

Abstract
Recent genetic screens of fly mutants and molecular analysis have revealed that the Hippo (Hpo) pathway controls both cell proliferation and cell death. Deregulation of its human counterpart (the MST pathway) has been implicated in human cancers. However, how this pathway is linked with the known tumor suppressor network remains to be established. RUNX3 functions as a tumor suppressor of gastric cancer, lung cancer, bladder cancer, and colon cancer. Here, we show that RUNX3 is a principal and evolutionarily conserved component of the MST pathway. SAV1/WW45 facilitates the close association between MST2 and RUNX3. MST2, in turn, stimulates the SAV1–RUNX3 interaction. In addition, we show that siRNA-mediated RUNX3 knockdown abolishes MST/Hpo-mediated cell death. By establishing that RUNX3 is an endpoint effector of the MST pathway and that RUNX3 is capable of inducing cell death in cooperation with MST and SAV1, we define an evolutionarily conserved novel regulatory mechanism loop for tumor suppression in human cancers.
In animal development, cell number and organ size are tightly controlled by mechanisms that regulate cell proliferation and cell death, and deregulation of these mechanisms is linked to cancer (Hanahan and Weinberg, 2000). Genetic screening of fly mutant libraries, aimed at genes that normally restrict cell growth and proliferation, identified three genes, Hippo (Hpo, Ste20 family Ser/Thr kinase), Salvador/sharpei (dSav) and Warts (Wts, also known as Large Tumor Suppressor (LATS), Ser/Thr kinase) (Tapon et al., 2002; Harvey et al., 2003). Molecular biological analysis revealed that dSav recruits Wts to Hpo to promote phosphorylation of Wts by Hpo, thereby constituting the Hpo-dSav-Wts pathway (Hpo pathway) that negatively regulates proliferation and promotes cell death (Hipfner and Cohen, 2004).
Recent studies discovered that Yorkie (Yki), a transcriptional co-activator, is a target of Wts (Huang et al., 2005). In addition, several genes (mob as tumor suppressor [mats], expanded [ex], fat [ft], merlin [mer], Ras Association Family [dRASSF] discs overgrown [dco] and dachs [d]) are known to regulate the Hpo pathway in Drosophila (Harvey and Tapon, 2007). However, the existence of yet unidentified components of the Hpo pathway has been suggested. For example, although Wts is a major target of Hpo and dSav, genetic analysis revealed Wts-independent functions of dSav, suggesting the existence of additional targets for Hpo and dSav (Tapon et al., 2002).
The Hpo pathway is evolutionarily conserved between fly and human (Callus et al., 2006; Dong et al., 2007). The mammalian proteins MST, SAV1/WW45, LATS, and YAP are the homologs for the Drosophila proteins Hpo, dSav, Wts, and Yki, respectively. The mammalian Hpo pathway (MST pathway) is a potent regulator of organ size, and its deregulation leads to tumorigenesis (Dong et al., 2007). Mammals have two genes, MST1 and MST2, which are homologous to Drosophila Hpo. MST1 and MST2 have an almost identical kinase domain, and these kinases are ubiquitously expressed in most tissues and cell lines examined (Harvey and Tapon, 2007). In contrast, mammals have a single gene, SAV1/WW45, which is homologous to Drosophila Sav, and disruption of the gene in mice resulted in hyperplasia accompanied by defects in epithelial terminal differentiation in various organs (Lee et al., 2008).
Runt and Lozenge (Lz) are two functionally expressed members of the Drosophila Runt domain transcription factor family that control various developmental processes, including segmentation, sex determination, eye development, and hematopoiesis (Canon and Banerjee, 2000). During eye development, different types of cells are derived from a common pool of undifferentiated precursors. Lz plays critical roles in the transition from uniformity to diversity and determines the fate of distinct cell types in the compound eye of the fly (Canon and Banerjee, 2000). Notably, mutation of Lz results in an increase in inter-ommatidial cells, a phenotype that is similar to that seen with mutations of Hpo pathway components (Hipfner and Cohen, 2004) that result in the retention of an excess number of undifferentiated precursor cells (Wildonger et al., 2005), whereas Wts promotes apoptosis by inhibiting Yki function, which, in turn, leads to a decrease in diap1 transcription (12). Lz induces apoptosis by activating pro-apoptotic genes (i.e., Reaper, Hid, and Grim) through induction of Klumpfuss and Argos (Wildonger et al., 2005).
Mammals have three evolutionally conserved Runt domain family genes, RUNX1/Aml1, RUNX2/Cbfa1, and RUNX3 (van Wijnen et al., 2004). RUNX1 is required for definitive hematopoiesis and is a frequent target of chromosome translocation in leukemia (Speck and Gilliland, 2002). RUNX2 is essential for osteogenesis (Ducy et al., 1997; Komori et al., 1997; Otto et al., 1997), and RUNX3 is involved in neurogenesis (Inoue et al., 2002) and thymopoiesis (Taniuchi et al., 2002) and functions as a tumor suppressor of gastric cancer (Li et al., 2002), bladder cancer (Kim et al., 2005), colon cancer (Ito et al., 2008), and lung cancer (Lee et al., 2010). The tumor suppressor activity of RUNX3 is associated with its ability to induce cell cycle arrest and programmed cell death by transcriptional up-regulation of the CDK inhibitor p21CIP/WAF1 (Chi et al., 2005) and the apoptotic regulator Bim (Bcl2L11) (Yano et al., 2006), respectively. Notably, RUNX3 is stabilized by Ras activation through the p14 ARF-MDM2 signaling pathway and plays an essential role in oncogenic Ras-induced apoptosis in HEK293 cells (Chi et al., 2009).
In this study, we found that RUNX3 forms a complex with SAV1 in an MST2-dependent manner. MST2 also physically interacts with RUNX3 through SAV1 and is co-localized with RUNX3 in the nucleus. Importantly, RUNX3 is required for MST pathway-mediated cell death in HEK293 cells and MCF-7 cells. We further show that the MST-SAV1-RUNX pathway is evolutionarily conserved between fly and human. Collectively, our results identify RUNX3/Lz as a component of the MST/Hpo pathway.
Materials and Methods
Plasmids
cDNA fragments spanning the coding regions of SAV1(NM_021818) and MST2 (NM_006281) were amplified by PCR and subcloned into pCS4-Myc or -Flag or -HA at the EcoRI, XhoI restriction sites. The MST2-K56R substitution mutant and deletion mutants were generated by site-directed mutagenesis and PCR. All mutants were verified by sequencing.
Antibodies
The RUNX3-specific mouse monoclonal antibody 5G4 was purchased from Abcam (Cambridge, UK). Mouse anti c-Myc-biotin (mouse) and streptavidin-conjugated horseradish peroxidase were purchased from Serotec (Oxford, UK). Biotin-conjugated rabbit anti-DYKDDDDK and Biotin-conjugated rabbit-anti-HA were purchased from ICL (Newberg), respectively. Epitope-tag antibodies against Myc (9E10; Delaware Avenue Santa Cruz, CA), HA (12CA5; Roche Applied Science, Mannheim, Germany), and Flag (M2; Sigma, MI) were purchased from the indicated vendors. Tubulin antibodies were obtained from Lab Frontier (Seoul, Korea). The following secondary antibodies were used: Alexa Fluor 594-conjugated goat anti-mouse IgG (Invitrogen, Carlsbad, CA) and horseradish peroxidase-conjugated goat anti-mouse IgG (Amersham Pharmacia Biotech, Buckinghamshire , UK).
Yeast two-hybrid screening
Yeast two-hybrid screening was performed using the DupLEX-ATM Yeast Two-Hybrid System in accordance with the method described in the Yeast Protocols Handbook (OriGene, Rockville, MD). pEG202-RUNX3 was used as a bait plasmid. A human fetal liver cDNA library was screened as suggested in the manufacturer’s manual (OriGene). The positive clones were selected by growth on SD/-His/-Ura/-Trp media and tested on X-gal plates for β-galactosidase activity.
Cell culture and transfection
HEK293 cells and MCF-7 cells were maintained in Dulbecco’s modified Eagle’s medium (Gibco BRL, Carlsbad, California) supplemented with 10% fetal bovine serum (Gibco BRL) and 100 unit/ml of penicillin–streptomycin (Sigma) at 37° C in a humidified atmosphere with 5% CO2. Transient transfection was carried out using Lipofectamine Plus reagent (Invitrogen). Cells were incubated for 24–48 h before harvest.
Phosphatase treatment of cell extracts
Dephosphorylation of RUNX3 was performed by incubating whole-cell extracts with calf intestinal phosphatase (CIP) (NEB, Bedford, MA) at 37° C for 1 h. The reactions were terminated by boiling in SDS sample buffer, followed by SDS–PAGE and Western blotting.
Immunoprecipitation and immunoblotting
Transfected cell lysates (500 μg) were incubated with monoclonal antibodies at 4° C for 2 h or overnight in the presence of protein G-Sepharose beads (Amersham Pharmacia Biotech). After extensive washing, the immunoprecipitates were resolved by SDS–PAGE and transferred to polyvinylidene difluoride membrane (Millipore, MA). The membrane was blocked in TBST buffer (20 mM Tris–HCl, pH 7.4, 150 mM NaCl, and 0.1% Tween 20) with 5% skim milk (Becton, Dickinson and Company, Franklin Lakes, NJ) at room temperature for 1 h. The membrane was incubated with the indicated primary antibodies for 1–2 h at room temperature or overnight at 4° C. After extensive rinsing, the membrane was incubated with horseradish peroxidase-conjugated secondary antibody for 2 h. After further rinsing with TBST buffer, the blot was visualized by AGFA film after treatment with ECL solution (Amersham Pharmacia Biotech).
In vitro kinase assay
His-tagged RUNX3 was expressed in bacteria and purified on a Ni-agarose column. Bacterially expressed SAV1 and GST-MST2 were purchased from OriGene and Anova (Taopei, Taiwan), respectively. These proteins were mixed together with kinase reaction buffer (25 mM Tris (pH 7.5), 5 mM beta-glycerophosphate, 2 mM DTT, 0.1 mM Na3VO4, and 10 mM MgCl2). γ32P-ATP (50 μM) was added and the reaction mixtures were incubated for 30 min at 30° C. The reactions were terminated by adding SDS-loading buffer and analyzed by SDS–PAGE electrophoresis followed by autoradiography.
Results
Isolation of SAV1 as a RUNX3-binding protein via yeast two-hybrid screening
To further our understanding of the biochemical mechanisms underlying RUNX3 function, we screened a human fetal liver cDNA library via yeast two-hybrid analysis to search for novel RUNX3-interacting proteins. One of the recovered clones encodes the fragment of SAV1 encoding the aa 51-260 region. The SAV1-interacting region of RUNX3 was roughly mapped by co-transfecting yeast cells with an N-terminal fragment (LexA-RX3-N) or C-terminal fragment (LexA-RX3-C) of RUNX3 and a construct of SAV1(51-260) fused to the B42 transactivation domain. Analysis of β-galactosidase (β-Gal) activity showed that the aa 51-260 rsegion of SAV1 interacts with the aa 1-187 region of RUNX3 that includes the Runt domain (Fig. 1A). This clone is of particular interest because disruption of Drosophila Sav causes a phenotype similar to that observed for Lz (a Drosophila homolog of mammalian RUNX3) mutants, which exhibit eye defects and an increased number of inter-ommatidial cells (Wildonger et al., 2005). To confirm the physical interaction of RUNX3 and SAV1 in mammalian cells, HEK293 cells were transfected with a fixed amount of HA-tagged RUNX3 and an increasing amount of Myc-tagged SAV1 and analyzed by immunoprecipitation (IP) with anti-HA antibody and immunoblotting (IB) with anti-Myc antibody. The result showed that RUNX3 and SAV1 interact weakly (Fig. 1B, lane 3), and that the interaction became stronger as the amount of SAV1 increased (lanes 3–5).
Fig. 1.

RUNX3 is a component of the MST pathway. A: A human fetal liver cDNA library was screened using a yeast two-hybrid system with full-length RUNX3 as bait. From 2 × 106 transformants, 32 positive clones encoding eight different genes were identified. Sequence analysis revealed that seven clones encoded CBFβ, a heterodimeric partner of RUNX3, and one of the positive clones encoded SAV1 (aa 51-260). Runt and WW indicate the Runt domain and the WW domain, respectively. B: HEK293 cells were transfected with a fixed amount of HA-tagged RUNX3 (0.5 μg) and increasing amounts of Flag-tagged SAV1 (0, 0.1, 0.2, and 0.4 μg), and were analyzed by immunoprecipitation (IP) with an anti-Flag antibody and by immunoblotting (IB) with anti-Flag or anti-HA antibodies. The expression levels of the transfected genes were detected by IB using the appropriate antibodies. Co-immunoprecipitated SAV1 and RUNX3 are indicated as RX3/SAV1. C: MST2 stimulated the interaction between RUNX3 and SAV1. HEK293 cells were transfected with an increasing amount of Myc-MST2 (0, 0.1, and 0.3 μg) and fixed amounts of HA-RUNX3 (0.5 μg)andFlag-SAV1(0.5 μg) as indicated. The association between SAV1 and RUNX3 was analyzed by IP with an anti-Flag antibody followed by IB with the appropriate antibodies. D: Endogenous RUNX3 and SAV1 formed a complex when MST2 was exogenously expressed. Lysates were obtained from HEK293 cells transfected with MST2 or vector, and the physical interaction between the endogenous RUNX3 and SAV1 was measured by IP with anti-SAV1 antibody followed by IB with anti-RUNX3 antibody (5G4). E: MST2 stimulated the interaction between SAV1 and endogenous RUNX3. Myc-SAV1 and HA-MST2 were expressed in HEK293 cells, and the interaction between exogenous SAV1 and endogenous RUNX3 was measured by IP (Myc) followed by IB with anti-RUNX3 antibody. Expression of Myc-SAV1, HA-MST2 and endogenous RUNX3 was detected by IB with the corresponding antibodies. F: MST2 interacted with RUNX3 through SAV1. HA-MST2, Myc-SAV1, and Flag-RUNX3 were expressedinHEK-293 cells as indicated, and the physical interaction between RUNX3 and MST2 was analyzed by IP followed by IB (top panel). The RUNX3–MST2 interaction was detected only when SAV1 was co-expressed.
MST2 stimulates SAV1–RUNX3 interaction
To investigate whether RUNX3 is involved in the MST pathway, cells were transfected with a fixed amount of SAV1 and RUNX3 and an increasing amount of MST2, and the levels of the proteins were measured. As reported previously, MST2 expression was associated with an increase in SAV1 levels (Fig. 1C, fifth panel) (Callus et al., 2006). When the physical interaction of SAV1 and RUNX3 was analyzed by IP followed by IB, only limited interaction between RUNX3 and SAV1 was observed in the absence of MST2; however, this dramatically increased upon coexpression of MST2 (Fig. 1C, top panel).
We then examined the physical interactions between endogenous RUNX3 and SAV1 in the presence or absence of exogenous MST2. The result showed that the endogenous RUNX3 and SAV1 physically interact only when MST2 is expressed (Fig. 1D). To obtain clearer evidence, we exogenously expressed SAV1 and measured the interaction between exogenous SAV1 and endogenous RUNX3. Endogenous RUNX3 interacted weakly with exogenous SAV1, and the interaction was significantly enhanced by co-expression of MST2 (Fig. 1E).
Next, the role played by SAV1 in the interaction between MST2 and RUNX3 was examined. Co-expression followed by IB analysis showed that MST2 interacted with RUNX3 only when co-expressed with SAV1, demonstrating that the interaction between MST2 and RUNX3 is not direct but, rather, is mediated by SAV1 (Fig. 1F, first panel).
Mapping of the regions responsible for the interaction between SAV1 and RUNX3
To narrow down the RUNX3 region responsible for the interaction with SAV1, serial deletion constructs of RUNX3 were co-expressed with full-length SAV1 and analyzed by IP and IB. The Runt domain (aa 54-187) interacted with SAV1 more strongly than did full-length RUNX3 (Fig. 2A,B), suggesting that the Runt domain is sufficient to interact with SAV1 and that the C-terminal region of RUNX3 inhibits the interaction. The marked increase in RUNX3-SAV1 interaction resulting from deletion of the aa 285-325 region from RUNX3 indicates that the region contains the inhibitory activity (Fig. 2B). The Runt domain deletion mutant (ΔRunt) interacted with SAV1 only very weakly compared to full-length RUNX3, even in the presence of MST2 (Fig. 2C). These results suggest that the Runt domain is essential for interaction with SAV1.
Fig. 2.

Mapping of the regions responsible for the interaction between SAV1 and RUNX3. A: Schematic diagram of RUNX3. The region required for the interaction with SAV1 is indicated by a solid line, and the inhibitory region is indicated by a dotted line. Runt and P indicate the Runt domain and the PPxY motif, respectively. BD and ID indicate regions required for the interaction with SAV1and inhibition of the interaction, respectively. B: To narrow down the RUNX3 regions responsible for the interaction with SAV1, serial deletion constructs of RUNX3 were co-expressed with SAV1 and then analyzed by IP and IB with indicated antibodies. The result shows that the aa 1-187 region of RUNX3, consisting mainly of the Runt domain, is sufficient for the interaction with SAV1 and that the aa 285-325 region inhibits the interaction. BD and ID indicate regions required for the interaction with RUNX3 and inhibition of the interaction, respectively. C: RUNX3 deletion constructs were co-expressed with SAV1 and MAT2 and then analyzed by IP and IB. The deletion constructs lacking the Runt domain (181-145 and ΔRunt) interacted with SAV1only very weakly, even when MST2 was co-expressed. D:Schematic diagram of SAV1. The region required for the interaction with RUNX3 is indicated by a bracket, and the inhibitory region is indicated by a dotted line. WW and CC indicate the WW domain and the coiled-coil domain, respectively. E: To narrow down the SAV1 regions responsible for the interaction with RUNX3, serial deletion constructs of SAV1 were co-expressed with MST2 and/or SAV1 and analyzed by IP and IB with the indicated antibodies. The result showed that the aa 201-235 protein segment of SAV1 is required for the interaction with RUNX3 (BD) and that the aa 50-99 region inhibits the interaction (ID). Co-expression of MST2 not only increases the affinity of the two proteins but also abolishes the effect of the inhibitory region. (F) Wild-type RUNX3 and PPxY motif mutants (ΔP and P1) were co-expressed withSAV1andMST2,andtheRUNX3–SAVinteractionwasanalyzedbyIPandIB.PPxYmotif-mutatedRUNX3interactedwithSAV1aseffectively as did wild-type RUNX3.
Similarly, the SAV1 region responsible for the interaction with RUNX3 was mapped by co-expressing serial deletion constructs of SAV1 and RUNX3 with or without MST2. Because the C-terminal coiled-coil domain of SAV1 is required for the MST–SAV1 interaction, our analysis is restricted to N-terminal deletion mutants of SAV1 (Fig. 2D,E). Co-IP experiments showed that SAV1(201-384) interacts with RUNX3 but SAV1(236-384) does not, suggesting that the aa 201-235 protein segment (the first WW domain) is required for the interaction (Fig. 2E). Removal of the aa 50-99 region enhanced the SAV1–RUNX3 interaction significantly in the absence of MST2, suggesting that this region inhibits the interaction. Interestingly, MST2 abolished the inhibition by aa 50-99 (Fig. 2E), suggesting a cooperative binding mechanism in which MST2 facilitates the SAV1–RUNX3 interaction by canceling the inhibitory region of SAV1 (aa 50-99).
RUNX3 contains the PPxY motif (aa 309-312, PPPY) in its C-terminal transactivation domain. Since the WW domain of SAV1 interacts with RUNX3 and the PPxY motif is known to be recognized by the WW domain, we analyzed possible involvement of this motif in the RUNX3–SAV1 interaction. Co-IP analysis revealed that deletion of the motif (ΔP) or point mutation of the critical proline residue (P1) in the RUNX3 did not alter the RUNX3–SAV1 interaction (Fig. 2F, compare lanes 4, 6, and 7). This result suggests that the PPxY motif of RUNX3 is not essential for the RUNX3–SAV1 interaction.
RUNX3 is a target of MST2 kinase activity
MST phosphorylates SAV1, and this phosphorylation generates a very heterogeneous migration pattern (smearing) of SAV1 during denaturing protein electrophoresis (SDS–PAGE) (Callus et al., 2006). In our experiments, we found that the presence of MST2 resulted in electrophoretic band shifting of RUNX3 (Fig. 3A, third panel). MST2-K56R, which lacks kinase enzyme activity, failed to induce alterations in the migration pattern of RUNX3 (Fig. 3A, third panel). Furthermore, the shifted migration pattern of RUNX3 induced by MST2 and SAV1 was markedly decreased upon phosphatase treatment (Fig. 3B). These results suggest that RUNX3 is a specific target of MST2 kinase activity and that SAV1 may support phosphorylation by facilitating this interaction.
Fig. 3.

RUNX3 is a target of MST2 kinase activity (A) HA-RUNX3, Flag-SAV1, Myc-MST2 (wild type), and Myc-MST2-K56R (kinase dead mutant of MST2) were expressed in HEK293 cells, and the physical interaction between RUNX3 and SAV1 was analyzed by IP followed by IB (top panel). Expression levels of transfected genes were detected by IB with the corresponding antibodies. B:Cells were transfected with HA-RUNX3,Flag-SAV1, and Myc-MST2, as indicated. Cell lysates were incubated with phosphatase (CIP) for 1 h at 30°C, and RUNX3 and SAV1 were detected by IB. C: In vitro kinaseassay.PhosphorylationofRUNX3byMST2 was examined using an in vitro kinase assay with bacterially expressedGST-MST2andHis-RUNX3 in the presence or absence of SAV1. Auto-phosphorylation of MST2 and phosphorylation of RUNX3 by MST2 were detected by autoradiography. D: Schematic diagram of MST2 phosphorylation sites in RUNX3. The crystal structure of the Runt domain/CBFβ/DNA (PDB accession code: 1IO4) was used for drawing. The amino acid number of the Runt domain was labeled according to that of RUNX3. E: Flag-RUNX3, Myc-SAV1, HA-MST2, and Flag-Smurf1 were expressed in HEK293 cells as indicated, and the levels of the expressed proteins measured by IB. The expression of Smurf1 decrease RUNX3 levels, which were rescued by additional expression of MST2 and SAV1.
To understand the role of MST2-mediated RUNX3 phosphorylation, we examined the effect of MST2 and the kinase-defective mutant MST2-K56R on the interaction between SAV1 and RUNX3. Although MST2-K56R is unable to phosphorylate RUNX3, the mutant MST2 protein was nevertheless able to increase the interaction between SAV1 and RUNX3 (Fig. 3A, top panel). These results demonstrate that binding of RUNX3 to SAV1 is stimulated by physical association with MST2 and does not require phosphorylation by MST2.
To confirm that MST2 directly phosphorylates RUNX3, in vitro phosphorylation was performed using bacterially expressed GST-MST2, SAV1, and His-RUNX3. The results showed that MST2 weakly phosphorylates RUNX3 and that phosphorylation is markedly increased by SAV1 (Fig. 3C, top panel).
The exact sites within RUNX3 that are phosphorylated by MST2 were determined using epitope-tagged and immuno-affinity-purified RUNX3 proteins and MALDI-TOF and ion-trap mass spectrometry. The results showed that Ser-17, Thr-69, Ser-71, Ser-77, Ser-81, and Thr-153 within RUNX3 are phosphorylated by MST2 (Fig. 4D, Supplementary Fig. 1). Except for Ser-17, all of these amino acids are located within the conserved Runt domain, and four (Thr-69, Ser-71, Ser-77, and Thr-153) are conserved between flies and humans.
Fig. 4.

Involvement of RUNX3 in MST pathway-mediated cell death. A,B: RUNX3 is indispensable for MST-mediated apoptosis of MCF-7 cells. MCF-7 cells were transfected with expression plasmids for MST2,S AV1, RUNX3,and siRNA, as indicated. Expression levels of transfected genes were detected by IB with the corresponding antibodies. The viability of the cells was measured by MTT assay (A) and the numbers of non-viable cells were counted by Nucleo Counter using the Nucleo Cassette kit (Chemo Metec, Allerød, Denmark). C, D:HEK293 cells were transfected, and the viability of the cells and the number of dead cells were measured as described in (A,B).
Also, RUNX3 levels were increased by the co-expression of MST2 (Fig. 3A). This increase was independent of MST2 kinase activity. To understand whether MST2 interferes with Smurf1-mediated RUNX3 degradation (a known mechanism of RUNX3 degradation (Jin et al., 2004), the effects of MST2 and Smurf1 on RUNX3 levels were examined. The results showed that Smurf-mediated decreases in RUNX3 levels were rescued by co-expression of MST2 and SAV1, strongly suggesting that MST2-SAV1 interferes with Smurf1-mediated RUNX3 degradation (Fig. 3E).
Involvement of RUNX3 in MST pathway-mediated cell death
It has been consistently observed that the MST pathway promotes cell death (Pantalacci et al., 2003); thus, we postulated that RUNX3 may be an MST1/SAV1-dependent effector that promotes apoptosis. Hence, we measured the cell viability of MCF-7 cells transiently transfected with a combination of MST2, SAV1, and RUNX3 by the MTT assay. Co-expression of MST2 with SAV1 or RUNX3 decreased cell viability to 70% and 58%, respectively (Fig. 4A). Notably, expression of all three genes further decreased the viability of the cells to 37%. To assess the functional roles of SAV1 and RUNX3 in regulating cell viability, we reduced the levels of both factors by RNA interference (Supplementary Fig. 2). Importantly, the depletion of endogenous RUNX3 using RUNX3 siRNA abolished the MST2- and SAV1-mediated decrease in viable cell number (Fig. 4A, compare lanes 3 and 7). Similarly, treatment with SAV1 siRNA abolished the decrease in viable cell numbers by MST2 and RUNX3 (Fig. 4A, compare lanes 4 and 6). Assessment of the number of dead cells consistently showed that RUNX3 synergistically induced cell death in cooperation with MST2 and SAV1 (Fig. 4B). Notably, MST pathway-mediated cell death was inhibited by siRNA-mediated depletion of RUNX, and RUNX3-mediated cell death was inhibited by knockdown of SAV1 (Fig. 4B). Synergistic cooperation among MST2, SAV1, and RUNX3 in mediating cell death was also observed in HEK293 cells (Fig. 4C,D). These observations suggest that RUNX3 is an indispensable component of MST pathway-mediated cell death in MCF-7 and HEK293 cells.
Effect of MST2, SAV1, and RUNX3 on the known target genes
RUNX3 induces apoptosis by inducing BIM (Yano et al., 2006); therefore, the effect of MST2, SAV1, and RUNX3 on the expression of endogenous BIM was examined. Coexpression analysis revealed that RUNX3 increases BIM expression and that this is not altered by the additional expression of MST2 and/or SAV1. This suggests that BIM is not the target of MST2-SAV1-RUNX3-mediated apoptosis.
Next, the effect of LATS2 (a known target of MST2) on RUNX3 expression was examined. Phosphorylation of RUNX3 by MST2 and SAV1 was clearly observed (Fig. 5B, fourth panel, lane 4). Interestingly, additional expression of LATS2 inhibited MST2-SAV1-mediated RUNX3 phosphorylation (Fig. 5B, fourth panel, lane 8).
Fig. 5.
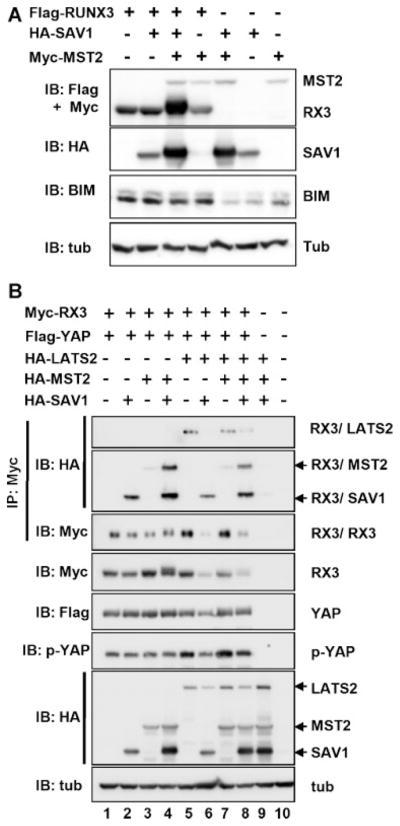
Effects of MST2, SAV1, and RUNX3 on known target genes (A) Flag-RUNX3, HA-SAV1, and Myc-MST2 were expressed in HEK293 cells as indicated and the levels of the expressed proteins and endogenous BIM were measured by IB. B: Myc-RUNX3, Flag-Yap, HA-LATS2, HA-MST2, and HA-SAV1 were expressed in HEK293 cells as indicated and the levels of the expressed proteins were measured by IB using the appropriate antibodies. Physical interactions between RUNX3-LATS2, RUNX3-MST2, and RUNX3-SAV1 were measured by IP with an anti-Myc antibody and IB with an anti-HA antibody.
Unexpectedly, however, RUNX3 also interacted with LATS2 (Fig. 5B, top panel, lane 4) and this interaction was disrupted by the co-expression of SAV1 (Fig. 5B, top panel, lane 6). Additional expression of MST2 rescued the RUNX3–LATS2 interaction (Fig. 5B, top panel, lane 8).
Nuclear co-localization of RUNX3, SAV1, and MST2
RUNX3 localizes primarily to the nucleus; SAV1, to the cytoplasm; and MST2, mainly to the cytoplasm, but with the ability to shuttle continuously between the cytoplasm and nucleus (Lee and Yonehara, 2002). Cytoplasmic localization of MST2 and SAV1 was not altered when both genes were co-expressed (data not shown). Co-expression with RUNX3 did not alter the cytoplasmic localization of SAV1 (Fig. 6A). Similarly, the localization of MST2 was not affected by RUNX3 in the absence of SAV1 (Fig. 6B). However, when MST2, SAV1, and RUNX3 were simultaneously expressed, the three proteins were all present in the nucleus (Fig. 6C). Remarkably, all cells with nuclear localization of the MST2-SAV1-RUNX3 complex showed evidence of cell death as reflected by membrane blebbing (Fig. 6C, DIC), which is known to be associated with MST-induced cell death (Lee et al., 2001). Essentially, the same result was observed in the cells with less condensed morphology (Fig. 3C, lower panels). This result is consistent with our finding that MST2, SAV1, and RUNX3 synergistically induce cell death and that RUNX3 is an essential effector of MST-mediated cell death. Importantly, the kinase-defective mutant, MST2-K56R, and SAV1 both co-localized with RUNX3 in the cytoplasm and membrane blebbing was not observed (Fig. 6D). Thus, the kinase activity of MST2 is required for nuclear localization of the MST2-SAV1-RUNX3 complex and for the induction of cell death.
Fig. 6.

Nuclear co-localization of RUNX3, SAV1, and MST2 (A) Myc-RUNX3 and/or Flag-SAV1 were expressed as indicated in HEK293 cells, and the subcellular localization of each protein was visualized by immunofluorescence microscopy. RUNX3 was probed with anti-Myc antibody emitting green fluorescence, and SAV1 was detected with an anti-Flag antibody emitting red fluorescence. SAV1 was mainly localized to the cytoplasm, whereas RUNX3 was localized to the nucleus, with limited signal overlap. B: Myc-RUNX3 and/or HA-MST2 were co-expressed as indicated. MST2 was probed with an anti-HA antibody emitting red fluorescence. MST2 was primarily detected in the cytoplasm, where as RUNX3 was mostly restricted to the nucleus. C: HA-MST2, Flag-SAV1, and Myc-RUNX3 were co-expressed. SAV1 was probed with anti-Flag antibody emitting blue fluorescence. Co-localization of the three proteins was detected in the nucleus (merged image, white color).Images of cells with less condensed morphology are shown in the lower panels. Cell death-associated membrane blebbing was observed only in cells expressing all three genes. DIC, differential interference contrast. D: HA-MST2-K56R, Flag-SAV1, and Myc-RUNX3 were co-expressed. Co-localization of the three proteins was detected in cytoplasm(merged image, white color). Because the K56R mutation inhibited MST2 kinase activity, cell death-associated blebbing of membranes was not observed, even in cells expressing all three genes.
RUNX3/Lz is an evolutionarily conserved component of the MST/Hpo pathway
To examine whether Lz, the fly ortholog of mammalian RUNX3, is also involved in the Hpo/MST pathway, we examined the physical interaction between Lz and dSav. Co-immunoprecipitation analysis revealed that Lz weakly interacted with dSav in the absence of Hpo (Fig. 7A, top panel). Interestingly, the Lz–dSav and Lz–Hpo interactions were dramatically increased by the co-expression of Hpo (Fig. 7A, top panel). The level of Lz protein was also increased by the co-expression of dSav and Hpo (Fig. 7A, second panel). To confirm the effects of Hpo on the interaction between Lz and dSav, fixed amounts of Lz and dSav were expressed along with serially increasing amounts of Hpo. Co-immunoprecipitation analysis showed that increasing amounts of Hpo induced further increases in the levels of Lz and dSav, and also increased the interaction between them (Fig. 7B). The dose-dependent effect was saturated at 30 ng of Hpo expression plasmid.
Fig. 7.

Physical interaction between Drosophila lozenge (Lz) and dSav. A: Myc-Lz, Flag-dSav, and Flag-Hpo were expressed in HEK293 cells as indicated and the physical interactions between the three proteins measured. Lz was co-precipitated with dSav and Hpo (top panel). Hpo increased the interaction between Lz and dSav, and Lz interacted with Hpo even in the absence of dSav (top panel). B: Myc-Lz, Flag-dSav, and an increasing amount of Flag-Hpo were expressed in HEK293 cells as indicated, and the Lz–dSav and Lz–Hpo interactions were measured. Lz was immunoprecipitated with an anti-Myc antibody, and dSav and Hpo were detected by IB with an anti-Flag antibody (top panel). The Lz–dSav interaction was markedly increased upon co-expression of Hpo. Lz also strongly interacted with dHpo. The expression levels of the transfected genes were analyzed by IB with the corresponding antibodies.
Interestingly, Hpo directly interacted with Lz in the absence of dSav (Fig. 7A), whereas the MST2–RUNX3 interaction required SAV1 (Fig. 1F). The interactions between Hpo, dSav, and Lz are analogous to those of their mammalian counter parts, except that dHpo and Lz are capable of interacting in the absence of dSav.
Discussion
The Hpo pathway is evolutionarily conserved from fly to human, and deregulation of this pathway has been implicated in human cancers (Hanahan and Weinberg, 2000; Saucedo and Edgar, 2007). However, much remains to be learned about the Hpo pathway. For example, it is not known whether the Hpo is linked with the known tumor suppressor network.
In this study, we identified RUNX3/Lz as a new downstream target of the MST/Hpo signaling pathway. SAV1 physically interacted with RUNX3, and the interaction was dramatically increased by MST2. MST2 indirectly interacted with RUNX3 through SAV1 and phosphorylated RUNX3. The phosphorylation of RUNX3 by MST2 and the stimulatory effects of SAV1 were confirmed by the in vitro kinase assay (Fig. 3C). Unexpectedly, MST2 was able to phosphorylate RUNX3 weakly, even in the absence of SAV1, which may suggest that MST2 and RUNX3 interact directly; however, the affinity is not strong enough to be detected by co-immunoprecipitation in the absence of SAV1.
The analysis of cell viability after transient transfection provided additional evidence for the essential role of RUNX3 in the MST pathway. MST-SAV-mediated cell death was abolished by RUNX3 siRNA in MCF-7 cells and HEK293 cells, strongly suggesting that RUNX3 is a component of the MST pathway required for MST pathway-mediated cell death.
We also showed that Drosophila Lz interacts with dSav and that the interaction is enhanced by Hpo, suggesting that the MST-SAV-RUNX (Hpo-dSav-Lz) pathway is evolutionarily conserved from fly to human. Lz determines whether cells are destined to become photoreceptor cells (R1, R6, and R7) or interommatidial cells (Canon and Banerjee, 2000). Lz also induces apoptosis of extra-interommatidial cells (about one-third of cells) by activating argos (aos) and klumpfus (klu) and modulating the pro-apoptotic factors hid and rpr (Wildonger et al., 2005). Notably, a phenotype of the Lz mutant fly, an increase in inter-ommatidial cells (Wildonger et al., 2005), is similar, although not identical, to that observed by mutation of Hpo pathway components (Hipfner and Cohen, 2004). This similar phenotype in mutant flies (Lz, Hpo, and dSav) strongly supports our observation that RUNX3/Lz is a component of the Hpo pathway. Although disruption of Hpo or dSav results in an increase in inter-ommatidial cells via failure of cell death and increased proliferation (Saucedo and Edgar, 2007), the Lz mutation results only in failure of cell death, the function of which is to remove an excess number of undifferentiated precursor cells (Wildonger et al., 2005). This result suggests that the Hpo pathway may stimulate apoptosis through the Hpo-dSav-Lz pathway, whereas it inhibits proliferation through the Hpo-dSav-Warts-Yki pathway.
It is worth mentioning that RUNX family members are known to interact with YAP (Yagi et al., 1999; Zaidi et al., 2004). In the present study, we showed that RUNX3 also interacts with LATS2, an upstream regulator of YAP, and that this interaction is inhibited by SAV1. Inhibition of the RUNX3–SAV1 interaction by SAV1 was cancelled out by additional expression of MST2. These results suggest that the connection between RUNX3 and the MST pathway is complex; RUNX3 may also be a target for LATS2, but the mechanism appears independent from that of the MST2-SAV1 pathway. It is worth mentioning that MST family members may be activated either by C-terminal cleavage (e.g., via the Fas pathway) or by other, unknown mechanisms that do not involve cleavage.
Importantly, C-terminally cleaved SAV1 is unable to interact with MST (Callus et al., 2006). Therefore, our results suggest that the activation of MST-LATS-YAP and MST-SAV-RUNX3 pathways could be dependent on the mechanism of MST activation, with or without Cpterminal cleavage of MST, respectively. Further studies will be required to fully understand the molecular networks connecting MST2, SAV1, LATS2, YAP, and RUNX3.
Taken together, our findings demonstrate that RUNX3 is an integral component of the MST pathway and an essential effector of MST pathway-mediated cell death. We also show that the molecular interactions between RUNX3/Lz and the MST/Hpo pathway have been functionally conserved from fly to human. Our findings establish a principal novel link between the apoptotic MST pathway and the tumor suppressor function of RUNX3.
Supplementary Material
Acknowledgments
This work was supported by the Creative Research Grant R16-2003-002-01001-02006 from the National Research Foundation of Korea to Suk-Chul. Bae and KRF-2006-311-E00194 to Yong-Hee Lee, as well as the National Institutes of Health grants P01 CA082834 to Gary S. Stein and R01 AR49069 to Andre J. van Wijnen.
Contract grant sponsor: Creative Research Grant;
Contract grant number: R16-2003-002-01001-02006.
Contract grant sponsor: National Research Foundation of Korea;
Contract grant number: KRF-2006-311-E00194.
Contract grant sponsor: National Institutes of Health;
Contract grant numbers: P01 CA082834 R01, AR49069. Jiyeon Kim’s present address is Department of Pharmacology and Cancer Biology, Duke University Medical Center, Durham, NC, USA.
Footnotes
Additional supporting information may be found in the online version of this article.
Literature Cited
- Callus BA, Verhagen AM, Vaux DL. Association of mammalian sterile twenty kinases, Mst1 and Mst2, with hSalvador via C-terminal coiled-coil domains, leads to its stabilization and phosphorylation. FEBS J. 2006;273:4264–4276. doi: 10.1111/j.1742-4658.2006.05427.x. [DOI] [PubMed] [Google Scholar]
- Canon J, Banerjee U. Runt and Lozenge function in Drosophila development. Semin Cell Dev Biol. 2000;11:327–336. doi: 10.1006/scdb.2000.0185. [DOI] [PubMed] [Google Scholar]
- Chi XZ, Yang JO, Lee KY, Ito K, Sakakura C, Li QL, Kim HR, Cha EJ, Lee YH, Kaneda A, Ushijima T, Kim WJ, Ito Y, Bae SC. RUNX3 suppresses gastric epithelial cell growth by inducing p21(WAF1/Cip1) expression in cooperation with transforming growth factor {beta}-activated SMAD. Mol Cell Biol. 2005;25:8097–8107. doi: 10.1128/MCB.25.18.8097-8107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi XZ, Kim J, Lee YH, Lee JW, Lee KS, Wee H, Kim WJ, Park WY, Oh BC, Stein GS, Ito Y, van Wijnen AJ, Bae SC. Runt-related transcription factor RUNX3 is a target of MDM2-mediated ubiquitination. Cancer Res. 2009;69:8111–8119. doi: 10.1158/0008-5472.CAN-09-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Harvey K, Tapon N. The Salvador-Warts-Hippo pathway—An emerging tumour-suppressor network. Nat Rev Cancer. 2007;7:182–191. doi: 10.1038/nrc2070. [DOI] [PubMed] [Google Scholar]
- Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114:457–467. doi: 10.1016/s0092-8674(03)00557-9. [DOI] [PubMed] [Google Scholar]
- Hipfner DR, Cohen SM. Connecting proliferation and apoptosis in development and disease. Nat Rev Mol Cell Biol. 2004;5:805–815. doi: 10.1038/nrm1491. [DOI] [PubMed] [Google Scholar]
- Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Inoue K, Ozaki S, Shiga T, Ito K, Masuda T, Okado N, Iseda T, Kawaguchi S, Ogawa M, Bae SC, Yamashita N, Itohara S, Kudo N, Ito Y. Runx3 controls the axonal projection of proprioceptive dorsal root ganglion neurons. Nat Neurosci. 2002;5:946–954. doi: 10.1038/nn925. [DOI] [PubMed] [Google Scholar]
- Ito K, Lim AC, Salto-Tellez M, Motoda L, Osato M, Chuang LS, Lee CW, Voon DC, Koo JK, Wang H, Fukamachi H, Ito Y. RUNX3 attenuates beta-catenin/T cell factors in intestinal tumorigenesis. Cancer Cell. 2008;14:226–237. doi: 10.1016/j.ccr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Jin YH, Jeon EJ, Li QL, Lee YH, Choi JK, Kim WJ, Lee KY, Bae SC. Transforming growth factor-beta stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation. J Biol Chem. 2004;279:29409–29417. doi: 10.1074/jbc.M313120200. [DOI] [PubMed] [Google Scholar]
- Kim WJ, Kim EJ, Jeong P, Quan C, Kim J, Li QL, Yang JO, Ito Y, Bae SC. RUNX3 inactivation by point mutations and aberrant DNA methylation in bladder tumors. Cancer Res. 2005;65:9347–9354. doi: 10.1158/0008-5472.CAN-05-1647. [DOI] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Lee KK, Yonehara S. Phosphorylation and dimerization regulate nucleocytoplasmic shuttling of mammalian STE20-like kinase (MST) J Biol Chem. 2002;277:12351–12358. doi: 10.1074/jbc.M108138200. [DOI] [PubMed] [Google Scholar]
- Lee KK, Ohyama T, Yajima N, Tsubuki S, Yonehara S. MST, a physiological caspase substrate, highly sensitizes apoptosis both upstream and downstream of caspase activation. J Biol Chem. 2001;276:19276–19285. doi: 10.1074/jbc.M005109200. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kim TS, Yang TH, Koo BK, Oh SP, Lee KP, Oh HJ, Lee SH, Kong YY, Kim JM, Lim DS. A crucial role of WW45 in developing epithelial tissues in the mouse. EMBO J. 2008;27:1231–1242. doi: 10.1038/emboj.2008.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Lee YS, Lee JM, Ito K, Cinghu S, Kim JH, Jang JW, Li YH, Goh YM, Chi XZ, Wee H, Lee HW, Hosoya A, Chung JH, Jang JJ, Kundu JK, Surh YJ, Kim WJ, Ito Y, Jung HS, Bae SC. Runx3 is required for the differentiation of lung epithelial cells and suppression of lung cancer. Oncogene. 2010;29:3349–3361. doi: 10.1038/onc.2010.79. [DOI] [PubMed] [Google Scholar]
- Li QL, Ito K, Sakakura C, Fukamachi H, Inoue K, Chi XZ, Lee KY, Nomura S, Lee CW, Han SB, Kim HM, Kim WJ, Yamamoto H, Yamashita N, Yano T, Ikeda T, Itohara S, Inazawa J, Abe T, Hagiwara A, Yamagishi H, Ooe A, Kaneda A, Sugimura T, Ushijima T, Bae SC, Ito Y. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002;109:113–124. doi: 10.1016/s0092-8674(02)00690-6. [DOI] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Pantalacci S, Tapon N, Leopold P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat Cell Biol. 2003;5:921–927. doi: 10.1038/ncb1051. [DOI] [PubMed] [Google Scholar]
- Saucedo LJ, Edgar BA. Filling out the Hippo pathway. Nat Rev Mol Cell Biol. 2007;8:613–621. doi: 10.1038/nrm2221. [DOI] [PubMed] [Google Scholar]
- Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer. 2002;2:502–513. doi: 10.1038/nrc840. [DOI] [PubMed] [Google Scholar]
- Taniuchi I, Osato M, Egawa T, Sunshine MJ, Bae SC, Komori T, Ito Y, Littman DR. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111:621–633. doi: 10.1016/s0092-8674(02)01111-x. [DOI] [PubMed] [Google Scholar]
- Tapon N, Harvey KF, Bell DW, Wahrer DC, Schiripo TA, Haber DA, Hariharan IK. salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell. 2002;110:467–478. doi: 10.1016/s0092-8674(02)00824-3. [DOI] [PubMed] [Google Scholar]
- van Wijnen AJ, Stein GS, Gergen JP, Groner Y, Hiebert SW, Ito Y, Liu P, Neil JC, Ohki M, Speck N. Nomenclature for Runt-related (RUNX) proteins. Oncogene. 2004;23:4209–4210. doi: 10.1038/sj.onc.1207758. [DOI] [PubMed] [Google Scholar]
- Wildonger J, Sosinsky A, Honig B, Mann RS. Lozenge directly activates argos and klumpfuss to regulate programmed cell death. Genes Dev. 2005;19:1034–1039. doi: 10.1101/gad.1298105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 1999;18:2551–2562. doi: 10.1093/emboj/18.9.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano T, Ito K, Fukamachi H, Chi XZ, Wee HJ, Inoue K, Ida H, Bouillet P, Strasser A, Bae SC, Ito Y. The RUNX3 tumor suppressor upregulates Bim in gastric epithelial cells undergoing transforming growth factor beta-induced apoptosis. Mol Cell Biol. 2006;26:4474–4488. doi: 10.1128/MCB.01926-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi SK, Sullivan AJ, Medina R, Ito Y, van Wijnen AJ, Stein JL, Lian JB, Stein GS. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 2004;23:790–799. doi: 10.1038/sj.emboj.7600073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.