

Abstract
RAS proteins are essential components of signalling pathways that emanate from cell surface receptors. Oncogenic activation of these proteins owing to missense mutations is frequently detected in several types of cancer. A wealth of biochemical and genetic studies indicates that RAS proteins control a complex molecular circuitry that consists of a wide array of interconnecting pathways. In this Review, we describe how RAS oncogenes exploit their extensive signalling reach to affect multiple cellular processes that drive tumorigenesis.
The realization in the late 1970s that RAS harboured transforming properties that were bestowed by gain-of-function mutations shaped our view of the molecular biology of cancer. These studies spearheaded the discovery of many more genes the functions of which were altered in tumours, and gave rise to the concept that the progressive transition from a normal to a malignant phenotype reflects the successive accumulation of genetic alterations that each confer a unique capability that cancer cells need to acquire in order to evade homeostatic barriers. These capabilities, which more than a decade ago were dubbed by Hanahan and Weinberg as the hallmarks of cancer, encompass self-sufficiency in growth signals, insensitivity to growth-inhibitory signals, evasion of programmed cell death, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis1. To accommodate the emergence of additional cancer-associated pathologies this list has been increased to include additional hallmarks such as metabolic fitness and genomic plasticity2. Within this paradigm, the transforming function of oncogenic RAS has been initially attributed to its capacity to endow cells with sufficiency in growth signals. As our understanding of the tumorigenic process and its underlying mechanisms is evolving, it is becoming clear that the net cast by oncogenic RAS is much wider and captures many of the original, as well as some of the newly established, hallmarks of cancer. This Review discusses the ensemble of oncogenic RAS functions that fuel the tumorigenic process.
Oncogenic activation – themes and variations
In humans, three RAS genes encode four distinct but highly homologous ~21 kDa RAS proteins: HRAS, NRAS, KRAS4A and KRAS4B (KRAS4A and KRAS4B are alternative splice variants of the KRAS gene). Serving as transducers that couple cell surface receptors to intracellular effector pathways, RAS proteins cycle between ‘on’ and ‘off ’ conformations that are conferred by the binding of GTP and GDP, respectively. Under physiological conditions, the transition between these two states is regulated by guanine nucleotide exchange factors (GEFs), which promote the activation of RAS proteins by stimulating GDP for GTP exchange, and by GTPase-activating proteins (GAPs), which accelerate RAS-mediated GTP hydrolysis. This inactivation of RAS activity by GAPs is the predominant target of the most common somatic mutations that are found in the oncogenic variants of RAS alleles. Accordingly, oncogenic mutations of Q61 impair the GTP hydrolysis reaction by interfering with the coordination of a water molecule that is required for the nucleophilic attack on the γ-phosphate3,4. Similarly, oncogenic substitutions in residues G12 and G13 prevent the formation of van der Waals bonds between RAS and the GAP through steric hindrance and so perturb the proper orientation of the catalytic glutamine (Q61) in RAS, which results in the pronounced attenuation of GTP hydrolysis5. The outcome of these substitutions is the persistence of the GTP-bound state of RAS and, as a consequence, the incessant activation of a multitude of RAS-dependent downstream effector pathways.
Although not all RAS mutants are created equal, the extent to which specific mutations affect the biological behaviour of RAS remains to be established (FIG. 1). Studies carried out in patients with leukaemia and bladder cancers failed to identify a correlation between the occurrence of specific RAS mutations and the aggressiveness of the disease, suggesting that the different RAS mutations may lead to a common pathophysiological end point6,7. Likewise, differences in the range of KRAS mutations observed in human tumours of the gastrointestinal tract seemingly reflect variations in the aetiology of RAS mutations as opposed to specific mutation-dependent disease characteristics8–10. By contrast, in colorectal and lung cancers, KRASG12V mutations have been associated with a worse prognosis than KRASG12D mutations, raising the possibility that particular amino acid substitutions might dictate specific transforming characteristics of oncogenic RAS alleles11,12. In support of this idea, HRASG12V exhibits weaker GTPase activity and stronger binding to GTP than HRASG12D (REFS 13,14), and it is also more potent in cell culture-based transformation assays15. A new perspective on the issue of the contributions of particular amino acid substitutions to the in vivo transforming capabilities of RAS has been added by recent findings that have documented previously uncharacterized RAS mutations in colorectal tumours and leukaemias16,17. In addition, certain RAS mutations may be associated with an altered response to therapy18. It thus seems that much remains to be learned about the link between sequence permutations and functional alterations of oncogenic forms of RAS.
Figure 1. Frequency of mutations at G12, G13 and Q61 in RAS isoforms.

The frequency of mutational substitution at G12, G13 or Q61 for a particular amino acid has been represented using pie charts. Percentages indicate the frequency with which a given residue is mutated within a particular isoform. Primary data source is the COSMIC database (see Further information).
Another unresolved question concerning the molecular principles of the oncogenic activation of RAS pertains to whether mutations in a particular RAS isoform dictate unique oncogenic outputs. This question has been predominantly instigated by the well-recognized non-random distribution pattern of activated isoforms of RAS among the range of cancer types19,20. Thus, KRAS mutations are most frequently detected in colorectal tumours, lung carcinomas (mostly non-small-cell lung cancer (NSCLC)) and in pancreatic carcinomas; HRAS mutations are associated with tumours of the skin and of the head and neck; and NRAS mutations are common in haematopoietic malignancies (TABLE 1). Earlier attempts to delineate the functional diversity of different oncogenic RAS isoforms mainly relied on the use of ectopic overexpression approaches21. However, as it became apparent that the expression levels of oncogenic RAS impinge on the phenotypic outcome, more recent attempts have explored the possible functional importance of tissue type-dependent isoform segregation using knockout and knock-in mouse models of oncogenic RAS alleles22–27 (TABLE 2). In a model where either Kras4b or Hras was expressed at endogenous levels from the Kras promoter, To et al.25 demonstrated that HRASQ61L was fully capable of substituting for KRASQ61L in carcinogen-induced lung tumorigenesis. Furthermore, carcinogen-induced oncogenic mutations were found to target the Hras allele knocked into the Kras locus in a lung tumorigenesis model, as well as the endogenous Hras allele in a skin tumorigenesis model. These observations suggest that the selective predisposition of RAS isoforms to mutagenesis in a particular tissue reflects gene-specific regulatory elements rather than distinct functional outputs. Examining the roles of KRASG12D and NRASG12D in colonic tumorigenesis and haematological malignancies, Haigis et al.26 and Li et al.27 have reached a somewhat different conclusion. Using knock-in mouse models that do not rely on chemical carcinogenesis and that feature intact endogenous regulatory elements, they have found that, of the two isoforms, only KRASG12D was capable of inducing hyperproliferation and enhanced tumorigenesis when expressed in the crypts of the colonic epithelium and that it induced an aggressive as opposed to an indolent myeloproliferative disorder when expressed in haematopoietic cells. In addition, germline mutations in KRAS and HRAS are associated with the acquisition of distinct developmental disorders (KRAS is associated with Noonan syndrome and cardio-facio-cutaneous syndrome, and HRAS is associated with Costello syndrome)28,29, thus suggesting that mutations in specific isoforms might be selected in accordance with their capacity to deregulate cellular homeostasis in vivo. Notably, a substitution of HRAS for KRAS in a knock-in model rescues the embryonic lethality that is caused by the loss of KRAS4B, thus implying a functional overlap between the two isoforms when expressed from the wild-type allele30.
Table 1.
Frequency of RAS mutations in human cancer.
| Tissue | HRAS* | KRAS* | NRAS* | Incidence Rate‡ | Mortality Rate‡ |
|---|---|---|---|---|---|
| Endocrine | 3% (535) | 0% (670) | 5% (570) | 0.7 | 0.3 |
| Biliary tract | 0% (153) | 31% (1679) | 1% (287) | NA§ | NA |
| Bone | 2% (199) | 1% (252) | 0% (207) | 0.9 | 0.4 |
| Breast | 1% (716) | 4% (782) | 2% (504) | 124 | 24 |
| Central nervous system | 0% (964) | 1% (1054) | 1% (1017) | 6.5 | 4.3 |
| Cervix | 9% (264) | 7% (637) | 2% (132) | 8.1 | 2.4 |
| Endometrium | 1% (291) | 14% (2251) | 0% (314) | 23.9 | 4.1 |
| Eye | 0% (33) | 4% (90) | 1% (106) | 0.8 | 0.1 |
| Haematopoietic and lymphoid tissue | 0% (3076) | 5% (5978) | 10% (8753) | 35.2 | 14.5 |
| Kidney | 0% (273) | 1% (704) | 0% (435) | 14.6 | 4.1 |
| Large intestine | 0% (617) | 33% (34013) | 2% (1570) | 47.2 | 17.6 |
| Liver | 0% (270) | 5% (461) | 3% (310) | 7.3 | 5.2 |
| Lung | 0% (2091) | 17% (16348) | 1% (3081) | 62 | 52.5 |
| Oesophagus | 1% (161) | 4% (375) | 0% (161) | 4.5 | 4.4 |
| Ovary | 0% (152) | 14% (3181) | 5% (191) | 12.8 | 8.6 |
| Pancreas | 0% (278) | 57% (5329) | 2% (305) | 12 | 10.7 |
| Pleura | 0% (19) | 0% (45) | 0% (30) | NA | NA |
| Prostate | 6% (558) | 8% (1184) | 2% (588) | 156 | 24.7 |
| Salivary gland | 15% (161) | 3% (170) | 0% (45) | NA | NA |
| Skin | 6% (2100) | 3% (1462) | 18% (4956) | 22.7 | 3.5 |
| Small intestine | 0% (5) | 20% (316) | 0% (5) | 2 | 0.4 |
| Stomach | 4% (384) | 6% (2793) | 2% (215) | 7.7 | 3.8 |
| Testis | 4% (130) | 4% (432) | 3% (283) | 5.5 | 0.2 |
| Thymus | 2% (46) | 2% (186) | 0% (46) | NA | NA |
| Thyroid | 3% (4137) | 2% (5166) | 8% (4662) | 11 | 0.5 |
| Upper aerodigestive tract | 9% (1083) | 3% (1582) | 3% (836) | 14 | 3.7 |
| Urinary tract | 11% (1765) | 5% (1099) | 2% (873) | 21.1 | 4.3 |
NA, not available.
Data from the COSMIC database (see Further information). Numbers in parentheses indicate total unique samples sequenced.
Data from the US National Cancer Institute SEER Cancer Statistics Review (see Further information). Rates are shown as per 100,000 people per year.
Tumour subtype for which data are unavailable in the SEER database.
Table 2.
Mouse models of oncogenic RAS activation
| Experimental approach | Targeted tissues and cell types | Tumorigenic phenotype |
|---|---|---|
| Conditional endogenous expression of Lox-STOP-Lox- KrasG12D or Lox-STOP-Lox- NrasG12D cassette | Lung (intranasal administration of adenoviral Cre), pancreas (Pdx1-Cre or Ptf1a-Cre), colon (Fabp1-Cre) and the haematopoietic system (Mx1-Cre) | Lung adenocarcinoma, pancreatic intraepithelial neoplasia, colonic hyperplasia (KrasG12D), resistance to apoptosis (NrasG12D) and aggressive myeloproliferative disorder (KrasG12D)26, 27, 222–224 |
| Endogenous expression of Lox-STOP-Lox-KrasG12V – IRES-β-galactosidase cassette | Whole-body (CMV-Cre) | Lung hyperplasia, adenocarcinoma and minor sarcoma lesions22 |
| Transgenic expression of KrasG12V | Gastric and pancreatic epithelium (Krt19 promoter-driven KrasG12V) | Gastric cell hyperplasia225 |
| Transgenic expression of HrasQ61L | Urothelium (Upk2 promoter-driven rabbit HrasQ61L) | Bladder tumorigenesis226 |
| Inducible transgenic expression of KrasG12D | Basal layer of stratified epithelium (Krt5 promoter-driven tetracycline-responsive KrasG12D) | Neoplastic squamous epithelium: skin, forestomach and oesophagus227 |
| Inducible transgenic expression of HrasG12V | Melanocytes (tyrosinase-driven tetracycline-responsive HrasG12V) | Melanoma78 |
| Spontaneous chemical carcinogenesis | Skin (DMBA-TPA-induced mutagenesis of Hras) and lung (urethane-induced mutagenesis of Kras) | Skin papillomas and lung tumours 25, 228 |
| Somatic oncogene transfer by RCAS-TVA gene delivery method | Brain (nestin-TVA-targeted expression of KrasG12D and Akt-myr) and pancreas (Lox-STOP-Lox-R26tva–LacZ/+-targeted expression of KrasG12D) | Glioblastoma and pancreatic intraepithelial neoplasia229, 230 |
| Spontaneous recombination that results in expression of activated KrasG12D | Whole-body | Lung hyperplasia and carcinoma, thymic lymphoma and skin papilloma231 |
CMV, cytomegalovirus promoter; Fabp1, fatty acid binding protein 1, liver; IRES, internal ribosome entry site; Krt, keratin; Mx1, myxovirus resistance 1; myr, myristoylated; Pdx1, pancreatic and duodenal homeobox 1; Ptfa1, pancreas transcription factor 1, a-subunit; R26, ROSA 26; TPA, 12-O-tetradecanoyl-phorbol-13-acetate; Upk2, uroplakin 2.
In summary, it seems that despite the remarkable progress made over the past three decades in understanding the mechanisms and outcomes of oncogenic RAS activation, fundamental issues concerning the role of distinct oncogenic RAS mutations and the contribution of different RAS isoforms to cancer aetiology remain. The recent emergence of sophisticated experimental tools to probe and to model oncogenic RAS-driven cancers clearly presents new opportunities to revisit these issues.
RAS and the transformed phenotype
Promotion of proliferation
The continual expansion of cancer cells relies on the erosion of the mechanisms that obligate cells to respond appropriately to mitogenic and anti-mitogenic factors that are present within the extracellular milieu. As RAS proteins are immediate arbitrators of mitogenic stimuli, it is perhaps not surprising that constitutive activation of RAS fuels cell proliferation (FIG. 2). Indeed, soon after the identification of RAS, pioneering studies revealed that overexpression of oncogenic HRAS is sufficient for driving the entry of G0 phase-arrested cells into the cell cycle in the absence of growth factors31,32. Adding to its function as a transducer of growth factor signalling, active RAS can also enhance the proliferative capacity of cells by inducing the transcriptional upregulation of growth factors such as heparin-binding epidermal growth factor-like growth factor (HBEGF), transforming growth factor-α (TGFα) and amphiregulin (AREG)33–35. Another output of oncogenic RAS signalling that contributes to the aberrant proliferation is the alteration in expression of growth factor receptors33,34. Additionally, oncogenic HRASG12V induces the upregulation of integrins that promote proliferation while downregulating the integrin subunits that could maintain cellular quiescence36,37. On the opposing side of the proliferative balance, oncogenic RAS can directly interfere with anti-proliferative signals by inhibiting signalling from TGFβ38–43.
Figure 2. RAS effects on proliferation.
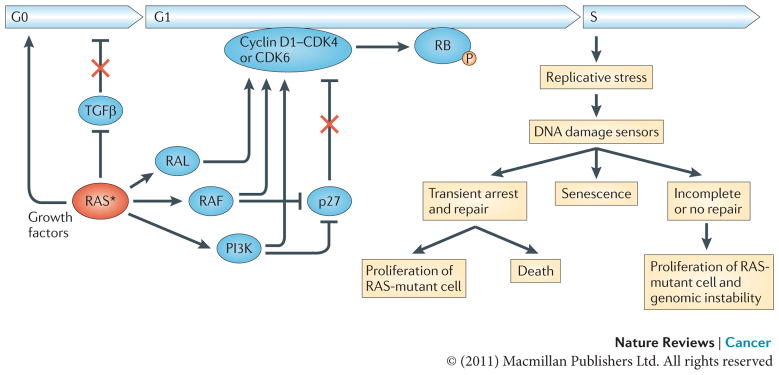
Oncogenic RAS establishes independence from extracellular growth factors and growth inhibitors, thereby promoting exit from the G0 phase of the cell cycle, progression through G1 and entry into S phase. RAS induces the transcriptional upregulation of growth factors and interferes with transforming growth factor-β (TGFβ) signalling through inhibition of TGFβ receptor expression or downstream signalling by downregulating the expression of SMAD3, as well as the nuclear accumulation of SMAD2 and SMAD3. RAS also upregulates the levels of cyclin D1 and suppresses the cyclin-dependent kinase inhibitor (CDKI) p27. The newly synthesized cyclin D1 associates with and activates the cyclin-dependent kinases CDK4 and CDK6, leading to the phosphorylation of RB and the subsequent dissolution of the RB–E2F transcription factor complexes. Once released, E2F transcription factors transactivate several genes that are required for cell cycle progression, including cyclin E (CCNE) and cyclin A (CCNA) that induce transition through the G1/S checkpoint (not shown). Hyperproliferative cues from activation of the RAS oncogene can result in replicative stress leading to DNA damage. In response to DNA damage cells can activate the DNA damage checkpoints to transiently arrest and restore the integrity of the genome, enter a state of irreversible arrest (senescence) or undergo apoptosis. Inaccurate repair of DNA damage can lead to mutations and chromosome aberrations, thereby contributing to tumorigenesis. The asterisk represents the mutational activation of RAS. P, phosphorylation.
The panoply of proliferative signals generated by oncogenic RAS culminates with the upregulation of several transcription factors that are required for cell cycle entry and progression, including FOS, serum response factor (SRF), the leucine zipper protein JUN, the ETS domain-containing transcription factor ELK1, activating transcription factor 2 (ATF2) and nuclear factor-κB (NF-κB)44–49. In turn, these factors trigger the expression of the G1 cyclin, cyclin D1 (REFS 50–52). Although initial studies attributed the stimulation of cyclin D1 transcription by oncogenic RAS to the activation of the RAF–MAPK pathway, it has become evident that additional levels of control are executed through RAS activation of PI3Ks, the RHO family GTPase RAC1, and the RAL-guanine nucleotide dissociation stimulator (GDS) family of GEFs50,51,53–57. In addition to stimulating cyclin D1 gene transcription, oncogenic HRASG12V also regulates the metabolic stability of the cyclin D1 protein through PI3K-dependent inhibition of glycogen synthase kinase 3β (GSK3β), which is the kinase that is responsible for the phosphorylation and the consequent ubiquitylation and proteasomal degradation of cyclin D1 (REF. 58).
Given the central role of cyclin D1 in RAS-induced self-sufficiency in growth signals it is perhaps not surprising that cyclin D1 is a crucial determinant of RAS-induced transformation. As such, cyclin D1-deficient mice are resistant to developing mammary cancers and squamous tumours that are induced by the HRAS oncogene59,60. Notably, dependency on cyclin D1 seems to be a unique property of RAS-induced tumours, as the absence of cyclin D1 had no effect on MYC- or WNT1-induced tumours. However, overexpression of cyclin D1 is not sufficient to transform cells without the activity of a cooperating oncogene, pointing to additional RAS-induced mechanisms to provoke a state of proliferative overdrive61,62. Indeed, oncogenic RAS can promote cell cycle progression by deregulating anti-growth signalling pathways through the suppression of cyclin-dependent kinase inhibitors (CKIs), such as p27 and p21, which would otherwise associate with and inhibit cyclin-dependent kinases (CDKs). This suppressive effect is mediated by multiple RAS effector pathways and is exerted at the transcriptional, translational and post-translational levels42,63–66.
An inevitable consequence of the persistent mitogenic stimulation that is imposed by RAS is the initiation of replicative stress, which is marked by increased numbers of active DNA replication origins and collapsed replication forks, which ultimately leads to DNA damage and the activation of the DNA damage response (DDR)67–70. Indeed, the presence of DNA damage in precancerous lesions has been reported in several cancers that harbour oncogenic RAS, such as cancers of the pancreas, colon and lung. The outcome of overworking the replicative machinery can vary depending on cellular and genetic context. In a cell that possesses the full complement of functional DNA damage checkpoints, the engagement of the DDR by oncogenic HRASG12V leads to an irreversible cell cycle arrest that is known as oncogene-induced senescence (OIS)67. Although OIS is a tumour-constraining response, its induction could contribute to the tumorigenic process by exerting a strong selective pressure in favour of cells in which crucial components of the DNA damage checkpoint have been lost (such as p53 and ARF (also known as p19))71. Conversely, in cells that are deficient in DNA damage checkpoints (through the loss of p53, for example) deregulated DNA replicative activity that is induced by oncogenic RAS results in the generation of chromosome aberrations such as dicentric chromosomes, acentric chromosomes and double-minute chromosomes, leading to the improper segregation of chromosomes and the consequent exclusion of chromosomes from daughter nuclei22,72–75. The documented capacity of oncogenic HRASG12V to induce accelerated transition through G2/M, to inhibit the activation of the G2 DNA damage checkpoint, as well as to induce defects in mitotic spindle checkpoints76, may also contribute to the genomic instability that is observed in RAS-driven cancers. With our understanding of genomic stress and its consequences in cancer initiation and progression still evolving, the contribution of oncogenic RAS to these processes will undoubtedly be an exciting avenue of cancer research in the coming years.
Suppression of apoptosis
Apoptotic cell death functions as a crucial defence mechanism against malignancy, and the corruption of the apoptotic machinery is a defining signature of cancer cells. A complex process, apoptosis is the result of balanced molecular actions that are initiated by a diverse range of signals, and it is regulated at several levels by both positive and negative modulators. Apoptotic cell death can be initiated extrinsically whereby extracellular cues such as growth factor withdrawal or matrix detachment induce the stimulation of death receptors, or it can be initiated intrinsically through the mitochondrion-mediated pathway, which is activated by cues such as DNA damage and nutrient deprivation. Although both the intrinsic and extrinsic signalling cascades converge at the point of caspase 3 activation, they are subject to different regulatory mechanisms, including the pro-apoptotic and anti-apoptotic members of the BCL-2 family acting on the mitochondrion-mediated pathway, the anti-apoptotic FADD-like molecule FLIP (also known as CFLAR) acting on the extrinsic pathway and the inhibitors of apoptosis (IAPs) acting on both pathways77. Oncogenic RAS-driven erosion of the apoptotic pathways and its contribution to cancer has been well documented77 (FIG. 3). For example, the elimination of inducible oncogenic HRASG12V expression results in the regression of melanomas accompanied by massive tumour cell apoptosis78. Additionally, withdrawal of doxycycline-inducible oncogenic KRASG12D expression from type II pneumocytes causes apoptosis and regression of both early proliferative lesions and lung cancers79.
Figure 3. RAS effects on apoptosis.

Oncogenic RAS may have both pro-apoptotic and anti-apoptotic functions depending on the status of RAS effector pathways and the apoptotic machinery. In many cases oncogenic RAS signalling through the RAF pathway engages an apoptotic response that is mediated by p53. Also, signalling through the RAS effectors RASSF1, NORE1, mammalian STE20-like protein kinase 1 (MST1) and JUN N-terminal kinase (JNK) can lead to apoptotic death via the activation of caspase 3 and the pro-apoptotic proteins BCL-2-associated X protein (BAX) and BCL-2-homologous antagonist/killer 1 (BAK1). Acquisition of a tumorigenic phenotype is marked by the suppression of such mediators of RAS-induced apoptosis. In this context, the anti-apoptotic activity of RAS takes a stronghold. The anti-apoptotic function of oncogenic RAS is mediated by several effector pathways, including the RAS–PI3K effector pathway, which regulates the levels of pro-apoptotic protein BAK1 and inhibitors of apoptosis (IAPs), and the RAS–RAF pathway, which downregulates the pro-apoptotic transcriptional repressor prostate apoptosis response 4 (PAR4) while upregulating the anti-apoptotic proteins BCL-2 and apoptosis repressor with caspase recruitment domain (ARC). Both pathways have been implicated in phosphorylating and inactivating the pro-apoptotic protein BCL-2-associated agonist of cell death (BAD). The mechanism through which RAS induces the epigenetic silencing of the pro-apoptotic CD95 gene remains to be uncovered. The asterisk represents the mutational activation of RAS protein. P, phosphorylation.
Mechanistically, the PI3K and RAF pathways activated by oncogenic RAS can downregulate pro-apoptotic mediators or upregulate anti-apoptotic molecules. Thus, activation of PI3K leads to the downregulation of the pro-apoptotic protein BCL-2-homologous antagonist/killer 1 (BAK1), and augments levels of IAPs through the activation of NF-κB80–83. RAF contributes to RAS-induced suppression of apoptosis by the downregulation of the pro-apoptotic transcriptional repressor prostate apoptosis response 4 (PAR4; also known as PAWR)84,85, and the upregulation of the anti-apoptotic proteins BCL-2 and apoptosis repressor with caspase recruitment domain (ARC; also known as NOL3)86,87. Additionally, both the RAS–PI3K–AKT and the RAS– RAF pathways have been shown to mediate the phosphorylation of the pro-apoptotic BCL-2 family member BCL-2-associated agonist of cell death (BAD) on serine 136 and serine 122. Phosphorylation of BAD in either site results in the preferential formation of an inactive complex with 14-3-3, and thereby prevents the heterodimerization with, and subsequent inactivation of, BCL-2 and BCL-XL88,89. Recent findings also implicate oncogenic RAS-induced epigenetic silencing of the pro-apoptotic CD95 (also known as TNFRSF6) gene as a potential anti-apoptotic mechanism that is induced by RAS90,91. The effector molecules operating downstream of RAS in this process remain to be elucidated.
In addition to its pro-survival function, cell type and context-specific oncogenic RAS signalling can also promote pro-apoptotic programmes92,93. For example, the preferential activation of the RAS–RAF–MAPKK–MAPK pathway exacerbates apoptosis94, and HRASQ61L-mediated activation of JUN N-terminal kinase (JNK) signalling has also been associated with a pro-apoptotic role95,96. Phosphorylation of oncogenic KRAS by protein kinase C (PKC) promotes translocation to the mitochondria and induces apoptosis in a BCL-XL-dependent manner97. Additional RAS-regulated molecules with a role in pro-apoptotic programmes include RASSF1 and NORE1 (also known as RASSF5)98. On overexpression, these proteins are found in a pre-existing complex with mammalian STE20-like protein kinase 1 (MST1; also known as STK4), which is an enhancer of caspase 3 activation, and co-transfection of oncogenic RAS results in increased binding of active RAS to this complex and increased apoptosis99,100. There is an indication that such a pro-apoptotic function of RAS might be selected against in cancer, as the expression of either RASSF1 or RASSF5 in tumours is frequently suppressed either owing to promoter hypermethylation or — in the case of RASSF1 — owing to the deletion of the chromosomal region that contains the gene98,101–105. Thus, although the context in which the pro-apoptotic capabilities of RAS are enacted remains unknown, it is the balance of pro-survival and pro-apoptotic signals that would ultimately determine whether the RAS-transformed cell will shift towards life or towards death. The sheer prevalence of oncogenic RAS in cancer is an indication that the pro-survival axis has a dominant role.
Metabolism
As a result of their high proliferative rates, cancer cells are crucially dependent on metabolic pathways that generate the building blocks that are needed to produce a new cell106. These unique metabolic needs were first described in the 1920s by Otto Warburg and are typified by an increase in glucose uptake and a shift from mitochondrial oxidative phosphorylation to aerobic glycolysis107,108. Although the full ramifications of this metabolic phenotype are yet to be deciphered, it has been postulated that — although it is less efficient in generating ATP — the catabolism of glucose through glycolysis is highly effective in providing the macromolecular precursors that are necessary for the replication of biomass (such as nucleotides, amino acids and lipids)109. Oncogenic RAS impinges on the metabolic reprogramming of cancer cells predominantly through the upregulation of hypoxia-inducible factor 1α (HIF1α), which forms the HIF transcription factor when bound to HIF1β (also known as ARNT), and is well recognized for its ability to stimulate a glycolytic shift. This is achieved through oncogenic RAS-induced concurrent activation of MAPK and PI3K effector pathways leading to the stimulation of mTOR activity and mTOR-mediated cap-dependent translation of HIF1α110,111. RAS-dependent upregulation of HIF1α has been implicated in enhancing both the transport and the glycolytic capture of glucose, as well as its processing to biosynthetic intermediates. With respect to the transport and capture of glucose, oncogenic RAS increases the transcription of the glucose transporter GLUT1 (also known as SLC2A1), thus conferring cells with an increased capacity to take up glucose112–117. With respect to the processing of biosynthetic intermediates , oncogenic RAS leads to an increase in the levels of key glycolytic enzymes, such as hexokinase, phosphofructokinase and lactate dehydrogenase117–121. Hence, oncogenic RAS directly contributes to metabolic reactions that promote the use of glucose as an anabolic substrate in generating building material for cellular growth106 (FIG. 4). Whether this contribution is solely attributable to the upregulation of HIF1α by oncogenic RAS or whether it might involve other RAS targets remains to be established.
Figure 4. Effect of RAS on energy metabolism in cancer cells: generating macromolecular precursors.

ERK and PI3K signalling downstream of oncogenic RAS converge to activate mTOR by inhibiting its negative regulators tuberin (TSC2) and liver kinase B1 (LKB1)–AMP-activated protein kinase (AMPK)116. TSC2 can be directly phosphorylated by both ERK and ERK-activated ribosomal protein S6 kinase (RSK) on S664 and S1798, respectively, as well as by AKT (on S939 and T1362), and, likewise, RAF–ERK1 or RAF–ERK2 signalling disrupts the LKB1–AMPK checkpoint200–204. This leads to mTOR–eukaryotic translation initiation factor 4 (eIF4)-dependent translation of hypoxia-inducible factor 1α (HIF1α). Activated RAS can also result in the transcriptional upregulation of HIF1A. Increased levels of HIF1α augment multiple steps in glycolytic metabolism (shown in blue). The upregulation of hexokinase (HK) facilitates the conversion of glucose to glucose-6-phosphate, a glycolytic intermediate that is used in pentose phosphate pathway-dependent nucleotide synthesis205. Higher levels of phosphofructokinase (PFK) lead to an enhanced glycolytic flux and the production of pyruvate, which, in conjunction with the oncogenic RAS-dependent increase in lactose dehydrogenase (LDH) levels, can allow glycolysis to persist by regenerating NAD+, a necessary cofactor for glycolytic reactions109,120,121. In addition, some of the pyruvate can enter the tricarboxylic acid (TCA) cycle where its conversion to citrate generates intermediates that are necessary for the synthesis of fatty acids and non-essential amino acids205. The asterisk represents the mutational activation of RAS. GLUT1, glucose transporter 1.
Another avenue through which oncogenic RAS interfaces with cellular metabolism is by affecting autophagy — a process of self-consumption that generates energy, as well as building blocks that are necessary for cellular survival, and that supports organelle homeostasis122. Although autophagy has been shown to have both tumour-suppressive and tumour-promoting qualities, recent studies have implicated oncogenic RAS in the upregulation of the autophagic processes, resulting in the upkeep of mitochondria, an increase in glycolytic rate and cellular viability, and ultimately supporting tumour growth in vivo123–125. Such dependency requires essential autophagy genes such as ATG5 and ATG7, as well as the autophagy cargo receptor p62 (REF. 125). With the rapid expansion of the field of cancer metabolism, we are likely to encounter an increasing number of molecular interactions that link metabolic nodes to oncogenic RAS signalling.
Remodeling the microenvironment
For decades RAS has been the prime example of a potent cell-autonomous oncogene. It is, however, becoming increasingly evident that the effects of oncogenic RAS stretch further to include non-cell-autonomous changes in the cellular microenvironment that have essential roles in tumour initiation and progression. A prime example of such an effect is the induction of the outgrowth of new blood vessels, or angiogenesis, which allows cells within the evolving tumour to accommodate the physiological need for an adequate supply of oxygen and nutrients126,127. The mechanisms by which RAS activation initiates and sustains pro-angiogenic processes are complex and impinge on the modulation of levels of endothelial growth factors and also increase local inflammation and stromal remodelling128–130 (FIG. 5).
Figure 5. RAS and angiogenesis.

a. The induction of pro-angiogenic growth factors (vascular endothelial growth factor A (VEGFA) and fibroblast growth factor 2 (FGF2)) by RAS in neoplastic cells is shown. RAS enhances the transcription of VEGFA by recruiting transcription factors such as SP1, SP2, AP2 and ETS to the VEGFA promoter. RAS also increases the stability of VEGFA mRNA and augments its translation206–213. The RAS–JUN N-terminal kinase (JNK) signalling axis is responsible for upregulating the transcription of prostaglandin-endoperoxide synthase 2 (PTGS2), which encodes COX2, by activating JUN, a component of the AP1 transcription complex, whereas the RAS–ERK1 and ERK2 pathway contributes to COX2 expression through the phosphorylation of CCAAT/enhancer binding protein-β (C/EBPβ) and ETS transcription factors such as PEA3 (REFS 214–217). Expression of COX2, in turn, increases the levels of VEGFA produced by RAS-transformed cells. b. The release of proteases by neoplastic cells cleaves components of the extracellular matrix (ECM) and releases VEGFA and FGF2, which are trapped in the ECM. Expression of proteases urokinase-type plasminogen activator (uPA), matrix metalloproteinase 2 (MMP2) and MMP9 in RAS-transformed cells is increased by the combined effects of ETS transcription factors (activated by the RAF–ERK pathway) and JUN (activated by the RAC–JNK pathway) binding to the promoters of PEA3–AP1 sites, as well as enhanced translation of polysome-associated MMP9 mRNA144,218,219. Stimulation of uPA expression is also dependent on RAS-mediated activation of RAL GTPase220,221. This induces neo-proliferation and sprouting of microvessels towards the tumour site. c. The recruitment of macrophages by neoplastic cells (through RAS-induced nuclear factor-κB (NF-κB)-dependent production of the cytokines interleukin-6 (IL-6) and IL-8) and subsequent promotion of endothelial proliferation and sprouting by newly recruited macrophages is shown.
Vascular endothelial growth factor A (VEGFA), which is a key player in the induction of endothelial cell proliferation and the sprouting of new blood vessels, is a well-recognized target of oncogenic RAS. In fact, the disruption of either KRASG12V or KRASG13D expression and the subsequent reduction in VEGFA levels leads to diminished tumour growth127,131. Oncogenic RAS-mediated upregulation of VEGFA involves the activation of multiple signalling cascades that eventually culminate in the stabilization of the pro-angiogenic transcription factor HIF1α, boosting its transactivation potential at the VEGFA promoter132–135. There is also another pathway by which RAS can upregulate VEGFA: via the pro-angiogenic enzyme cyclooxygenase 2 (COX2), which, through the production of prostaglandins, leads to the enhancement of the cyclic AMP-dependent transcription of VEGFA129. COX2 can also increase the levels of a plethora of other endothelial growth factors, such as basic fibroblast growth factor (bFGF; also known as FGF2) and platelet-derived growth factor (PDGF), and is required for integrin-mediated endothelial cell spreading and migration136,137.
Oncogenic HRASG12V-mediated production of pro-inflammatory cytokines has emerged as another contributor to the induction of angiogenesis130. Several cytokines have been implicated in this response, including interleukin-8 (IL-8), IL-6 and GRO1 (also known as CXCL1), and their RAS-dependent upregulation is predominantly mediated by the activation of signalling pathways that impinge on the transcriptional machinery controlling their expression130,138. Once produced, the pro-inflammatory cytokines recruit immune cells, such as neutrophils and macrophages, which produce angiogenic growth factors138–140.
Cancer cells and the pro-angiogenic growth factors that they produce are often physically trapped by the extensive network of the extracellular matrix (ECM). Hence, the modification of the surrounding ECM is necessary both for the growth factors to reach the target endothelium and for the migration of the newly generated endothelium into the tumour129,141,142. Oncogenic HRASG12V-mediated upregulation of matrix metalloproteinase 2 (MMP2), MMP9 and urokinase-type plasminogen activator (uPA) has been shown to be instrumental in removing the physical confines of the basement membrane, with upregulation of uPA especially important in potentiating endothelial cell migration and vessel sprouting129,143–148. Oncogenic RAS can also promote the angiogenic process by restricting the expression of negative regulators of neo-vascularization, such as thrombospondin 1 (TSP1; also known as THBS1) and TSP2 (also known as THBS2), in tumour cells149,150. These extracellular glycoproteins interact with components of the ECM to restrict the availability of endothelial growth factors and chemokines to the vascular system. In addition, they directly affect the viability of endothelial cells151,152. Overall, the role of oncogenic RAS in the development of tumour vasculature represents a compendium of cell-autonomous and non-cell-autonomous functions that are engaged together to activate the pro-angiogenic programme.
Evasion of the immune response
The emergence of a tumour in spite of an immune system that in principle should be able to recognize it as a foreign entity raises the question of how a cancer cell evades such surveillance. Thus far, two mechanisms by which oncogenic RAS can subvert antitumour immunity have surfaced.
First, oncogenic activation of RAS reduces the surface expression of antigen-presenting major histocompatibility complexes (MHC) on cancer cells, and such downregulation results in decreased immunogenicity of the RAS-transformed cells153–157. The oncogenic RAS-mediated downregulation of MHC was shown to be independent of the promoter activity at MHC loci, suggesting that defects in the components of antigen-processing machinery could be responsible for the compromised antigenic peptide transport and loading154,158,159. Indeed, transformation by RAS reduced the levels and functionality of the antigen peptide transporters TAP1 and TAP2 and proteasome subunits LMP2 (also known as PSMB9) and LMP7 (also known as PSMB8), resulting in decreased MHC expression160,161.
Second, data from human cancers and transgenic mouse models indicate that RAS-driven cancers possess the capacity to overcome host-protecting adaptive immune responses162. Thus, although T cells specific for the mutated RAS antigens can be found in patients with melanoma, pancreatic and colon cancers, they are often anergic and so inactive towards the tumour158,163–166. This concept is supported by recent experimental evidence from a mouse model of oncogenic KRASG12D-induced lung cancer, demonstrating that the initial immune response becomes substantially attenuated, eventually leading to a full escape from immune surveillance167. One possible mechanism by which oncogenic RAS expression may lead to a compromised antitumour immune response is through the recruitment of immunosuppressive regulatory T cells (TRegs) and myeloid-derived suppressor cells (MDSCs) to the tumour site168,169. The potential importance of this mode of immune modulation for the tumorigenic process is suggested by the observation that TRegs are required for cancer formation in the mouse model of KRASG12V-initiated lung tumorigenesis170. Whether RAS-transformed cells influence the immune response through direct recruitment of immunosuppressive cells or in conjunction with the induction of an inflammatory response remains to be elucidated171,172.
Metastasis
Among the most threatening aspects of an evolving tumour is the acquisition of metastatic properties, whereby the cancer cells spread to the surrounding and distant organs. Many metastatic tumours (such as lung, pancreas and colon tumours) contain RAS mutations. This fact, along with the demonstration that oncogenic mutants of RAS could confer metastatic properties to mouse cells in culture, served as a foundation for a large body of work that aimed to understand the role of oncogenic RAS in metastatic tumour spread. Although far from complete, the picture that has emerged so far implicates RAS in multiple cellular processes that endow cells with metastatic potential.
The initial step in the metastatic cascade is the establishment of local tumour cell invasion, a process that relies on the ability of tumour cells to break away from the primary tumour. Oncogenic RAS contributes to this process by inducing alterations in cell–cell and cell–matrix interactions and the acquisition of a migratory phenotype. The perturbation of cell–cell contacts by oncogenic RAS is accomplished through the targeting of the molecular machinery that maintains intercellular adhesion junctions, which includes the calcium-dependent E-cadherin receptor and its associated cytoplasmic protein β-catenin173–176. Thus, the expression of oncogenic RAS reduces the levels of E-cadherin through the upregulation of the E-cadherin transcriptional repressors SNAIL (also known as SNAI1) and SLUG (also known as SNAI2), the stimulation of E-cadherin proteolytic degradation and the induction of E-cadherin promoter methylation177–179. RAS activation has also been shown to induce the destabilization of E-cadherin–β-catenin complexes and the relocalization of β-catenin174,180–182. Along with the weakening of cell–cell interactions, oncogenic RAS expression reduces attachments to the ECM by downregulating integrin subunits (such as integrin α5β1) that facilitate the maintenance of stable adhesion complexes183–187. Finally, oncogenic RAS directly contributes to the enhanced motility of cancer cells by affecting pronounced changes in the polymerization, organization and contraction of actin; the polymerization and/or stability of microtubules; and the transcriptional regulation of mitogenic gene products188. Collectively, these changes establish the front–rear asymmetry that is required for cell migration.
Progression through the metastatic process requires the cancer cell to leave the confines of the primary tumour and to enter the blood or lymphatic system (intravasation). Crucial for the execution of this step is the capacity to invade through the physical barrier that is imposed by the basement membrane. The link between oncogenic RAS expression and the acquisition of the invasive phenotype has been attributed to alterations in cellular activities that control ECM degradation. Specifically, signalling pathways that are downstream of constitutively activated RAS can increase the expression and/or activity of various ECM proteases and in parallel can decrease the expression of protease inhibitors. Oncogenic RAS is also thought to contribute to the capacity of tumour cells to migrate through the circulatory system by protecting them from matrix deprivation-induced apoptosis, or anoikis77,80,127,174,189–191.
Given the multitude of cellular activities on which tumour metastasis relies, it is not surprising that oncogenic RAS promotes this aspect of the transformed phenotype by engaging a diverse and broad platform of effector mechanisms. RAS-dependent signalling pathways that have been demonstrated to have an essential role in metastatic progression include the RAS–MAPK, RAS–PI3K, RAS–RAL GTPase and RAS–RHO GTPase pathways182,188. Each of these pathways can promote the metastatic process at multiple steps. For example, the activation of RHO GTPases leads to concurrent alterations in cell adhesion and cell motility. In addition, the identity of RAS-dependent signals that promote metastasis has been shown to vary substantially depending on tissue type and genetic background192–196. Furthermore, in some settings, the induction of metastasis is the product of cooperation between oncogenic RAS and other metastasis-promoting pathways, such as the TGFβ pathway177,197. Thus, defining the precise modes by which RAS-responsive pathways affect metastatic capacity awaits an improved understanding of the context-dependent outcome of their coordinated activation.
The road ahead
As research on oncogenic RAS is entering its fourth decade, the information it has generated thus far serves as a rich and instructive backdrop for the challenges and opportunities that lie ahead. A unifying concept that emerges from the large number of genetic, biochemical and cell biological studies is that the oncogenic potential of RAS manifests in a context-dependent manner. Thus, the subcellular, cellular and tissue environments within which oncogenic RAS operates crucially determine its functional output. In addition, depending on the genetic landscape of an individual cell, different RAS-dependent oncogenic activities might become more or less important during tumour evolution. Although the task of developing a mechanistic understanding of how these determinants dictate a specific pathological outcome may seem daunting, the outpouring over the past few years of highly refined experimental tools to address these questions holds promise for considerable advances.
At the subcellular level, RAS proteins have been shown to reside in distinct compartments within the cell, with each compartment eliciting differential signalling outputs that may control various aspects of oncogenic transformation198,199. Recent advancements in the development of multi-parameter fluorescent reporters and biosensors, along with improved access to high-sensitivity real-time imaging techniques, should provide important insights into the spatiotemporal coordination of oncogenic RAS signalling in live cells. At the cellular and tissue level, our capacity to probe the in vivo ramifications of the expression of oncogenic RAS has been continuously improving owing to an ever-growing collection of sophisticated genetically engineered mouse models that feature activating mutations in RAS. By affording tissue- and cell-specific expression in a time-controlled and reversible manner, these models often recapitulate the genetic and biological evolution of human cancers. As such, their future use could not only augment the understanding of the crucial mediators of RAS-driven oncogenesis but could also be instrumental in testing and developing novel targeting strategies directed at RAS. Finally, signalling networks that are triggered by oncogenic RAS within the cell are complex and highly dynamic. Computational approaches that are designed to model how the integration of multi-pathway networks determines their biological output will clearly be an area of intense investigation in the years to come. Equipped with these tools we might be in a unique position to translate major advances in basic research on the RAS oncogenes into meaningful clinical benefits.
Acknowledgments
The authors’ work was supported by the US National Institutes of Health Grants CA055360 and GM078266 (D.B.-S.), the Ruth L. Kirschstein National Service Award 1F32CA13922 (E.G.) and the Irvington Institute Fellowship Program of the Cancer Research Institute (Y.P.-G). The authors would like to apologize to all their colleagues whose work was not included owing to space constraints.
Footnotes
Competing interests statement The authors declare no competing financial interests.
FURTHER INFORMATION COSMIC database:
http://www.sanger.ac.uk/genetics/CGP/cosmic/
SEER Cancer Statistics Review: http://seer.cancer.gov/csr/1975_2008/index.html
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Scheidig AJ, Burmester C, Goody RS. The pre-hydrolysis state of p21(ras) in complex with GTP: new insights into the role of water molecules in the GTP hydrolysis reaction of ras-like proteins. Structure. 1999;7:1311–1324. doi: 10.1016/s0969-2126(00)80021-0. [DOI] [PubMed] [Google Scholar]
- 4.Buhrman G, Holzapfel G, Fetics S, Mattos C. Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc Natl Acad Sci USA. 2010;107:4931–4936. doi: 10.1073/pnas.0912226107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheffzek K, et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. This article described the first three-dimensional structure of the RAS–RASGAP complex, providing insight into the mechanism of GTP hydrolysis and the structural basis for the oncogenicity of RAS mutants. [DOI] [PubMed] [Google Scholar]
- 6.Kompier LC, et al. FGFR3, HRAS, KRAS, NRAS and PIK3CA mutations in bladder cancer and their potential as biomarkers for surveillance and therapy. PLoS ONE. 2010;5:e13821. doi: 10.1371/journal.pone.0013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perentesis JP, et al. RAS oncogene mutations and outcome of therapy for childhood acute lymphoblastic leukemia. Leukemia. 2004;18:685–692. doi: 10.1038/sj.leu.2403272. [DOI] [PubMed] [Google Scholar]
- 8.Burmer GC, Rabinovitch PS, Loeb LA. Frequency and spectrum of c-Ki-ras mutations in human sporadic colon carcinoma, carcinomas arising in ulcerative colitis, and pancreatic adenocarcinoma. Environ Health Perspect. 1991;93:27–31. doi: 10.1289/ehp.919327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Capella G, Cronauer-Mitra S, Pienado MA, Perucho M. Frequency and spectrum of mutations at codons 12 and 13 of the c-K-ras gene in human tumors. Environ Health Perspect. 1991;93:125–131. doi: 10.1289/ehp.9193125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riely GJ, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res. 2008;14:5731–5734. doi: 10.1158/1078-0432.CCR-08-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keohavong P, et al. Detection of K-ras mutations in lung carcinomas: relationship to prognosis. Clin Cancer Res. 1996;2:411–418. [PubMed] [Google Scholar]
- 12.Andreyev HJ, Norman AR, Cunningham D, Oates JR, Clarke PA. Kirsten ras mutations in patients with colorectal cancer: the multicenter “RASCAL” study. J Natl Cancer Inst. 1998;90:675–684. doi: 10.1093/jnci/90.9.675. [DOI] [PubMed] [Google Scholar]
- 13.Goody RS, et al. Studies on the structure and mechanism of H-ras p21. Philos Trans R Soc Lond B Biol Sci. 1992;336:3–10. doi: 10.1098/rstb.1992.0037. discussion 10–11. [DOI] [PubMed] [Google Scholar]
- 14.Al-Mulla F, Milner-White EJ, Going JJ, Birnie GD. Structural differences between valine-12 and aspartate-12 Ras proteins may modify carcinoma aggression. J Pathol. 1999;187:433–438. doi: 10.1002/(SICI)1096-9896(199903)187:4<433::AID-PATH273>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 15.Seeburg PH, Colby WW, Capon DJ, Goeddel DV, Levinson AD. Biological properties of human c-Ha-ras1 genes mutated at codon 12. Nature. 1984;312:71–75. doi: 10.1038/312071a0. [DOI] [PubMed] [Google Scholar]
- 16.Edkins S, et al. Recurrent KRAS codon 146 mutations in human colorectal cancer. Cancer Biol Ther. 2006;5:928–932. doi: 10.4161/cbt.5.8.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tyner JW, et al. High-throughput sequencing screen reveals novel, transforming RAS mutations in myeloid leukemia patients. Blood. 2009;113:1749–1755. doi: 10.1182/blood-2008-04-152157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Roock W, et al. Association of KRAS p. G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304:1812–1820. doi: 10.1001/jama.2010.1535. [DOI] [PubMed] [Google Scholar]
- 19.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 20.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nature Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bar-Sagi D. A Ras by any other name. Mol Cell Biol. 2001;21:1441–1443. doi: 10.1128/MCB.21.5.1441-1443.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guerra C, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–120. doi: 10.1016/s1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 23.Tuveson DA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 24.Braun BS, et al. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci USA. 2004;101:597–602. doi: 10.1073/pnas.0307203101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.To MD, et al. Kras regulatory elements and exon 4A determine mutation specificity in lung cancer. Nature Genet. 2008;40:1240–1244. doi: 10.1038/ng.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haigis KM, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nature Genet. 2008;40:600–608. doi: 10.1038/ngXXXX. In this study, the authors engineered a knock-in system for the endogenous expression of KRAS or NRAS oncogenic mutants to document, for the first time, that the different RAS isoforms have distinct biological functions during tumorigenesis in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Q, et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood. 2011;117:2022–2032. doi: 10.1182/blood-2010-04-280750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schubbert S, et al. Germline KRAS mutations cause Noonan syndrome. Nature Genet. 2006;38:331–336. doi: 10.1038/ng1748. This study identified novel germline activating KRAS mutations in patients with Noonan and cardio-facio-cutaneous syndromes. These mutations lead to milder biochemical activation of RAS than seen in cancer, suggesting why such alterations might be tolerated during development. [DOI] [PubMed] [Google Scholar]
- 29.Tidyman WE, Rauen KA. Noonan, Costello and cardio-facio-cutaneous syndromes: dysregulation of the Ras-MAPK pathway. Expert Rev Mol Med. 2008;10:e37. doi: 10.1017/S1462399408000902. [DOI] [PubMed] [Google Scholar]
- 30.Potenza N, et al. Replacement of K-Ras with H-Ras supports normal embryonic development despite inducing cardiovascular pathology in adult mice. EMBO Rep. 2005;6:432–437. doi: 10.1038/sj.embor.7400397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stacey DW, Kung HF. Transformation of NIH 3T3 cells by microinjection of Ha-ras p21 protein. Nature. 1984;310:508–511. doi: 10.1038/310508a0. [DOI] [PubMed] [Google Scholar]
- 32.Feramisco JR, Gross M, Kamata T, Rosenberg M, Sweet RW. Microinjection of the oncogene form of the human H-ras (T-24) protein results in rapid proliferation of quiescent cells. Cell. 1984;38:109–117. doi: 10.1016/0092-8674(84)90531-2. References 31 and 32 were the first studies to demonstrate that microinjection of purified oncogenic RAS induced dramatic morphological changes and stimulated the proliferation of quiescent cells. [DOI] [PubMed] [Google Scholar]
- 33.McCarthy SA, Samuels ML, Pritchard CA, Abraham JA, McMahon M. Rapid induction of heparin-binding epidermal growth factor/diphtheria toxin receptor expression by Raf and Ras oncogenes. Genes Dev. 1995;9:1953–1964. doi: 10.1101/gad.9.16.1953. [DOI] [PubMed] [Google Scholar]
- 34.Gangarosa LM, et al. A raf-independent epidermal growth factor receptor autocrine loop is necessary for Ras transformation of rat intestinal epithelial cells. J Biol Chem. 1997;272:18926–18931. doi: 10.1074/jbc.272.30.18926. [DOI] [PubMed] [Google Scholar]
- 35.Schulze A, Lehmann K, Jefferies HB, McMahon M, Downward J. Analysis of the transcriptional program induced by Raf in epithelial cells. Genes Dev. 2001;15:981–994. doi: 10.1101/gad.191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woods D, et al. Induction of β3-integrin gene expression by sustained activation of the Ras-regulated Raf-MEK-extracellular signal-regulated kinase signaling pathway. Mol Cell Biol. 2001;21:3192–3205. doi: 10.1128/MCB.21.9.3192-3205.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dajee M, Tarutani M, Deng H, Cai T, Khavari PA. Epidermal Ras blockade demonstrates spatially localized Ras promotion of proliferation and inhibition of differentiation. Oncogene. 2002;21:1527–1538. doi: 10.1038/sj.onc.1205287. [DOI] [PubMed] [Google Scholar]
- 38.Filmus J, Zhao J, Buick RN. Overexpression of H-ras oncogene induces resistance to the growth-inhibitory action of transforming growth factor β-1 (TGF-β 1) and alters the number and type of TGF-β1 receptors in rat intestinal epithelial cell clones. Oncogene. 1992;7:521–526. [PubMed] [Google Scholar]
- 39.Zhao J, Buick RN. Regulation of transforming growth factor β receptors in H-ras oncogene-transformed rat intestinal epithelial cells. Cancer Res. 1995;55:6181–6188. [PubMed] [Google Scholar]
- 40.Massague J. How cells read TGF-β signals. Nature Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 41.Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFβ/ Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kfir S, et al. Pathway- and expression level-dependent effects of oncogenic N-Ras: p27(Kip1) mislocalization by the Ral-GEF pathway and Erk-mediated interference with Smad signaling. Mol Cell Biol. 2005;25:8239–8250. doi: 10.1128/MCB.25.18.8239-8250.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daly AC, Vizan P, Hill CS. Smad3 protein levels are modulated by Ras activity and during the cell cycle to dictate transforming growth factor-β responses. J Biol Chem. 2010;285:6489–6497. doi: 10.1074/jbc.M109.043877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stacey DW, Watson T, Kung HF, Curran T. Microinjection of transforming ras protein induces c-fos expression. Mol Cell Biol. 1987;7:523–527. doi: 10.1128/mcb.7.1.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gutman A, Wasylyk C, Wasylyk B. Cell-specific regulation of oncogene-responsive sequences of the c-fos promoter. Mol Cell Biol. 1991;11:5381–5387. doi: 10.1128/mcb.11.10.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Urich M, Senften M, Shaw PE, Ballmer-Hofer K. A role for the small GTPase Rac in polyomavirus middle-T antigen-mediated activation of the serum response element and in cell transformation. Oncogene. 1997;14:1235–1241. doi: 10.1038/sj.onc.1200982. [DOI] [PubMed] [Google Scholar]
- 47.Westwick JK, et al. Oncogenic Ras activates c-Jun via a separate pathway from the activation of extracellular signal-regulated kinases. Proc Natl Acad Sci USA. 1994;91:6030–6034. doi: 10.1073/pnas.91.13.6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finco TS, et al. Oncogenic Ha-Ras-induced signaling activates NF-κB transcriptional activity, which is required for cellular transformation. J Biol Chem. 1997;272:24113–24116. doi: 10.1074/jbc.272.39.24113. [DOI] [PubMed] [Google Scholar]
- 49.Malumbres M, Pellicer A. RAS pathways to cell cycle control and cell transformation. Front Biosci. 1998;3:D887–D912. doi: 10.2741/a331. [DOI] [PubMed] [Google Scholar]
- 50.Filmus J, et al. Induction of cyclin D1 overexpression by activated ras. Oncogene. 1994;9:3627–3633. [PubMed] [Google Scholar]
- 51.Albanese C, et al. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- 52.Winston JT, Coats SR, Wang YZ, Pledger WJ. Regulation of the cell cycle machinery by oncogenic ras. Oncogene. 1996;12:127–134. [PubMed] [Google Scholar]
- 53.Liu JJ, et al. Ras transformation results in an elevated level of cyclin D1 and acceleration of G1 progression in NIH 3T3 cells. Mol Cell Biol. 1995;15:3654–3663. doi: 10.1128/mcb.15.7.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gille H, Downward J. Multiple ras effector pathways contribute to G(1) cell cycle progression. J Biol Chem. 1999;274:22033–22040. doi: 10.1074/jbc.274.31.22033. [DOI] [PubMed] [Google Scholar]
- 55.Westwick JK, et al. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol Cell Biol. 1997;17:1324–1335. doi: 10.1128/mcb.17.3.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takuwa N, Fukui Y, Takuwa Y. Cyclin D1 expression mediated by phosphatidylinositol 3-kinase through mTOR-p70(S6K)-independent signaling in growth factor-stimulated NIH 3T3 fibroblasts. Mol Cell Biol. 1999;19:1346–1358. doi: 10.1128/mcb.19.2.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science. 1993;260:1658–1661. doi: 10.1126/science.8503013. This is the first publication describing RAF as a direct effector of RAS and providing a structural basis for their interaction. [DOI] [PubMed] [Google Scholar]
- 58.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robles AI, et al. Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 1998;12:2469–2474. doi: 10.1101/gad.12.16.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–1021. doi: 10.1038/35082500. [DOI] [PubMed] [Google Scholar]
- 61.Quelle DE, et al. Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 1993;7:1559–1571. doi: 10.1101/gad.7.8.1559. [DOI] [PubMed] [Google Scholar]
- 62.Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S. phase transition by expression of cyclins D1 and E with an inducible system. Mol Cell Biol. 1994;14:1669–1679. doi: 10.1128/mcb.14.3.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rivard N, Boucher MJ, Asselin C, L’Allemain G. MAP kinase cascade is required for p27 downregulation and S. phase entry in fibroblasts and epithelial cells. Am J Physiol. 1999;277:C652–C664. doi: 10.1152/ajpcell.1999.277.4.C652. [DOI] [PubMed] [Google Scholar]
- 64.Sa G, Stacey DW. P27 expression is regulated by separate signaling pathways, downstream of Ras, in each cell cycle phase. Exp Cell Res. 2004;300:427–439. doi: 10.1016/j.yexcr.2004.07.032. [DOI] [PubMed] [Google Scholar]
- 65.Leone G, DeGregori J, Sears R, Jakoi L, Nevins JR. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature. 1997;387:422–426. doi: 10.1038/387422a0. [DOI] [PubMed] [Google Scholar]
- 66.Pruitt K, Pestell RG, Der CJ. Ras inactivation of the retinoblastoma pathway by distinct mechanisms in NIH 3T3 fibroblast and RIE-1 epithelial cells. J Biol Chem. 2000;275:40916–40924. doi: 10.1074/jbc.M006682200. [DOI] [PubMed] [Google Scholar]
- 67.Di Micco R, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 68.Bartkova J, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 69.Gorgoulis VG, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 70.Koorstra JB, et al. Widespread activation of the DNA damage response in human pancreatic intraepithelial neoplasia. Mod Pathol. 2009;22:1439–1445. doi: 10.1038/modpathol.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 72.Hagag N, Diamond L, Palermo R, Lyubsky S. High expression of ras p21 correlates with increased rate of abnormal mitosis in NIH3T3 cells. Oncogene. 1990;5:1481–1489. [PubMed] [Google Scholar]
- 73.Denko N, Stringer J, Wani M, Stambrook P. Mitotic and post mitotic consequences of genomic instability induced by oncogenic Ha-ras. Somat Cell Mol Genet. 1995;21:241–253. doi: 10.1007/BF02255779. This study provides the first evidence for an association between oncogenic RAS-induced chromosome aberrations and disruption of the mitotic machinery in the process of cellular transformation. [DOI] [PubMed] [Google Scholar]
- 74.Denko NC, Giaccia AJ, Stringer JR, Stambrook PJ. The human Ha-ras oncogene induces genomic instability in murine fibroblasts within one cell cycle. Proc Natl Acad Sci USA. 1994;91:5124–5128. doi: 10.1073/pnas.91.11.5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wani MA, Denko NC, Stambrook PJ. Expression of Rap 1 suppresses genomic instability of H-ras transformed mouse fibroblasts. Somat Cell Mol Genet. 1997;23:123–133. doi: 10.1007/BF02679971. [DOI] [PubMed] [Google Scholar]
- 76.Knauf JA, et al. Oncogenic RAS induces accelerated transition through G2/M and promotes defects in the G2 DNA damage and mitotic spindle checkpoints. J Biol Chem. 2006;281:3800–3809. doi: 10.1074/jbc.M511690200. [DOI] [PubMed] [Google Scholar]
- 77.Cox AD, Der CJ. The dark side of Ras: regulation of apoptosis. Oncogene. 2003;22:8999–9006. doi: 10.1038/sj.onc.1207111. [DOI] [PubMed] [Google Scholar]
- 78.Chin L, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–472. doi: 10.1038/22788. This study showed that downregulation of HRASG12V leads to apoptosis of tumour cells and host-derived endothelial cells, and consequently, regression of primary and explanted melanomas, indicating the need for RAS in tumour maintenance. [DOI] [PubMed] [Google Scholar]
- 79.Fisher GH, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–3262. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rosen K, et al. Downregulation of the pro-apoptotic protein Bak is required for the ras-induced transformation of intestinal epithelial cells. Curr Biol. 1998;8:1331–1334. doi: 10.1016/s0960-9822(07)00564-7. [DOI] [PubMed] [Google Scholar]
- 81.Sulciner DJ, et al. rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-κB activation. Mol Cell Biol. 1996;16:7115–7121. doi: 10.1128/mcb.16.12.7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Joneson T, Bar-Sagi D. Suppression of Ras-induced apoptosis by the Rac GTPase. Mol Cell Biol. 1999;19:5892–5901. doi: 10.1128/mcb.19.9.5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mayo MW, Baldwin AS. The transcription factor NF-κB: control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta. 2000;1470:M55–M62. doi: 10.1016/s0304-419x(00)00002-0. [DOI] [PubMed] [Google Scholar]
- 84.Nalca A, Qiu SG, El-Guendy N, Krishnan S, Rangnekar VM. Oncogenic Ras sensitizes cells to apoptosis by Par-4. J Biol Chem. 1999;274:29976–29983. doi: 10.1074/jbc.274.42.29976. [DOI] [PubMed] [Google Scholar]
- 85.Ahmed MM, et al. Downregulation of PAR-4, a pro-apoptotic gene, in pancreatic tumors harboring K-ras mutation. Int J Cancer. 2008;122:63–70. doi: 10.1002/ijc.23019. [DOI] [PubMed] [Google Scholar]
- 86.Kinoshita T, Yokota T, Arai K, Miyajima A. Regulation of Bcl-2 expression by oncogenic Ras protein in hematopoietic cells. Oncogene. 1995;10:2207–2212. [PubMed] [Google Scholar]
- 87.Wu L, Nam YJ, Kung G, Crow MT, Kitsis RN. Induction of the apoptosis inhibitor ARC by Ras in human cancers. J Biol Chem. 2010;285:19235–19245. doi: 10.1074/jbc.M110.114892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Datta SR, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 89.Fang X, et al. Regulation of BAD phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncogene. 1999;18:6635–6640. doi: 10.1038/sj.onc.1203076. [DOI] [PubMed] [Google Scholar]
- 90.Peli J, et al. Oncogenic Ras inhibits Fas ligand-mediated apoptosis by downregulating the expression of Fas. EMBO J. 1999;18:1824–1831. doi: 10.1093/emboj/18.7.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature. 2007;449:1073–1077. doi: 10.1038/nature06251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arber N. Janus faces of ras: anti or pro-apoptotic? Apoptosis. 1999;4:383–388. doi: 10.1023/a:1009651406017. [DOI] [PubMed] [Google Scholar]
- 93.Vermeulen K, Berneman ZN, Van Bockstaele DR. Cell cycle and apoptosis. Cell Prolif. 2003;36:165–175. doi: 10.1046/j.1365-2184.2003.00267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kauffmann-Zeh A, et al. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. Oncogenic RAS can suppress MYC-induced apoptosis in a PKB–AKT-dependent manner and can promote apoptosis through activation of the RAF pathway, thus RAS is capable of eliciting contradictory signals that modulate cell viability. [DOI] [PubMed] [Google Scholar]
- 95.Kennedy NJ, et al. Suppression of Ras-stimulated transformation by the JNK signal transduction pathway. Genes Dev. 2003;17:629–637. doi: 10.1101/gad.1062903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lei K, et al. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol Cell Biol. 2002;22:4929–4942. doi: 10.1128/MCB.22.13.4929-4942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bivona TG, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21:481–493. doi: 10.1016/j.molcel.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 98.Richter AM, Pfeifer GP, Dammann RH. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim Biophys Acta. 2009;1796:114–128. doi: 10.1016/j.bbcan.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 99.Vos MD, Ellis CA, Bell A, Birrer MJ, Clark GJ. Ras uses the novel tumor suppressor RASSF1 as an effector to mediate apoptosis. J Biol Chem. 2000;275:35669–35672. doi: 10.1074/jbc.C000463200. [DOI] [PubMed] [Google Scholar]
- 100.Khokhlatchev A, et al. Identification of a novel Ras-regulated proapoptotic pathway. Curr Biol. 2002;12:253–265. doi: 10.1016/s0960-9822(02)00683-8. [DOI] [PubMed] [Google Scholar]
- 101.Patra SK. Ras regulation of DNA-methylation and cancer. Exp Cell Res. 2008;314:1193–1201. doi: 10.1016/j.yexcr.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 102.Dammann R, et al. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nature Genet. 2000;25:315–319. doi: 10.1038/77083. [DOI] [PubMed] [Google Scholar]
- 103.Dammann R, et al. The tumor suppressor RASSF1A in human carcinogenesis: an update. Histol Histopathol. 2005;20:645–663. doi: 10.14670/HH-20.645. [DOI] [PubMed] [Google Scholar]
- 104.Burbee DG, et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst. 2001;93:691–699. doi: 10.1093/jnci/93.9.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fullwood P, et al. Detailed genetic and physical mapping of tumor suppressor loci on chromosome 3p in ovarian cancer. Cancer Res. 1999;59:4662–4667. [PubMed] [Google Scholar]
- 106.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–548. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 108.Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem. 1997;272:22776–22780. doi: 10.1074/jbc.272.36.22776. [DOI] [PubMed] [Google Scholar]
- 109.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Johannessen CM, et al. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci USA. 2005;102:8573–8578. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071–14077. doi: 10.1074/jbc.R109.094003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem. 2001;276:9519–9525. doi: 10.1074/jbc.M010144200. [DOI] [PubMed] [Google Scholar]
- 113.Blum R, Jacob-Hirsch J, Amariglio N, Rechavi G, Kloog Y. Ras inhibition in glioblastoma down-regulates hypoxia-inducible factor-1α, causing glycolysis shutdown and cell death. Cancer Res. 2005;65:999–1006. [PubMed] [Google Scholar]
- 114.Flier JS, Mueckler MM, Usher P, Lodish HF. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science. 1987;235:1492–1495. doi: 10.1126/science.3103217. [DOI] [PubMed] [Google Scholar]
- 115.Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci. 1999;24:68–72. doi: 10.1016/s0968-0004(98)01344-9. [DOI] [PubMed] [Google Scholar]
- 116.Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 117.Semenza GL. Hypoxia and cancer. Cancer Metastasis Rev. 2007;26:223–224. doi: 10.1007/s10555-007-9058-y. [DOI] [PubMed] [Google Scholar]
- 118.Kole HK, Resnick RJ, Van Doren M, Racker E. Regulation of 6-phosphofructo-1-kinase activity in ras-transformed rat-1 fibroblasts. Arch Biochem Biophys. 1991;286:586–590. doi: 10.1016/0003-9861(91)90084-v. [DOI] [PubMed] [Google Scholar]
- 119.Ramanathan A, Wang C, Schreiber SL. Perturbational profiling of a cell-line model of tumorigenesis by using metabolic measurements. Proc Natl Acad Sci USA. 2005;102:5992–5997. doi: 10.1073/pnas.0502267102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chiaradonna F, et al. Ras-dependent carbon metabolism and transformation in mouse fibroblasts. Oncogene. 2006;25:5391–5404. doi: 10.1038/sj.onc.1209528. This paper demonstrates that the known sensitivity of RAS-transformed cells to glucose levels is a consequence of RAS-induced global transcriptomic changes in genes associated with the shift of carbon metabolism towards glycolysis. [DOI] [PubMed] [Google Scholar]
- 121.Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/ fructose-2, 6-bisphosphatases in cancer. Exp Mol Pathol. 2009;86:174–179. doi: 10.1016/j.yexmp.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 122.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Guo JY, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lock R, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2010;22:165–178. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim MJ, et al. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem. 2011;286:12924–12932. doi: 10.1074/jbc.M110.138958. References 123–125 show that RAS-mediated upregulation of autophagy increases cell viability and tumorigenic potential through facilitation of glycolysis and mitochondrial metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. 1989;339:58–61. doi: 10.1038/339058a0. [DOI] [PubMed] [Google Scholar]
- 127.Rak J, et al. Mutant ras oncogenes upregulate VEGF/ VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res. 1995;55:4575–4580. [PubMed] [Google Scholar]
- 128.Rak J, Yu JL. Oncogenes and tumor angiogenesis: the question of vascular “supply” and vascular “demand”. Semin Cancer Biol. 2004;14:93–104. doi: 10.1016/j.semcancer.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 129.Kranenburg O, Gebbink MF, Voest EE. Stimulation of angiogenesis by Ras proteins. Biochim Biophys Acta. 2004;1654:23–37. doi: 10.1016/j.bbcan.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 130.Ancrile BB, O’Hayer KM, Counter CM. Oncogenic ras-induced expression of cytokines: a new target of anti-cancer therapeutics. Mol Interv. 2008;8:22–27. doi: 10.1124/mi.8.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tokunaga T, et al. Ribozyme-mediated inactivation of mutant K-ras oncogene in a colon cancer cell line. Br J Cancer. 2000;83:833–839. doi: 10.1054/bjoc.2000.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jung F, et al. Hypoxic induction of the hypoxia-inducible factor is mediated via the adaptor protein Shc in endothelial cells. Circ Res. 2002;91:38–45. doi: 10.1161/01.res.0000024412.24491.ca. [DOI] [PubMed] [Google Scholar]
- 133.Blancher C, Moore JW, Robertson N, Harris AL. Effects of ras and von Hippel-Lindau (VHL) gene mutations on hypoxia-inducible factor (HIF)-1αl, HIF-2α, and vascular endothelial growth factor expression and their regulation by the phosphatidylinositol 3′-kinase/Akt signaling pathway. Cancer Res. 2001;61:7349–7355. [PubMed] [Google Scholar]
- 134.Lee E, Yim S, Lee SK, Park H. Two transactivation domains of hypoxia-inducible factor-1α regulated by the MEK-1/p42/p44 MAPK pathway. Mol Cells. 2002;14:9–15. [PubMed] [Google Scholar]
- 135.Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1α (HIF-1α) and enhance the transcriptional activity of HIF-1. J Biol Chem. 1999;274:32631–32637. doi: 10.1074/jbc.274.46.32631. [DOI] [PubMed] [Google Scholar]
- 136.Tsujii M, et al. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- 137.Dormond O, Foletti A, Paroz C, Ruegg C. NSAIDs inhibit α V β 3 integrin-mediated and Cdc42/ Rac-dependent endothelial-cell spreading, migration and angiogenesis. Nature Med. 2001;7:1041–1047. doi: 10.1038/nm0901-1041. [DOI] [PubMed] [Google Scholar]
- 138.Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–458. doi: 10.1016/j.ccr.2004.09.028. This study demonstrates that oncogenic RAS-mediated production of the cytokine IL-8 has a vital role in neo-angiogenesis and tumour growth by instigating inflammatory reactions in the neoplastic microenvironment. [DOI] [PubMed] [Google Scholar]
- 139.Borrello MG, Degl’Innocenti D, Pierotti MA. Inflammation and cancer: the oncogene-driven connection. Cancer Lett. 2008;267:262–270. doi: 10.1016/j.canlet.2008.03.060. [DOI] [PubMed] [Google Scholar]
- 140.Feng Y, Santoriello C, Mione M, Hurlstone A, Martin P. Live imaging of innate immune cell sensing of transformed cells in zebrafish larvae: parallels between tumor initiation and wound inflammation. PLoS Biol. 2010;8:e1000562. doi: 10.1371/journal.pbio.1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bergers G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Engelse MA, Hanemaaijer R, Koolwijk P, van Hinsbergh VW. The fibrinolytic system and matrix metalloproteinases in angiogenesis and tumor progression. Semin Thromb Hemost. 2004;30:71–82. doi: 10.1055/s-2004-822972. [DOI] [PubMed] [Google Scholar]
- 143.Testa JE, Medcalf RL, Cajot JF, Schleuning WD, Sordat B. Urokinase-type plasminogen activator biosynthesis is induced by the EJ-Ha-ras oncogene in CL26 mouse colon carcinoma cells. Int J Cancer. 1989;43:816–822. doi: 10.1002/ijc.2910430513. [DOI] [PubMed] [Google Scholar]
- 144.Gum R, et al. Stimulation of 92-kDa gelatinase B promoter activity by ras is mitogen-activated protein kinase kinase 1-independent and requires multiple transcription factor binding sites including closely spaced PEA3/ets and AP-1 sequences. J Biol Chem. 1996;271:10672–10680. doi: 10.1074/jbc.271.18.10672. [DOI] [PubMed] [Google Scholar]
- 145.Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;21:1104–1117. doi: 10.1161/hq0701.093685. [DOI] [PubMed] [Google Scholar]
- 146.Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nature Rev Mol Cell Biol. 2002;3:932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 147.Carmeliet P, Collen D. Development and disease in proteinase-deficient mice: role of the plasminogen, matrix metalloproteinase and coagulation system. Thromb Res. 1998;91:255–285. doi: 10.1016/s0049-3848(98)00122-4. [DOI] [PubMed] [Google Scholar]
- 148.Brodsky S, et al. Plasmin-dependent and -independent effects of plasminogen activators and inhibitor-1 on ex vivo angiogenesis. Am J Physiol Heart Circ Physiol. 2001;281:H1784–H1792. doi: 10.1152/ajpheart.2001.281.4.H1784. [DOI] [PubMed] [Google Scholar]
- 149.Zabrenetzky V, Harris CC, Steeg PS, Roberts DD. Expression of the extracellular matrix molecule thrombospondin inversely correlates with malignant progression in melanoma, lung and breast carcinoma cell lines. Int J Cancer. 1994;59:191–195. doi: 10.1002/ijc.2910590209. [DOI] [PubMed] [Google Scholar]
- 150.Rak J, et al. Oncogenes and tumor angiogenesis: differential modes of vascular endothelial growth factor up-regulation in ras-transformed epithelial cells and fibroblasts. Cancer Res. 2000;60:490–498. [PubMed] [Google Scholar]
- 151.Volpert OV, et al. Inhibition of angiogenesis by thrombospondin-2. Biochem Biophys Res Commun. 1995;217:326–332. doi: 10.1006/bbrc.1995.2780. [DOI] [PubMed] [Google Scholar]
- 152.Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med. 2002;6:1–12. doi: 10.1111/j.1582-4934.2002.tb00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Maudsley DJ, Bateman WJ, Morris AG. Reduced stimulation of helper T cells by Ki-ras transformed cells. Immunology. 1991;72:277–281. [PMC free article] [PubMed] [Google Scholar]
- 154.Lohmann S, Wollscheid U, Huber C, Seliger B. Multiple levels of MHC class I down-regulation by ras oncogenes. Scand J Immunol. 1996;43:537–544. doi: 10.1046/j.1365-3083.1996.d01-73.x. [DOI] [PubMed] [Google Scholar]
- 155.Seliger B, et al. Suppression of MHC class I antigens in oncogenic transformants: association with decreased recognition by cytotoxic T lymphocytes. Exp Hematol. 1996;24:1275–1279. [PubMed] [Google Scholar]
- 156.Ehrlich T, et al. The effect of H-ras expression on tumorigenicity and immunogenicity of Balb/c 3T3 fibroblasts. Immunol Lett. 1993;39:3–8. doi: 10.1016/0165-2478(93)90156-v. [DOI] [PubMed] [Google Scholar]
- 157.Testorelli C, et al. Dacarbazine-induced immunogenicity of a murine leukemia is attenuated in cells transfected with mutated K-ras gene. J Exp Clin Cancer Res. 1997;16:15–22. [PubMed] [Google Scholar]
- 158.Weijzen S, Velders MP, Kast WM. Modulation of the immune response and tumor growth by activated Ras. Leukemia. 1999;13:502–513. doi: 10.1038/sj.leu.2401367. [DOI] [PubMed] [Google Scholar]
- 159.Sers C, et al. Down-regulation of HLA Class I and NKG2D ligands through a concerted action of MAPK and DNA methyltransferases in colorectal cancer cells. Int J Cancer. 2009;125:1626–1639. doi: 10.1002/ijc.24557. [DOI] [PubMed] [Google Scholar]
- 160.Seliger B, et al. Down-regulation of the MHC class I antigen-processing machinery after oncogenic transformation of murine fibroblasts. Eur J Immunol. 1998;28:122–133. doi: 10.1002/(SICI)1521-4141(199801)28:01<122::AID-IMMU122>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 161.Delp K, Momburg F, Hilmes C, Huber C, Seliger B. Functional deficiencies of components of the MHC class I antigen pathway in human tumors of epithelial origin. Bone Marrow Transplant. 2000;25 (Suppl 2):S88–S95. doi: 10.1038/sj.bmt.1702363. [DOI] [PubMed] [Google Scholar]
- 162.Clark CE, Beatty GL, Vonderheide RH. Immunosurveillance of pancreatic adenocarcinoma: insights from genetically engineered mouse models of cancer. Cancer Lett. 2009;279:1–7. doi: 10.1016/j.canlet.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 163.Kubuschok B, et al. Naturally occurring T-cell response against mutated p21 ras oncoprotein in pancreatic cancer. Clin Cancer Res. 2006;12:1365–1372. doi: 10.1158/1078-0432.CCR-05-1672. [DOI] [PubMed] [Google Scholar]
- 164.Fossum B, Olsen AC, Thorsby E, Gaudernack G. CD8+ T cells from a patient with colon carcinoma, specific for a mutant p21-Ras-derived peptide (Gly13-->Asp), are cytotoxic towards a carcinoma cell line harbouring the same mutation. Cancer Immunol Immunother. 1995;40:165–172. doi: 10.1007/BF01517348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Qin H, et al. CD4+ T-cell immunity to mutated ras protein in pancreatic and colon cancer patients. Cancer Res. 1995;55:2984–2987. [PubMed] [Google Scholar]
- 166.Gjertsen MK, Gaudernack G. Mutated Ras peptides as vaccines in immunotherapy of cancer. Vox Sang. 1998;74 (Suppl 2):489–495. doi: 10.1111/j.1423-0410.1998.tb05462.x. [DOI] [PubMed] [Google Scholar]
- 167.DuPage M, et al. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell. 2011;19:72–85. doi: 10.1016/j.ccr.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Clark CE, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 169.Tran Thang NN, et al. Immune infiltration of spontaneous mouse astrocytomas is dominated by immunosuppressive cells from early stages of tumor development. Cancer Res. 2010;70:4829–4839. doi: 10.1158/0008-5472.CAN-09-3074. [DOI] [PubMed] [Google Scholar]
- 170.Granville CA, et al. A central role for Foxp3+ regulatory T cells in K-Ras-driven lung tumorigenesis. PLoS ONE. 2009;4:e5061. doi: 10.1371/journal.pone.0005061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ji H, et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene. 2006;25:2105–2112. doi: 10.1038/sj.onc.1209237. [DOI] [PubMed] [Google Scholar]
- 172.Soudja SM, et al. Tumor-initiated inflammation overrides protective adaptive immunity in an induced melanoma model in mice. Cancer Res. 2010;70:3515–3525. doi: 10.1158/0008-5472.CAN-09-4354. [DOI] [PubMed] [Google Scholar]
- 173.Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nature Rev Mol Cell Biol. 2003;4:657–665. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- 174.Smakman N, Borel Rinkes IH, Voest EE, Kranenburg O. Control of colorectal metastasis formation by K-Ras. Biochim Biophys Acta. 2005;1756:103–114. doi: 10.1016/j.bbcan.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 175.Muschel RJ, Williams JE, Lowy DR, Liotta LA. Harvey ras induction of metastatic potential depends upon oncogene activation and the type of recipient cell. Am J Pathol. 1985;121:1–8. [PMC free article] [PubMed] [Google Scholar]
- 176.Bondy GP, Wilson S, Chambers AF. Experimental metastatic ability of H-ras-transformed NIH3T3 cells. Cancer Res. 1985;45:6005–6009. [PubMed] [Google Scholar]
- 177.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 178.Schmidt CR, et al. E-cadherin is regulated by the transcriptional repressor SLUG during Ras-mediated transformation of intestinal epithelial cells. Surgery. 2005;138:306–312. doi: 10.1016/j.surg.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 179.Horiguchi K, et al. Role of Ras signaling in the induction of snail by transforming growth factor-β. J Biol Chem. 2009;284:245–253. doi: 10.1074/jbc.M804777200. [DOI] [PubMed] [Google Scholar]
- 180.Oft M, Heider KH, Beug H. TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998;8:1243–1252. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- 181.Fujimoto K, Sheng H, Shao J, Beauchamp RD. Transforming growth factor-β1 promotes invasiveness after cellular transformation with activated Ras in intestinal epithelial cells. Exp Cell Res. 2001;266:239–249. doi: 10.1006/excr.2000.5229. [DOI] [PubMed] [Google Scholar]
- 182.Giehl K. Oncogenic Ras in tumour progression and metastasis. Biol Chem. 2005;386:193–205. doi: 10.1515/BC.2005.025. [DOI] [PubMed] [Google Scholar]
- 183.Plantefaber LC, Hynes RO. Changes in integrin receptors on oncogenically transformed cells. Cell. 1989;56:281–290. doi: 10.1016/0092-8674(89)90902-1. [DOI] [PubMed] [Google Scholar]
- 184.Danen EH, Yamada KM. Fibronectin, integrins, and growth control. J Cell Physiol. 2001;189:1–13. doi: 10.1002/jcp.1137. [DOI] [PubMed] [Google Scholar]
- 185.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nature Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 186.Schramm K, et al. Activated K-ras is involved in regulation of integrin expression in human colon carcinoma cells. Int J Cancer. 2000;87:155–164. doi: 10.1002/1097-0215(20000715)87:2<155::aid-ijc1>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 187.Pollock CB, Shirasawa S, Sasazuki T, Kolch W, Dhillon AS. Oncogenic K-RAS is required to maintain changes in cytoskeletal organization, adhesion, and motility in colon cancer cells. Cancer Res. 2005;65:1244–1250. doi: 10.1158/0008-5472.CAN-04-1911. [DOI] [PubMed] [Google Scholar]
- 188.Campbell PM, Der CJ. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin Cancer Biol. 2004;14:105–114. doi: 10.1016/j.semcancer.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 189.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. This study documents the capacity of oncogenic HRAS to abrogate the apoptotic barrier to malignancy initiated by matrix detachment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhang YA, Nemunaitis J, Scanlon KJ, Tong AW. Anti-tumorigenic effect of a K-ras ribozyme against human lung cancer cell line heterotransplants in nude mice. Gene Ther. 2000;7:2041–2050. doi: 10.1038/sj.gt.3301331. [DOI] [PubMed] [Google Scholar]
- 191.Rosen K, et al. Activated Ras prevents downregulation of Bcl-X(L) triggered by detachment from the extracellular matrix. A mechanism of Ras-induced resistance to anoikis in intestinal epithelial cells. J Cell Biol. 2000;149:447–456. doi: 10.1083/jcb.149.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zondag GC, et al. Oncogenic Ras downregulates Rac activity, which leads to increased Rho activity and epithelial-mesenchymal transition. J Cell Biol. 2000;149:775–782. doi: 10.1083/jcb.149.4.775. These authors show that oncogenic RAS signalling is capable of downregulating the activity of RAC and promoting epithelial-to-mesenchymal transition through enhancing signalling through RHO GTPase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Gupta S, Plattner R, Der CJ, Stanbridge EJ. Dissection of Ras-dependent signaling pathways controlling aggressive tumor growth of human fibrosarcoma cells: evidence for a potential novel pathway. Mol Cell Biol. 2000;20:9294–9306. doi: 10.1128/mcb.20.24.9294-9306.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Quinlan MP. Rac regulates the stability of the adherens junction and its components, thus affecting epithelial cell differentiation and transformation. Oncogene. 1999;18:6434–6442. doi: 10.1038/sj.onc.1203026. [DOI] [PubMed] [Google Scholar]
- 195.Braga VM, Betson M, Li X, Lamarche-Vane N. Activation of the small GTPase Rac is sufficient to disrupt cadherin-dependent cell-cell adhesion in normal human keratinocytes. Mol Biol Cell. 2000;11:3703–3721. doi: 10.1091/mbc.11.11.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nature Rev Cancer. 2002;2:133–142. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 197.Turley EA, Veiseh M, Radisky DC, Bissell MJ. Mechanisms of disease: epithelial-mesenchymal transition--does cellular plasticity fuel neoplastic progression? Nature Clin Pract Oncol. 2008;5:280–290. doi: 10.1038/ncponc1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rocks O, Peyker A, Bastiaens PI. Spatio-temporal segregation of Ras signals: one ship, three anchors, many harbors. Curr Opin Cell Biol. 2006;18:351–357. doi: 10.1016/j.ceb.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 199.Henis YI, Hancock JF, Prior IA. Ras acylation, compartmentalization and signaling nanoclusters (Review) Mol Membr Biol. 2009;26:80–92. doi: 10.1080/09687680802649582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Manning BD, Cantley LC. United at last: the tuberous sclerosis complex gene products connect the phosphoinositide 3-kinase/Akt pathway to mammalian target of rapamycin (mTOR) signalling. Biochem Soc Trans. 2003;31:573–578. doi: 10.1042/bst0310573. [DOI] [PubMed] [Google Scholar]
- 201.Ballif BA, et al. Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors. Proc Natl Acad Sci USA. 2005;102:667–672. doi: 10.1073/pnas.0409143102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 203.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA. 2004;101:13489–13494. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Esteve-Puig R, Canals F, Colome N, Merlino G, Recio JA. Uncoupling of the LKB1-AMPKα energy sensor pathway by growth factors and oncogenic BRAF. PLoS ONE. 2009;4:e4771. doi: 10.1371/journal.pone.0004771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Vizan P, et al. K-ras codon-specific mutations produce distinctive metabolic phenotypes in NIH3T3 mice [corrected] fibroblasts. Cancer Res. 2005;65:5512–5515. doi: 10.1158/0008-5472.CAN-05-0074. [DOI] [PubMed] [Google Scholar]
- 206.Milanini J, Vinals F, Pouyssegur J, Pages G. p42/ p44 MAP kinase module plays a key role in the transcriptional regulation of the vascular endothelial growth factor gene in fibroblasts. J Biol Chem. 1998;273:18165–18172. doi: 10.1074/jbc.273.29.18165. [DOI] [PubMed] [Google Scholar]
- 207.Milanini-Mongiat J, Pouyssegur J, Pages G. Identification of two Sp1 phosphorylation sites for p42/p44 mitogen-activated protein kinases: their implication in vascular endothelial growth factor gene transcription. J Biol Chem. 2002;277:20631–20639. doi: 10.1074/jbc.M201753200. [DOI] [PubMed] [Google Scholar]
- 208.Merchant JL, Du M, Todisco A. Sp1 phosphorylation by Erk 2 stimulates DNA binding. Biochem Biophys Res Commun. 1999;254:454–461. doi: 10.1006/bbrc.1998.9964. [DOI] [PubMed] [Google Scholar]
- 209.Oikawa T. ETS transcription factors: possible targets for cancer therapy. Cancer Sci. 2004;95:626–633. doi: 10.1111/j.1349-7006.2004.tb03320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.White FC, Benehacene A, Scheele JS, Kamps M. VEGF mRNA is stabilized by ras and tyrosine kinase oncogenes, as well as by UV radiation—evidence for divergent stabilization pathways. Growth Factors. 1997;14:199–212. doi: 10.3109/08977199709021520. [DOI] [PubMed] [Google Scholar]
- 211.Berra E, Pages G, Pouyssegur J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev. 2000;19:139–145. doi: 10.1023/a:1026506011458. [DOI] [PubMed] [Google Scholar]
- 212.Kevil CG, et al. Translational regulation of vascular permeability factor by eukaryotic initiation factor 4E: implications for tumor angiogenesis. Int J Cancer. 1996;65:785–790. doi: 10.1002/(SICI)1097-0215(19960315)65:6<785::AID-IJC14>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 213.Mamane Y, et al. eIF4E–from translation to transformation. Oncogene. 2004;23:3172–3179. doi: 10.1038/sj.onc.1207549. [DOI] [PubMed] [Google Scholar]
- 214.Xie W, Herschman HR. Transcriptional regulation of prostaglandin synthase 2 gene expression by platelet-derived growth factor and serum. J Biol Chem. 1996;271:31742–31748. doi: 10.1074/jbc.271.49.31742. [DOI] [PubMed] [Google Scholar]
- 215.Sheng H, et al. Induction of cyclooxygenase-2 by activated Ha-ras oncogene in Rat-1 fibroblasts and the role of mitogen-activated protein kinase pathway. J Biol Chem. 1998;273:22120–22127. doi: 10.1074/jbc.273.34.22120. [DOI] [PubMed] [Google Scholar]
- 216.Reddy ST, Wadleigh DJ, Herschman HR. Transcriptional regulation of the cyclooxygenase-2 gene in activated mast cells. J Biol Chem. 2000;275:3107–3113. doi: 10.1074/jbc.275.5.3107. [DOI] [PubMed] [Google Scholar]
- 217.Subbaramaiah K, Norton L, Gerald W, Dannenberg AJ. Cyclooxygenase-2 is overexpressed in HER-2/neu-positive breast cancer: evidence for involvement of AP-1 and PEA3. J Biol Chem. 2002;277:18649–18657. doi: 10.1074/jbc.M111415200. [DOI] [PubMed] [Google Scholar]
- 218.Lengyel E, Stepp E, Gum R, Boyd D. Involvement of a mitogen-activated protein kinase signaling pathway in the regulation of urokinase promoter activity by c-Ha-ras. J Biol Chem. 1995;270:23007–23012. doi: 10.1074/jbc.270.39.23007. [DOI] [PubMed] [Google Scholar]
- 219.Jiang Y, Muschel RJ. Regulation of matrix metalloproteinase-9 (MMP-9) by translational efficiency in murine prostate carcinoma cells. Cancer Res. 2002;62:1910–1914. [PubMed] [Google Scholar]
- 220.Shapiro RL, et al. Induction of primary cutaneous melanocytic neoplasms in urokinase-type plasminogen activator (uPA)-deficient and wild-type mice: cellular blue nevi invade but do not progress to malignant melanoma in uPA-deficient animals. Cancer Res. 1996;56:3597–3604. [PubMed] [Google Scholar]
- 221.Wolthuis RM, Bos JL. Ras caught in another affair: the exchange factors for Ral. Curr Opin Genet Dev. 1999;9:112–117. doi: 10.1016/s0959-437x(99)80016-1. [DOI] [PubMed] [Google Scholar]
- 222.Jackson EL, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. This paper was the first to feature conditional activation of an endogenous KRASG12D allele in an animal model, and showed that this is sufficient to drive progression from pulmonary hyperplasia to adenocarcinoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Hingorani SR, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 224.Chan IT, et al. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J Clin Invest. 2004;113:528–538. doi: 10.1172/JCI20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Brembeck FH, et al. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003;63:2005–2009. [PubMed] [Google Scholar]
- 226.Mo L, et al. Hyperactivation of Ha-ras oncogene, but not Ink4a/Arf deficiency, triggers bladder tumorigenesis. J Clin Invest. 2007;117:314–325. doi: 10.1172/JCI30062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Vitale-Cross L, Amornphimoltham P, Fisher G, Molinolo AA, Gutkind JS. Conditional expression of K-ras in an epithelial compartment that includes the stem cells is sufficient to promote squamous cell carcinogenesis. Cancer Res. 2004;64:8804–8807. doi: 10.1158/0008-5472.CAN-04-2623. [DOI] [PubMed] [Google Scholar]
- 228.Balmain A, Ramsden M, Bowden GT, Smith J. Activation of the mouse cellular Harvey-ras gene in chemically induced benign skin papillomas. Nature. 1984;307:658–660. doi: 10.1038/307658a0. [DOI] [PubMed] [Google Scholar]
- 229.Holland EC, et al. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nature Genet. 2000;25:55–57. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- 230.Seidler B, et al. A Cre-loxP-based mouse model for conditional somatic gene expression and knockdown in vivo by using avian retroviral vectors. Proc Natl Acad Sci USA. 2008;105:10137–10142. doi: 10.1073/pnas.0800487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Johnson L, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. This study featured activation of oncogenic KRAS transgene by spontaneous somatic recombination events in the whole animal, resulting in the acquisition of a broad range of tumour types. [DOI] [PubMed] [Google Scholar]