

Summary
Stress facilitates reinstatement of addictive drug-seeking in animals and promotes relapse in humans. Acute stress has marked and long-lasting effects on plasticity at both inhibitory and excitatory synapses on dopamine neurons in the ventral tegmental area (VTA), a key region necessary for drug reinforcement. Stress blocks long-term potentiation at GABAergic synapses on dopamine neurons in the VTA (LTPGABA), potentially removing a normal brake on activity. Here we show that blocking kappa opioid receptors (KORs) prior to forced-swim stress rescues LTPGABA. In contrast, blocking KORs does not prevent stress-induced potentiation of excitatory synapses nor morphine-induced block of LTPGABA. Using a kappa receptor antagonist as a selective tool to test the role of LTPGABA in vivo, we find that blocking KORs within the VTA prior to forced-swim stress prevents reinstatement of cocaine-seeking. These results suggest that KORs may represent a useful therapeutic target for treatment of stress-triggered relapse in substance abuse.
Introduction
Despite the health risks associated with drug abuse, addicted individuals have difficulty abstaining, and relapse rates remain high. Exposure to stress is associated with drug addiction in humans and can induce relapse and craving (Brown et al., 1995; Doherty et al., 1995; Sinha et al., 1999; Sinha, 2001; Sinha and Li, 2007). Similarly, a variety of stressful stimuli can reinstate drug seeking in animal models (Erb et al., 1996; Ahmed and Koob, 1997; Shaham et al., 2000; Sorge and Stewart, 2005), even when stressors are separated both in time and in context from the drug-taking environment (Conrad et al., 2010). This behavior relies on the mesolimbic reward system, but the mechanisms that underlie stress-induced reinstatement remain unclear. By understanding the influence of stress on the mesolimbic reward system, it may be possible to develop valid treatments addressing stress-related drug craving, a major contributor to relapse.
Acute and sustained exposure to drugs of abuse alters synaptic plasticity in vulnerable brain areas (Wolf, 2002; Kauer and Malenka, 2007; Lüscher and Malenka, 2011). VTA dopamine neurons are part of the circuit contributing to the reinforcing effects of addictive drugs (Wise, 2004; Everitt and Robbins, 2005). Furthermore, acute exposure to either addictive drugs or stress causes parallel changes in plasticity at excitatory and inhibitory synapses on VTA dopamine neurons. At glutamatergic inputs, a single exposure to psychostimulants, morphine, nicotine, ethanol or to acute stress increases the AMPAR/NMDAR ratio, a measure of long-term potentiation (LTP) (Ungless et al., 2001; Saal et al., 2003; Kauer and Malenka, 2007). The potentiation of excitatory VTA synapses following cocaine or acute stress is blocked by NMDAR antagonists and requires insertion of GluA1 receptors (Saal et al., 2003; Dong et al., 2004). Stress activates the hypothalamic-pituitary-adrenal (HPA) axis leading to an increase in circulating glucocorticoid hormone, thus activating glucocorticoid receptors initiating transcription. The potentiation of VTA excitatory synapses by stress is prevented when glucocorticoid receptors are blocked and can be mimicked by direct activation of glucocorticoid receptors in VTA slices (Saal et al., 2003; Daftary et al., 2009). At inhibitory synapses on VTA dopamine neurons, one injection of morphine, nicotine, cocaine, ethanol, or a single exposure to acute stress blocks long-term potentiation (LTPGABA) at GABAA receptor synapses on dopamine neurons (Nugent et al., 2007; Nugent et al., 2009; Guan and Ye, 2010; Niehaus et al., 2010). Glucocorticoid receptor activation is required for stress to block LTPGABA, but in general much less is known about the mechanisms controlling plasticity of inhibitory synapses in the VTA (Niehaus et al., 2010).
Previous studies suggested that opioid receptor activation blocks LTP at GABAergic synapses (Nugent et al., 2007; Guan and Ye, 2010). Here we have tested the idea that stress may block LTP through release of endogenous opioids and activation of opioid receptors. Stress is often accompanied by release of the endogenous opioid peptide, dynorphin and subsequent activation of its receptor, the kappa opioid receptor (KOR) (Nabeshima et al., 1992). There is strong evidence indicating that the kappa opioid system regulates neuronal activity in the VTA. KORs are functionally expressed in the VTA (Speciale et al., 1993; Arvidsson et al., 1995; Mansour et al., 1996), and multiple dynorphin expressing regions innervate the VTA (Fallon et al., 1985; Meredith, 1999; Dong and Swanson, 2003; Chartoff et al., 2009; Poulin et al., 2009). Pharmacological activation of kappa opioid receptors in the VTA causes a subset of dopamine neurons to hyperpolarize and decrease their firing rate (Margolis et al., 2003; Margolis et al., 2006a; Margolis et al., 2008). KOR activation also reduces the somatodendritic release of dopamine in the VTA (Ford et al., 2007).
The dynorphin-kappa opioid receptor system is also strongly implicated in stress-related behaviors (Bals-Kubik et al., 1993; Beardsley et al., 2005; Valdez et al., 2007; Land et al., 2009; Beardsley et al., 2010; Bruchas et al., 2010). Conditioned aversion, elicited when a place or an odor is paired with stress, is absent in mice lacking dynorphin and in animals treated with kappa antagonists (Land et al., 2008), suggesting that KOR activation is a necessary step in the development of aversion to stressful experiences. Kappa opioid receptors are also involved in behaviors at the nexus between stress and drug-seeking (Mclaughlin et al., 2003; Beardsley et al., 2005; McLaughlin et al., 2006; Carey et al., 2007; Redila and Chavkin, 2008; Beardsley et al., 2010; Sperling et al., 2010). These findings suggest that the kappa opioid system could modulate the compulsion to seek cocaine (or other drugs of abuse) after stress (Bruchas et al., 2010)
We tested the hypothesis that kappa opioid receptor activation blocks LTPGABA in VTA dopaminergic neurons following acute cold water swim stress. A similar stressor has recently been shown to induce reinstatement to drug seeking in rats, an effect that lasts for several days after stress (Conrad et al., 2010). We report here that a KOR antagonist given prior to acute stress rescues LTPGABA. We also find that delivery of the same antagonist directly into the VTA prevents stress-induced reinstatement of cocaine-seeking behavior in a self-administration paradigm. Together our results suggest a link between the GABAergic control of VTA neurons and cocaine seeking after exposure to stress, and provide a rationale for the use of KOR antagonists in therapeutic treatment of stress-induced relapse.
Results
Stress blocks LTPGABA
GABAergic synapses on dopamine neurons are persistently potentiated by an increase in postsynaptic Ca2+, which activates nitric oxide (NO) synthase in turn generating NO. NO then travels retrogradely to potentiate GABA release from nearby presynaptic GABAergic nerve terminals through a cGMP-dependent signaling cascade (Nugent et al., 2007), thus inducing LTPGABA (Figure 7). Our previous work demonstrated that brief exposure to stress blocks LTPGABA at these synapses 24 hours later (Niehaus et al., 2010). Confirming our earlier results, after a 5 minute cold water forced swim stress (FSS), when midbrain slices were prepared 24 hours later, exposure to the NO donor, SNAP, blocked LTPGABA in VTA dopamine neurons. In contrast, SNAP triggered robust LTPGABA in slices from control animals that did not experience forced-swim stress (Figure 1A-C).
Figure 7. Proposed model of signaling molecules involved in LTPGABA.

(A), an in vivo exposure to stress blocks LTPGABA through activation of kappa opioid receptors (KORs) with subsequent modulation of the NO signaling pathway at a point downstream of NO production. Presynaptic terminals expressing LTPGABA may originate from local interneurons or extrinsic GABAergic afferents. (B), in vivo exposure to morphine alters LTPGABA through activation of μ-opioid receptors (μOR), inhibiting the cGMP pathway at the level of sGC (soluble guanylate cyclase)(Niehaus et al., 2010). (C), Forskolin potentiates GABAergic synapses by activating adenylyl cyclase (AC), which in turn activates the cAMP/PKA pathway. This pathway is unaffected by in vivo manipulations with morphine or stress (Nugent et al., 2009).
Figure 1. Stress blocks LTPGABA; forskolin can still potentiate GABAergic synapses.

(A), Single experiment illustrating SNAP-induced LTPGABA in a dopamine cell from a naïve animal, or (B) from an animal exposed to five min cold water forced swim. SNAP was bath-applied as indicated by the bar. (C), LTPGABA in slices from naïve or stressed animals (IPSC amplitudes, naïve rats: 128.2 ± 7.9% of control values, n = 3, 24 hours after stress, 95.4 ± 2.0%, n = 3; p < 0.05). (D), Single experiment illustrating forskolin-induced LTPGABA in slices from naïve animals or (E), in slices from animals exposed to forced swim stress. Forskolin (10 μM) was bath-applied as indicated by the bar. (F), Averaged data showing that forskolin potentiates IPSCs in dopamine neurons from naive and stressed animals. Insets: IPSCs before (Control) and 30 minutes after drug application (SNAP, 400 μM). Scale bars: 20 ms, 100 pA. Insets for this and all figures are averages of ten IPCSs.
We next wanted to identify the mechanisms by which stress blocked LTPGABA. The lack of LTPGABA could reflect a stress-induced loss of the synapse’s ability to potentiate. Alternatively, if GABAergic synapses were maximally potentiated following the stressful experience, no further potentiation would be possible in vitro (occlusion). We carried out three experiments to determine whether acute stress blocks or occludes LTPGABA. We bath applied the adenylate cyclase activator, forskolin, to slices from animals stressed 24 hrs earlier. Forskolin potentiates GABAergic synapses on VTA dopamine neurons via cAMP/PKA signaling, and this pathway intersects the NO\cGMP-induced potentiation of GABAA synapses as the effects of SNAP and forskolin are not additive (Nugent et al., 2009). We found that in slices from both naïve animals and animals exposed to forced swim, forskolin potentiated GABAergic synapses. These data indicate that acute stress does not itself induce LTP, but instead blocks the ability of GABAergic synapses to potentiate; forskolin can overcome this block (Figure 1D-F) (IPSC amplitudes: naïve animal, 131 ± 7% of pre-forskolin values, n = 9; stressed animal, 136 ± 5% of pre-forskolin values, n = 5). We also recorded miniature IPSCs (mIPSCs) in dopamine neurons from stressed animals. Since GABAergic synapses on VTA dopamine neurons are potentiated via increased presynaptic GABA release (Nugent et al., 2007), we examined paired pulse ratios and mIPSC frequency in animals after stress. Because LTPGABA is associated with a decrease in the paired pulse ratio (PPR), a lower PPR in stressed animals vs. controls would indicate that LTPGABA was occluded by stress. Instead, paired-pulse ratios (PPR) in dopamine neurons from stressed animals were not significantly different from those in slices from naïve animals (PPR: non-stressed animals, 0.61 ± 0.04, n = 28; stressed animals, 0.65 ± 0.06, n = 21, p > 0.05, Student’s t-test). Similarly, if potentiation at GABAergic synapses were maximally induced by stress in vivo, we would expect an increased mIPSC frequency. Instead, there was no significant difference in the mIPSC frequency between control and saline-injected stressed animals (control, 5.65 ± 1.0 Hz, n = 10; saline + forced swim stress, 5.49 ± 1.2 Hz, n = 10, p > 0.05, Student’s t-test) (Figure S1). Together these experiments support a stress-induced block of LTPGABA rather than occlusion.
Blocking kappa opioid receptors rescues the stress-induced block of LTPGABA
How might stress block LTPGABA? Our previous work demonstrated that LTPGABA was blocked 24 hours after an in vivo injection of morphine (Nugent et al., 2007), suggesting the possibility that stress might act via the release of endogenous opioids. To test this idea, animals were pretreated with either saline or the opioid receptor antagonist, naloxone (10 mg/kg), prior to forced swim stress, and midbrain slices were cut 24 hours later. Stress blocked LTPGABA in slices from saline treated animals (Figure 2B), but this block was rescued in animals pretreated with naloxone (Figure 2C) (IPSC amplitudes: saline + FSS, 112 ± 5%, n = 14; naloxone + FSS, 129 ± 5%, n = 9; p < 0.05). These data support our hypothesis that opioid receptor activation is required for stress-induced block of LTPGABA.
Figure 2. Blocking KORs rescues the stress-induced block of LTPGABA.

(A), Injection protocol for naloxone treated animals. (B) LTPGABA in a dopamine cell from an animal administered saline 15 minutes prior to swim stress; slices were prepared 24 hrs after stress. Insets, representative IPSCs before (Control) and 30 minutes after SNAP application (SNAP). (C) Rescue of SNAP-induced LTPGABA in a slice from an animal treated with naloxone prior to stress. (D) Averaged experiments (saline-treated rats, n = 17; naloxone-treated rats, n = 9). (E) Injection protocol for nor-BNI treated animals; nor-BNI is effective for over a week following injection (Endoh et al., 1992). (F) Absence of LTPGABA in a dopamine cell from an animal administered saline 24 hrs prior to stress and (G) from an animal pretreated with nor-BNI 24 hours prior to stress. (H) Averaged experiments (saline-treated rats, n = 8; nor-BNI-treated rats, n = 8). Scale bars: 20 ms, 100 pA.
Naloxone at the concentration used (10 mg/kg) blocks μ, δ, and κ opioid receptors (Lewanowitsch and Irvine, 2003). Kappa opioid receptors are activated following exposure to stressful stimuli (Takahashi et al., 1990; Bruchas et al., 2007; Land et al., 2008), so we next tested the effects of a selective kappa receptor antagonist on LTPGABA. Nor-BNI (10 mg/kg) was injected 24 hours prior to forced swim stress. As with naloxone pretreatment, dopamine neurons from nor-BNI pretreated animals exhibited robust LTPGABA after stress, while those from saline pretreated animals had strongly attenuated LTPGABA (Figure 2F and G) (saline + FSS, 100 ± 10%, n = 8; nor-BNI + FSS, 132 ± 8%, n = 6; p < 0.05). Animals injected with either naloxone (10 mg/kg) or nor-BNI (10 mg/kg) alone, without forced swim stress exposure, also exhibited normal potentiation of GABAergic synapses (IPSC amplitudes: naloxone, 128 ± 2%, n = 3; nor-BNI, 130 ± 6%, n = 4)( Figure S2). Furthermore, there was no change in mIPSC frequency between controls and nor-BNI injected stressed animals (control, 5.65 ± 1.0 Hz, n = 10; nor-BNI + forced swim stress, 4.98 ± 0.8 Hz, n = 10, p > 0.05, Student’s t-test) (Figure S2). These data suggest that kappa opioid receptors are required for the stress-induced block of LTPGABA, and that blocking these receptors rescues LTPGABA from the effects of stress.
KOR activation in vitro blocks LTPGABA in VTA dopamine neurons
Systemic injection of naloxone and nor-BNI could influence VTA synapses either directly or via activation of other brain regions. To test whether activation of kappa opioid receptors (KOR) blocks LTPGABA locally in the VTA, we bath-applied the KOR agonist U69593 (1 μM) to midbrain slices from naïve rats. The KOR agonist entirely blocked LTPGABA in slices from naïve animals (Figure 3A-C) (vehicle, 135 ± 7%, n = 5; U69593, 114 ± 5%, n = 5; p < 0.05). Thus, activation of kappa opioid receptors locally within the VTA can block LTPGABA. Moreover, since in these experiments we elicited LTP using an NO donor, our data demonstrate that a KOR agonist prevents the potentiation downstream of GABAergic synapses to nitric oxide. U69593 (1 μM) did not alter GABAA receptor mediated synaptic currents on its own (IPSC amplitude: 103± 5%, n = 11) (Figure S3). Paired-pulse ratios were also unaffected by U69593 (1 μM) perfusion (PPR before U69593, 0.54 ± 0.09, n = 11; PPR after U69593, 0.49 ± 0.08, n = 11, p > 0.05, Student’s t-test).
Figure 3. Kappa opioid receptor activation in vitro blocks LTPGABA.
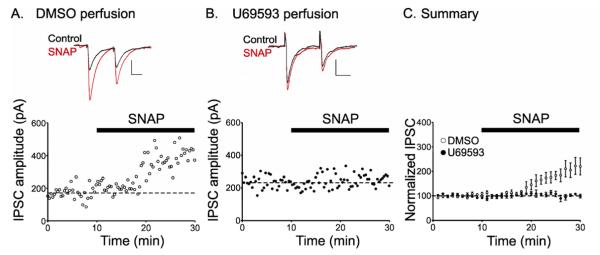
(A), LTPGABA in a dopamine cell from a naïve rat perfused with DMSO. (B), SNAP does not induce LTPGABA in a dopamine neuron perfused with the kappa agonist, U69593 (1 μM) 10 min prior to SNAP application. (C), Averaged experiments; U69593 (n = 6), DMSO (n = 5). Insets, representative IPSCs before (Control) and 30 min after SNAP (SNAP). Scale bars: 20 ms, 100 pA.
Kappa opioid receptor activation is not required for the stress-induced potentiation of excitatory synapses
Stress not only blocks LTPGABA, but also triggers potentiation of excitatory synapses on VTA dopamine neurons (Saal et al., 2003; Dong et al., 2004; Daftary et al., 2009), and we hypothesized that KOR activation mediates both effects. We next tested this idea, using nor-BNI pretreatment before forced swim stress, and then measuring AMPA/NMDA receptor ratios in slices from these animals 24 hours later. Although excitatory synapses from animals that had undergone forced swim stress were significantly potentiated compared with those from naïve animals (Figure 4A-C) (AMPA/NMDA ratio: naïve animals, 0.43 ± 0.06, n = 10; stressed animals, 0.76 ± 0.14, n = 10, p < 0.05), dopamine neurons from both saline injected and nor-BNI injected animals undergoing forced swim stress had similarly potentiated excitatory synapses (AMPA/NMDA ratios: saline injected + stress, 0.84 ± 0.11, n = 19; nor-BNI injected + stress, 0.74 ± 0.11, n = 19, p > 0.05). Thus, while kappa opioid receptor activation is necessary to block LTP at GABAAR synapses on dopamine neurons, excitatory synapses are potentiated via a distinct mechanism (Figure 4D-F).
Figure 4. Kappa opioid receptor activation is not required for long-term potentiation at excitatory synapses on DA neurons in the VTA.

(A) AMPAR- (red) and NMDAR-mediated (black) EPSCs recorded from a dopamine neuron from a naïve animal (naïve). (B) AMPAR- and NMDAR-mediated EPSCs 24 hours after stress. (C) AMPA/NMDA ratios recorded in dopamine neurons from naïve animals and animals 24 hours after stress. Asterisk, p < 0.05, t-test. (D) AMPAR and NMDAR EPSCs from an animal injected with saline or (E) nor-BNI prior to forced swim stress. (F) Summary of AMPA/NMDA ratios recorded in dopamine neurons from saline and nor-BNI pretreated animals 24 hours after swim stress. Scale bars: 50 pA, 20 ms.
Morphine and stress block LTPGABA through distinct mechanisms
As previously shown, a single treatment with morphine blocks LTPGABA in VTA dopamine neurons (Nugent et al., 2007). We had not previously defined the involvement of specific opioid receptors, although morphine can activate KORs directly (Mignat et al., 1995). To investigate which opioid receptors are required, animals were administered either saline (control) or nor-BNI (10 mg/kg) 24 hours prior to morphine injection. We confirmed that morphine blocked LTPGABA both in slices from animals pre-treated with saline (Figure 5B) (Nugent et al., 2007; Niehaus et al., 2010) and in animals pretreated with nor-BNI (Figure 5C), indicating that the morphine-induced block of LTPGABA is not mediated by KOR activation (morphine, 118 ± 11%, n = 12; nor-BNI + morphine, 111 ± 7.4%, n = 8, ANOVA [F2,39=31.49, p<0.05, Tukey’s]). Morphine binds with highest affinity to μ-opioid receptors, and so we next pretreated rats with cyprodime (10 mg/kg), a μ-opioid receptor antagonist, 30 min prior to morphine injection, and LTPGABA was tested in slices cut 24 hrs later. Cyprodime pretreatment effectively rescued LTPGABA in animals treated with morphine (Figure 5E) (LTPGABA measured 15-20 min after SNAP application: morphine, 118 ± 11%, n = 12; cyprodime + morphine, 161 ± 11%, n = 5, ANOVA [F2,39=31.49, p<0.05, Tukey’s]). Together our data demonstrate that morphine’s block of LTPGABA is, in fact, mediated through the activation of μ-opioid receptors and not KORs.
Figure 5. Morphine and stress block LTPGABA through distinct mechanisms.

(A) Diagram of injection protocol for B-C. (B) SNAP fails to induce LTPGABA in a dopamine neuron from an animal administered saline prior to morphine. Slices were prepared 24 h after the animal was exposed to morphine. Insets, representative IPSCs before (Control) and 30 min after SNAP application (SNAP). (C) SNAP also fails to induce LTPGABA in a slice from an animal treated with nor-BNI prior to morphine. (D) Injection protocol for E-F. (E) Rescue of SNAP-induced LTPGABA in a dopamine cell from an animal treated with cyprodime prior to morphine. (F) Averaged experiments showing that LTPGABA is blocked in dopamine cells from animals treated with saline (black symbols, n = 15) or with nor-BNI (gray symbols, n=9) before morphine; LTPGABA is rescued in dopamine cells from animals pretreated with cyprodime (open symbols, n = 6). No significant differences were found between groups of saline pretreated animals, so the data were pooled. (G) Injection protocol prior to stress (H-I). (H) Lack of LTPGABA in a slice from an animal treated with cyprodime prior to stress. (I) SNAP does not induce LTPGABA in cyprodime pretreated stressed animals (n=8). Scale bars: 20 ms, 100 pA.
We also tested for the involvement of μ-opioid receptors in the stress-induced block of LTPGABA. Rats were pretreated with cyprodime 30 min prior to forced swim stress and LTPGABA was tested in slices cut 24 hrs after stress. GABAergic synapses on VTA dopamine neurons from stressed animals failed to potentiate despite pretreatment with cyprodime (Figure 5H and I) (cyprodime + stress, 102 ± 12%, n = 8). Thus, although morphine and stress both inhibit synaptic potentiation of VTA GABAergic synapses, they act through distinct mechanisms.
VTA kappa receptors regulate stress-induced reinstatement of drug seeking
Our in vitro data suggested that kappa receptors control stress-induced synaptic changes in the VTA. Moreover, the kappa receptor antagonist blocked only LTP at GABAergic synapses without preventing stress-induced potentiation of excitatory synapses. Previous work has implicated kappa receptors in stress-induced reinstatement of drug-seeking (Mclaughlin et al., 2003; Beardsley et al., 2005; McLaughlin et al., 2006; Carey et al., 2007; Redila and Chavkin, 2008; Beardsley et al., 2010). We therefore asked whether blocking kappa receptors in the VTA, which we know rescues the stress-induced loss of LTPGABA, influences the reinstatement of stress-induced cocaine seeking. Rats were trained to self-administer cocaine for nineteen days, and then cocaine was withdrawn until responding was extinguished. LTPGABA in VTA dopamine neurons was present in slices from animals at the end of the extinction period (IPSC amplitude: 126 ± 10%; n=4)(Figure S4). Either nor-BNI or saline were locally microinjected into the VTA (Figure S4); 24 hours later, animals underwent swim stress. Reinstatement responding was measured 24 hours after the swim stress. Robust reinstatement of cocaine seeking was observed in the vehicle-stress group. The stress-induced reinstatement response was strongly attenuated by pretreatment with intra-VTA nor-BNI; in fact, in these animals there was no difference between responding at the end of the extinction period and responding 24 hours after stress (Figure 6). Total active and inactive lever press data were analyzed with a two-way repeated-measures ANOVA, which revealed a significant main effect of lever [F(1,12)= 46.42, p<.0001] and treatment [F(1,12)= 19.80, p<.001] as well as a treatment x lever interaction [F(1,12)= 10.23, p<.01]. Subsequent pair wise comparisons showed that the active lever response rate in the nor-BNI treatment was significantly less than the vehicle treatment (Bonferonni, p<.001). There was no significant difference between treatments in terms of inactive lever responding. These data support the hypothesis that kappa receptors in the VTA play a significant role in reinstatement to cocaine-seeking.
Figure 6. Blocking kappa opioid receptors in the VTA blocks stress-induced reinstatement of cocaine seeking.

(A) Training protocol: 19 d of intravenous cocaine self-administration (2 h per day) were followed by daily extinction sessions until lever pressing decreased to <15% of responding on the last day of cocaine self-administration. Rats required ≤ 8 days to achieve this criterion. Twenty-four hours following the attainment of extinction criterion, animals were given a microinfusion of nor-BNI or vehicle into the VTA. 24 hours later, they were exposed to a cold swim stressor and tested for reinstatement responding 24 hours later. (B) Total active and inactive lever presses during the reinstatement phase are shown from animals administered vehicle (n = 7) or 2.5 μg (n = 7) nor-BNI into the ventral tegmental area prior to stress. Unlike the vehicle controls, animals that received nor-BNI infusions into the VTA did not show significant reinstatement of cocaine responding as compared to their extinction responding.
Discussion
This study provides three key findings. First, acute stress activates kappa opioid receptors and blocks nitric oxide-triggered long-term potentiation of GABAergic synapses on VTA dopamine neurons. Second, although acute stress also potentiates excitatory synapses on dopamine neurons, this potentiation is not prevented by a kappa opioid receptor antagonist. Finally, blocking kappa opioid receptors in the ventral tegmental area prevents stress-induced reinstatement of cocaine-seeking.
LTPGABA is blocked when kappa opioid receptors are activated
LTPGABA is a mechanism normally available to control VTA dopamine neurons, and may play an essential role in modulating their firing rates (Johnson and North, 1992; Paladini and Tepper, 1999; Tan et al., 2012; van Zessen et al., 2012). Our results show that the kappa opioid receptor system mediates the stress-induced block of LTPGABA, as systemic treatment with naloxone or a selective kappa receptor antagonist entirely rescued LTPGABA 24 hours later. A kappa receptor agonist also blocked LTPGABA in VTA slices in vitro, indicating that the kappa receptors within the VTA itself can control the induction of LTPGABA (Figure 7). Projections from the BNST, nucleus accumbens, amygdala, and hypothalamus all contain dynorphin that could be released in the VTA and block LTPGABA following acute stress (Fallon et al., 1985; Meredith, 1999; Dong and Swanson, 2003; Poulin et al., 2009). The precise step at which kappa opioid receptors inhibit LTPGABA is not yet known. We favor a presynaptic effect on GABAergic nerve terminals (downstream of nitric oxide production), since in our experiments, exogenously added nitric oxide fails to potentiate GABAA synapses after KOR activation.
Kappa opioid receptors are not required for potentiation of glutamatergic synapses
Following a brief stressful stimulus, excitatory synapses on VTA dopamine neurons are potentiated (Saal et al., 2003). In other brain regions, e.g. the hippocampus and the amygdala, decreased GABAergic inhibition increases the likelihood of LTP at neighboring excitatory synapses (Wigstrom and Gustafsson, 1986; Mott et al., 1990; Shaban et al., 2006), so the loss of LTPGABA could promote LTP at excitatory synapses on dopamine cells (Niehaus et al., 2010). Instead, we found that the loss of LTPGABA is not required for potentiation of excitatory VTA synapses, as nor-BNI blocked the former but not the latter. Thus, kappa opioid receptor activation is not required for stress-induced LTP at excitatory synapses.
Previous work demonstrated that glucocorticoid receptor (GR) activation is required both for stress-induced LTP of excitatory synapses and for the block of LTPGABA. A GR antagonist blocks both forms of plasticity (Saal et al., 2003; Dong et al., 2004; Daftary et al., 2009; Niehaus et al., 2010), and an in vivo treatment or an in vitro bath application of a glucocorticoid receptor agonist increases the AMPA/NMDA ratio (Daftary et al., 2009), similarly to what is seen after exposure to stress (Saal et al., 2003; Dong et al., 2004; Daftary et al., 2009). It is reasonable then to speculate that glucocorticoids released after exposure to stress set into motion plasticity at both inhibitory and excitatory VTA synapses, but ultimately act through independent downstream mechanisms. Our ability to use nor-BNI as a selective tool to rescue stress effects on LTPGABA allowed us to test the role of LTPGABA in stress-induced reinstatement of drug-seeking even when excitatory synapses were allowed to potentiate (discussed below).
How might the loss of LTPGABA influence dopamine neuron firing?
LTPGABA represents a mechanism to increase the magnitude of GABAergic inhibition of dopamine neurons, so its loss after stress is expected to increase cell firing or bursting. Changes in GABAA inhibition have particularly significant consequences in dopamine neurons, which are relatively depolarized and therefore at or very close to the threshold for firing action potentials (Johnson and North, 1992). Optogenetic activation of VTA GABA neurons drastically decreases the firing rate of dopamine neurons (Tan et al., 2012; van Zessen et al., 2012), and local pressure application of GABAA receptor antagonists powerfully shifts the firing pattern of dopamine neurons from tonic to bursting (Paladini and Tepper, 1999). Since burst firing activity in dopamine neurons is more effective than tonic firing at releasing dopamine in target sites, stress-induced loss of LTPGABA also is expected to increase dopamine release in target sites (Gonon, 1988; Manley et al., 1992). Consistent with this idea, in vivo recordings show that burst firing of VTA dopamine neurons increases with acute restraint stress and persists for at least 24 hours (Anstrom and Woodward, 2005), and administration of the GABAA receptor agonist, muscimol, into the VTA blocks footshock-induced reinstatement (McFarland et al., 2004). We speculate that the dynorphin/KOR system could regulate dopamine neuron firing by potently reducing GABAA-mediated inhibition of dopamine cells over many hours, increasing drug-seeking behavior.
Blocking kappa receptors within the VTA attenuates cocaine reinstatement
We reported earlier that LTPGABA is blocked in slices 24 hours after in vivo exposure to morphine (Nugent et al., 2007; Niehaus et al., 2010); indeed, this result first suggested that endogenous opioids could in theory mediate the effects of stress exposure on synaptic function. One hypothesis we entertained was that both addictive drugs and stress might block LTPGABA through a common mechanism. Instead, however, we report here that morphine and stress block LTPGABA through distinct signaling pathways: while stress acts via the kappa opioid receptor, morphine acts instead via the μ-opioid receptor. Consistent with this model, blocking μ-receptors fails to rescue the stress-induced block of LTPGABA, while blocking KORs does not rescue the morphine-induced block. Similarly, others have shown that ethanol inhibits LTPGABA, and this inhibition is rescued by prior naloxone and was attributed to μ-opioid receptor block (although at a concentration not entirely selective for μ receptors) (Guan and Ye, 2010). Much like an acute stressor, drug administration can robustly trigger reinstatement of drug seeking (Stewart, 2000). However, the mechanisms appear to be different. In fact, μ-opioid receptors may be required for drug-triggered reinstatement of drug seeking (Comer et al., 1993; Shaham and Stewart, 1996; Le et al., 1999; Ciccocioppo et al., 2002; Redila and Chavkin, 2008; Simmons and Self, 2009). Moreover, kappa receptor agonists appear to reduce cocaine-primed reinstatement, with no observed effect of nor-BNI, although age and cannula placements may also contribute to differences (Sun et al., 2010). Although our data cannot directly address drug-primed reinstatement, it will be important in future work to compare differences in corticotrophin releasing factor and noradrenergic systems, which may be selectively activated in stress-induced reinstatement (Stewart, 2000; Briand and Blendy, 2010).
We found that stress-induced reinstatement of cocaine seeking was significantly reduced in animals that had received intra-VTA injections of nor-BNI. Previous work showed that kappa receptor antagonists block stress-induced reinstatement of cocaine or ethanol conditioned place preference as well as stress-induced reinstatement of cocaine or ethanol self-administration (Beardsley et al., 2005; Carey et al., 2007; Redila and Chavkin, 2008; Sperling et al., 2010). Additionally, mice lacking either kappa opioid receptors or prodynorphin fail to exhibit footshock stress-induced reinstatement of cocaine place preference (Redila and Chavkin, 2008), and prodynorphin knockout mice fail to exhibit swim stress-induced reinstatement of ethanol place preference (Sperling et al., 2010). Conversely, pharmacological activation of KORs is sufficient to induce robust reinstatement of place preference to drugs of abuse in unstressed animals (Redila and Chavkin, 2008; Sperling et al., 2010). Taken together, these studies support the involvement of kappa opioid receptors in animal models of drug relapse. However, the relevant site of kappa opioid action was unknown, as these studies utilized systemic drug administration. Our results indicate that the kappa receptors within the VTA critically control stress-induced drug seeking in animals trained to self-administer cocaine.
It is worth noting that nor-BNI’s effect on drug reinstatement is not likely to result from a reduction in the perceived desirability of cocaine, as nor-BNI does not affect acquisition or maintenance of cocaine self-administration (Glick et al., 1995; Negus et al., 1997; Negus, 2004) or place preference (Carlezon et al., 1998). KOR antagonists also fail to attenuate cocaine-primed reinstatement, suggesting that KORs exert a selective effect on stress-induced behavior (Beardsley et al., 2005; Carey et al., 2007; Redila and Chavkin, 2008).
Stress alters synaptic plasticity in the VTA, contributing to drug-seeking
Our in vitro results demonstrate that pretreatment with nor-BNI rescued LTPGABA after stress, with no effect on LTP at glutamatergic synapses in stressed animals. Yet in animals that had learned and then extinguished responding for cocaine, delivery of nor-BNI into the VTA blocked cocaine-seeking after stress; there was no significant difference between lever pressing for cocaine after extinction versus after stress in the nor-BNI treated animals. These data are consistent with a model in which the rescue of LTPGABA in the VTA blunts stress-induced reinstatement even when excitatory synapses on dopamine neurons are potentiated. Previous work has suggested that NMDAR-dependent processes in the VTA play an important role in reinforcement. Administration of an NMDAR antagonist directly into the VTA (which would be expected to block LTP at glutamatergic synapses; (Bonci and Malenka, 1999)) decreases the total number of cocaine reinforcements on a progressive ratio schedule following social defeat stress, and also significantly reduces the effect of social defeat stress on cocaine intake during a 24-hour binge access period (Covington et al., 2008). However, excitatory synapses remain potentiated in animals that have self-administered cocaine even after an extinction period, suggesting that further potentiation of these synapses is unlikely to contribute to the reinstatement observed after stress (Chen et al., 2008). Taken together, the evidence suggests that the block of LTP at GABAergic synapses contributes to stress-induced reinstatement. Our experiments cannot rule out other effects of KORs in the VTA, such as transient dopamine neuron hyperpolarization and inhibition of glutamatergic transmission (Margolis et al., 2003; Margolis et al., 2006a). However, these effects are reversed rapidly upon removal of the agonist, while the long time course over which LTPGABA is blocked following stress is consistent with the persistent increase in cocaine-seeking reported after an acute stressor (Conrad et al., 2010).
Exposure to either an aversive stimulus, (such as stress or KOR activation), or to an appetitive stimulus such as a priming dose of drug robustly induces reinstatement (Piazza and Le Moal, 1998; McLaughlin et al., 2005; Carey et al., 2007; Redila and Chavkin, 2008; Conrad et al., 2010). Both stimuli also block LTPGABA on VTA dopamine neurons (Niehaus et al., 2010; Luscher and Malenka, 2011). The similar synaptic responses to both stress and drugs are compelling, suggesting that the synaptic responses may make an important contribution to relapse to drug seeking. Several studies suggest that kappa receptor agonists do not reverse cocaine-primed reinstatement of drug-seeking or conditioned place preference, suggesting that KORs exert a selective effect on stress-induced behavior (Beardsley et al., 2005; Carey et al., 2007; Redila and Chavkin, 2008), or perhaps that additional mechanisms are involved in drug-primed reinstatement.
Brain circuitry underlying stress-induced relapse
Stress is strongly associated with relapse to compulsive drug use in drug addiction (Sinha, 2001; Cleck and Blendy, 2008; Koob, 2009). The kappa opioid system in the VTA is likely to be component of a larger circuit, including the bed nucleus of the stria terminalis (BNST), dorsal raphe and amygdala (Erb et al., 2001; Leri et al., 2002; Wang et al., 2007; Land et al., 2009; Bruchas et al., 2011; Nawata et al., 2012; Smith et al., 2012). Swim stress triggers c-fos activation in neurons that project to the VTA and this activation in many regions is dependent upon CREB (Briand and Blendy, 2010). CREB regulates dynorphin expression, and overexpression of CREB increases dynorphin mRNA (Cole et al., 1995; Carlezon et al., 1998). One region that is both activated by stress and necessary for swim stress-induced reinstatement of conditioned place preference is the BNST (Briand and Blendy, 2010). Dynorphin-containing neurons in the BNST project to the VTA (Dong and Swanson, 2003), and it will be of interest to test whether this projection is required for the observed block of LTPGABA seen in our experiments.
Kappa receptor antagonists in treating substance abuse
Kappa opioid receptors have been implicated previously in human substance abuse. Polymorphisms in the prodynorphin gene promoter region are associated with the risk of cocaine dependence and abuse (Chen et al., 2002; Dahl et al., 2005; Williams et al., 2007). Prodynorphin gene expression levels are higher in individuals with a history of psychostimulant abuse, although this may have been true even before the drug abuse (Hurd and Herkenham, 1993). Our data show that kappa receptors in the VTA modulate stress-induced reinstatement, and as discussed above, there is a growing preclinical literature supporting this hypothesis. There is also considerable evidence that KORs contribute to depression, and perhaps most strongly to depression resulting from repeated stress exposure (Knoll and Carlezon Jr, 2010). Kappa opioid receptors are an important regulatory control on dopamine neurotransmission during stress, and may provide a useful strategy for the prevention of stress-induced relapse (Kreek et al., 2009; Shippenberg, 2009; Bruchas et al., 2010; Wee and Koob, 2010).
Experimental Procedures
Animals for in vitro experiments
For morphine and stress experiments, one brain slice per rat was used for each experiment (n= number of animals). The acute stress was a modified Porsolt forced cold-water swim task (Niehaus et al., 2010). Naloxone was administered 15 minutes prior to forced swim except as noted; cyprodime 30 min prior to cold swim or morphine, and norbinaltorphimine 24 h prior to forced swim stress or morphine injection; at this time point nor-BNI is selective for KORs (Endoh et al., 1992; Bruchas et al., 2007; Redila and Chavkin, 2008). Nor-BNI was given 15 min prior to forced swim stress when testing AMPA/NMDA ratios, to block μ- and κ-opioid receptors (Endoh et al., 1992). Midbrain slices were prepared 24 h after drug administration or stress exposure.
Self-administration procedures
Identical to those described previously (Famous et al., 2008); briefly: Rats were anesthetized with ketamine (80 mg/kg, i.p.; Sigma-Aldrich, St. Louis, MO) and xylazine (12 mg/kg, i.p.; Sigma-Aldrich, St. Louis, MO). An indwelling silastic catheter was inserted into the right jugular vein and was sutured in place. Two stainless steel guide cannulae for microinjections were implanted bilaterally 2 mm dorsal to the ventral tegmental area in each animal (−5.6 mm A/P, ± 0.6 mm M/L and −6.2 mm D/V; Paxinos and Watson). Cocaine Self-Administration. Rats (200-225g) were allowed to lever press for cocaine over 5 s, 6 days a week on a FR1 schedule with a time-out of 20 s and a maximum of 30 infusions per session. Once an animal achieved at least 20 infusions in a single daily session, the subject was switched to a fixed-ratio 5 (FR5) schedule with the same maximum.
Extinction & Reinstatement
Following 19 days of cocaine self-administration, cocaine was replaced with saline, and extinction phase continued until responding on the active lever was < 15% of the response rate maintained by cocaine self-administration under the FR5 schedule (approximately 8 days). Next, an additional extinction session was given and 1 hour later animals were exposed to a 3-minute cold-water swim stress. The reinstatement session occurred 24 hours later (Conrad et al., 2010).
Microinjections
Rats were bilaterally microinjected with nor-BNI (2.5μg) or vehicle in the VTA over a 120 s time period 24 hours prior to the stress exposure.
Preparation of brain slices
Midbrain slices (250 μm) were prepared as previously described from deeply anesthetized Sprague-Dawley rats (Nugent et al., 2007; Niehaus et al., 2010). Slices were stored in oxygenated ACSF (Nugent et al., 2007; Niehaus et al., 2010), or (in mM): 86 NaCl, 2.5 KCl, 1.2 NaH2PO4, 35 NaHCO3, 20 HEPES, 25 glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 1 MgSO4.7H2O, 2 CaCl2.2H2O (Zhao et al., 2011).
Analysis
Data are presented as means ± SEM of the percent change in LTP. Significance was determined using a two-tailed Student’s t-test or a one-way ANOVA with a significance level of p < 0.05.
Supplementary Material
Acknowledgements
We would like to thank Drs. Jane Sullivan, Jonathan Ting, Pavel Ortinski and Pierce and Kauer lab members for their help and suggestions. This research was supported by DA011289 (JAK), MH019118 (NMG and AMP), AA007459 (AMP) DA026660 (LAB), DA15214 (RCP), and DA18678 (RCP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed SH, Koob GF. Cocaine-but not food-seeking behavior is reinstated by stress after extinction. Psychopharmacol. 1997;132:289–295. doi: 10.1007/s002130050347. [DOI] [PubMed] [Google Scholar]
- Anstrom KK, Woodward DJ. Restraint increases dopaminergic burst firing in awake rats. Neuropsychopharmacol. 2005;30:1832–1840. doi: 10.1038/sj.npp.1300730. [DOI] [PubMed] [Google Scholar]
- Arvidsson U, Riedl M, Chakrabarti S, Vulchanova L, Lee JH, Nakano AH, Lin X, Loh HH, Law PY, Wessendorf MW, et al. The kappa-opioid receptor is primarily postsynaptic: combined immunohistochemical localization of the receptor and endogenous opioids. Proc. Natl. Acad. Sci. USA. 1995;92:5062–5066. doi: 10.1073/pnas.92.11.5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bals-Kubik R, Ableitner A, Herz A, Shippenberg TS. Neuroanatomical sites mediating the motivational effects of opioids as mapped by the conditioned place preference paradigm in rats. J. Pharmacol.Exp. Ther. 1993;264:489–495. [PubMed] [Google Scholar]
- Beardsley P, Howard J, Shelton K, Carroll F. Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacol. 2005;183:118–126. doi: 10.1007/s00213-005-0167-4. [DOI] [PubMed] [Google Scholar]
- Beardsley PM, Pollard GT, Howard JL, Carroll FI. Effectiveness of analogs of the kappa opioid receptor antagonist (3R)-7-hydroxy-N-((1S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl ]methyl}-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide (JDTic) to reduce U50,488-induced diuresis and stress-induced cocaine reinstatement in rats. Psychopharmacol. 2010;210:189–198. doi: 10.1007/s00213-010-1846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Malenka RC. Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J. Neurosci. 1999;19:3723–3730. doi: 10.1523/JNEUROSCI.19-10-03723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand LA, Blendy JA. Molecular and genetic substrates linking stress and addiction. Brain Res. 2010;1314:219–234. doi: 10.1016/j.brainres.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SA, Vik PW, Patterson TL, Grant I, Schuckit MA. Stress, vulnerability and adult alcohol relapse. J. Stud. Alcohol. 1995;56:538–545. doi: 10.15288/jsa.1995.56.538. [DOI] [PubMed] [Google Scholar]
- Bruchas M, Land B, Aita M, Xu M, Barot S, Li S, Chavkin C. Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J. Neurosci. 2007;27:11614–11623. doi: 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas M, Land B, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, Lemos JC, Hagan CE, Neumaier JF, Quintana A, et al. Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron. 2011;71:498–511. doi: 10.1016/j.neuron.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey A, Borozny K, Aldrich J, McLaughlin J. Reinstatement of cocaine place-conditioning prevented by the peptide kappa-opioid receptor antagonist arodyn. Eur. J. Pharmacol. 2007;569:84–89. doi: 10.1016/j.ejphar.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr., Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- Chartoff EH, Papadopoulou M, MacDonald ML, Parsegian A, Potter D, Konradi C, Carlezon WA., Jr Desipramine reduces stress-activated dynorphin expression and CREB phosphorylation in NAc tissue. Mol. Pharmacol. 2009;75:704–712. doi: 10.1124/mol.108.051417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AC, LaForge KS, Ho A, McHugh PF, Kellogg S, Bell K, Schluger RP, Leal SM, Kreek MJ. Potentially functional polymorphism in the promoter region of prodynorphin gene may be associated with protection against cocaine dependence or abuse. Am. J. Med. Genet. 2002;114:429–435. doi: 10.1002/ajmg.10362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008;59:288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccocioppo R, Martin-Fardon R, Weiss F. Effect of selective blockade of mu(1) or delta opioid receptors on reinstatement of alcohol-seeking behavior by drug-associated stimuli in rats. Neuropsychopharmacol. 2002;27:391–399. doi: 10.1016/S0893-133X(02)00302-0. [DOI] [PubMed] [Google Scholar]
- Cleck JN, Blendy JA. Making a bad thing worse: adverse effects of stress on drug addiction. J. Clin. Invest. 2008;118:454–461. doi: 10.1172/JCI33946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole RL, Konradi C, Douglass J, Hyman SE. Neuronal adaptation to amphetamine and dopamine: molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron. 1995;14:813–823. doi: 10.1016/0896-6273(95)90225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer SD, Lac ST, Curtis LK, Carroll ME. Effects of buprenorphine and naltrexone on reinstatement of cocaine-reinforced responding in rats. J. Pharmacol. Exp. Ther. 1993;267:1470–1477. [PubMed] [Google Scholar]
- Conrad K, James EM, Lindsay MC, Kerstin AF, Mitch B, Michela M. Persistent increases in cocaine-seeking behavior after acute exposure to cold swim stress. Biol. Psychiatry. 2010;68:303–305. doi: 10.1016/j.biopsych.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, 3rd, Tropea TF, Rajadhyaksha AM, Kosofsky BE, Miczek KA. NMDA receptors in the rat VTA: a critical site for social stress to intensify cocaine taking. Psychopharmacol. 2008;197:203–216. doi: 10.1007/s00213-007-1024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daftary S, Panksepp J, Dong Y, Saal D. Stress-induced, glucocorticoid-dependent strengthening of glutamatergic synaptic transmission in midbrain dopamine neurons. Neurosci. Lett. 2009;452:273–276. doi: 10.1016/j.neulet.2009.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JP, Weller AE, Kampman KM, Oslin DW, Lohoff FW, Ferraro TN, O’Brien CP, Berrettini WH. Confirmation of the association between a polymorphism in the promoter region of the prodynorphin gene and cocaine dependence. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005;139B:106–108. doi: 10.1002/ajmg.b.30238. [DOI] [PubMed] [Google Scholar]
- Doherty K, Kinnunen T, Militello F, Garvey A. Urges to smoke during the first month of abstinence: relationship to relapse and predictors. Psychopharmacol. 1995;119:171–178. doi: 10.1007/BF02246158. [DOI] [PubMed] [Google Scholar]
- Dong HW, Swanson LW. Projections from the rhomboid nucleus of the bed nuclei of the stria terminalis: implications for cerebral hemisphere regulation of ingestive behaviors. J. Comp. Neurol. 2003;463:434–472. doi: 10.1002/cne.10758. [DOI] [PubMed] [Google Scholar]
- Dong Y, Saal D, Thomas M, Faust R, Bonci A, Robinson T, Malenka RC. Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA(-/-) mice. Proc Natl. Acad. Sci. U S A. 2004;101:14282–14287. doi: 10.1073/pnas.0401553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh T, Matsuura H, Tanaka C, Nagase H. Nor-binaltorphimine: a potent and selective kappa-opioid receptor antagonist with long-lasting activity in vivo. Arch. Int. Pharmacodyn. Ther. 1992;316:30–42. [PubMed] [Google Scholar]
- Erb S, Salmaso N, Rodaros D, Stewart J. A role for the CRF-containing pathway from central nucleus of the amygdala to bed nucleus of the stria terminalis in the stress-induced reinstatement of cocaine seeking in rats. Psychopharmacol. 2001;158:360–365. doi: 10.1007/s002130000642. [DOI] [PubMed] [Google Scholar]
- Erb S, Shaham Y, Stewart J. Stress reinstates cocaine-seeking behavior after prolonged extinction and a drug-free period. Psychopharmacol. 1996;128:408–412. doi: 10.1007/s002130050150. [DOI] [PubMed] [Google Scholar]
- Everitt B, Robbins T. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat. Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Fallon JH, Leslie FM, Cone RI. Dynorphin-containing pathways in the substantia nigra and ventral tegmentum: a double labeling study using combined immunofluorescence and retrograde tracing. Neuropeptides. 1985;5:457–460. doi: 10.1016/0143-4179(85)90053-8. [DOI] [PubMed] [Google Scholar]
- Famous KR, Kumaresan V, Sadri-Vakili G, Schmidt HD, Mierke DF, Cha JH, Pierce RC. Phosphorylation-dependent trafficking of GluR2-containing AMPA receptors in the nucleus accumbens plays a critical role in the reinstatement of cocaine seeking. J. Neurosci. 2008;28:11061–11070. doi: 10.1523/JNEUROSCI.1221-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Beckstead MJ, Williams JT. Kappa opioid inhibition of somatodendritic dopamine inhibitory postsynaptic currents. J. Neurophysiol. 2007;97:883–891. doi: 10.1152/jn.00963.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J. Neurosci. 2006;26:2788–97. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick SD, Maisonneuve IM, Raucci J, Archer S. Kappa opioid inhibition of morphine and cocaine self-administration in rats. Brain Res. 1995;681:147–152. doi: 10.1016/0006-8993(95)00306-b. [DOI] [PubMed] [Google Scholar]
- Gonon FG. Nonlinear relationship between impulse flow and dopamine released by rat midbrain dopaminergic neurons as studied by in vivo electrochemistry. Neurosci. 1988;24:19–28. doi: 10.1016/0306-4522(88)90307-7. [DOI] [PubMed] [Google Scholar]
- Guan Y, Ye J. Ethanol blocks long-term potentiation of GABAergic synapses in the ventral tegmental area involving mu-opioid receptors. Neuropsychopharmacol. 2010;35:1841–1849. doi: 10.1038/npp.2010.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan P, Taylor J, Yamamura HI, Porreca F. Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J. Pharmacol. Exp. Ther. 1992;260:1237–1243. [PubMed] [Google Scholar]
- Hurd YL, Herkenham M. Molecular alterations in the neostriatum of human cocaine addicts. Synapse. 1993;13:357–369. doi: 10.1002/syn.890130408. [DOI] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J. Neurosci. 1992;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer J, Malenka R. Synaptic plasticity and addiction. Nat. Rev. Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Knoll A, Carlezon WA., Jr Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacol. 2009;56(Suppl 1):18–31. doi: 10.1016/j.neuropharm.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreek MJ, Zhou Y, Butelman ER, Levran O. Opiate and cocaine addiction: from bench to clinic and back to the bench. Curr. Opin. Pharmacol. 2009;9:74–80. doi: 10.1016/j.coph.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land B, Bruchas M, Lemos J, Xu M, Melief E, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J. Neurosci. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, Hnasko TS, Palmiter RD, Chavkin C. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc. Natl. Acad. Sci. 2009;106:19168–19173. doi: 10.1073/pnas.0910705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le AD, Poulos CX, Harding S, Watchus J, Juzytsch W, Shaham Y. Effects of naltrexone and fluoxetine on alcohol self-administration and reinstatement of alcohol seeking induced by priming injections of alcohol and exposure to stress. Neuropsychopharmacol. 1999;21:435–444. doi: 10.1016/S0893-133X(99)00024-X. [DOI] [PubMed] [Google Scholar]
- Leri F, Flores J, Rodaros D, Stewart J. Blockade of stress-induced but not cocaine-induced reinstatement by infusion of noradrenergic antagonists into the bed nucleus of the stria terminalis or the central nucleus of the amygdala. J. Neurosci. 2002;22:5713–5718. doi: 10.1523/JNEUROSCI.22-13-05713.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewanowitsch T, Irvine RJ. Naloxone and its quaternary derivative, naloxone methiodide, have differing affinities for μ, δ, and κ opioid receptors in mouse brain homogenates. Brain Res. 2003;964:302–305. doi: 10.1016/s0006-8993(02)04117-3. [DOI] [PubMed] [Google Scholar]
- Luscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Malenka Robert C. Drug-evoked synaptic plasticity in addiction: From molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley LD, Kuczenski R, Segal DS, Young SJ, Groves PM. Effects of frequency and pattern of medial forebrain bundle stimulation on caudate dialysate dopamine and serotonin. J. Neurochem. 1992;58:1491–1498. doi: 10.1111/j.1471-4159.1992.tb11369.x. [DOI] [PubMed] [Google Scholar]
- Mansour A, Burke S, Pavlic RJ, Akil H, Watson SJ. Immunohistochemical localization of the cloned kappa 1 receptor in the rat CNS and pituitary. Neurosci. 1996;71:671–690. doi: 10.1016/0306-4522(95)00464-5. [DOI] [PubMed] [Google Scholar]
- Margolis E, Hjelmstad G, Bonci A, Fields H. Kappa-opioid agonists directly inhibit midbrain dopaminergic neurons. J. Neurosci. 2003;23:9981–9986. doi: 10.1523/JNEUROSCI.23-31-09981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Kappa-opioid agonists directly inhibit midbrain dopamine neurons. J. Neurosci. 2005;23:9981–9986. doi: 10.1523/JNEUROSCI.23-31-09981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, Fields HL. κ opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proc. Natl. Acad. Sci. USA. 2006a;103:2938–2942. doi: 10.1073/pnas.0511159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J. Physiol. 2006b;577:907–924. doi: 10.1113/jphysiol.2006.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Mitchell JM, Ishikawa J, Hjelmstad GO, Fields HL. Midbrain dopamine neurons: projection target determines action potential duration and dopamine D(2) receptor inhibition. J. Neurosci. 2008;28:8908–8913. doi: 10.1523/JNEUROSCI.1526-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Davidge S, Lapish C, Kalivas P. Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J. Neurosci. 2004;24:1551–1560. doi: 10.1523/JNEUROSCI.4177-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J. Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Land BB, Li S, Pintar JE, Chavkin C. Prior activation of kappa opioid receptors by U50,488 mimics repeated forced swim stress to potentiate cocaine place preference conditioning. Neuropsychopharmacol. 2005;31:787–794. doi: 10.1038/sj.npp.1300860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Land BB, Li S, Pintar JE, Chavkin C. Prior activation of kappa opioid receptors by U50,488 mimics repeated forced swim stress to potentiate cocaine place preference conditioning. Neuropsychopharmacol. 2006;31:787–794. doi: 10.1038/sj.npp.1300860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith GE. The synaptic framework for chemical signaling in nucleus accumbens. Ann. N. Y. Acad Sci. 1999;877:140–156. doi: 10.1111/j.1749-6632.1999.tb09266.x. [DOI] [PubMed] [Google Scholar]
- Mignat C, Wille U, Ziegler A. Affinity profiles of morphine, codeine, dihydrocodeine and their glucuronides at opioid receptor subtypes. Life Sci. 1995;56:793–799. doi: 10.1016/0024-3205(95)00010-4. [DOI] [PubMed] [Google Scholar]
- Mott DD, Lewis DV, Ferrari CM, Wilson WA, Swartzwelder HS. Baclofen facilitates the development of long-term potentiation in the rat dentate gyrus. Neurosci. Lett. 1990;113:222–226. doi: 10.1016/0304-3940(90)90307-u. [DOI] [PubMed] [Google Scholar]
- Nabeshima T, Katoh A, Wada M, Kameyama T. Stress-induced changes in brain Met-enkephalin, Leu-enkephalin and dynorphin concentrations. Life Sci. 1992;51:211–217. doi: 10.1016/0024-3205(92)90077-3. [DOI] [PubMed] [Google Scholar]
- Nawata Y, Kitaichi K, Yamamoto T. Increases of CRF in the amygdala are responsible for reinstatement of methamphetamine-seeking behavior induced by footshock. Pharmacol. Biochem. Behav. 2012;101:297–302. doi: 10.1016/j.pbb.2012.01.003. [DOI] [PubMed] [Google Scholar]
- Negus SS. Effects of the kappa opioid agonist U50,488 and the kappa opioid antagonist nor-binaltorphimine on choice between cocaine and food in rhesus monkeys. Psychopharmacol. 2004;176:204–213. doi: 10.1007/s00213-004-1878-7. [DOI] [PubMed] [Google Scholar]
- Negus SS, Mello NK, Portoghese PS, Lin CE. Effects of kappa opioids on cocaine self-administration by rhesus monkeys. J. Pharmacol. Exp. Ther. 1997;282:44–55. [PubMed] [Google Scholar]
- Niehaus J, Murali M, Kauer J. Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. Eur. J. Neurosci. 2010;32:108–117. doi: 10.1111/j.1460-9568.2010.07256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent F, Penick E, Kauer J. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007;446:1086–1090. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- Nugent FS, Niehaus JL, Kauer JA. PKG and PKA Signaling in LTP at GABAergic Synapses. Neuropsychopharmacol. 2009;34:1829–1842. doi: 10.1038/npp.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini CA, Tepper JM. GABA(A) and GABA(B) antagonists differentially affect the firing pattern of substantia nigra dopaminergic neurons in vivo. Synapse. 1999;32:165–176. doi: 10.1002/(SICI)1098-2396(19990601)32:3<165::AID-SYN3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Piazza PV, Le Moal M. The role of stress in drug self-administration. Trends Pharmacol. Sci. 1998;19:67–74. doi: 10.1016/s0165-6147(97)01115-2. [DOI] [PubMed] [Google Scholar]
- Poulin JF, Arbour D, Laforest S, Drolet G. Neuroanatomical characterization of endogenous opioids in the bed nucleus of the stria terminalis. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2009;33:1356–1365. doi: 10.1016/j.pnpbp.2009.06.021. [DOI] [PubMed] [Google Scholar]
- Redila V, Chavkin C. Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacol. 2008;200:59–70. doi: 10.1007/s00213-008-1122-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka R. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Shaban H, Humeau Y, Herry C, Cassasus G, Shigemoto R, Ciocchi S, Barbieri S, van der Putten H, Kaupmann K, Bettler B, Luthi A. Generalization of amygdala LTP and conditioned fear in the absence of presynaptic inhibition. Nat. Neurosci. 2006;9:1028–1035. doi: 10.1038/nn1732. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Erb S, Stewart J. Stress-induced relapse to heroin and cocaine seeking in rats: a review. Brain Res. Rev. 2000;33:13–33. doi: 10.1016/s0165-0173(00)00024-2. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Stewart J. Effects of opioid and dopamine receptor antagonists on relapse induced by stress and re-exposure to heroin in rats. Psychopharmacol. 1996;125:385–391. doi: 10.1007/BF02246022. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS. The dynorphin/kappa opioid receptor system: a new target for the treatment of addiction and affective disorders? Neuropsychopharmacol. 2009;34:247. doi: 10.1038/npp.2008.165. [DOI] [PubMed] [Google Scholar]
- Simmons D, Self DW. Role of mu- and delta-opioid receptors in the nucleus accumbens in cocaine-seeking behavior. Neuropsychopharmacol. 2009;34:1946–1957. doi: 10.1038/npp.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R. How does stress increase risk of drug abuse and relapse? Psychopharmacol. 2001;158:343–359. doi: 10.1007/s002130100917. [DOI] [PubMed] [Google Scholar]
- Sinha R, Catapano D, O’Malley S. Stress-induced craving and stress response in cocaine dependent individuals. Psychopharmacol. 1999;142:343–351. doi: 10.1007/s002130050898. [DOI] [PubMed] [Google Scholar]
- Sinha R, Li C. Imaging stress- and cue-induced drug and alcohol craving: association with relapse and clinical implications. Drug. Alcohol.Rev. 2007;26:25–31. doi: 10.1080/09595230601036960. [DOI] [PubMed] [Google Scholar]
- Smith JS, Schindler AG, Martinelli E, Gustin RM, Bruchas MR, Chavkin C. Stress-induced activation of the dynorphin/kappa-opioid receptor system in the amygdala potentiates nicotine conditioned place preference. J. Neurosci. 2012;32:1488–1495. doi: 10.1523/JNEUROSCI.2980-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge RE, Stewart J. The contribution of drug history and time since termination of drug taking to footshock stress-induced cocaine seeking in rats. Psychopharmacol. 2005;183:210–217. doi: 10.1007/s00213-005-0160-y. [DOI] [PubMed] [Google Scholar]
- Speciale SG, Manaye KF, Sadeq M, German DC. Opioid receptors in midbrain dopaminergic regions of the rat. II. Kappa and delta receptor autoradiography. J. Neural Transm. Gen. Sect. 1993;91:53–66. doi: 10.1007/BF01244918. [DOI] [PubMed] [Google Scholar]
- Sperling RE, Gomes SM, Sypek EI, Carey AN, McLaughlin JP. Endogenous kappa-opioid mediation of stress-induced potentiation of ethanol-conditioned place preference and self-administration. Psychopharmacol. 2010;210:199–209. doi: 10.1007/s00213-010-1844-5. [DOI] [PubMed] [Google Scholar]
- Stewart J. Pathways to relapse: the neurobiology of drug- and stress-induced relapse to drug-taking. J. Psychiatry Neurosci. 2000;25:125–136. [PMC free article] [PubMed] [Google Scholar]
- Sun W, Xue Y, Huang Z, Steketee J. Regulation of cocaine-reinstated drug-seeking behavior by κ-opioid receptors in the ventral tegmental area of rats. Psychopharmacol. 2010;210:179–188. doi: 10.1007/s00213-010-1812-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Senda T, Tokuyama S, Kaneto H. Further evidence for the implication of a kappa-opioid receptor mechanism in the production of psychological stress-induced analgesia. Jpn. J. Pharmacol. 1990;53:487–494. doi: 10.1254/jjp.53.487. [DOI] [PubMed] [Google Scholar]
- Tan KR, Yvon C, Turiault M, Mirzabekov JJ, Doehner J, Labouebe G, Deisseroth K, Tye KM, Luscher C. GABA neurons of the VTA drive conditioned place aversion. Neuron. 2012;73:1173–1183. doi: 10.1016/j.neuron.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez G, Platt D, Rowlett J, Ruedi-Bettschen D, Spealman R. Kappa agonist-induced reinstatement of cocaine seeking in squirrel monkeys: a role for opioid and stress-related mechanisms. J. Pharmacol. Exp. Ther. 2007;323:525–533. doi: 10.1124/jpet.107.125484. [DOI] [PubMed] [Google Scholar]
- van Zessen R, Phillips JL, Budygin EA, Stuber GD. Activation of VTA GABA neurons disrupts reward consumption. Neuron. 2012;73:1184–1194. doi: 10.1016/j.neuron.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, You ZB, Rice KC, Wise RA. Stress-induced relapse to cocaine seeking: roles for the CRF(2) receptor and CRF-binding protein in the ventral tegmental area of the rat. Psychopharmacol. 2007;193:283–294. doi: 10.1007/s00213-007-0782-3. [DOI] [PubMed] [Google Scholar]
- Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacol. 2010;210:121–135. doi: 10.1007/s00213-010-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigstrom H, Gustafsson B. Postsynaptic control of hippocampal long-term potentiation. J. Physiol. (Paris) 1986;81:228–236. [PubMed] [Google Scholar]
- Williams TJ, LaForge KS, Gordon D, Bart G, Kellogg S, Ott J, Kreek MJ. Prodynorphin gene promoter repeat associated with cocaine/alcohol codependence. Addict. Biol. 2007;12:496–502. doi: 10.1111/j.1369-1600.2007.00069.x. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat. Rev. Neurosc.i. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Wolf ME. Addiction: making the connection between behavioral changes and neuronal plasticity in specific pathways. Mol. Interv. 2002;2:146–157. doi: 10.1124/mi.2.3.146. [DOI] [PubMed] [Google Scholar]
- Yu W, Miller RF. NBQX, an improved non-NMDA antagonist studied in retinal ganglion cells. Brain Res. 1995;692:190–194. doi: 10.1016/0006-8993(95)00665-d. [DOI] [PubMed] [Google Scholar]
- Zhao S, Ting JT, Atallah HE, Qiu L, Tan J, Gloss B, Augustine GJ, Deisseroth K, Luo M, Graybiel AM, Feng G. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat. Methods. 2011;8:745–752. doi: 10.1038/nmeth.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.