

Abstract
Background and Purpose
Prasugrel is a third-generation thienopyridine prodrug and ticagrelor is a non-competitive P2Y12 receptor antagonist. In their phase 3 studies, both agents reduced rates of ischemic events relative to treatment with clopidogrel.
Experimental Approach
The pharmacodynamic profile of anti-platelet effects of prasugrel was compared with that of ticagrelor in rats.
Key Results
The active metabolite of prasugrel was less potent than ticagrelor and its active metabolite on platelet aggregation in vitro. In contrast, prasugrel was a more potent antiplatelet agent than ticagrelor on ex vivo platelet aggregation: their ED50 values at peak for ADP 20 μmol·L−1 were 1.9 and 8.0 mg·kg−1, respectively. Prasugrel's inhibition of platelet aggregation was maintained for up to 24 h after administration, but ticagrelor's duration of action was substantially shorter. Prasugrel and ticagrelor significantly inhibited thrombus formation with ED50 values of 1.8 and 7.7 mg·kg−1, respectively. Both agents also prolonged bleeding times (ED200 values of 3.0 and 13 mg·kg−1 respectively) suggesting that at equivalent levels of inhibition of platelet aggregation, the agents would show comparable antithrombotic activity with similar bleeding risk. Platelet transfusion significantly increased blood platelet numbers similarly in prasugrel- and ticagrelor-treated rats. In the prasugrel-treated group, platelet transfusion caused significant shortening of bleeding time, while in the ticagrelor-treated group, platelet transfusion showed no influence on bleeding time under the experimental conditions employed.
Conclusions and Implications
Prasugrel and ticagrelor showed several differences in their pharmacological profiles and these disparities may reflect their differing reversibility and/or pharmacokinetic profiles.
Keywords: P2Y12 receptor antagonist, thrombosis, haemostasis, prasugrel, ticagrelor, platelet
Introduction
Prasugrel is a third generation thienopyridine antiplatelet prodrug (Niitsu et al., 2005; Jakubowski et al., 2007), requiring in vivo metabolism to generate the active metabolite R-138727 that is a specific and irreversible antagonist of the platelet P2Y12 ADP receptor (Sugidachi et al., 2001; 2007; receptor nomenclature follows Alexander et al., 2011). Prasugrel has the potential to provide both more consistent and greater blockade of P2Y12 receptors than clopidogrel (Dobesh, 2009). The better pharmacokinetics and pharmacodynamics of prasugrel compared with clopidogrel result not only in more effective platelet inhibition, but greater clinical benefits in acute coronary syndrome (ACS) patients undergoing percutaneous coronary intervention (PCI; Wiviott et al., 2007; Li et al., 2009).
Ticagrelor, a cyclopentyl-triazolo-pyrimidine, is a new chemical class of non-competitive and reversible P2Y12 receptor antagonist (van Giezen et al., 2009). Ticagrelor was recently approved for use in ACS patients (Wijeyeratne et al., 2012) based on its phase 3 study, (PLATO, platelet inhibition and patient outcomes) which showed a significant benefit compared with clopidogrel (Wallentin et al., 2009). As with the thienopyridines, ticagrelor undergoes CYP-mediated metabolism to produce an active metabolite, AR-C124910XX (Teng and Butler, 2010), but in contrast to the thienopyridines, both the parent drug and active metabolite exhibit platelet inhibitory activity (van Giezen and Humphries, 2005; Teng and Butler, 2010). Pharmacological assessment of treatment with ticagrelor must therefore consider the pharmacokinetics and pharmacodynamics of both ticagrelor and its active metabolite.
Both prasugrel and ticagrelor were more effective than clopidogrel and may therefore be preferred in ACS patients. However, to date, no large-scale clinical study has directly compared the efficacy and safety of prasugrel and ticagrelor (Alber et al., 2011), although a small clinical pharmacology study in ACS patients with high on-treatment platelet reactivity (Alexopoulos et al., 2012) and an adjusted indirect meta-analysis comparing both agents (Biondi-Zoccai et al., 2011) have been reported. Moreover, there is, so far, no detailed non-clinical comparison of the pharmacological profile of prasugrel and ticagrelor.
In the present study, we determined the relative pharmacological profiles of the anti-platelet activities of prasugrel and ticagrelor, using experimental models in rats, and found several differences. To our knowledge, this is the first formal, detailed non-clinical study to compare the pharmacological profiles of these novel P2Y12 receptor antagonists.
Methods
Experimental animals
All animal care and experimental procedures complied with the institutional ‘Animal experiment ethical code’ (NISSEI BILIS Co., Ltd. and the Safety Research Institute for Chemical Compounds Co., Ltd) and the guidelines of the Institutional Animal Care and Use Committee (Daiichi Sankyo Co., Ltd). All studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). A total of 457 animals were used in the experiments described here. Male Sprague-Dawley rats (Japan SLC, Inc., Shizuoka, Japan; 7 weeks old at receipt) were used in the present study. Rats were housed in animal quarters set at a constant temperature, humidity and 12 h light/dark cycle. Rats were maintained with free access to water and food and used after an acclimatization period of at least 6 days.
Preparation of platelet-rich plasma (PRP) and platelet-poor plasma (PPP)
In ex vivo studies, blood was collected from the abdominal aorta under anaesthesia with pentobarbital sodium (35 mg·kg−1, i.p.) at 1, 2, 4, 8, 12 and 24 h after the administration of the test agent. 4.5 mL of blood was drawn into a disposable syringe containing 0.5 mL of 3.8% sodium citrate (pH 7.4). For in vitro studies, 6 mL of blood was collected in a similar manner. The anti-coagulated blood was centrifuged (150–200× g for 10 min at room temperature) to obtain PRP. After obtaining PRP, the remaining blood was centrifuged (1300–1500× g for 15 min at room temperature) to obtain PPP. Platelet counts in the PRP were obtained using an automated blood cell counter (F-800 or XT-2000 iV, Sysmex Corporation, Hyogo, Japan), and PRP containing 50 ± 5 × 104 platelets·μL−1 was prepared by diluting with PPP.
Platelet aggregation
In the in vitro studies, PRP was incubated with the vehicle or the test agents for 30 min at room temperature before measuring aggregation to ADP. In the ex vivo studies, 240 μL of the PRP prepared was stirred for 1 min at 37°C, and 10 μL of agonist (ADP or collagen) was subsequently added to induce platelet aggregation. Platelet aggregation was monitored for 10 min after agonist addition and recorded as maximum platelet aggregation using a 12-channel automated platelet aggregometer (MCM HEMA TRACER 313 or 712, MC Medical, Inc., Tokyo, Japan).
Arterio-venous (AV) shunt thrombosis model
The ability of the agents to prevent thrombus formation was assessed using an AV shunt model described previously by Sugidachi et al. (2000) with slight modifications. After anaesthesia with pentobarbital sodium (50 mg·kg−1, i.p.), the jugular vein and contralateral carotid artery of rats were exposed and were cannulated with a tube that contained a 10 cm long silk thread in its lumen and filled with heparinized saline (30 units·kg−1). Blood circulation was started through the shunt, initiating thrombus formation on the silk thread 4 h after administration of test agents. After blood circulation for 30 min, the shunt was removed from the vein, the rat killed by exsanguination and the thread was carefully removed from the tube. After blotting the thread with a filter paper, the wet weight of the thread was determined, and the thrombus weight was calculated by subtracting the original weight of the silk thread. An electronic analytical balance (AX200; Shimadzu Corporation, Kyoto, Japan) was used for measuring the weight of the thrombus.
Measurement of bleeding time
Four hours after administration of vehicle, prasugrel or ticagrelor, rats were placed in a rodent restrainer, their tails sterilized with an alcohol swab and dried with tissue paper. A 21-G needle was advanced 1 cm into the tail vein at approximately 3 cm from the tail end and immediately withdrawn. Issuing blood was carefully blotted every 5 s using the rough side of a filter paper (Qualitative filter paper No. 2; Advantec Toyo Kaisha, Ltd., Tokyo, Japan). When no further blood appeared on the filter paper, the bleeding was judged to have ceased and the measurement concluded. The number of bloodstains on the filter paper was counted, and bleeding time (s) was calculated by multiplying the total number of blood stains by 5.
Platelet transfusion experiment
Prasugrel (10 mg·kg−1, p.o.) and ticagrelor (30 mg·kg−1, p.o.), doses that produced similar levels of inhibition of platelet aggregation, were administered to rats 4 h before the bleeding time measurements. Fresh, washed platelets (1 × 1010 platelets·mL−1) were prepared from other rats and suspended in Hank's balanced salt solution. Platelets (1 × 1010 platelets/rat) were transfused via the jugular vein to rats 1 h before the bleeding time measurements and the bleeding time was determined as described earlier. Red blood cell and platelet numbers in whole blood were measured just before platelet transfusion and after bleeding time measurements using an automated blood cell counter (KX-21N, Sysmex Corporation).
Data analysis
Platelet aggregation, thrombus weight and bleeding time were expressed as means ± SEM. Dunnett's multiple comparison test was carried out for platelet aggregation, thrombus weight and bleeding time at each time point using the vehicle group as control. In platelet transfusion experiments, a t-test was performed for the prasugrel and ticagrelor groups without platelet transfusion using the vehicle group as a control, and was also performed between the groups with and without platelet transfusion in prasugrel- and ticagrelor-treated rats. In all analyses, statistical significance was defined as P < 0.05. IC50, ED50 and ED200 values were calculated from the regression line for dose–response relationship for inhibition of platelet aggregation, thrombus weight and bleeding time for each test article. SAS 8.2 and 9.2 for Windows (SAS Institute Inc., Cary, NC, USA) and EXSUS Ver. 7.1.6 and 7.7.1 (Arm Systex Co., Ltd., Osaka, Japan) or GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA, USA) were used to test significance and calculate ED50 or IC50 values.
Materials
Prasugrel hydrochloride and R-138727 were synthesized by Ube Industries, Ltd. (Yamaguchi, Japan). Ticagrelor was synthesized by Chemtech Labo, Inc. (Tokyo, Japan). AR-C124910XX was synthesized by Daiichi Sankyo RD Novare Co., Ltd. (Tokyo, Japan). Prasugrel and ticagrelor were suspended in 5% (w/v) solution of gum Arabic (Wako Pure Chemical Industries, Osaka, Japan). Prasugrel, ticagrelor and vehicle (5% gum Arabic solution) were orally administered to non-fasted rats in a volume of 1 mL·kg−1. The source of other reagents was as follows: ADP sodium salt and collagen (LMS Co., Ltd., Tokyo, Japan).
Results
ADP-induced platelet aggregation in vitro
In the vehicle group of each test agent, mean platelet aggregations ranged from 52 to 59% for 5 μmol·L−1 ADP and 67–74% for 20 μmol·L−1 ADP. R-138727 (prasugrel's active metabolite, 0.30–300 μmol·L−1), ticagrelor (0.030–30 μmol·L−1) and AR-C124910XX (ticagrelor's active metabolite, 0.030–30 μmol·L−1) inhibited ADP- (5 and 20 μmol·L−1) induced platelet aggregation in a concentration-related manner. The IC50 values (with 95% confidence intervals) are summarized in Table 1. In the present study, the in vitro effect of prasugrel was not tested because prasugrel is a prodrug and thus has no in vitro effect on platelet aggregation (Sugidachi et al., 2000).
Table 1.
IC50 values (95% confidence intervals) for each P2Y12 receptor antagonist for in vitro platelet aggregation in rat PRP, induced by ADP
| IC50 (μmol·L−1) with 95% confidence interval | ||
|---|---|---|
| Agents | ADP 5 μmol·L−1 | ADP 20 μmol·L−1 |
| R-138727 | 18 (16–20) | 24 (22–27) |
| Ticagrelor | 0.82 (0.75–0.91) | 1.5 (0.99–2.1) |
| AR-C124910XX | 0.34 (0.14–0.50) | 0.52 (0.39–0.67) |
Test agents were incubated with rat PRP for 30 min in vitro, and platelet aggregation induced by ADP was assessed. R-138727 is the active metabolite of prasugrel and AR-C124910XX is an active metabolite of ticagrelor.
Time course of ex vivo platelet aggregation induced by ADP
Ex vivo platelet aggregation was used to measure the effects of single oral doses of prasugrel and ticagrelor on platelet aggregation induced by 5 and 20 μmol·L−1 ADP and were determined in blood samples taken at 1, 2, 4, 8, 12 and 24 h after administration. For platelet aggregation induced by 20 μmol·L−1 ADP, single oral administration of prasugrel (0.3–3 mg·kg−1) caused dose-related inhibitory effects (Figure 1A). With 1 and 3 mg·kg−1 of prasugrel, significant inhibition was observed from 1 to 2 h after dosing. This inhibitory effect peaked at 4 h and lasted for 24 h after dosing, indicating a long duration of action by prasugrel. Single oral administration of ticagrelor (1–10 mg·kg−1) also caused dose-related inhibitory effect on platelet aggregation (Figure 1B). Ticagrelor, at the highest dose (10 mg·kg−1) significantly inhibited platelet aggregation at 1 h after dosing and the peak inhibition was observed at 4 h after dosing. The inhibitory effect on platelet aggregation lasted for 12 h after the dosing, but had returned to control values by 24 h, indicating a shorter duration of antiplatelet action than that of prasugrel. For platelet aggregation induced by 5 μmol·L−1 ADP, similar time courses of inhibition of platelet aggregation were observed for both prasugrel- and ticagrelor-treated rats (data not shown), although the level of inhibition of platelet aggregation was slightly greater compared with that observed with 20 μmol·L−1 ADP. The ED50 values for prasugrel and ticagrelor at 4 h after the dosing were 1.5 mg·kg−1 and 6.0 mg·kg−1, respectively, for 5 μmol·L−1 ADP and 1.9 mg·kg−1 and 8.0 mg·kg−1, respectively, for 20 μmol·L−1 ADP.
Figure 1.
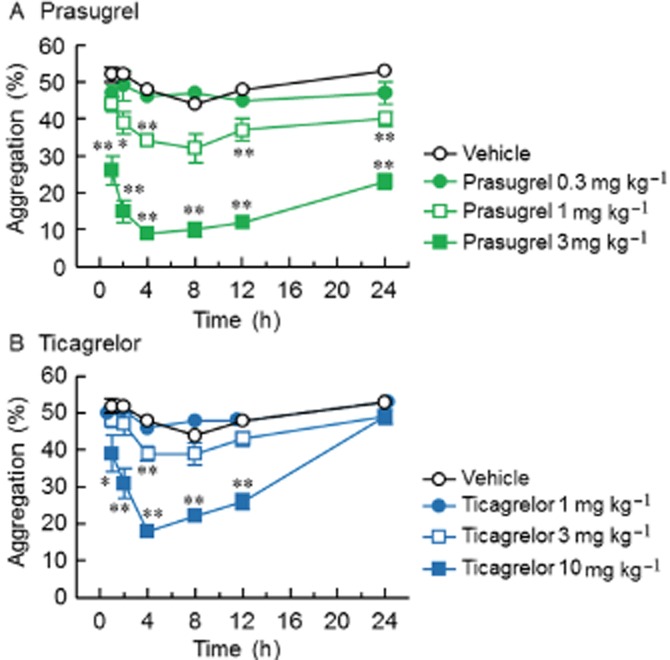
Ex vivo effects of single doses of prasugrel (A) or ticagrelor (B) on platelet aggregation induced by ADP in rats. Agents were orally administered to rats and blood was collected 1, 2, 4, 8, 12 and 24 h after dosing. Ex vivo platelet aggregation in PRP was induced by 20 μmol·L−1 ADP. Results are presented as the mean ± SEM (n = 5). *P < 0.05, **P < 0.01, significantly different from vehicle (Dunnett's test).
ADP concentration-response for ex vivo platelet aggregation
The inhibitory effects of prasugrel and ticagrelor on platelet aggregation induced by higher concentrations of ADP (50 and 200 μmol·L−1) were also determined at the time of peak effect, 4 h after dosing. Prasugrel (1 or 3 mg·kg−1) significantly inhibited platelet aggregation induced by ADP at all concentrations tested in a dose-related manner, and the effect was not reversed by increasing the concentration of ADP (Figure 2). Ticagrelor (3 or 10 mg·kg−1) also showed significant inhibition of platelet aggregation induced by ADP at all concentrations used, and, similarly, the effect of ticagrelor on ADP-induced aggregation appeared to be insurmountable, with no tested concentration of ADP completely overcoming the inhibition (Figure 2).
Figure 2.
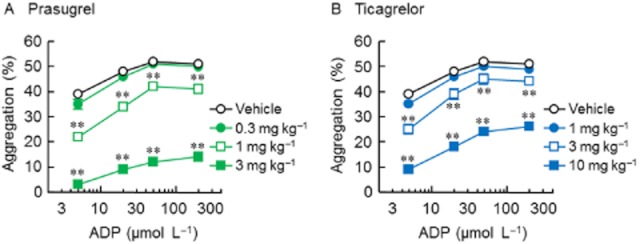
Concentration-response curve for ADP-induced platelet aggregation in prasugrel- (A) and ticagrelor-treated rats (B). Prasugrel and ticagrelor were orally administered to rats 4 h before blood collection. Ex vivo platelet aggregation in PRP was induced by 5, 20, 50 and 200 μmol·L−1 ADP. Results are presented as the mean ± SEM (n = 5). **P < 0.01, significantly different from vehicle group (Dunnett's test).
Collagen-induced ex vivo platelet aggregation
In addition to ADP-induced platelet aggregation, inhibitory effects of prasugrel and ticagrelor on collagen-induced platelet aggregation (5 μg·mL−1) were determined 4 h after dosing. Both prasugrel (0.3–3 mg·kg−1) and ticagrelor (1–10 mg·kg−1) inhibited collagen-induced platelet aggregation in a dose-related manner, with significant inhibitory effects being observed after 1 or 3 mg·kg−1 prasugrel and 3 or 10 mg·kg−1 ticagrelor (Figure 3).
Figure 3.
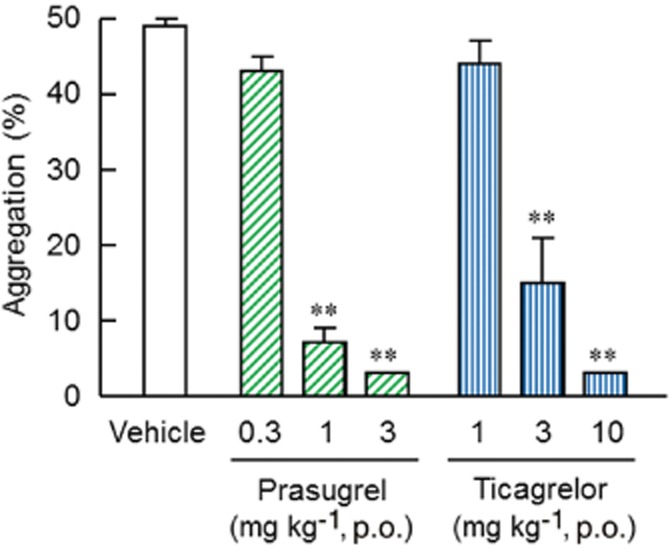
Ex vivo effects of prasugrel or ticagrelor on collagen-induced platelet aggregation in rats. Prasugrel and ticagrelor were orally administered to rats 4 h before blood collection. Ex vivo platelet aggregation in PRP was induced by 5 μg·mL−1 collagen. Results are presented as the mean + SEM (n = 5). **P < 0.01, significantly different from vehicle group (Dunnett's test).
AV shunt thrombosis
The anti-thrombotic effects of prasugrel and ticagrelor in vivo were assessed in the rat AV shunt thrombosis model. Prasugrel (0.3–3 mg·kg−1) and ticagrelor (1–10 mg·kg−1) were orally administered, and blood was allowed to circulate through the shunt 4 h after dosing. Thrombus weight in the vehicle-treated group was 45.9 ± 1.3 mg. Prasugrel significantly reduced thrombus weight at doses of 1 and 3 mg·kg−1 in a dose-related manner, compared with that in the vehicle-treated group with an ED50 value of 1.8 mg·kg−1. Ticagrelor also significantly reduced the thrombus weight at doses of 3 and 10 mg·kg−1 with an ED50 value of 7.7 mg·kg−1 (Figure 4A).
Figure 4.
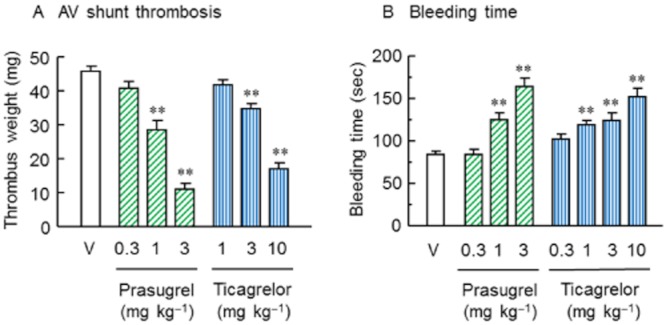
Effects of prasugrel or ticagrelor on AV shunt thrombosis (A) and bleeding time (B) in rats. Prasugrel and ticagrelor were orally administered to rats 4 h before circulation of blood through the AV shunt or bleeding time measurements. Results are presented as the mean + SEM (n = 10). V = vehicle control. **P < 0.01, significantly different from vehicle group (Dunnett's test).
Bleeding time
Effects of prasugrel (0.3–3 mg·kg−1) and ticagrelor (0.3–10 mg·kg−1) on the bleeding time in rats were determined 4 h after administration of vehicle, prasugrel or ticagrelor (Figure 4B). Prasugrel and ticagrelor each significantly prolonged bleeding time at 1 mg·kg−1 or higher doses, compared with the vehicle group (84 ± 4 s). Doses causing a twofold increase of the bleeding time in the vehicle group were calculated by linear regression analysis; the dose was estimated without logarithmic transformation of the bleeding time. These values (ED200) were 3.0 mg·kg−1 for prasugrel and 13 mg·kg−1 for ticagrelor.
Platelet transfusion study
Both prasugrel (10 mg·kg−1) and ticagrelor (30 mg·kg−1) significantly prolonged the bleeding time compared with vehicle-treated control groups (P < 0.001 and P < 0.01, respectively) (Figure 5). Platelet transfusion at 3 h after prasugrel or ticagrelor dosing resulted in similar significant increases (1.61- and 1.56-fold, respectively) in blood platelet numbers in prasugrel- and ticagrelor-treated rats (data not shown). In contrast, red blood cell numbers were not changed by platelet transfusion in either group (data not shown). In the prasugrel-treated group, platelet transfusion resulted in significant shortening of bleeding time (P < 0.05, Figure 5). In the ticagrelor-treated group, by contrast, platelet transfusion did not change bleeding time (P > 0.05, Figure 5). Thus, while the prolongation of bleeding time induced by high-dose of prasugrel could be significantly reversed by platelet transfusion, this was not the case for ticagrelor.
Figure 5.
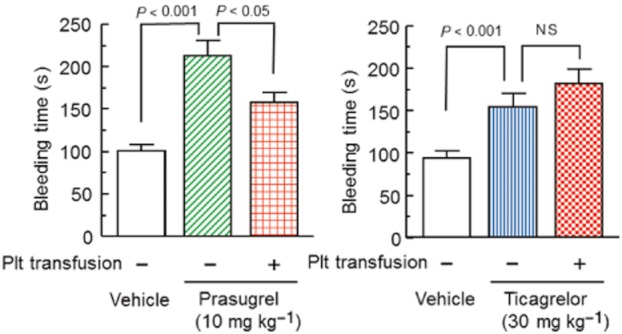
Effects of platelet transfusion on prolongation of bleeding time by prasugrel or ticagrelor in rats. Prasugrel and ticagrelor were orally administered 4 h before bleeding time measurements. Platelets (Plt) were infused i.v. 1 h before bleeding time measurements. Bleeding times are shown as the means + SEM (n = 13–14). Plt, platelets; NS, not significant (t-test).
Discussion and conclusions
Thienopyridines including prasugrel are antiplatelet prodrugs and their action is mediated by their active metabolites generated in vivo (Savi et al., 2000; Sugidachi et al., 2000). In contrast, ticagrelor itself has antiplatelet activity (Springthorpe et al., 2007) and Sillén et al. (2010) have reported recently that ticagrelor also has an in vivo active metabolite AR-C124910XX, with potency similar to that of ticagrelor (van Giezen and Humphries, 2005; Teng and Butler, 2010). To our knowledge, however, there is no detailed report describing the antiplatelet activity of ticagrelor's active metabolite. The present study is the first report showing antiplatelet activity of AR-C124910XX, which was 2.4- to 2.9 times more potent than its parent, ticagrelor, in rat PRP. In addition, our preliminary experiment showed similarly potent activity of AR-C124910XX in human PRP (data not shown). The pharmacokinetic profile of AR-C124910XX in rats is not known, but, in humans, the AUC0–∞ of AR-C124910XX is about half to one-fifth of ticagrelor's AUC0–∞ (Teng and Butler, 2010; Husted et al., 2012). Taken together, therefore, these results suggest that clinically, both ticagrelor and AR-C124910XX would have approximately equivalent effects on in vivo platelet inhibition following ticagrelor dosing.
In the present study, prasugrel's active metabolite (R-138727) inhibited in vitro platelet aggregation, and its in vitro antiplatelet activity was less potent than ticagrelor and AR-C124910XX. Nevertheless, as reported in clinical studies (Jernberg et al., 2006; Gurbel et al., 2010), on a mg·kg−1 basis, orally administered prasugrel was more potent than ticagrelor on ex vivo platelet aggregation induced by ADP. In addition, we found prasugrel's inhibition of ex vivo platelet aggregation was sustained for up to 24 h after the administration, while ticagrelor demonstrated a shorter duration of action and the antiplatelet effect was lost 24 h after dosing. Possible explanations for these differences between in vitro and ex vivo effects may be due to differences in pharmacokinetic profiles and their different irreversible (Sugidachi et al., 2000; 2001) and reversible antiplatelet actions (Wijeyeratne et al., 2012). Irreversible inhibition of P2Y12 receptors by prasugrel is also an explanation of the longer duration of antiplatelet action. In addition, a recent report showed that ticagrelor may have effects on adenosine transporters in human platelets, which may contribute to its antiplatelet action (Iyú et al., 2011). Thus, inhibition of adenosine transporters may contribute to the more potent in vitro activity of ticagrelor. However, another report suggested that any effect of ticagrelor on adenosine uptake was not enough to amplify the antiplatelet effects of any adenosine generated in the presence of P2Y12 receptor antagonists (van Giezen et al., 2012). Furthermore, the ADP concentration/response aggregation profiles of prasugrel and ticagrelor were compared at peak inhibition of platelet aggregation, and found to be similar with both agents, as previously reported (Sugidachi et al., 2000; van Giezen et al., 2009), acting as non-competitive antagonists.
Antithrombotic therapy is a cornerstone of treatment in patients with cardiovascular disease with bleeding being the most worrisome complication. Indeed, while prasugrel and ticagrelor provide greater inhibition of platelet aggregation and clinical efficacy than clopidogrel, both agents were associated with more bleeding compared with clopidogrel (Wiviott et al., 2007; Becker et al., 2011). In the present study, the relationship among antiplatelet, antithrombotic and bleeding activities of both prasugrel and ticagrelor were compared in rats. Both agents inhibited platelet aggregation, thrombus formation and haemostasis in a dose-related manner with a similar potency ratio of about four times among all the parameters tested at the time of peak inhibition of platelet aggregation (4h). These data suggest that at equivalent levels of platelet inhibition, the two agents would show similar antithrombotic activity with similar bleeding risk despite their distinct modes of actions at the P2Y12 receptor level, that is reversible (ticagrelor) or irreversible (prasugrel) antagonism.
There are several reversal strategies available for patients on antiplatelet therapy who present with an acute haemorrhage or require urgent surgery, including platelet transfusion (Lemmer, 2000; McMillian and Rogers, 2009), although some potentially deleterious effects have been observed (Bassand, 2009). Indeed, Vilahur et al. (2007) reported that in vitro platelet concentrates could restore haemostatic potential in the face of clopidogrel-induced platelet dysfunction. The present in vivo result showed that platelet transfusion significantly shortened prasugrel-induced prolongation of bleeding time in rats. In contrast, in ticagrelor-treated rats, platelet transfusion failed to reverse prolongation of bleeding time. This unexpected finding may depend on the different reversibility profile and/or different pharmacokinetic profile of the agent: It would appear that antiplatelet action of ticagrelor correlates with blood levels of ticagrelor and its active metabolite (Teng and Butler, 2010; Husted et al., 2012). Thus, newly transfused platelets would be readily inhibited by the presence of free ticagrelor and/or its active metabolite in plasma. In contrast, prasugrel is an irreversible antiplatelet agent with only transient exposure of platelets to its active metabolite needed for sustained platelet inhibition. At 4 h after the dosing, when maximum inhibition of platelet aggregation was observed in rats, the blood concentration of prasugrel's active metabolite is significantly lower than its peak (Cmax) level (Sugidachi et al., 2007; Hagihara et al., 2009), thereby allowing transfused platelets to remain functional and provide haemostatic potential. Furthermore, this pharmacokinetic / pharmacodynamic relationship in prasugrel-treated rats is similar to that observed in humans (Jakubowski et al., 2007).
In conclusion, both prasugrel and ticagrelor inhibited platelet aggregation and thrombus formation while prolonging bleeding with a similar potency ratio of approximately four times among these activities. In addition, prasugrel showed longer duration of antiplatelet action compared with ticagrelor. The present study also suggested that ticagrelor and its active metabolite might play equal roles in providing in vivo antiplatelet activity. Although the platelet inhibitory effect of prasugrel was reversed by platelet transfusion, that of ticagrelor was not. The disparity in findings between prasugrel- and ticagrelor-treated rats may reflect the different reversibility and/or pharmacokinetic profiles of the two agents.
Acknowledgments
We thank Ms Naomi Kasanuki, Yuki Yokouchi and Chiemi Koide for their expert technical assistance, and also thank NISSEI BILIS Co., Ltd. and Safety Research Institute for Chemical Compounds Co., Ltd. for their expert experiments.
Glossary
- ACS
acute coronary syndrome
- PCI
percutaneous coronary intervention
- PRP
platelet-rich plasma
Conflict of interest
J. A. Jakubowski is an employee of Eli Lilly and Company. Other authors are employees of Daiichi Sankyo Co., Ltd.
References
- Alber HF, Huber K, Pachinger O, Frick M. Prasugrel vs. ticagrelor in acute coronary syndromes: which one to choose? Wien Klin Wochenschr. 2011;123:468–476. doi: 10.1007/s00508-011-0027-7. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulos D, Galati A, Xanthopoulou I, Mavronasiou E, Kassimis G, Theodoropoulos KC, et al. Ticagrelor versus prasugrel in acute coronary syndrome patients with high on-clopidogrel platelet reactivity following percutaneous coronary intervention: a pharmacodynamic study. J Am Coll Cardiol. 2012;60:193–199. doi: 10.1016/j.jacc.2012.03.050. [DOI] [PubMed] [Google Scholar]
- Bassand JP. Acute Coronary Syndromes and Percutaneous Coronary Interventions: impact of bleeding and blood transfusion. Hämostaseologie. 2009;29:381–387. [PubMed] [Google Scholar]
- Becker RC, Bassand JP, Budaj A, Wojdyla DM, James SK, Cornel JH, et al. Bleeding complications with the P2Y12 receptor antagonists clopidogrel and ticagrelor in the PLATelet inhibition and patient Outcomes (PLATO) trial. Eur Heart J. 2011;32:2933–2944. doi: 10.1093/eurheartj/ehr422. [DOI] [PubMed] [Google Scholar]
- Biondi-Zoccai G, Lotrionte M, Agostoni P, Abbate A, Romagnoli E, Sangiorgi G, et al. Adjusted indirect comparison meta-analysis of prasugrel versus ticagrelor for patients with acute coronary syndromes. Int J Cardiol. 2011;150:325–331. doi: 10.1016/j.ijcard.2010.08.035. [DOI] [PubMed] [Google Scholar]
- Dobesh PP. Pharmacokinetics and pharmacodynamics of prasugrel, a thienopyridine P2Y12 inhibitor. Pharmacotherapy. 2009;29:1089–1102. doi: 10.1592/phco.29.9.1089. [DOI] [PubMed] [Google Scholar]
- van Giezen JJ, Humphries RG. Preclinical and clinical studies with selective reversible direct P2Y12 antagonists. Semin Thromb Hemost. 2005;31:195–204. doi: 10.1055/s-2005-869525. [DOI] [PubMed] [Google Scholar]
- van Giezen JJ, Nilsson L, Berntsson P, Wissing BM, Giordanetto F, Tomlinson W, et al. Ticagrelor binds to human P2Y12 independently from ADP but antagonizes ADP-induced receptor signaling and platelet aggregation. J Thromb Haemost. 2009;7:1556–1565. doi: 10.1111/j.1538-7836.2009.03527.x. [DOI] [PubMed] [Google Scholar]
- van Giezen JJ, Sidaway J, Glaves P, Kirk I, Björkman JA. Ticagrelor inhibits adenosine uptake in vitro and enhances adenosine-mediated hyperemia responses in a canine model. J Cardiovasc Pharmacol Ther. 2012;17:164–172. doi: 10.1177/1074248411410883. [DOI] [PubMed] [Google Scholar]
- Gurbel PA, Bliden KP, Butler K, Antonino MJ, Wei C, Teng R, et al. Response to ticagrelor in clopidogrel nonresponders and responders and effect of switching therapies: the RESPOND study. Circulation. 2010;121:1188–1199. doi: 10.1161/CIRCULATIONAHA.109.919456. [DOI] [PubMed] [Google Scholar]
- Hagihara K, Kazui M, Ikenaga H, Nanba T, Fusegawa K, Takahashi M, et al. Comparison of formation of thiolactones and active metabolites of prasugrel and clopidogrel in rats and dogs. Xenobiotica. 2009;39:218–226. doi: 10.1080/00498250802650077. [DOI] [PubMed] [Google Scholar]
- Husted SE, Storey RF, Bliden K, Tantry US, Høimark L, Butler K, et al. Pharmacokinetics and pharmacodynamics of ticagrelor in patients with stable coronary artery disease: results from the ONSET-OFFSET and RESPOND studies. Clin Pharmacokinet. 2012;51:397–409. doi: 10.2165/11599830-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Iyú D, Glenn JR, White AE, Fox SC, Heptinstall S. Adenosine derived from ADP can contribute to inhibition of platelet aggregation in the presence of a P2Y12 antagonist. Arterioscler Thromb Vasc Biol. 2011;31:416–422. doi: 10.1161/ATVBAHA.110.219501. [DOI] [PubMed] [Google Scholar]
- Jakubowski JA, Winters KJ, Naganuma H, Wallentin L. Prasugrel: a novel thienopyridine antiplatelet agent. A review of preclinical and clinical studies and the mechanistic basis for its distinct antiplatelet profile. Cardiovasc Drug Rev. 2007;25:357–374. doi: 10.1111/j.1527-3466.2007.00027.x. [DOI] [PubMed] [Google Scholar]
- Jernberg T, Payne CD, Winters KJ, Darstein C, Brandt JT, Jakubowski JA, et al. Prasugrel achieves greater inhibition of platelet aggregation and a lower rate of non-responders compared with clopidogrel in aspirin-treated patients with stable coronary artery disease. Eur Heart J. 2006;27:1166–1173. doi: 10.1093/eurheartj/ehi877. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG NC3Rs Reporting Guidelines Working Group. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmer JH., Jr Clinical experience in coronary bypass surgery for abciximab-treated patients. Ann Thorac Surg. 2000;70:S33–S37. doi: 10.1016/s0003-4975(00)01605-2. [DOI] [PubMed] [Google Scholar]
- Li YG, Ni L, Brandt JT, Small DS, Payne CD, Ernest CS, 2nd, et al. Inhibition of platelet aggregation with prasugrel and clopidogrel: an integrated analysis in 846 subjects. Platelets. 2009;20:316–327. doi: 10.1080/09537100903046317. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillian WD, Rogers FB. Management of prehospital antiplatelet and anticoagulant therapy in traumatic head injury: a review. J Trauma. 2009;66:942–950. doi: 10.1097/TA.0b013e3181978e7b. [DOI] [PubMed] [Google Scholar]
- Niitsu Y, Jakubowski JA, Sugidachi A, Asai F. Pharmacology of CS-747 (prasugrel, LY640315), a novel, potent antiplatelet agent with in vivo P2Y12 receptor antagonist activity. Semin Thromb Hemost. 2005;31:184–194. doi: 10.1055/s-2005-869524. [DOI] [PubMed] [Google Scholar]
- Savi P, Pereillo JM, Uzabiaga MF, Combalbert J, Picard C, Maffrand JP, et al. Identification and biological activity of the active metabolite of clopidogrel. Thromb Haemost. 2000;84:891–896. [PubMed] [Google Scholar]
- Sillén H, Cook M, Davis P. Determination of ticagrelor and two metabolites in plasma samples by liquid chromatography and mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:2299–2306. doi: 10.1016/j.jchromb.2010.06.018. [DOI] [PubMed] [Google Scholar]
- Springthorpe B, Bailey A, Barton P, Birkinshaw TN, Bonnert RV, Brown RC, et al. From ATP to AZD6140: the discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis. Bioorg Med Chem Lett. 2007;17:6013–6018. doi: 10.1016/j.bmcl.2007.07.057. [DOI] [PubMed] [Google Scholar]
- Sugidachi A, Asai F, Ogawa T, Inoue T, Koike H. The in vivo pharmacological profile of CS-747, a novel antiplatelet agent with platelet ADP receptor antagonist properties. Br J Pharmacol. 2000;129:1439–1446. doi: 10.1038/sj.bjp.0703237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugidachi A, Asai F, Yoneda K, Iwamura R, Ogawa T, Otsuguro K, et al. Antiplatelet action of R-99224, an active metabolite of a novel thienopyridine-type G(i)-linked P2T antagonist, CS-747. Br J Pharmacol. 2001;132:47–54. doi: 10.1038/sj.bjp.0703761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugidachi A, Ogawa T, Kurihara A, Hagihara K, Jakubowski JA, Hashimoto M, et al. The greater in vivo antiplatelet effects of prasugrel as compared to clopidogrel reflect more efficient generation of its active metabolite with similar antiplatelet activity to that of clopidogrel's active metabolite. J Thromb Haemost. 2007;5:1545–1551. doi: 10.1111/j.1538-7836.2007.02598.x. [DOI] [PubMed] [Google Scholar]
- Teng R, Butler K. Pharmacokinetics, pharmacodynamics, tolerability and safety of single ascending doses of ticagrelor, a reversibly binding oral P2Y12 receptor antagonist, in healthy subjects. Eur J Clin Pharmacol. 2010;66:487–496. doi: 10.1007/s00228-009-0778-5. [DOI] [PubMed] [Google Scholar]
- Vilahur G, Choi BG, Zafar MU, Viles-Gonzalez JF, Vorchheimer DA, Fuster V, et al. Normalization of platelet reactivity in clopidogrel-treated subjects. J Thromb Haemost. 2007;5:82–90. doi: 10.1111/j.1538-7836.2006.02245.x. [DOI] [PubMed] [Google Scholar]
- Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045–1057. doi: 10.1056/NEJMoa0904327. [DOI] [PubMed] [Google Scholar]
- Wijeyeratne YD, Joshi R, Heptinstall S. Ticagrelor: a P2Y12 antagonist for use in acute coronary syndromes. Expert Rev Clin Pharmacol. 2012;5:257–269. doi: 10.1586/ecp.12.17. [DOI] [PubMed] [Google Scholar]
- Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001–2015. doi: 10.1056/NEJMoa0706482. [DOI] [PubMed] [Google Scholar]