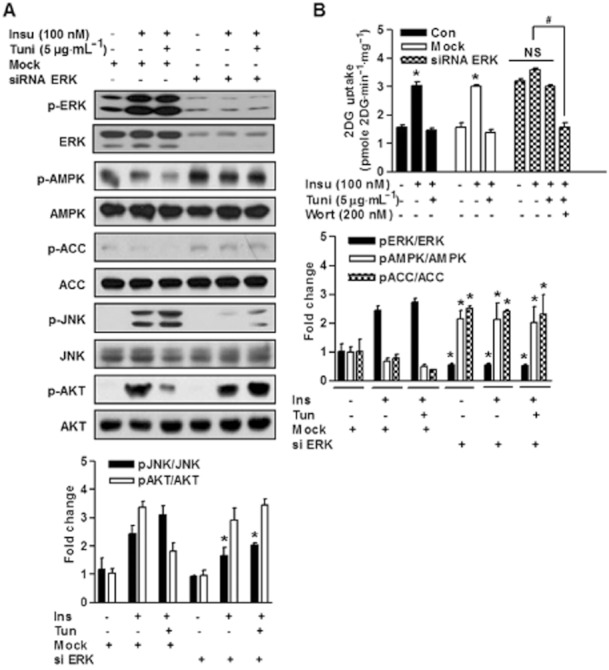
Inhibition of ERK phosphorylation using ERK siRNA abolishes ER stress-induced insulin resistance. (A and B) L6 myotubes were transfected with control siRNA (mock) or ERK siRNA for 48 h and then stimulated with tunicamycin (5 μg·mL−1) or left untreated for 3 h, and then stimulated for 10 min with 100 nM insulin. Tunicamycin reduced AMPK activity as well as insulin-stimulated Akt phosphorylation and glucose uptake (A and B) after 3 h of treatment. Short interfering RNA directed towards ERK (ERK siRNA) inhibited increases in ERK and JNK phosphorylation as well as suppression of AMPK phosphorylation and insulin-stimulated Akt phosphorylation (A) and glucose uptake (B) by tunicamycin (5 μg·mL−1). All blots are representative of at least four to six blots from four to six independent experiments. *P < 0.05 compared with siRNA ERK treated cells (Student's t-test). Glucose uptake was measured. Results are expressed as means ± SEMs (n = 4). *P < 0.05 versus untreated control (Student's t-test) and #P < 0.05 for the indicated comparisons (Student's t-test). NS: not significant.
Knockdown of AMPKα2 promotes the development of insulin resistance in L6 myotubes
To determine whether AMPK modulates ERK activity and function, we used AMPKα2-specific siRNA to reduce AMPKα2 levels in L6 myotubes. siRNA-mediated knockdown of AMPKα2 led to a marked decrease in basal levels of AMPKα2 (Figure 5A). Furthermore, AMPK was highly phosphorylated in U0126-treated cells, and its knockdown using AMPKα2 siRNA abolished its constitutive phosphorylation, whereas treatment with control siRNA did not (Figure 5A). Moreover, AMPKα2 knockdown completely abolished the increase in glucose uptake elicited by U0126 (Figure 5B). These observations suggest that AMPK activation is required for the U0126-induced increases in glucose uptake. In addition, U0126 treatment induced insulin-stimulated IRS-1 tyrosine phosphorylation and glucose uptake under ER stress, indicating enhanced insulin sensitivity (Figure 3), and furthermore, ERK knockdown significantly induced Akt phosphorylation and glucose uptake under ER stress (Figure 4), and this glucose uptake was completely blocked by AMPKα2 knockdown (Figure 5A and B). These findings strongly suggest that AMPK activation plays a key role in the insulin-sensitizing effect of U0126.
Figure 5.
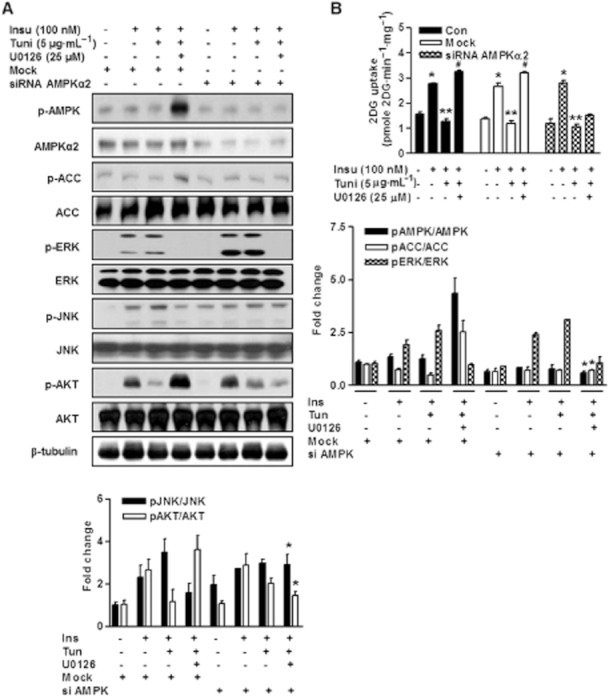
AMPK mediates cross-talk between ER stress and insulin action (A) L6 myotubes were transfected with control siRNA (mock) or AMPKα2 siRNA for 48 h, pretreated with or without U0126 for 30 min, and then treated with 5 μg·mL−1 of tunicamycin for 3 h and stimulated with or without 100 nM insulin for 10 min. Under these conditions, the phosphorylated (p-AMPK, p-ACC, p-ERK, p-JNK, p-Akt) and total forms (ACC, Akt, β-tubulin) of each protein were examined via immunoblotting using specific antibodies. All blots are representative of at least four to six blots from four to six independent experiments. *P < 0.05 versus siRNA AMPK treated cells (Student's t-test). (B) Next, glucose uptake was determined as in (A) L6 myotubes were transfected with mock or AMPKα2 siRNA for 48 h, pretreated with or without U0126 for 30 min, and then treated with tunicamycin for 3 h and stimulated with or without insulin for 10 min. Glucose uptake was examined as described in ‘the Experimental section’. Results are expressed as means ± SEMs (n = 4). *P < 0.05 versus untreated control (Student's t-test) and **P < 0.05 versus insulin-treated cells (Student's t-test) and #P < 0.05 versus untreated control and/or insulin-treated cells (Student's t-test or anova).
The inhibition of ERK decreases blood glucose levels in db/db mice
To examine the metabolic effects of U0126 on insulin resistance, db/db mice were treated with U0126 (10 mg·kg−1, i.p.) daily for 2 weeks (from 8 to 10 weeks of age). Mean food intakes and body weights were similar for U0126-treated, rosiglitazone-treated or untreated db/db mice (Figure 6A and B), but non-fasting blood glucose and insulin levels were markedly lower in mice treated with U0126 and rosiglitazone than in untreated db/db mice (Figure 6C and D). Next, the effects of U0126 on serum triglycerides and cholesterol levels were investigated in db/db mice. U0126 was found to decrease serum triglycerides levels versus untreated db/db mice (Table 1), reduced serum LDL cholesterol levels and increased serum HDL cholesterol (Table 1). Serum adiponectin concentrations are known to be closely related to systemic insulin sensitivity, and in the present study, serum adiponectin levels were slightly elevated in U0126-treated mice than in untreated controls (Figure 6E). Taken together, these results show that U0126 improves plasma glucose and lipid profiles in db/db mice.
Figure 6.
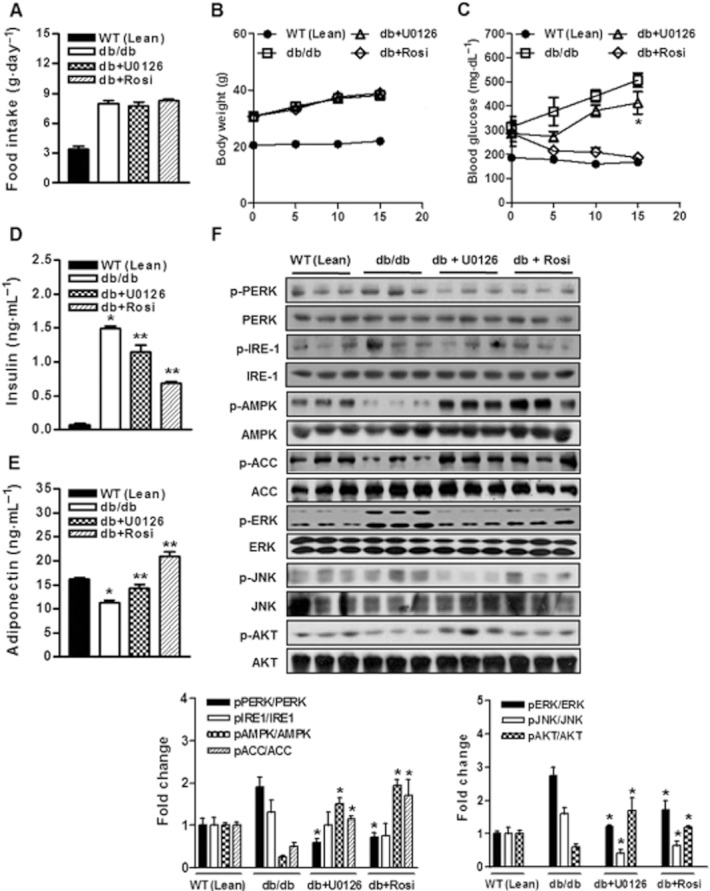
Effect of U0126 on systemic glucose metabolism and insulin sensitivity in db/db mice A and B, 8-week-old lean WT and db/db mice were injected with U0126 (10 mg·kg−1 day−1, i.p.) or rosiglitazone (5 mg·kg−1 day−1, i.p.) for 14 days. (A) Effect of U0126 on food intake. WT (Lean), db/db, db/db + U0126, db/db + rosi. (B) Effect of U0126 or rosiglitazone on body weight.  , WT (Lean);
, WT (Lean);  , db/db;
, db/db;  , db/db + U0126;
, db/db + U0126;  , db/db + rosi. (C) Effect of U0126 on non-fasting blood glucose levels. , WT (Lean); , db/db; , db/db + U0126; , db/db + rosi. Results are presented as means ± SEMs (n = 8). *P < 0.05 vs. db/db untreated controls (Student's t-test). (D) Effect of U0126 on non-fasting serum insulin levels. Results are presented as means ± SEMs (n = 8). *P < 0.05 vs. WT (Lean) (Student's t-test) and **P < 0.05 vs. WT (Lean) and/or db/db untreated controls (Student's t-test or anova). (E) Effect of U0126 on serum adiponectin levels. Results are presented as means ± SEMs (n = 8). *P < 0.05 vs. WT (Lean) (Student's t-test) and **P < 0.05 vs. WT (Lean) and/or db/db untreated controls (Student's t-test or anova). (F) Effect of U0126 treatment on ER stress parameters, JNK activation, and on the insulin signal transduction pathway in the skeletal muscles of db/db mice. Total protein was obtained from db/db soleus muscle tissue after administering U0126 i.p. for 14 days, and lysates were immunoblotted with corresponding antibodies. Phosphorylated AMPK, ACC, Akt, ERK, and JNK levels were examined in soleus muscle extracts by direct immunoblotting. All blots are representative of at least four to six blots from four to six independent experiments. *P < 0.05 vs. untreated db/db controls (Student's t-test).
, db/db + rosi. (C) Effect of U0126 on non-fasting blood glucose levels. , WT (Lean); , db/db; , db/db + U0126; , db/db + rosi. Results are presented as means ± SEMs (n = 8). *P < 0.05 vs. db/db untreated controls (Student's t-test). (D) Effect of U0126 on non-fasting serum insulin levels. Results are presented as means ± SEMs (n = 8). *P < 0.05 vs. WT (Lean) (Student's t-test) and **P < 0.05 vs. WT (Lean) and/or db/db untreated controls (Student's t-test or anova). (E) Effect of U0126 on serum adiponectin levels. Results are presented as means ± SEMs (n = 8). *P < 0.05 vs. WT (Lean) (Student's t-test) and **P < 0.05 vs. WT (Lean) and/or db/db untreated controls (Student's t-test or anova). (F) Effect of U0126 treatment on ER stress parameters, JNK activation, and on the insulin signal transduction pathway in the skeletal muscles of db/db mice. Total protein was obtained from db/db soleus muscle tissue after administering U0126 i.p. for 14 days, and lysates were immunoblotted with corresponding antibodies. Phosphorylated AMPK, ACC, Akt, ERK, and JNK levels were examined in soleus muscle extracts by direct immunoblotting. All blots are representative of at least four to six blots from four to six independent experiments. *P < 0.05 vs. untreated db/db controls (Student's t-test).
Table 1.
Effects of U0126 on the plasma lipid levels in db/db mice
| Group | Total cholesterol (mg·dL−1) | Triglyceride (mg·dL−1) | HDL cholesterol (mg·dL−1) | LDL cholesterol (mg·dL−1)a |
|---|---|---|---|---|
| WT (Lean) | 136.6 ± 8.9 | 70.0 ± 4.5 | 62.0 ± 0.8 | 60.6 ± 9.2 |
| db | 209.3 ± 7.3 | 147.5 ± 10.3 | 54.0 ± 7.0 | 129.3 ± 11.1 |
| db+U0126 | 210.9 ± 10.0 | 109.1 ± 6.7* | 75.9 ± 1.8* | 113.1 ± 9.7 |
| db+Rosi | 307.4 ± 29.1* | 83.0 ± 13.6** | 79.6 ± 1.8* | 181.6 ± 19.0* |
Each value is mean ± SE for six mice. *P < 0.05, **P < 0.01 compared to db control.
Total cholesterol – HDL cholesterol – Triglyceride/5.
To determine whether lowered glucose levels in the mice treated with U0126 were due directly to increased skeletal muscle insulin signalling, muscle tissues were isolated 2 weeks after a vehicle or U0126 (10 mg·kg−1, i.p.) treatment measured by immunoblotting. Akt phosphorylation was markedly lower in the muscle tissues of saline-treated db/db mice than in lean (WT) control mice (Figure 6F). In contrast, insulin signalling was significantly increased in the soleus muscles of U0126-treated db/db mice than in vehicle-treated db/db controls (Figure 6F). Furthermore, U0126-stimulated decreases in glucose levels occurred concomitantly with significant increases in the phosphorylation levels of AMPK and ACC. Furthermore, U0126 treatment markedly reduced ERK phosphorylation in the muscle tissues of the U0126-treated db/db mice (Figure 6F). Recently, it was demonstrated that ER stress plays a central role in the development of insulin resistance and diabetes by impairing insulin signalling via JNK activation (9). As shown in Figure 6F, JNK phosphorylation, a marker of ER stress, was increased in the muscle tissues of db/db mice versus lean mice, whereas U0126 reduced JNK activity. Indeed, PERK phosphorylation was decreased by U0126 in muscle tissues (Figure 6F). Consistent with the results obtained for ERK, and JNK, PERK phosphorylation was also markedly suppressed in muscle tissue after rosiglitazone treatment (Figure 6F). These finding show that U0126 suppressed ER stress, and suggest that ERK activity plays a key role in ER stress-induced insulin resistance.
Discussion
This study, demonstrates that the hyper-phosphorylation of ERK is promoted under ER stress and that this contributes to the inhibition of insulin-stimulated IRS-1 tyrosine phosphorylation and glucose uptake, at least in part, via ERK-mediated insulin signalling. If hyper-phosphorylation of ERK mediates ER stress-induced insulin resistance, then inhibition of this process should prevent the inhibitory effect of ER stress-induced insulin resistance. In the present study, we found that ER stress-induced ERK hyper-phosphorylation was inhibited by the specific ERK inhibitor U0126. Furthermore, we provide in vitro and in vivo pharmacological evidence that specific loss of ERK activity can reverse increases in insulin resistance.
We found that treatment of L6 myotubes with an ER stressor (tunicamycin or thapsigargin) inhibited the ability of insulin to stimulate IRS-1 tyrosine phosphorylation and glucose uptake, and thus, drastically reduced endogenous AMPK activity. In contrast, ER stress significantly increased ERK phosphorylation, and ER stress-induced ERK phosphorylation was diminished by the MEK inhibitor U0126 and by the siRNA silencing of ERK. Importantly, AMPK phosphorylation and glucose uptake were recovered by U0126 or ERK siRNA in ER-stressed L6 myotubes. Furthermore, siRNA-mediated knockdown of AMPKα2 markedly abolished recovered insulin-sensitizing Akt phosphorylation and glucose uptake via ERK inhibition under ER stress. Taken together, these findings demonstrate that AMPK might have protective effects against ER stress-induced insulin resistance. To confirm this possibility, we compared glucose homeostasis in U0126-injected db/db mice and rosiglitazone (an antidiabetic)-injected db/db mice. U0126-injected and rosiglitazone-injected db/db mice were found to exhibit significantly lower blood glucose concentrations than control db/db mice, possibly due to the specific activity of AMPK in muscle tissue. We hypothesize that a deficiency of ERK activity with a subsequent increase in AMPK phosphorylation might be beneficial for attenuating insulin resistance.
In a recent study, we described a pathway mediated by ER stress involving the subsequent suppression of AMPK phosphorylation and glucose uptake (Hwang et al., 2010). The experiments performed in the present study show that the selective inactivation of ERK by U0126 or siRNA knockdown significantly increased AMPK phosphorylation under ER stress. In skeletal muscle, the action of AMPK is believed to be mediated mainly through the AMPKα2 isoform (Vavvas et al., 1997; Wojtaszewski et al., 2002), whereas in other tissues, the physiological role of this isoform is less clear. In a recent study, it was shown that AMPKα2–/– mice exhibit glucose intolerance associated with low plasma insulin levels and reduced muscle glycogen synthesis during hyperinsulinemic clamp conditions, and that reductions in insulin sensitivity are not dependent on muscle AMPK activity, since mice expressing a dominant-negative AMPK mutant in skeletal muscle were not insulin resistant (Viollet et al., 2003). However, in the present study, the U0126-induced activation of AMPK and glucose uptake were blocked by the inhibitions of α1 or α2-AMPK in L6 myotubes. These observations strongly suggest that the insulin-sensitizing effect of U0126 triggers the activations of α1 and α2-AMPK in L6 myotubes.
A previous study demonstrated that ER stress leads to the activations of UPR signalling pathways that play dominant roles in the development of obesity-induced insulin resistance and type 2 diabetes (Ozcan et al., 2004). In another, the reversal of ER stress using chemical chaperone agents that enhance the ER folding machinery increased insulin sensitivity and reversed type 2 diabetes in obese mice (Ozcan et al., 2006). Insulin resistance is a major metabolic feature in the obesity and an important pathophysiological factor in the development of metabolic abnormalities, including hypertension, dyslipidaemias, cardiovascular disease and type 2 diabetes mellitus (Permutt et al., 2005). Furthermore, recently, ER stress was found to play a major role in the pathogeneses of insulin resistance and type 2 diabetes due to its inhibition of insulin signalling (Ozcan et al., 2004; Um et al., 2004).
Precise details of the molecular mechanisms underlying the attenuation of insulin resistance in U0126-injected mice remain to be elucidated. Recently, it has been reported that ERK1-deficient mice are resistant to diet-induced obesity and protected from the development of insulin resistance (Bost et al., 2005), and furthermore, ER stress has been reported to attenuate IR tyrosine phosphorylation via an ATF6-ERK dependent pathway (Tang et al., 2011). Importantly, several inducers of insulin resistance, such as, IKKβ, JNK, ERK and S6K, which phosphorylate IRS-1 protein at inhibitory sites in an uncontrolled manner, also activate several kinases (Taniguchi et al., 2006; Thirone et al., 2006). These inhibitory phosphorylations provide negative feedback to insulin signalling and serve as mechanisms for cross-talk with other pathways that produce insulin resistance.
In particular, a pathway involving ERK (a MAPK) is known to be deregulated in the obese, and thus, ERK is suspected to play a major role in insulin resistance. Indeed, ERK activity is abnormally elevated in human and rodent adipose tissues in the diabetic state (Bouzakri et al., 2003; Bashan et al., 2007), which may explain, in part, the enhanced insulin sensitivity observed after U0126 administration in the present study. In the present study, the muscle tissues of db/db mice exhibited characteristics of ER stress, such as, elevated phosphorylated ERK levels and JNK activity as compared with lean animals. Our findings further indicate that the ERK pathway may have contributed to insulin resistance in type 2 diabetes by acting on insulin signalling. In addition, we examined the effect of low dose U0126 on severe insulin resistance in db/db mice, and found that at 10 weeks of age, U0126-injected db/db mice were partially protected against insulin resistance, demonstrating that ERK inhibition influenced insulin resistance in this model of diabetes. In addition, U0126 reduced serum LDL and triglycerides levels and increased serum HDL levels in db/db mice. We also evaluated phosphorylation statuses of AMPK in muscle tissues; it has been previously shown that the small molecule-mediated activation of AMPK improves insulin resistance in db/db mice (Lee et al., 2006) and in hepatic cells (Lin and Lin, 2008). We found a lack of ERK activity resulted in a significant increase in AMPK phosphorylation, which could have contributed to the observed ERK-mediated alleviation of insulin resistance. These findings suggest that ER stress-induced ERK activation could play an important role in ER stress-induced decreases in AMPK activity and insulin sensitivity. In addition, we found that ERK is constitutively associated with AMPK in L6 myotubes and that ER stress significantly increases this association. Furthermore, we found that U0126 reduces the strength of the ERK-AMPK interaction.
Although we cannot demonstrate the effects of tunicamycin in vivo, our findings are consistent with the notions that tunicamycin directly affects the ERK pathway, AMPK signalling and glucose uptake in skeletal muscle. Overall, these findings suggest that improved insulin resistance after U0126 treatment is due to increased activations of the insulin and AMPK signalling pathways.
Our results are consistent with a model in which loss of ERK results in enhanced AMPK activity and decreased insulin resistance in vitro and in vivo. Furthermore, our findings show that ER stress-induced insulin resistance in muscle cells is prevented by pharmacological or genetic inhibition of the ERK pathway, which suggests a role for ERK in insulin resistance and diabetes. However, the molecular mechanism whereby ER stress activates the ERK pathway in muscle cells is not well understood. Overall, our results reveal novel physiological functions of ERK, which plays important roles in diabetes development and associated insulin resistance by negatively modulating AMPK. Therefore, ERK inhibitors and AMPK modulators are being developed for the treatment of insulin resistance and type 2 diabetes. Furthermore, our findings suggest that the action mechanisms of these agents overlap.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology 2011-0010798 (to H. W. C.) and a grant of the Korea Health Technology R&D Project, Ministry of Health & Welfare Grant A111345, WCU (World Class University) program through the National Research Foundation of Korea funded by Ministry of Education, Science and Technology Grant R32-10064 (to I-K. L.) and in part by the 2011 Yeungnam University Research Grant (to S. L. H.).
Glossary
- ACC
acetyl-CoA carboxylase
- Ad-DN-AMPK
AMPK adenovirus dominant negative
- AMPK
AMP-activated protein kinase
- ER
endoplasmic reticulum
- PERK
RNA-activated protein kinase-like ER resident kinase
- IRE-1
inositol-requiring kinase-1
- siRNA
small interfering RNA
- UPR
unfolded protein response
- IRS-1
insulin receptor substrate-1
- WT
wild type
Conflicts of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 AMPKα1 activity modifies U0126 sensitivity to glucose uptake. (A) L6 myotubes infected with control vector (Vector) or vector expressing dominant negative AMPKα1 and then were treated with U0126 for 30 min, and the level of phospho-AMPK and phospho-ACC was examined by Western blot. Expression of myc-tagged Ad-DN-AMPKα1 was detected using anti-myc antibody. (B) Cells were transfected with control vector (Vector) or vector expressing dominant negative AMPKα1 and then exposed to U0126 for 30 min. Glucose uptake measurement was determined as described in the ‘Experimental’. Data are expressed as means ± SEM (n = 4). *P < 0.05 compared with untreated control (Student's t-test).
Figure S2 L6 myotubes were pretreated with U0126 (25 μmol·L−1) for 30 min and then stimulated or not with tunicamycin (5 μg·mL−1) for 3 h and then stimulated for 10 min with 100 nM insulin. AMPK phosphorylation, ACC phosphorylation and their total protein levels were examined by direct immunoblotting. Immunoprecipitates of ERK were analysed by immunoblotting with anti-AMPK antibody, followed by reprobing of the blots with anti-ERK antibody.
References
- Barbosa-Tessmann IP, Chen C, Zhong C, Siu F, Schuster SM, Nick HS, et al. Activation of the human asparagine synthetase gene by the amino acid response and the endoplasmic reticulum stress response pathways occurs by common genomic elements. J Biol Chem. 2000;275:26976–26985. doi: 10.1074/jbc.M000004200. [DOI] [PubMed] [Google Scholar]
- Bashan N, Dorfman K, Tarnovscki T, Harman-Boehm I, Liberty IF, Bluher M, et al. Mitogen-activated protein kinases, inhibitory-kappaB kinase, and insulin signaling in human omental versus subcutaneous adipose tissue in obesity. Endocrinology. 2007;148:2955–2962. doi: 10.1210/en.2006-1369. [DOI] [PubMed] [Google Scholar]
- Bost F, Aouadi M, Caron L, Even P, Belmonte N, Prot M, et al. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes. 2005;54:402–411. doi: 10.2337/diabetes.54.2.402. [DOI] [PubMed] [Google Scholar]
- Bouzakri K, Roques M, Gual P, Espinosa S, Guebre-Egziabher F, Riou JP, et al. Reduced activation of phosphatidylinositol-3 kinase and increased serine 636 phosphorylation of insulin receptor substrate-1 in primary culture of skeletal muscle cells from patients with type 2 diabetes. Diabetes. 2003;52:1319–1325. doi: 10.2337/diabetes.52.6.1319. [DOI] [PubMed] [Google Scholar]
- Carlson CJ, Koterski S, Sciotti RJ, Poccard GB, Rondinone CM. Enhanced basal activation of mitogen-activated protein kinases in adipocytes from type 2 diabetes: potential role of p38 in the downregulation of GLUT4 expression. Diabetes. 2003;52:634–641. doi: 10.2337/diabetes.52.3.634. [DOI] [PubMed] [Google Scholar]
- Chawla A, Chakrabarti S, Ghosh G, Niwa M. Attenuation of yeast UPR is essential for survival and is mediated by IRE1 kinase. J Cell Biol. 2011;193:41–50. doi: 10.1083/jcb.201008071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deivanayagam S, Mohammed BS, Vitola BE, Naguib GH, Keshen TH, Kirk EP, et al. Nonalcoholic fatty liver disease is associated with hepatic and skeletal muscle insulin resistance in overweight adolescents. Am J Clin Nutr. 2008;88:257–262. doi: 10.1093/ajcn/88.2.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaster M, Rustan AC, Aas V, Beck-Nielsen H. Reduced lipid oxidation in skeletal muscle from type 2 diabetic subjects may be of genetic origin: evidence from cultured myotubes. Diabetes. 2004;53:542–548. doi: 10.2337/diabetes.53.3.542. [DOI] [PubMed] [Google Scholar]
- Gregor MF, Hotamisligil GS. Thematic review series: adipocyte biology. Adipocyte stress: the endoplasmic reticulum and metabolic disease. J Lipid Res. 2007;48:1905–1914. doi: 10.1194/jlr.R700007-JLR200. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ. Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism. Diabetes. 2000;49:527–531. doi: 10.2337/diabetes.49.4.527. [DOI] [PubMed] [Google Scholar]
- Hwang SL, Chang HW, Lee IK, Yang BK, Magae J, Chang YC. Ascofuranone prevents ER stress-induced insulin resistance via activation of AMP-activated protein kinase in L6 myotube cells. Biochem Biophys Res Commun. 2010;396:967–972. doi: 10.1016/j.bbrc.2010.05.034. [DOI] [PubMed] [Google Scholar]
- Jager J, Corcelle V, Gremeaux T, Laurent K, Waget A, Pages G, et al. Deficiency in the extracellular signal-regulated kinase 1 (ERK1) protects leptin-deficient mice from insulin resistance without affecting obesity. Diabetologia. 2011;54:180–189. doi: 10.1007/s00125-010-1944-0. [DOI] [PubMed] [Google Scholar]
- Jorgensen SB, Nielsen JN, Birk JB, Olsen GS, Viollet B, Andreelli F, et al. The alpha2-5′AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes. 2004;53:3074–3081. doi: 10.2337/diabetes.53.12.3074. [DOI] [PubMed] [Google Scholar]
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- Kelley DE, Simoneau JA. Impaired free fatty acid utilization by skeletal muscle in non-insulin-dependent diabetes mellitus. J Clin Invest. 1994;94:2349–2356. doi: 10.1172/JCI117600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korenblat KM, Fabbrini E, Mohammed BS, Klein S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology. 2008;134:1369–1375. doi: 10.1053/j.gastro.2008.01.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortum RL, Costanzo DL, Haferbier J, Schreiner SJ, Razidlo GL, Wu MH, et al. The molecular scaffold kinase suppressor of Ras 1 (KSR1) regulates adipogenesis. Mol Cell Biol. 2005;25:7592–7604. doi: 10.1128/MCB.25.17.7592-7604.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Pfluger PT, Kim JY, Nogueiras R, Duran A, Pages G, et al. A functional role for the p62-ERK1 axis in the control of energy homeostasis and adipogenesis. EMBO Rep. 2010;11:226–232. doi: 10.1038/embor.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes. 2006;55:2256–2264. doi: 10.2337/db06-0006. [DOI] [PubMed] [Google Scholar]
- Lin CL, Lin JK. Epigallocatechin gallate (EGCG) attenuates high glucose-induced insulin signaling blockade in human hepG2 hepatoma cells. Mol Nutr Food Res. 2008;52:930–939. doi: 10.1002/mnfr.200700437. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- Neschen S, Morino K, Hammond LE, Zhang D, Liu ZX, Romanelli AJ, et al. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab. 2005;2:55–65. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Permutt MA, Wasson J, Cox N. Genetic epidemiology of diabetes. J Clin Invest. 2005;115:1431–1439. doi: 10.1172/JCI24758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–1388. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- Savage DB, Choi CS, Samuel VT, Liu ZX, Zhang D, Wang A, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. 2006;116:817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Ellis RE, Lee K, Liu CY, Yang K, Solomon A, et al. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell. 2001;107:893–903. doi: 10.1016/s0092-8674(01)00612-2. [DOI] [PubMed] [Google Scholar]
- Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, et al. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18:7499–7509. doi: 10.1128/mcb.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Taylor SI, Tan SL, Sonenberg N. When translation meets metabolism: multiple links to diabetes. Endocr Rev. 2003;24:91–101. doi: 10.1210/er.2002-0018. [DOI] [PubMed] [Google Scholar]
- Tang X, Shen H, Chen J, Wang X, Zhang Y, Chen LL, et al. Activating transcription factor 6 protects insulin receptor from ER stress-stimulated desensitization via p42/44 ERK pathway. Acta Pharmacol Sin. 2011;32:1138–1147. doi: 10.1038/aps.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signaling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Tanti JF, Jager J. Cellular mechanisms of insulin resistance: role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr Opin Pharmacol. 2009;9:753–762. doi: 10.1016/j.coph.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Thirone AC, Huang C, Klip A. Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol Metab. 2006;17:72–78. doi: 10.1016/j.tem.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- Vavvas D, Apazidis A, Saha AK, Gamble J, Patel A, Kemp BE, et al. Contraction-induced changes in acetyl-CoA carboxylase and 5′-AMP-activated kinase in skeletal muscle. J Biol Chem. 1997;272:13255–13261. doi: 10.1074/jbc.272.20.13255. [DOI] [PubMed] [Google Scholar]
- Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszewski JF, Jørgensen SB, Hellsten Y, Hardie DG, Richter EA. Glycogen-dependent effects of 5-aminoimidazole-4-carboxamide (AICA)-riboside on AMP-activated protein kinase and glycogen synthase activities in rat skeletal muscle. Diabetes. 2002;51:284–292. doi: 10.2337/diabetes.51.2.284. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Zhang W, Pendleton E, Leng S, Wu J, Chen R, et al. Improved insulin sensitivity by calorie restriction is associated with reduction of ERK and p70S6K activities in the liver of obese Zucker rats. J Endocrinol. 2009;203:337–347. doi: 10.1677/JOE-09-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.