

Abstract
Background and Purpose
Malignant gliomas, the most common primary brain tumours, are highly invasive and neurologically destructive neoplasms with a very bad prognosis due to the difficulty in removing the mass completely by surgery and the limited activity of current therapeutic agents. PHA-848125 is a multi-kinase inhibitor with broad anti-tumour activity in pre-clinical studies and good tolerability in phase 1 studies, which could affect two main pathways involved in glioma pathogenesis, the G1-S phase progression control pathway through the inhibition of cyclin-dependent kinases and the signalling pathways mediated by tyrosine kinase growth factor receptors, such as tropomyosin receptors. For this reason, we tested PHA-848125 in glioma models.
Experimental Approach
PHA-848125 was tested on a panel of glioma cell lines in vitro to evaluate inhibition of proliferation and mechanism of action. In vivo efficacy was evaluated on two glioma models both as single agent and in combination with standard therapy.
Key Results
When tested on a subset of representative glioma cell lines, PHA-848125 blocked cell proliferation, DNA synthesis and inhibited both cell cycle and signal transduction markers. Relevantly, PHA-848125 was also able to induce cell death through autophagy in all cell lines. Good anti-tumour efficacy was observed by oral route in different glioma models both with s.c. and intracranial implantation. Indeed, we demonstrate that the drug is able to cross the blood–brain barrier. Moreover, the combination of PHA-848125 with temozolomide resulted in a synergistic effect, and a clear therapeutic gain was also observed with a triple treatment adding PHA-848125 to radiotherapy and temozolomide.
Conclusions and Implications
All the pre-clinical data obtained so far suggest that PHA-848125 may become a useful agent in chemotherapy regimens for glioma patients and support its evaluation in phase 2 trials for this indication.
Keywords: glioblastoma, CDK, TRKA, signal transduction, anti-tumour efficacy, animal models, combination therapy
Introduction
Malignant gliomas are the most common type of primary brain cancer, the most malignant being glioblastoma multiforme (GBM) (Kleihues et al., 2002). Approximately 10 000 GBMs are diagnosed each year in the United States (Reardon and Wen, 2006). GBMs are anaplastic, highly proliferating and invasive, neurologically destructive tumours with a very bad prognosis (median survival in the range of 12–15 months) due to the difficulty in removing them completely by surgery and the limited activity of therapeutic agents (Louis and Cavenee, 2005). Surgery followed by radiotherapy with concomitant adjuvant chemotherapy with temozolomide (TMZ) is the standard of care in patients with glioblastoma (Stupp et al., 2005). In 2009, the Food and Drug Administration granted bevacizumab (Avastin®, Genetech Roche, Basel, Switzerland) accelerated approval for the treatment of glioblastoma in progressed disease after standard therapy. Tumour response was observed in 26% of Avastin monotherapy patients, with a median duration response of 4.2 months: rates significantly higher than historical control data (Chamberlain, 2010), but still very disappointing.
Multiple genetic changes are involved in the development of GBMs (Zhu and Parada, 2002). Two main pathways aberrantly activated in GBM pathogenesis are the cyclin-dependent kinase (CDK) circuit and the signal transduction cascades. Overall, mutations in INK4A/CDK/RB pathway are detected in more than 80% of GBMs: homozygous deletions of p16INK4a and p18INK4c are found in more than 60%, loss of RB are identified in 14–33% and amplifications of individual cyclins and CDKs are detected in another 5–10% (Zhu and Parada, 2002; Rich and Bigner, 2004). Furthermore, the Ras-MAP kinase cascade is activated in nearly all GBMs, mainly through genetic alteration at the receptor level (Joensuu et al., 2005; Puputti et al., 2006), such as EGFR amplification, whereas AKT pathway is activated in approximately 70% of GBMs, mainly due to PTEN mutation or deletion (Ohgaki and Kleihues, 2007; Parsons et al., 2008). Both MAPK and AKT pathways are involved in tropomyosin receptor kinases (TRK) signalling activated by neurotrophin in gliomas (Assimakopoulou et al., 2007).
Given the complexity of glioblastoma development, the use of a compound able to block simultaneously two main pathways controlling its growth could represent a relevant approach for the treatment of this aggressive tumour.
PHA-848125 (N,1,4,4-tetramethyl-8-{[4-(4-methylpiperazin-1-yl)phenyl]amino}-4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline-3-carboxamide) is a potent inhibitor of CDKs and tyrosine kinases (Brasca et al., 2009; Albanese et al., 2010), with demonstrated anti-tumour activity in many pre-clinical tumour models and good tolerability in phase I (Weiss et al., 2012), currently in phase II clinical trials. Based on its selectivity profile affecting two main pathways involved in glioblastoma pathogenesis, we investigated the pre-clinical activity of PHA-848125 in brain tumours as a potential clinical indication for this drug.
Methods
Chemical
PHA-848125 was synthesized at Nerviano Medical Sciences, S.r.l., as previously reported (Brasca et al., 2009). TMZ was purchased from Sequoia Research Products Limited (Pangbourne, UK). [14C]-PHA-848125 AC (batch 18653/A/82, specific activity of 5 μCi·mg−1) was prepared at Nerviano Medical Sciences, Isotope Chemistry.
Cell culture
Human cancer cell lines were obtained either from the American Type Culture Collection (Manassas, VA, USA) or from NCI (Bethesda, MD, USA). Cells were maintained in the conditions suggested by the suppliers and routinely characterized using AmpFlSTR Identifiler PCR amplification kit (Applied Biosystems, Paisley, UK). Cells were also characterized, when needed, for mRNA expression by RT-PCR as previously described (Albanese et al., 2010). Proliferation after compound treatment was evaluated as previously reported (Albanese et al., 2010). Briefly, cells were seeded into 96 or 384 well plates at densities ranging from 10.000 to 30.000 cells·cm–2 in appropriate medium plus 10% FCS. After 24 h, cells were treated in duplicate with serial dilutions of PHA-848125, and 72 h later, viable cell number was assessed using the CellTiter-Glo Assay (Promega, San Luis Obispo, CA, USA). Concentrations inhibiting cell growth by 50% (IC50) were calculated using a Sigmoidal fitting algorithm (Assay Explorer MDL). Experiments were performed independently at least twice.
Cell-based mechanism assays
PHA-848125 mechanism of action was investigated using four different human glioma cells (SF539, SF268, U251 and U87MG) treated with the compound at several doses for different times depending on the assay. Cells were analysed either by Western blot or flow cytometry, evaluating signal transduction, DNA synthesis and autophagy.
Western blot analysis
The effect of compound treatment on signalling pathways was evaluated after 1 h of treatment. Cell extracts were prepared and SDS-PAGE was performed as previously described (Albanese et al., 2010). For immunoblotting, standard procedures were used and staining was carried out with the following antibodies against: P-p44/42 MAPK(Thr202/Tyr204), p44/42 MAPK, P-AKT(Thr308), P-AKT(Ser473), P-IGF-1Rβ(Tyr1131), P-Src(Tyr416), P-EGFR(Tyr 1068) and P-S6(Ser240/244) (Cell Signaling, Danvers, MA, USA), P-c-Yes(Tyr537) (BD Transduction Laboratories, Franklin Lakes, NJ, USA), pRb, cyclin B1 and cyclin A (Pharmingen-BD Biosciences, Franklin Lakes, NJ, USA), P-Rb(Thr 821) (Biosource, Paisley, UK), Cdc6 (NeoMarkers, Fremont, CA, USA), P-FGFR3(Tyr724), TrkA, P-TrkA(Tyr490), GAPDH (Santa Cruz Biotechnology, Dallas, TX, USA), ATG4A (Millipore, Billerica, MA, USA) and actin (Sigma, St Louis, MO, USA).
Analysis of bromodeoxyuridine incorporation
During the last 30 min of incubation with the compound (24 h), 33 μg·mL−1 bromodeoxyuridine (BrdU) (Sigma) was added to the culture medium and then the cells were washed and fixed in 70% ethanol overnight at −20°C. After denaturation with 2 N HCl for 20 min, the cells were stained with anti-BrdU monoclonal antibody (Becton Dickinson, Franklin Lakes, NJ, USA) according to the datasheet procedure and then analysed by flow cytometry (FACScalibur, Becton Dickinson).
Evaluation of autophagy
During the last 15 min of incubation with the compound (72 h), 1 μg·mL−1 acridine orange (Sigma) was added to the cells, which were then collected, washed with PBS and analysed by flow activated cell sorter (FACS) for their fluorescence emission as previously described (Kanzawa et al., 2004). In acridine orange-stained cells, the cytoplasm and nucleolus fluoresce bright green and dim red, while the acidic vesicular organelles formed during autophagy fluoresce bright red, so the intensity of red fluorescence is proportional to the volume and acidity degree of autophagosomic compartment (Louis and Cavenee, 2005).
Evaluation of apoptosis
For both the assays performed, after 24 h of treatment, the cells were harvested, washed, fixed in 1% formaldehyde in PBS on ice for 15 min, then rinsed, post-fixed in 70% cold ethanol and stored at –20°C. For TUNEL assay, samples were processed as reported in the datasheet of APO-BRDU kit (Becton Dickinson). For cleaved caspase-3 assay, fixed cells were permeabilized in 0.2% Triton X-100 in PBS containing 1% FCS for 30 min, incubated overnight at 4°C with anti-cleaved caspase-3 polyclonal Ab (Cell Signaling), labelled with FITC-conjugated goat anti-rabbit immunoglobulin (Jackson Laboratories, West Grove, PA, USA) for 1 h at room temperature (RT), counterstained with propidium iodide (5 μg·mL−1 + 12.5 μg·mL−1 DNase-free RNase) for 30 min at RT and analysed by FACS.
Animal studies
All procedures adopted for housing and handling of animals were in strict compliance with European and Italian Guidelines for Laboratory Animal Welfare. The animals were maintained in Nerviano Medical Sciences animal facilities, in cages using steam autoclaved (sterile) bedding, γ-radiated diet and acidified mineral water. Animals were identified by a uniquely numbered ear-tag, which appears on the datasheets. ARRIVE guidelines were followed for reporting these experiments (Kilkenny et al., 2010; McGrath et al., 2010).
Brain distribution evaluation by autoradioluminography
[14C]-PHA-848125 was administered by single oral administration (20 mg·kg−1, approximately 100 μCi·kg−1) to six male Sprague-Dawley albino rats (Charles River, Calco, Italy) killed 2, 6 and 24 h (two animals per time) after administration. The radioactivity distribution of compound-related material was evaluated using sections of 30 μm thick. One head per time was sectioned sagittally or transversally. Sections were placed in Fuji BAS 2040 exposure cassettes along with calibrated 14C-standard sets (ARC) and exposed to a phosphor imaging plate (IP BAS IIISR Fuji) for 168 h. Quantitative evaluation of the autoradioluminograms was performed using AIDA (version 2.43, Raytest, Straubenhardt, Germany) software.
Efficacy studies
Subcutaneous injection of human glioblastomas
U87MG or U251 cell lines were transplanted s.c. in BALB/c athymic nu/nu male mice (Harlan, Udine, Italy). Mice randomization, treatment start, tumour measurements and calculation of tumour growth inhibition (TGI) were conducted as previously described (Albanese et al., 2010). Briefly, dimensions of the tumours were measured regularly by caliper during the experiments and tumour masses were calculated as follows: Tumour weight (mg) = length (mm) × width2 (mm2)/ 2 × ρ, assuming density ρ = 1 mg·mm–3 for tumour tissue. TGI was determined according to the equation % TGI = 100 − (mean tumour weight of treated group/mean tumour weight of control group) × 100.
The effect of treatment was also expressed as the T-C value, defined as the difference in median times (in days) required for the treatment and control group tumours to reach the predetermined size of 1 g (Bissery et al., 1996). Tumour-free animals at 90 days after tumour implant were considered cured.
Both PHA-848125 and TMZ were dissolved in 5% glucose solution immediately before use and administered to 8–9 mice per group orally in a volume of 10 mL·kg−1 at doses and schedules indicated in figures. In the first combination experiment, different sequential schedules were tested: one dose of TMZ (25 mg·kg−1) was administered for 5 consecutive days, followed by a 7-day treatment with PHA-848125 at two doses (20 and 40 mg·kg−1) or the 7-day treatment with PHA-848125 at two doses (20 and 40 mg·kg−1) was followed by 5-day treatment with TMZ (25 mg·kg−1). In the second experiment, treatments were contemporaneous: a single radiotherapy treatment (4 Gy on day 3) was administered with PHA-848125 [20 mg·kg−1 twice a day (BID), days 1–10] or TMZ (16 mg·kg−1, days 1–5) or the combination of the two compounds.
Ex vivo autophagy evaluation
Five days after the last treatment with 40 mg·kg−1 PHA-848125 BID for 10 consecutive days, U87MG tumours from control and treated animals were excised and divided into two pieces; one placed in liquid nitrogen for subsequent Western blot analysis and the other formalin fixed and paraffin embedded for immunohistochemistry (IHC). Western blot was performed as above-described loading 100μg protein per lane. IHC was carried out following standard procedures (Albanese et al., 2010). With both techniques, the expression of ATG4A was revealed using anti-ATG4A, clone EPR4122, rabbit monoclonal antibody (Millipore).
Intracranial injection
U251 cells (2 × 105 in 5 μL or 1 × 105 in 2 μL of PBS) were implanted in the caudate nuclei region of BALB/c athymic nu/nu mice (Harlan) by the stereotactic apparatus as previously described (Radaelli et al., 2009). Avertin (100 mg·kg–1, i.p.) was used as anaesthetic. Mice were monitored daily for clinical signs of disease and deaths were recorded for calculation of the median survival time (MST).
Statistical analysis
The Mann–Whitney U-test was used to test differences in tumour weight, and the log-rank (Mantel–Cox) test was used to test differences in survival between treatment groups. These analyses were carried out using GraphPad Prism software (GraphPad Software, Inc., San Diego, CA, USA). A P-value <0.05 was considered statistically significant.
MRI
MRI was performed at different time points to follow tumour growth in control (n = 10) and treated mice (n = 8). Treatment (PHA-848125 40 mg·kg–1 OS BID for 20 days) started 15 days after cell implantation. A Bruker Pharmascan instrument (Ettlingen, Germany) operating at 7.0 T was used. Anaesthetized animals (isoflurane 2–3% with air 0.5 L·min−1) were positioned prone in the animal bed and inserted in the radiofrequency coil (38 mm i.d.) inside the magnet. A pneumatic sensor was positioned underneath the mouse for respiratory monitoring. Low resolution axial and sagittal images were acquired as pilot scans for correct positioning of brain region. A fast T2-weighted sequence (Bruker RARE) was used in both axial and coronal orientations to better delineate tumours after 3 μM treatment. Eight or ten adjacent 1-mm-thick slices were acquired all across the area to cover the mass. The whole acquisition, including induction of anaesthesia, positioning and setup, took around 20 min per animal. A macro (VolumeCalc, Bruker) was used to calculate tumour volumes (in mm3) from the tumour areas in each slice and the slice thickness.
Results
Effect on glioma cell lines
In order to characterize the effect of PHA-848125 on glioma cells, four representative cell lines were selected: SF539, Rb null, and U251, U87MG, SF268 with p16 deletion. All cell lines present signalling over-activation: SF268 with oncogenic EGFR variant (A289V or EGFRvIII), others with PTEN deletion and all express TRK-family members (Singer et al., 1999) (Table 1).
Table 1.
Effect of PHA-848125 on cell proliferation and autophagy or apoptosis induction
| Cell line | Mutations | Cell proliferation IC50 (μM) | BrdU incorporation inhibition (%)a | Autophagic cellsb (%)a | Cleaved caspase-3 positive cellsb (%)a | TUNEL positive cellsb (%)a |
|---|---|---|---|---|---|---|
| SF268 | EGFRvIII; p53; P16INK4a | 2.5 ± 0.6 | 78 | 70 | nt | nt |
| SF539 | PTEN; RB1 | 1.4 ± 0.5 | 39 | 35 | 8.7 | 10.3 |
| U251 | PTEN; p53; P16INK4a | 2.1 ± 0.8 | 74 | 58 | 0 | 0 |
| U87MG | PTEN; P16INK4a | 1.6 ± 0.8 | 92 | 52 | 0 | 0 |
Net values (subtracted of the value of control cells), obtained with a 3 μM treatment for 24 h.
Positive control were treated with 100 μM temozolomide for autophagy (positive cells: 19–27%) and with 1 μM camptothecin for apoptosis (positive cells: 23–86% for activated caspase and 30–87% for TUNEL).
PHA-848125 inhibited cell proliferation of all tested cell lines (IC50 = 1.5–2.5 μM) (Table 1). After 24-h treatment, a reduction of DNA synthesis (39–92%) was measured in all cell lines. As expected for a CDK inhibitor, inhibition of Rb phosphorylation was observed in the three cell lines expressing Rb and a decreased expression of cdc6, cyclin A and cyclin B was observed in all cell lines after 24-h treatment at 1 μM (Figure 1). TRKA activation could be evaluated only in SF539 cells because they had the highest TRKA level, as shown in Figure 2A, which reports TRKA (ntrk1) mRNA expression evaluated by RT-PCR. In this cell line, a strong inhibition of TRKA, MAPK and AKT activation was observed after 1-h treatment at 3 μM (Figure 2B). A clear reduction of MAPK, AKT and c-YES activation was also evident in the other three cell lines (Figure 3), together with inhibition of EGFR phosphorylation in SF-268 cells and inhibition of the phosphorylation of both IGF-1R and FGFR3 in U251 and U87-MG cells. None of these receptors was found inhibited in SF539 cells, as well as c-Yes. No inhibition of P-Src was observed in any cell line. As we already reported that PHA-848125 is poorly or not active on the kinase activity of MAPK (IC50 > 10 μM), AKT (IC50 > 10 μM) and tyrosine kinase receptors such as EGFR (IC50 = 3.069 μM), FGFR (IC50 = 1.207 μM) and IGF1R (IC50 > 10 μM) (Albanese et al., 2010), we consider that the observed reduced activation of these pathways could be a consequence of a direct inhibition of c-Yes. Therefore, to confirm this hypothesis as an explanation of these unexpected data, compound activity was tested in a bioassay against c-Yes. A strong inhibitory activity was observed, being 4 and 0% the residual kinase activity in the presence of the compound at 100 and 1000 nM respectively (Millipore KinaseProfiler™).
Figure 1.
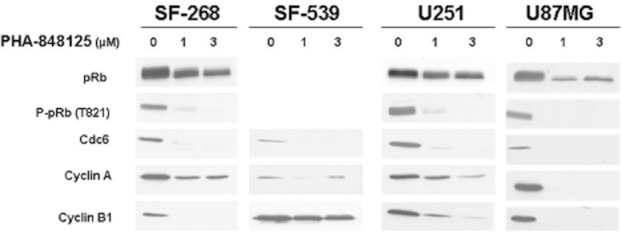
Effect of PHA-848125 on signal transduction markers of CDK/pRb pathway. Four different glioma cell lines were treated with scalar concentration of PHA-848125 for 24 h. Total cell lysates were immunoblotted with specific antibodies.
Figure 2.
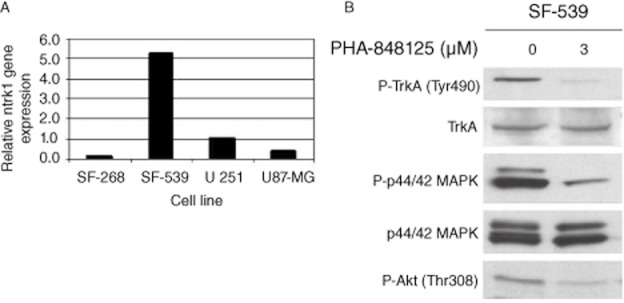
Evaluation of TRK expression and its modulation by PHA-848125. (A) Ntrk1 gene expression was evaluated in mRNA of four different glioma cell lines. SF-539 showed the highest level of expression. (B) SF-539 cells were treated with 3 μM PHA-848125 for 1 h. Total cell lysates were immunoblotted with specific antibodies.
Figure 3.
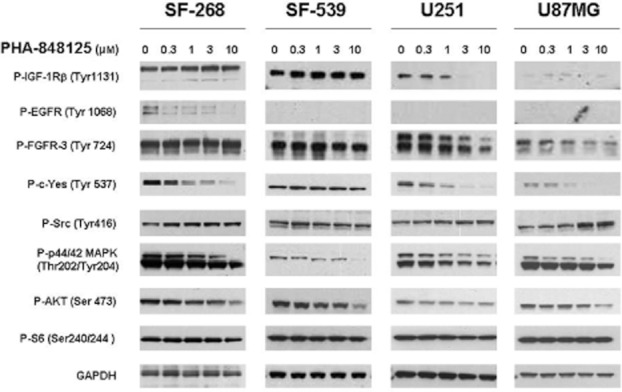
Effect of PHA-848125 on signal transduction markers of tyrosine receptor pathways. Four different glioma cell lines were treated with scalar concentration of PHA-848125 for 1 h. Total cell lysates were immunoblotted with specific antibodies.
Finally, the compound resulted more potent than TMZ in inducing cell death through autophagy in all cell lines, whereas apoptosis was activated only in the Rb null cell line SF539 (Table 1).
Distribution of PHA-848125 in rat brain post-oral administration
Following a single oral administration of [14C]-PHA-848125, total radioactivity levels in the brain were measured and found to be higher than the plasma levels at all time points tested (Figure 4A). The calculated brain concentrations indicated that the compound is present in the brain at levels higher than the concentration active in proliferation assay (mean IC50 = 1.9 μM; Table 1) at least up to 12 h. As shown in the autoradiograms of Figure 4B, where the different brain areas are indicated, the compound distribution was homogeneous, with no specific site accumulation. Therefore, PHA-848125 was able to pass the blood–brain barrier and could reach brain tumours at efficacious dosage.
Figure 4.
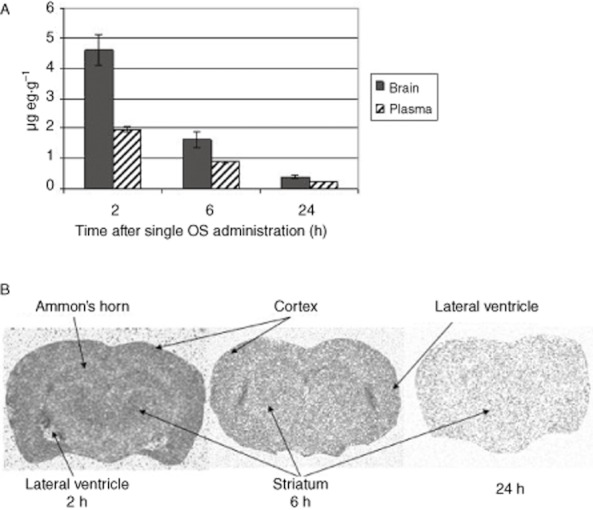
Brain distribution of PHA-848125. [14C]-PHA-848125 was administered at 20 mg·kg−1 by single oral administration, rats were killed 2, 6 and 24 h (two animals per time-point) after administration. (A) Comparison of brain and plasma compound concentrations. (B) Transversal section autoradiograms.
Single agent efficacy on glioma models
The in vivo efficacy of PHA-848125 was tested on two s.c. glioma models at the best schedule [40 mg·kg−1 BID for 10 consecutive days (Albanese et al., 2010)]. Tumour stabilization or regression with a maximal TGI of 80% (P = 0.0009 on day 24) and 71% (P = 0.0002 on day 18) was observed in U251 (Table 2; Figure 5A) and U87MG model (Table 2; Figure 5B) respectively. In both cases, only a marginal body weight loss was reported (≤10%). After being killed, U87MG tumours from control and treated animals were analysed either by Western blot (Figure 5C) and IHC (Figure 5D) and a strong (>10×) increase in autophagy, evaluated as expression of ATG4A (a cysteine protease required for autophagy), was observed upon PHA-848125 treatment. In the corresponding H&E stained tumours, no morphological sign of apoptosis was observed (Figure 5D), whereas the mitotic index was inhibited by 90% in the treated samples. Western blot analysis evidenced also a clear inhibition of AKT activation induced by the compound (Figure 5C). Anti-tumour efficacy of PHA-848125 was also tested in intracranially (i.c.) implanted U251 model (Table 2). In a first experiment, treatment started the day after the inoculum of tumoural cells (not established model) and lasted for 10 days. Treated animals presented a statistically significant increase in MST (28 days vs. 22 days of the control mice, P-value <0.0001) (Table 2; Figure 6A). In a second experiment, a lower cell number implantation permitted to assess the efficacy of the compound starting the treatment with an established tumour mass (day 15) and prolonging it for 20 days: with this scheduling, the MST was 42 days for treated versus 33 days for the control mice (Table 2; Figure 6B). Also, in this case, the increase in survival was statistically significant (P-value = 0.005). MR images (Figure 6C) clearly showed presence of tumour in the right hemisphere. Viable tumour appeared surrounded by an oedematous hyper-intense region, and hyper-intense internal necrosis was also clearly distinguishable, these being typical features of GBM growth in humans (Radaelli et al., 2009). A TGI of 71% was obtained at the end of treatment (P = 0.0061) (Table 2).
Table 2.
Activity of PHA-848125 as single agent on different human glioma models
| Model | Inoculum | Schedule | No. of mice per group | Treatment start after inoculum | Average tumour weight at start (g) | TGI (measured the day after end of treatment) | MST treated (MST control) |
|---|---|---|---|---|---|---|---|
| U251 | s.c. | 40 mg·kg−1 | 8 | Day 14 | 0.163 | 80% (day 24) | n.a. |
| 1–10 OS bid | |||||||
| U87MG | s.c. | 40 mg·kg−1 | 8 | Day 8 | 0.224 | 71% (day 18) | n.a. |
| 1–10 OS bid | |||||||
| U251 | i.c. 2 × 105 cells per mouse | 40 mg·kg−1 | 9 | Day 1 | 0 | – | 28 (22) |
| 1–10 OS bid | |||||||
| U251 | i.c. 1 × 105 cells per mouse | 40 mg·kg−1 | 9 | Day 15 | 0.003 | 71% (day 35) | 42 (33) |
| 1–20 OS bid |
n.a., not applicable.
Figure 5.

In vivo anti-tumour efficacy in s.c. implanted models. Animals were treated with vehicle or with PHA-848125 at 40 mg·kg−1 BID for 10 consecutive days. (A) Tumour growth curves of U251 glioma model, treatment started on day 14; (B) tumour growth curves of U87MG glioma model, treatment started on day 8. For ex vivo analysis, 5 days after the last treatment, U87MG tumours from four control and four treated animals were excised. (C) Evaluation of ATG4A and P-AKT expression by Western blot analysis; (D) evaluation of ATG4A expression by immunohistochemistry. Images of the same tumours stained with H&E are reported. Morphological analysis allowed counting mitotic figures (indicated by arrows): CTR > 100/field 200×; PHA 848125 < 10/field 200×.
Figure 6.
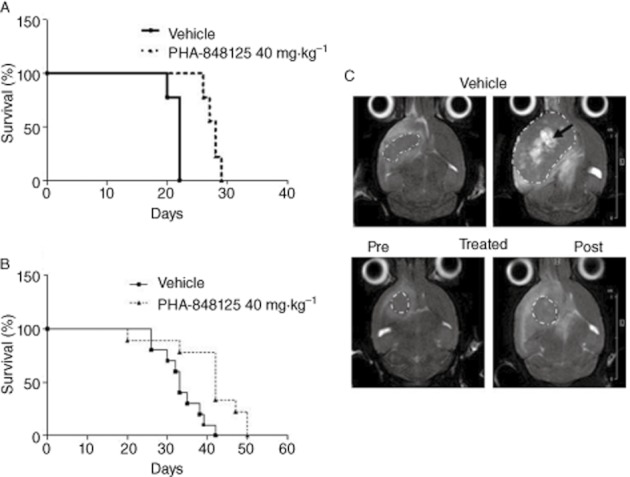
In vivo anti-tumour efficacy in intracranially (i.c.) implanted tumour. (A) Survival curves of mice with U251 cells i.c. implanted, treatment for 10 consecutive days starting from the day after cell injection; (B) survival curves of mice with U251 cells i.c. implanted, treatment for 20 consecutive days starting from day 15 after cell injection; (C) MR images of one representative control and one treated mouse pre-treatment (day 15) and the day after the last treatment (day 35). Tumours (encircled by white dotted lines) are surrounded by a hyper-intense oedematous area; big and fast growing tumours also present necrotic areas (black arrow). Significantly smaller tumours (P = 0.0061) are found in the treated cohort (0.056 ± 0.023 cm3 vs. 0.195 ± 0.05 cm3 in the vehicle group).
Efficacy in combination with standard therapy for GBM
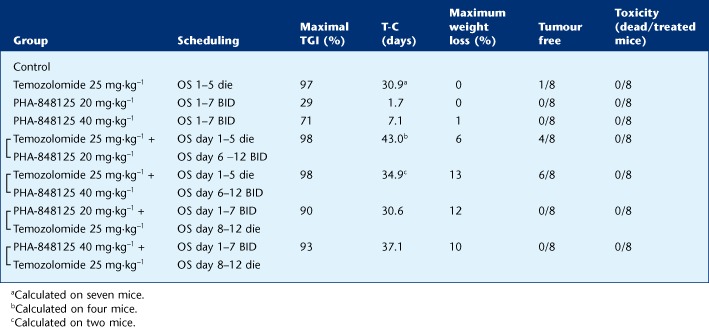
We tested PHA-848125 in combination with TMZ directly in vivo in the U87MG xenograft model (Table 3) because the in vitro IC50 values of TMZ were very high in all cell lines (>100 μM with 72-h treatment) and this did not permit an accurate evaluation of the combinatory effect. Sequential treatments of the two compounds were tested and the number of tumour-free animals clearly showed that a synergistic effect is obtained with the administration of TMZ before PHA-848125: indeed 4/8 and 6/8 tumour-free mice were observed in these combination groups treated with PHA-848125 at 20 and 40 mg·kg−1, respectively, whereas 0/8 and 1/8 tumour-free animals were obtained using PHA-848125 or TMZ, respectively, as single agent and no tumour-free mice were reported in the groups treated with PHA-848125 followed by TMZ. No toxicity was observed and only marginal body weight loss (<20%) was reported in all combination groups.
Table 3.
Activity of PHA-848125 in combination with temozolomide on U87MG human glioblastoma
 |
To evaluate the effect of the addition of a third treatment, radiotherapy, we decided to apply a contemporaneous schedule. In fact, in a second experiment on the same model, the administration of PHA-848125 was concomitant with TMZ and a single radiotherapy treatment was also added. Statistical analysis of the tumour weight on day 35 (Figure 7) confirmed the improvement obtained with all combination groups in comparison with groups treated with the corresponding single agents and showed that addition of PHA-848125 to radiotherapy and TMZ treatments resulted in a clear therapeutic gain (P = 0.037).
Figure 7.
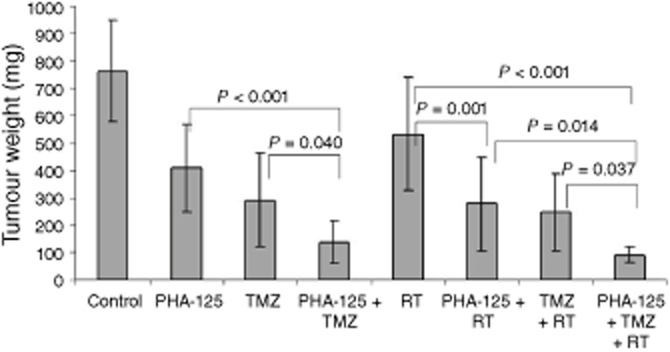
Efficacy of combination therapies. Mice bearing U87MG human glioma xenograft model were treated with PHA-848125 (20 mg·kg−1 bid days 1–10) or temozolomide (16 mg·kg−1 days 1–5) or a single radiotherapy treatment (4 Gy on day 3) or with all the possible combinations. Animals were killed on day 35 and tumour weight was evaluated. Statistical analysis was performed, comparing the different groups and the resulting P-values are reported.
Discussion and conclusions
Malignant gliomas are aggressive tumours, with an extremely unfavourable prognosis. They are heterogeneous tumours in terms of pathology and gene expression, but despite the variability, they display common alterations in specific cellular signal transduction pathways or cellular functions that are concurrently altered, such as p53, pRB and growth factor receptor pathways (Rich and Bigner, 2004). Recent genomic surveys have confirmed these core pathways as the most relevantly mutated and identified also previously unrecognized alterations such as NF1 loss and IDH1 mutation (Parsons et al., 2008; The Cancer Genome Atlas Research Network, 2008).
Currently available treatment options are poorly active and limited to agents that specifically kill cells or block angiogenesis. None of the new targeted compound has demonstrated efficacy in these patients so far. Despite the compelling biological plausibility, the EGFR inhibitors erlotinib and gefitinib and the mTOR complex inhibitor rapamycin, which target key proteins within mutated core pathways, failed to demonstrate clinical benefit for GBM patients, resulting in only a few clinical responses of very short duration (Haas-Kogan et al., 2005; Mellinghoff et al., 2005; Cloughesy et al., 2008). These disappointing results could be justified by the high frequency of PTEN loss in these tumors that can cause resistance (Guillamo et al., 2009), but could also suggest the necessity of simultaneously blocking more than one pathway involved in glioma pathogenesis (Huse and Holland, 2010) to obtain some therapeutic success.
Whereas pharmacological inhibition of CDK4 and 6 has been reported to be sufficient for the growth arrest of Rb-proficient glioma models (Michaud et al., 2010), we believe that the concomitant inhibition of CDK and tyrosine kinase signalling by a compound such as PHA-848125, active on both families, could result in a more efficacious effect. Indeed, PHA-848125 is able to block glioma proliferation not only in cells with functional RB but also in RB null ones and affects MAPK and AKT cell signalling, also displaying a strong inhibition of c-YES, which is known to mediate the activation of PI3K in response to extracellular signals in glioblastoma models (Kleber et al., 2008). The precocity of this cascade inhibition demonstrates that the compound acts directly on receptor signal transduction. Therefore, PHA-848125 can hit glioma cells in two of the pathways aberrantly activated during their development, blocking both cell cycle progression and cell signalling. This dual inhibition causes not only a block in cell proliferation but also induction of cell death. The compound induces apoptosis only in the cell line not expressing RB; however, in all cell lines, it causes autophagy, which is the programmed cell death pathway preferentially activated by these cells in response to stress agents (Kanzawa et al., 2004; Lefranc and Kiss, 2006) and to cytotoxic drug such as TMZ (Lefranc et al., 2007). In this subset of cell lines, PHA-848125 was more potent than TMZ in promoting autophagy.
The feature that renders the compound particularly attractive for this pathology is its ability to cross the blood–brain barrier, with active concentration levels maintained for a long period, demonstrated by a distribution assay with the radiolabelled compound in healthy rats where the blood–brain barrier is intact. The maximal drug concentration reached in brain with PHA-848125 is higher than the Cmax reported for TMZ in brain and tumour of rats bearing a human glioma model (Zhou et al., 2012). As a consequence, we could observe a good anti-tumour efficacy upon compound treatment not only in glioma xenograft models subcutaneously implanted but also in the i.c. implanted model. Although a complete response was not obtained, tumour regression was observed during the treatment in the U251, the model that better recapitulates the most salient pathobiological features reported for human GBM (Radaelli et al., 2009).
Combination experiments with currently applied front-line therapies showed that the combination with TMZ produced an evident synergism, resulting in a very high number of tumour-free animals at the end of the experiment. Moreover, we demonstrated that the addition of PHA-848125 to the standard therapy, which includes also radiotherapy, produced a further improvement in terms of anti-tumour efficacy without increased toxicity.
In conclusion, all the pre-clinical data, particularly the results obtained in vivo in combination with standard therapy, indicate that PHA-848125 may become a useful agent in chemotherapy regimens for glioma patients and support its evaluation for this indication.
Acknowledgments
We thank L. Bassignani, F. Caprera, A. Ferrario and G. Stortini for their contribution to the experimental activities in animal models, particularly for conducting the intracranial xenograft therapy response experiments, and we are indebted to A. Ghiglieri for determination of radioactivity distribution in the brain.
Glossary
- Ab
antibody
- BID
twice a day
- BrdU
bromodeoxyuridine
- FACS
flow activated cell sorter
- GBM
glioblastoma multiforme
- H&E
haematoxylin & eosin
- i.c
intracranially
- IHC
immunohistochechemistry
- MST
median survival time
- OS
orally
- RT
radiotherapy
- T-C
tumour growth delay (treated – control)
- TGI
tumour growth inhibition
- TMZ
temozolomide
Conflict of interest
None declared.
References
- Albanese C, Alzani R, Amboldi N, Avanzi N, Ballinari D, Brasca MG, et al. Dual targeting of CDK and tropomyosin receptor kinase families by the oral inhibitor PHA-848125, an agent with broad-spectrum antitumor efficacy. Mol Cancer Ther. 2010;9:2243–2254. doi: 10.1158/1535-7163.MCT-10-0190. [DOI] [PubMed] [Google Scholar]
- Assimakopoulou M, Kondyli M, Gatzounis G, Maraziotis T, Varakis J. Neurotrophin receptors expression and JNK pathway activation in human astrocytomas. BMC Cancer. 2007;7:202. doi: 10.1186/1471-2407-7-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissery MC, Vrignaud P, Lavelle F, Chabot GG. Experimental antitumor activity and pharmacokinetics of the camptothecin analog irinotecan (CPT-11) in mice. Anticancer Drugs. 1996;7:437–460. doi: 10.1097/00001813-199606000-00010. [DOI] [PubMed] [Google Scholar]
- Brasca MG, Amboldi N, Ballinari D, Cameron A, Casale E, Cervi G, et al. Identification of N,1,4,4-tetramethyl-8-{[4-(4-methylpiperazin-1-yl)phenyl]amino}-4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline-3-carboxamide (PHA-848125), a potent, orally available cyclin dependent kinase inhibitor. J Med Chem. 2009;52:5152–5163. doi: 10.1021/jm9006559. [DOI] [PubMed] [Google Scholar]
- Chamberlain MC. Emerging clinical principles on the use of bevacizumab for the treatment of malignant gliomas. Cancer. 2010;116:3988–3999. doi: 10.1002/cncr.25256. [DOI] [PubMed] [Google Scholar]
- Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, et al. Antitumor activity of rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastoma. PloS Med. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. 0139–0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillamo JS, de Boüard S, Valable S, Marteau L, Leuraud P, Marie Y, et al. Molecular mechanisms underlying effects of epidermal growth factor receptor inhibition on invasion, proliferation, and angiogenesis in experimental glioma. Clin Cancer Res. 2009;15:3697–3704. doi: 10.1158/1078-0432.CCR-08-2042. [DOI] [PubMed] [Google Scholar]
- Haas-Kogan DA, Prados MD, Tihan T, Eberhard, DA, Jelluma, N, Arvold, ND, et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97:880–887. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer. 2010;10:319–331. doi: 10.1038/nrc2818. [DOI] [PubMed] [Google Scholar]
- Joensuu H, Puputti M, Sihto H, Tynninen O, Nupponen NN. Amplification of genes encoding KIT, PDGFR alpha and VEGFR2 receptor tyrosine kinases is frequent in glioblastoma multiforme. J Pathol. 2005;207:224–231. doi: 10.1002/path.1823. [DOI] [PubMed] [Google Scholar]
- Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11:448–457. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleber S, Sancho-Martinez I, Wiestler B, Beisel A, Gieffers C, Hill O, et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell. 2008;13:235–248. doi: 10.1016/j.ccr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61:215–225. doi: 10.1093/jnen/61.3.215. [DOI] [PubMed] [Google Scholar]
- Lefranc F, Kiss R. Autophagy, the Trojan horse to combat glioblastomas. Neurosurg Focus. 2006;20:E7. doi: 10.3171/foc.2006.20.4.4. 1-6. [DOI] [PubMed] [Google Scholar]
- Lefranc F, Facchini V, Kiss R. Proautophagic drugs: a novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist. 2007;12:1395–1403. doi: 10.1634/theoncologist.12-12-1395. [DOI] [PubMed] [Google Scholar]
- Louis DN, Cavenee WK. Neoplasms of the central nervous system. In: DeVita VT, Hellman S, Rosenberg SA, editors. Cancer: Principles & Practice of Oncology. 7th edn. Philadelphia, PA: Lippincott Williams & Wilkins; 2005. pp. 1827–1834. [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD, et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010;70:3228–3238. doi: 10.1158/0008-5472.CAN-09-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puputti M, Tynninen O, Sihto H, Blom T, Mäenpää H, Isola J, et al. Amplification of KIT, PDGFRA, VEGFR2, and EGFR in gliomas. Mol Cancer Res. 2006;4:927–934. doi: 10.1158/1541-7786.MCR-06-0085. [DOI] [PubMed] [Google Scholar]
- Radaelli E, Ceruti R, Patton V, Russo M, Degrassi A, Croci V, et al. Immunohistopathological and neuroimaging characterization of murine orthotopic xenograft models of glioblastoma multiforme recapitulating the most salient features of human disease. Histol Histopathol. 2009;24:879–891. doi: 10.14670/HH-24.879. [DOI] [PubMed] [Google Scholar]
- Reardon DA, Wen PY. Therapeutic advances in the treatment of glioblastoma: rationale and potential role of targeted agents. Oncologist. 2006;11:152–164. doi: 10.1634/theoncologist.11-2-152. [DOI] [PubMed] [Google Scholar]
- Rich JN, Bigner DD. Development of novel targeted therapies in the treatment of malignant glioma. Nat Rev Drug Discov. 2004;3:430–446. doi: 10.1038/nrd1380. [DOI] [PubMed] [Google Scholar]
- Singer HS, Hansen B, Martinie D, Karp CL. Mitogenesis in glioma multiforme cell lines: a role for NGF and its TrkA receptors. J Neurooncol. 1999;45:1–8. doi: 10.1023/a:1006323523437. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, Van Den Bent M, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss GJ, Hidalgo M, Borad MJ, Laheru D, Tibes R, Ramanathan RK, et al. Phase I study of the safety, tolerability and pharmacokinetics of PHA-848125AC, a dual tropomyosin receptor kinase A and cyclin-dependent kinase inhibitor, in patients with advanced solid malignancies. Invest New Drugs. 2012;30:2334–2343. doi: 10.1007/s10637-011-9774-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Guo P, Kruh GD, Vicini P, Wang X, Gallo JM. Predicting human tumor drug concentrations from a preclinical pharmacokinetic model of temozolomide brain disposition. Clin Cancer Res. 2007;13:4271–4279. doi: 10.1158/1078-0432.CCR-07-0658. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Parada LF. The molecular and genetic basis of neurological tumours. Nat Rev Cancer. 2002;2:616–626. doi: 10.1038/nrc866. [DOI] [PubMed] [Google Scholar]