

Abstract
Background
Single Cell Network Profiling (SCNP) is used to simultaneously measure the effects of modulators on signaling networks at the single cell level. SCNP-based biomarker assays predictive of response to induction therapy and relapse risk in AML patients are being developed. Such assays have typically utilized bonemarrow (BM) as the sample source of blasts. Since circulating peripheral blasts are detectable in ~65% of AML patients and peripheral blood (PB) sampling is less invasive than BM sampling, this study was performed to assess the effect of sample source on AML blasts signaling as measured in SCNP assay.
Methods
SCNP using multiparametric flow cytometry was used to evaluate the activation state of intracellular signaling molecules in leukemic blasts under basal conditions and after treatment with modulators in 46 pairs of BM mononuclear cells/PB mononuclear cells. The relationship between readouts of modulated intracellular proteins (“nodes”) was measured using linear regression, Bland-Altman method and Lin’s concordance correlation coefficient.
Results
The majority (156/161) of signaling nodes show strong correlations between paired PB and BM samples independently from the statistical method used. Notable exceptions were two PB samples with almost undetectable levels of circulating blasts compared to paired BM samples.
Conclusions
Our results demonstrate that specimen source (BM or PB) does not significantly affect proteomic signaling in patients with AML and circulating blasts. The ability to use PB as a sample source will facilitate the monitoring of cellular signaling effects following administration of targeted therapies and at time-points when BM aspirates are not clinically justifiable.
Keywords: cell signaling, acute myeloid leukemia, multiparameter analysis, flow cytometry, standardization, single cell network profiling, diagnostic, specimen source
INTRODUCTION
Acute myeloid leukemia (AML) traditionally has been classified based on cellular morphology after examination of bone marrow (BM) aspirate and biopsy specimens, immunophenotypic characteristics, cytochemistry staining patterns and cytogenetics (1–4). More recently, molecular markers, such as the presence of either isolated nucleophosmin-1 (NPM1) gene mutations or internal duplications of the FLT3 gene (FLT3ITD) in cytogenetically normal AML, or the presence of c-kit mutation in the core binding factor AML subgroup, have been used to further classify AML and inform on the intensity of consolidation therapy post-remission (5–15). Although these markers offer directionally predictive information at a population level, their degree of accuracy at the individual patient level is still suboptimal. Since a wide range of chromosomal, genetic, epigenetic and other molecular alterations ultimately converge at the level of protein function and cell signaling pathways, a functional biological characterization of AML based on the analysis of single cell signaling networks could ultimately provide increased accuracy for identifying disease heterogeneity and prognosis and for treatment selection.
Single cell network profiling (SCNP) using multiparametric flow cytometry measures changes in intracellular cell signaling upon exposure of live cells to extracellular modulators revealing network properties that would not be seen in resting cells (16–19) or in assays performed on fixed tissues. The potential usefulness of this technology to generate novel and clinically relevant information has been previously demonstrated (17,20–21). More recently, SCNP assay was used to predict the likelihood of response to standard induction chemotherapy in acute myelogenous leukemia (AML) patients (22–24). All together, these studies demonstrated that functional pathway analysis of leukemic blasts can provide additional information distinct from other known AML prognostic factors such as age, secondary AML, cytogenetics, and molecular alterations, and is potentially combinable with the latter to improve clinical decision making. Therefore, SCNP using multiparametric flow cytometry is emerging as a valuable tool to inform on therapy outcomes or relapse risks in both pediatric and adult AML.
The application of SCNP to clinical decision making requires the generation of high-content SCNP assays with robust, accurate and reproducible results across operators and instruments. In addition, the assays must be characterized by longitudinal stability, as data will be compared across sequential pilot studies, training studies, validation studies, and in clinical patient care as results are delivered to physicians. The pre-analytic, analytic and post-analytic technical variables likely to play a significant role in SCNP assay performance and reproducibility have been previously reviewed (25) and include patient specimen source such as bone marrow mononuclear cells (BMMC) or peripheral blood mononuclear cells (PBMC).
In approximately two-thirds of AML patients, circulating blasts can be detected in PB making this sample source an attractive option due to its less invasive acquisition process compared to BM sampling. However, it is unknown whether or not certain biologic characteristics of AML blasts (such as cell signaling pathways and sensitivity to cytokine, DNA damage inducing or apoptosis causing modulators) are different in blasts collected from the BM samples versus those from the matching PB samples.
Here we discuss the specific methodology and processes that were implemented to control for technical assay variability in SCNP with the main objective of the study to examine the effect of specimen source on proteomic signaling by comparing signaling readouts for viable paired BMMC and PBMC in a relatively large set of non-M3 AML samples.
MATERIALS AND METHODS
Sample Disposition
This study used cryopreserved samples collected at the time of original AML diagnosis from subjects enrolled in the Phase 3 trial E3999 conducted by the Eastern Cooperative Oncology Group (ECOG) between 2003 and 2005. The sample set used for the present study represents a subset of the subjects enrolled in E3999 which was selected based on the availability of stored aliquots of PB and BM samples.
In order to be included in the experimental analysis, each of the matched BMMC and PBMC specimens had to contain a minimum of 2 million viable cells post-thaw. Specimen quality could only be assessed upon thawing of the sample at assay initiation. Therefore, inclusion (or exclusion) of samples in the study was determined post thawing and processing.
Supplemental Figure S1 illustrates patient enrollment and sample disposition. The original protocol stated 49 donors were to be included in the study. Paired BM and PB samples, with associated clinical data, were available for 46 of the donors. Table 1 summarizes the baseline characteristics of all 46 donors included in the analysis. After thawing, each sample was divided into two aliquots which were separately processed to assess assay reproducibility. Samples from all 46 donors were thawed and processed. To be included in this analysis, a donor was required to have at least one SCNP assay readout in paired BM and PB samples. All 46 donors met this criteria and were included in this analysis.
Table 1.
Baseline characteristics for patients.
| Characteristic | All Patients (n=46) | |
|---|---|---|
| Number (Percent) of Donors | 46 (100.0) | |
| ECOG: Best Confirmed Induction Response | CR | 14 (30.4) |
| mCR | 4 (8.7) | |
| PD | 20 (43.5) | |
| Unevaluable | 8 (17.4) | |
| Last Known Survival Status | Dead | 46 (100.0) |
| ECOG Performance Status | 0 (Fully active | 6 (13.0) |
| 1 (Ambulatory and able to do light work) | 24 (52.2) | |
| 2 (Ambulatory, 100% self-care but cannot work) | 12 (26.1) | |
| 3 (Limited self-care, confined to bed or chair >50% of awake time) | 4 (8.7) | |
| Disease Type | De novo AML | 30 (65.2) |
| RAEB-t | 2 (4.3) | |
| Secondary AML | 14 (30.4) | |
| Cytogenics | Missing/Indeterminate | 13 (28.3) |
| Intermediate | 22 (47.8) | |
| Unfavorable | 11 (23.9) | |
| Age at Diagnosis (years) | Mean (Std Dev) | 69.5 (5.67) |
| Sex | Male | 28 (60.9) |
| Female | 18 (39.1) | |
| Race | White | 44 (95.7) |
| Black | 2 (4.3) | |
| Treatment Received | AC+ Zosuquidar | 26 (56.5) |
| AC+ Placebo | 20 (43.5) |
Ethics
In accordance with the Declaration of Helsinki, all patients provided written informed consent for the collection and use of their samples for research purposes. Institutional Review Board approval was obtained from Independent Review Consulting, Inc. (Approval No. 09068-01) on August 31, 2009. Clinical data were de-identified in compliance with Health Insurance Portability and Accountability Act regulations.
Study Design
This study investigated paired cryopreserved BMMC and PBMC samples from patients at least 60 years of age who had received cytarabine-based induction chemotherapy for newly diagnosed non-M3 AML and was performed in a blinded fashion to clinical outcomes. A paired sample was defined as BMMC and PBMC specimens collected from the same patient on the same day and prior to the initiation of induction therapy.
The assay measured cell signaling and apoptosis in separate 96-well plates. Figures 1A and 1B illustrate the signaling and apoptosis plate layouts, respectively. Paired BM and PB samples were tested on the same plate, along with instrument and laboratory work flow controls (see Experimental Controls below). Plates were processed in duplicate and repeatability data was analyzed (Figure 1C). Experiments were performed on two instruments, LSRII (signaling plates) and FACS Canto II (apoptosis plates). Nine donors were processed per batch, with two samples (BM and PB) per donor. In total, 140 96-well plates were processed during the 3 week experiment, and data was collected from over 8,024 wells. The signaling and apoptosis plates that were run in duplicate are the focus of this analysis and correspond to 161 node-metric calculations (Table 2). The high-throughput nature of this experiment is illustrated schematically in Figure 1C.
Figure 1. Experimental setup, including signaling and apoptosis plate layouts.
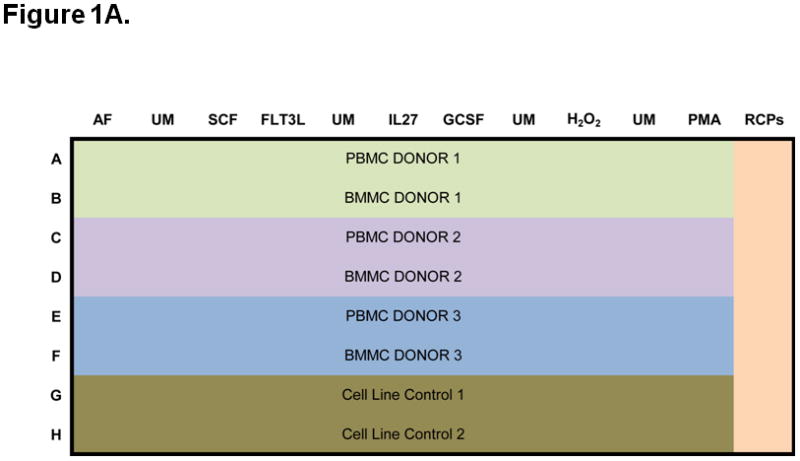
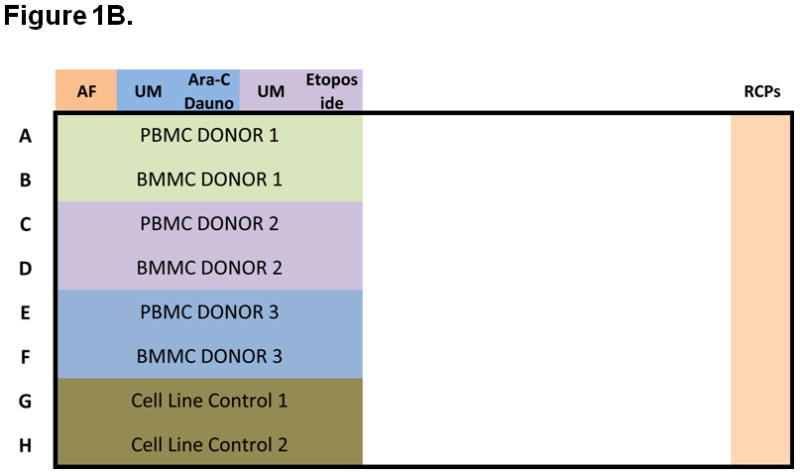
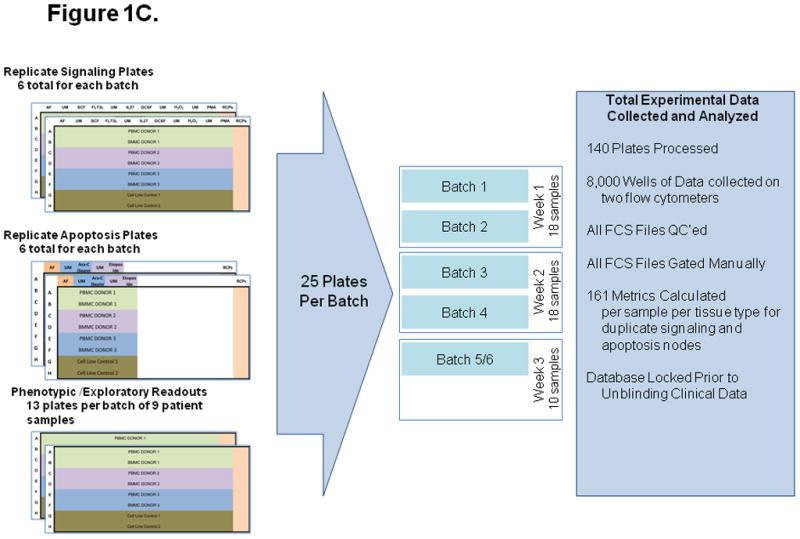
A) Signaling plates layout. Each plate contains PB and BM samples from 3 AML patients (rows A to F). Experiment conditions are shown across the top, with an autofluorescence well (AF), followed by an unmodulated reference well (UM) and the associated modulated wells, shown color-coded. A total of six modulated conditions and four associated unmodulated conditions are tested in this plate format. Column 12 contains rainbow control particles (RCPs) used to calibrate the fluorescence readouts on a plate-by-plate basis. Rows G and H contain cell line controls (GDM-1/U937 mix and RS4;11) used to monitor the modulation, fixation, permeabilization, and staining processes. Each plate is setup using a laboratory management system which populates sample, reagent, antibody cocktails, modulators, cytometer and other settings used to track information during the conduct of the experiment. B) Apoptosis plates layout. As in the signaling plates, each apoptosis plates contain a total of 3 paired PB and BM samples, cell line controls, and rainbow control particles (RCPs) in column 12. One autofluorescence column is included as a reference. Unmodulated (UM) and modulated columns are used to assess response to Ara-C/Daunorubicin (Ara-C Dauno) and Etoposide. C) Each signaling and each apoptosis plate was run in duplicate, setup with the laboratory management system and data tracking was managed throughout the experiment.
Table 2.
Comparison of BMMC and PBMC measurements acquired on the flow cytometer using the Bland-Altman methodology.
| Metric1 | Measured Population | Reference State | Biology Investigated | Total Nodes | Nodes where measurements of BMMC and PBMC are | ||
|---|---|---|---|---|---|---|---|
| Equivalent3 | Lin Equivalent4 | Other5 | |||||
| ERF2 | Calibrated Instrument Fluorescence Intensities | None | 41 | 15 (37%) | 23 (56%) | 3 (7%) | |
| Basal = log2(ERFu/ERFa) | Unmodulated | Autofluorescence | Basal Pathway Activity | 16 | 15 (94%) | 1 (6%) | 0 |
| log2 Fold = log2(ERFm/ERFu) | Modulated | Unmodulated | Induced Pathway Activity | 22 | 18 (82%) | 3 (14%) | 1 (5%) |
| Total Phospho = log2(ERFm/ERFa) | Modulated | Autofluorescence | Total Pathway Activity | 22 | 21 (95%) | 1 (5%) | 0 |
| Ua | Modulated | Autofluorescence | Basal Pathway Activity | 38 | 22 (58%) | 15 (39%) | 1 (3%) |
| Uu | Modulated | Unmodulated | Induced Pathway Activity | 22 | 19 (86%) | 3 (14%) | 0 |
| Total Node-Metrics | 161 | 110 (68%) | 46 (29%) | 5 (3%) | |||
The subscripts represent the following: a=auto fluorescence, m=modulated, u=unmodulated.
Equivalent Number of Reference Fluorophores
The variance does not depend on the size of the observations; either the difference between BMMC and PBMC was not statistically significant at the 5% level, or BMMC is a simple linear transformation of PBMC with R2 >= 80%.
For nodes that did not satisfy the requirements of the Bland-Altman method, Lin’s condordance correlation coefficient was applied to partition variation into precision (Pearson correlation) and accuracy(Cb). BM and PB were judged “Lin equivalent” if Pearson’s correlation >= 0.6 and Lin’s accuracy measure was >= 0.80 and the concordance correlation was at least 0.60.
Test for independence indicates standard deviation depends on the size of the observation and Lin’s concordance correlation coefficient was less than 0.60. Additional analysis is needed to verify the equivalence of BM and PB.
Sample Processing
SCNP Assay
Three groups of measurements were performed for each sample: cell surface marker expression, intracellular signaling and induced apoptosis (Table 3). Modulators, antibodies, and reagents were selected based on biological relevance and performance in previously conducted training studies (data not shown) (22).
Table 3.
List of nodes tested.
| Category | Node: Modulator | Node: Readout (antibody specificity) |
|---|---|---|
| Apoptosis | Dauno+Ara-C | Cleaved PARP p-Chk2 |
| Apoptosis | Etoposide | Cleaved PARP p-Chk2 |
| Apoptosis | No Modulator | Cleaved PARP p-Chk2 |
| Expression | No Modulator | CD117 CD135 CXCR4 |
| Signaling | FLT-3 Ligand | p-Akt p-ERK p-S6 |
| Signaling | G-CSF | p-STAT1 p-STAT3 p-STAT5 |
| Signaling | Hydrogen peroxide | p-Akt p-PLCγ2 p-SLP-76 |
| Signaling | IL-27 | p-STAT1 p-STAT3 p-STAT5 |
| Signaling | PMA | p-CREB p-ERK p-S6 |
| Signaling | SCF | p-Akt p-ERK p-S6 |
| Signaling | No Modulator | p-Akt p-CREB p-ERK p-PLCγ2 p-S6 p-SLP-76 p-STAT1 p-STAT3 p-STAT5 |
SCNP assays were performed as described previously (22). Cryopreserved samples were thawed at 37°C, washed in RPMI 60% FCS, and purified through ficoll density centrifugation. Mononuclear cells were washed in RPMI 1% FBS before staining with Aqua Viability Dye to distinguish non-viable cells. Cells were then counted, re-suspended in RPMI 10% FBS, aliquoted to 100,000 cells/condition, and rested for 2 hours at 37°C. For apoptosis assays, cells were incubated for 24 hours with cytotoxic drugs (e.g., etoposide or Ara-C and daunorubicin) and re-stained with Aqua Viability Dye. For signaling assays, cells were incubated with modulators (Table 4A) at 37°C for 15 minutes. After exposure to modulators, cells were fixed with 1.6% paraformaldehyde (final concentration) for 10 minutes at 37°C, pelleted and permeabilized with 100% ice-cold methanol, and stored at −80°C overnight. Subsequently, cells were washed with FACS buffer (PBS, 0.5% BSA, 0.05% NaN3), pelleted, and stained with cocktails of fluorochrome–conjugated antibodies (Table 4B). These cocktails included antibodies against 2 to 5 phenotypic markers for cell population gating (e.g., CD45, CD34, CD11b, CD15) and up to 3 antibodies against intracellular signaling molecules or against surface markers for an 8-color flow cytometry assay. Isotype controls or phosphopeptide blocking experiments were performed to characterize each phospho-antibody (data not shown). The laboratory process was carried out in parallel for all BMMC and PBMC paired samples.
Table 4A.
Modulators and technical conditions.
| Modulator | Final Concentration | Modulator Treatment Duration | Manufacturer (Location) |
|---|---|---|---|
| Ara-C | 0.5 μg/mL | 24 hours | Sigma Aldrich (St Louis, MO) |
| Daunorubicin | 100 ng/mL | 24 hours | Sigma Aldrich (St Louis, MO) |
| Etoposide | 30 μg/mL | 24 hours | Sigma Aldrich (St Louis, MO) |
| FLT3L | 50 ng/mL | 15 mins | R&D (Minneapolis, MN) |
| G-CSF | 50 ng/mL | 15 mins | R&D (Minneapolis, MN) |
| Hydrogen Peroxide | 3.0 mM | 15 mins | JT Baker (Phillipsburg, NJ) |
| IL-27 | 50 ng/mL | 15 mins | R&D (Minneapolis, MN) |
| PMA | 400 nM | 15 mins | Sigma Aldrich (St Louis, MO) |
| SCF | 20 ng/mL | 15 mins | R&D (Minneapolis, MN) |
Table 4B.
Antibodies and reagents.
| Antibody | Species & Isotype | Manufacturer (Location) | Clone |
|---|---|---|---|
| CD11b | Mouse IgG1 | Beckman (Miami, FL) | Bear1 |
| CD15 | Mouse IgG1 | Biolegend (San Diego, CA) | W6D3 |
| CD34 | Mouse IgG1 | BD (San Jose, CA) | 8G12 |
| CD45 | Mouse IgG1 | Beckman (Miami, FL) | J33 |
| cKit (CD117) | Mouse IgG1 | DAKO (Carpinteria, CA) | 104D2 |
| c-PARP(Asp214) | Mouse IgG1, k | BD (San Jose, CA) | F21-852 |
| Control Ig | Rat IgG1 | MBL (Woburn, MA) | 1H5 |
| Control Ig | Mouse IgG1, k | BD (San Jose, CA) | MOPC-21 |
| CXCR4 (CD184) | Rat IgG1 | MBL (Woburn, MA) | A145 |
| FLT3 Receptor (CD135) | Mouse IgG1 | BD (San Jose, CA) | 4G8 |
| p-Akt (S473) | Rabbit IgG | CST (Danvers, MA) | 193H12 |
| p-Chk2 (T68) | Rabbit IgG | CST (Danvers, MA) | Polyclonal |
| p-CREB (pS133) | Mouse IgG1, k | BD (San Jose, CA) | J151-21 |
| p-Erk 1/2 (T202/204) | Mouse IgG1 | BD (San Jose, CA) | 20A |
| p-Lck (Y505) | Mouse IgG1 | BD (San Jose, CA) | 4/Lck-Y505 |
| p-PLCγ2 (Y759) | Mouse IgG1, k | BD (San Jose, CA) | K86-689.37 |
| p-S6 (S235/236) | Rabbit IgG | CST (Danvers, MA) | 2F9 |
| p-SLP-76 (pY128) | Mouse IgG1, k | BD (San Jose, CA) | J141-668.36.58 |
| p-STAT1 (pY701) | Mouse IgG2a | BD (San Jose, CA) | 4a |
| p-STAT3 (pY705) | Mouse IgG2a, k | BD (San Jose, CA) | 4/P-STAT3 |
| p-STAT5 (pY694) | Mouse IgG1 | BD (San Jose, CA) | 47 |
| Non-Antibody Stains | n/a | Manufacturer (Location) | n/a |
| Amine Aqua Viability Dye | n/a | Invitrogen (Carlsbad, CA) | n/a |
| Streptavidin-Qdot 605 | n/a | Invitrogen (Carlsbad, CA) | n/a |
Flow Cytometry Data Acquisition and Analysis
Flow cytometry data was acquired on an LSR II and/or CANTO II flow cytometer using the FACS DIVA software (BD Biosciences, San Jose, CA) and standardized using methods described previously (26). All flow cytometry data were gated with WinList (Verity House Software, Topsham, ME). Dead cells and debris were excluded by forward scatter, side scatter, and Amine Aqua Viability Dye measurement. Leukemic cells were identified as cells that fit the CD45 versus right-angle light-scatter characteristics consistent with myeloid leukemia blasts and that lacked the characteristics of mature lymphocytes, as described previously (22,27).
Metrics
In SCNP assay terminology a “signaling node” is used to refer to a proteomic readout in the presence or absence of a specific modulator. For example, the response to FLT3L treatment can be measured using p-Akt as a readout. That signaling node is designated “FLT3L → p-Akt”. The normalized assay readouts for surface and intracellular markers are measured using two broad classes of calculated metrics that are applied to interpret the functionality and biology of each signaling node. They are referenced following the node e.g. “FLT3L→p-Akt | Fold”, “G-CSF→p-STAT5 | Total” or “p-STAT5 | Basal” and defined in detail below.
Supplemental Figure S2A shows the workflow for calculation of the metrics used to quantify the assay readouts. In all cases, the raw instrument fluorescence intensities are converted to calibrated intensity metrics (ERFs, Equivalent Number of Reference Fluorophores) (26,28–29). The calibration is applied on a plate-by-plate basis using the rainbow calibration particles as shown in Figures 1A and 1B. This correction ensures that data across the plate and between plates are calibrated to the same values, regardless of the instrument used for acquisition.
Two broad classes of metrics were developed to measure two distinct functional aspects of signaling proteins. The Fold Metric Class measures the magnitude of the responsiveness of a cell population to modulation relative to the same cell population in the reference well (i.e. unmodulated) by comparing the median fluorescence values of the responsive cell population to that of the reference population on a log2 scale. A value of zero would indicate overlapping populations and a value different from zero indicates the responsive population has shifted to higher fluorescence (positive values) or to lower fluorescence (negative values). The “U” Metric Class measures the fraction or proportion of a cell population that is responsive to modulation relative to the same population in the reference well by comparing the overlap of the responsive cellular population relative to the reference population evaluated on a cell-by-cell basis. This class is mathematically equivalent to an AUC metric (which is a scaled Mann-Whitney U metric) and is scaled to range from zero to one. For overlapping populations, roughly half of the cells in the modulated population have higher intensities than those in the reference population, so the U metric has a value of 0.5. A value different from 0.5 indicates the responsive population has shifted to higher fluorescence (values >0.5) or to lower fluorescence (values <0.5). This metric has an upper limit of 1.0, which represents the situation in which there is no overlap between the modulated and reference populations.
Examples of both metric classes are presented in Supplemental Figure S2B to highlight the different and complementary information that the use of both metrics provides. The annotations in each of the plots show both metric values for different types of responses routinely seen in SCNP assay results. A summary of the various metrics showing the experimental and reference populations used for calculations and the relevant biology captured is shown in Table 2. For the current analysis, a total of 6 metrics were calculated, resulting in a total of 161 node-metrics.
In addition, for each sample the cell health (Percent Healthy, PH) is measured as the fraction of leukemic cells that are live and non-apoptotic (i.e., Aqua and c-PARP negative) as a fraction of the total number of intact cells.
Experimental Controls
Instrument Controls
Linearity verification was performed daily for all fluorescence detectors on the cytometer using 8 Peak SPHERO™ Rainbow Calibration Particles (Spherotech, Lake Forest, IL). The slope, intercept and R squared values obtained from the linear regression were used to standardize, qualify and monitor the instrument during setup. They were also used to calibrate the raw fluorescence intensity readouts to control for instrument variability. For this experiment, the CVs of the rainbow particle bead peaks on both the LSRII and the CANTOII were under 2.1%. The dimmest peaks were not included in this calculation as they were considered to be near the baseline variance in the instrument.
Cell Line Controls
Three cell lines, GDM-1, U937 and RS4;11 were included as controls on each plate to ensure consistency in assay performance. Data from previous experiments show that the modulators used in this study, other than FLT3 ligand, induce robust signaling in GDM-1 and/or U937 cell lines. FLT3 ligand induces signaling in the RS4;11 cell line. In combination, the three cell lines provided plate-based controls for all modulator/antibody readout combinations used in this study. Therefore, these cell lines were used to identify potential technical variability at the modulation, fixation and staining steps in the laboratory work flow. The assay performance was measured on the Uu metric (see above for definition). In this study, CVs below 10% were observed for 49 of 54 (91%) signaling nodes and 5 of 8 (62%) apoptosis nodes. Higher variance in the remaining nodes was attributable to either technical issues such as cytometer clogs or an inherent lack of cell line responsiveness to specific modulators or poor signaling due to high levels of induced apoptosis and cell death.
Process Controls
Several data analysis tools were developed to facilitate experimental setup and data tracking to ensure verification of data integrity at each step in the sample processing workflow (30). These tools were used to manage the sample, reagent, and instrument data from initial study design through processing and to un-blinding of clinical data. Additionally, a set of internal software tools were developed to perform high-volume gating, to calculate all metrics employed and to allow for data analysis, interpretation, and visualization. Together these represent significant process efficiency improvements in SCNP assays.
Sample Quality Assessment
Sample quality was determined by calculation of the percent of cell recovery and percentage of healthy cells. Samples were thawed and ficolled on the day of the experiment and total cell number and leukocyte count were determined and compared to the expected number of cells in the vials, as reported in the sample manifest. The fraction of live, non-apoptotic cells (Percent Healthy, see metrics) was calculated from the unmodulated wells at the 6 hour timepoint.
SCNP Assay Repeatability in AML Samples
Assay repeats were run on different plates on the same day. Teams of operators were fixed, but individual operators could vary for each step in the laboratory work flow. Following laboratory processing, linear regression and Bland-Altman analyses (31–32) were performed to assess assay repeatability.
Determining Equivalence between PB and BM
Three analysis methods were utilized to investigate equivalence between paired PB and BM readouts for signaling nodes, apoptosis nodes and surface markers.
Linear Regression
Linear regression with the BM result as the dependent variable and the PB result as the independent variable was performed.
Bland-Altman
Using the Bland-Altman method (31–32), the relationship between the readouts difference (BM−PB) and the readouts average (BM+PB)/2 was determined by inspecting two plots of the data and by testing the hypothesis that the correlation of (BM−PB) and (BM+PB)/2 is equal to zero. The difference between BM and PB readouts was plotted, and the mean difference ± 2 standard deviations was calculated. The fitted regression line, the identity line (BM=PB), a 95% prediction ellipse about the point defined by the means of BM and PB sample readouts, and the 95% confidence band about the regression line were also computed.
Lin’s Concordance Correlation Coefficient
Lin’s concordance correlation coefficient (33), a measure that incorporates accuracy (distance from the identity line) and precision (distance of each pair (PB, BM) readout from the best-fit linear regression line), was computed.
Identification of Outliers
Outliers were identified by review of linear regression plots. Results that were outside the 95% confidence interval for the linear regression fit were identified as outliers and subject to follow-up analysis.
Evaluation of the Impact of Sample Quality on Paired BM and PB Readouts Concordance
In order to evaluate the impact of sample quality on paired PB and BM readouts concordance, regression analysis was performed to include the PH as a covariate as follows:
where:
PB = Node-metric values for PB samples
BM = Node-metric values for BM samples
dPH = Difference in PH between the two tissue types at 6 hours
This analysis used the 6 hour timepoint because PH data was not collected at 0 hours in this experiment and, based on previous studies (data not shown), the 6 hour timepoint is an acceptable measure of sample quality (i.e. shows a good correlation with intrinsic cell capability to respond to modulation). Unfortunately, due to cell number limitation, data for PH at 6 hours was not available for all samples and therefore, this analysis was performed only on the subset of samples with paired 6 hour PH data (N=37).
RESULTS
Sample Quality Assessment
We assessed the sample quality as determined by the percent of cell recovery after thawing (N=46 paired samples) and PH after 6 hours(N=37 paired samples, see metrics section for definition). A total of 96% of the samples contained sufficient cells to analyze the nodes listed in Table 3. The median percent recovery, based on reported cryopreserved cell numbers from the sample manifest, was 51.3%, with a minimum of 1.56% and maximum of 462%. The median PH was 41.6%, with a minimum of 0% and maximum of 83.2%. Of the 37 samples, 6 PB and 9 BM samples had PH<25%. The importance of this observation is related to the association observed between baseline level of apoptosis and cell signaling ability; specifically samples with low PH (high levels of apoptosis) displayed poor induced signaling potential while samples with high PH were shown to display high signaling potential. Figure 2 shows the relationship between PH and FLT3L-induced signaling of p-S6 using the Fold metric. Samples with low percentage health showed little or no signaling. In addition, PH at 6 hours is correlated between matched PB and BM samples (Figure 3) suggesting that similar biology and/or pre-analytical variables influenced samples of both tissue types for each donor. Overall, these results show the appropriateness of these methods to measure and control sample quality for inclusions in SCNP assay.
Figure 2.

Comparison of the FLT3L-induced signaling changes in p-S6 on the Fold metric (y-axis) as a function of the percentage of healthy cells (PH) (x-axis).
Figure 3.

Correlation of PH between BM and PB samples at 6 hour time point.
A number of outliers with lower correlation between PB and BM sample quality were observed (Figure 3). Regression analysis was performed to evaluate the impact of sample quality on concordance between paired PB and BM signaling (see below).
SCNP Assay Repeatability in AML Samples
Linear regression analysis demonstrated excellent correlation between sample repeats. 78% (PB) and 68% (BM) of nodes displayed R2>0.80 for the Uu metric (Tables 5A and 5B, respectively), and 73% (PB) and 68% (BM) of nodes displayed R2>0.80 for the Fold metric (data not shown). Equivalent readouts from the two repeats (Slope = 1.0 and Intercept = 0.0) were observed for 38 out of 44 (87%) nodes.
Table 5A.
Repeatability study in peripheral blood samples.
Uu metric
| Modulator | Antibody | Color | N* | R2 | Intercept | p (Intercept=0.0)*** | Slope | p (Slope=1.0)*** | p (Slope=0.0)** |
|---|---|---|---|---|---|---|---|---|---|
| AraC+Daunorubicin | cPARP | FITC-A | 34 | 0.91 | 0.05 | 0.201 | 0.94 | 0.210 | <0.0001 |
| p-Chk2 | Alexa Fluor 647-A | 34 | 0.89 | 0.05 | 0.242 | 0.95 | 0.251 | <0.0001 | |
| Etoposide | cPARP | FITC-A | 38 | 0.69 | 0.15 | 0.002 | 0.71 | 0.001 | <0.0001 |
| p-Chk2 | Alexa Fluor 647-A | 38 | 0.74 | 0.06 | 0.291 | 0.91 | 0.225 | <0.0001 | |
| FLT-3 Ligand | p-Akt | Alexa Fluor 647-A | 41 | 0.77 | 0.06 | 0.218 | 0.89 | 0.157 | <0.0001 |
| p-ERK | PE-A | 41 | 0.68 | 0.13 | 0.008 | 0.76 | 0.008 | <0.0001 | |
| p-S6 | Alexa Fluor 488-A | 41 | 0.97 | 0.03 | 0.121 | 0.96 | 0.118 | <0.0001 | |
| G-CSF | p-STAT1 | Alexa Fluor 488-A | 39 | 0.86 | 0.06 | 0.110 | 0.92 | 0.148 | <0.0001 |
| p-STAT3 | PE-A | 39 | 0.97 | 0.01 | 0.770 | 1.00 | 0.394 | <0.0001 | |
| p-STAT5 | Alexa Fluor 647-A | 39 | 0.97 | 0.00 | 0.933 | 1.01 | 0.383 | <0.0001 | |
| Hydrogen Peroxide | p-Akt | Alexa Fluor 488-A | 38 | 0.94 | −0.01 | 0.867 | 1.01 | 0.388 | <0.0001 |
| p-PLCγ2 | PE-A | 38 | 0.98 | 0.00 | 0.822 | 1.00 | 0.396 | <0.0001 | |
| p-SLP-76 | Alexa Fluor 647-A | 38 | 0.97 | 0.02 | 0.224 | 0.96 | 0.180 | <0.0001 | |
| IL-27 | p-STAT1 | Alexa Fluor 488-A | 41 | 0.95 | 0.01 | 0.625 | 0.99 | 0.390 | <0.0001 |
| p-STAT3 | PE-A | 41 | 0.94 | 0.00 | 0.905 | 1.01 | 0.390 | <0.0001 | |
| p-STAT5 | Alexa Fluor 647-A | 41 | 0.82 | 0.04 | 0.301 | 0.93 | 0.224 | <0.0001 | |
| PMA | p-CREB | PE-A | 37 | 0.95 | 0.00 | 0.996 | 1.00 | 0.394 | <0.0001 |
| p-ERK | Alexa Fluor 647-A | 37 | 0.98 | 0.00 | 0.944 | 0.99 | 0.387 | <0.0001 | |
| p-S6 | Alexa Fluor 488-A | 37 | 0.98 | −0.02 | 0.270 | 1.02 | 0.317 | <0.0001 | |
| SCF | p-Akt | Alexa Fluor 647-A | 41 | 0.96 | 0.02 | 0.276 | 0.97 | 0.242 | <0.0001 |
| p-ERK | PE-A | 41 | 0.72 | 0.12 | 0.008 | 0.79 | 0.016 | <0.0001 | |
| p-S6 | Alexa Fluor 488-A | 41 | 0.91 | 0.08 | 0.015 | 0.89 | 0.030 | <0.0001 |
The number of samples varies due to insufficient cells to test all the conditions in a prioritized order or due to insufficient cells in the myeloblast gate (e.g. in apoptosis not enough viable cells remain in a well after 24 hour drug treatment).
p(Slope=0) shows the probability that the correlation between BM and PB samples is random.
The combination of p(Intercept=0) and p(Slope=1) indicate if an equivalent relationship between BM and PB can be ruled out. If both of these p-values are above 0.05, an equivalent relationship (slope=1.0, intercept=0) is within the 95% confidence limits of the linear fit.
Table 5B.
Repeatability study in bone marrow samples.
Uu metric
| Modulator | Antibody | Color | N* | R2 | Intercept | p (Intercept=0.0)** | Slope | p (Slope=1.0)** |
|---|---|---|---|---|---|---|---|---|
| AraC+Daunorubicin | cPARP | FITC-A | 29 | 0.91 | 0.06 | 0.149 | 0.93 | 0.178 |
| p-Chk2 | Alexa Fluor 647-A | 29 | 0.84 | 0.01 | 0.828 | 0.98 | 0.387 | |
| Etoposide | cPARP | FITC-A | 34 | 0.82 | 0.12 | 0.002 | 0.78 | 0.002 |
| p-Chk2 | Alexa Fluor 647-A | 34 | 0.78 | 0.12 | 0.033 | 0.83 | 0.041 | |
| FLT-3 Ligand | p-Akt | Alexa Fluor 647-A | 41 | 0.71 | 0.16 | 0.000 | 0.72 | 0.001 |
| p-ERK | PE-A | 41 | 0.49 | 0.12 | 0.125 | 0.77 | 0.076 | |
| p-S6 | Alexa Fluor 488-A | 41 | 0.94 | 0.02 | 0.577 | 0.97 | 0.315 | |
| G-CSF | p-STAT1 | Alexa Fluor 488-A | 41 | 0.78 | 0.05 | 0.266 | 0.93 | 0.265 |
| p-STAT3 | PE-A | 41 | 0.86 | 0.06 | 0.129 | 0.91 | 0.110 | |
| p-STAT5 | Alexa Fluor 647-A | 41 | 0.91 | 0.02 | 0.504 | 0.97 | 0.308 | |
| Hydrogen Peroxide | p-Akt | Alexa Fluor 488-A | 36 | 0.93 | 0.07 | 0.047 | 0.92 | 0.062 |
| p-PLCγ2 | PE-A | 36 | 0.98 | 0.01 | 0.376 | 0.99 | 0.358 | |
| p-SLP-76 | Alexa Fluor 647-A | 36 | 0.96 | 0.04 | 0.007 | 0.93 | 0.029 | |
| IL-27 | p-STAT1 | Alexa Fluor 488-A | 42 | 0.95 | 0.05 | 0.058 | 0.95 | 0.111 |
| p-STAT3 | PE-A | 42 | 0.77 | 0.09 | 0.081 | 0.85 | 0.053 | |
| p-STAT5 | Alexa Fluor 647-A | 42 | 0.70 | 0.05 | 0.329 | 0.89 | 0.194 | |
| PMA | p-CREB | PE-A | 35 | 0.93 | 0.06 | 0.070 | 0.93 | 0.114 |
| p-ERK | Alexa Fluor 647-A | 35 | 0.99 | 0.04 | 0.006 | 0.94 | 0.004 | |
| p-S6 | Alexa Fluor 488-A | 35 | 0.98 | 0.04 | 0.066 | 0.95 | 0.078 | |
| SCF | p-Akt | Alexa Fluor 647-A | 41 | 0.92 | 0.02 | 0.526 | 0.98 | 0.362 |
| p-ERK | PE-A | 41 | 0.86 | 0.01 | 0.885 | 0.99 | 0.392 | |
| p-S6 | Alexa Fluor 488-A | 41 | 0.97 | 0.02 | 0.411 | 0.98 | 0.263 |
The number of samples varies due to insufficient cells to test all the conditions in a prioritized order or due to insufficient cells in the myeloblast gate (e.g. in apoptosis not enough viable cells remain in a well after 24 hour drug treatment).
The combination of p(Intercept=0) and p(Slope=1) indicate if an equivalent relationship between BM and PB can be ruled out. If both of these p-values are above 0.05, an equivalent relationship (slope=1.0, intercept=0) is within the 95% confidence limits of the linear fit.
A technical outlier was identified during the repeatability analysis based on low reproducibility metrics. Specifically, low reproducibility for FLT3L (R2=0.49, Table 5B) was attributed to a cytometer clog. Overall, only two wells out of the 8,024 wells processed had a cytometer error.
Effect of Specimen Source on Proteomic Signaling
Equivalence was shown for the majority of signaling nodes (>90%) between matched PBMC and BMMC samples (Table 2 and Supplemental Tables S1A–S1C, S2A–S2C). Simple linear regression models and bias estimates from the Bland-Altman analysis between BMMC and PBMC are shown in Tables 6 and 7 (unmodulated and modulated, respectively). The Bland-Altman plots for the Ua and Uu metrics are shown in Supplemental Figures S3 and S4.
Table 6.
Comparison of bone marrow and peripheral blood with respect to each node for the Ua1 metric comparing the modulated and auto fluorescence wells (No modulator)
| Readout | Simple linear regression BM = intercept + slope*PB |
Bland-Altman Analysis Difference3 (BM − PB) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Modulator | Antibody | Color | R2 | Slope | p-value2 | Intercept | Bias estimate | Std Dev | p-value4 | Classification5 |
| No Modulator | p-Akt | Alexa Fluor 488-A | 0.58 | 0.79 | <.0001 | 0.13 | 0.004 | 0.044 | 0.5649 | Equivalent |
| No Modulator | p-Akt | Alexa Fluor 647-A | 0.74 | 0.93 | <.0001 | 0.09 | 0.042 | 0.046 | <.0001 | Other |
| No Modulator | p-CREB | PE-A | 0.65 | 0.76 | <.0001 | 0.21 | −0.006 | 0.043 | 0.3769 | Equivalent |
| No Modulator | p-ERK | Alexa Fluor 647-A | 0.76 | 0.85 | <.0001 | 0.11 | 0.015 | 0.033 | 0.0049 | Other |
| No Modulator | p-ERK | PE-A | 0.63 | 0.61 | <.0001 | 0.37 | −0.005 | 0.035 | 0.3049 | Other |
| No Modulator | p-PLCγ2 | PE-A | 0.69 | 0.95 | <.0001 | 0.03 | −0.011 | 0.029 | 0.0201 | Other |
| No Modulator | p-S6 | Alexa Fluor 488-A | 0.47 | 0.63 | <.0001 | 0.34 | −0.01 | 0.043 | 0.1057 | Equivalent |
| No Modulator (DMSO only) | p-S6 | Alexa Fluor 488-A | 0.58 | 1.01 | <.0001 | −0.02 | −0.013 | 0.032 | 0.0085 | Other |
| No Modulator | p-SLP-76 | Alexa Fluor 647-A | 0.77 | 0.88 | <.0001 | 0.1 | 0.02 | 0.038 | 0.0012 | Other |
| No Modulator | p-STAT1 | Alexa Fluor 488-A | 0.71 | 0.88 | <.0001 | 0.06 | −0.031 | 0.068 | 0.0037 | Other |
| No Modulator | p-STAT3 | PE-A | 0.61 | 0.77 | <.0001 | 0.19 | −0.013 | 0.051 | 0.0908 | Equivalent |
| No Modulator | p-STAT5 | Alexa Fluor 647-A | 0.73 | 0.82 | <.0001 | 0.17 | 0.027 | 0.049 | 0.0005 | Other |
Ua=Mann-Whitney U comparing modulated and auto fluorescence wells (No modulator)
p-value of the test that the slope equals 0.
When (BM-PB) and (BM+PB)/2 are not correlated, the difference between the BM and PB mean values is an unbiased estimate of the bias, the difference one would expect on average between BM and PB flow readouts.
When the p-value is marked in dark green, the bias is 0 and BM may be substituted for PB and vice-versa. When the p-value is marked in light green, the substition can be made with the addition (subtraction) of the bias.
Classification of the comparison of BM and PB for each nodemetric following the method of Altman and Bland (1983). Nodemetrics where the BM and PB values are judged equivalent are classified as “Equivalent” and can be used interchangeably on average. Nodemetrics classified as “Other” require further investigation before they can be used interchangeably.
Table 7.
Comparison of bone marrow and peripheral blood with respect to each node for the Uu1 metric comparing the modulated and auto fluorescence wells
| Readout | Simple linear regression BM = intercept + slope*PB |
Bland-Altman Analysis Difference3 (BM − PB) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Biological Effect | Modulator | Antibody | Color | R2 | Slope | p-value2 | Intercept | Bias estimate | Std Dev | p-value4 | Classification5 |
| Apoptosis | |||||||||||
|
| |||||||||||
| Dauno + Ara-C | Cleaved PARP | FITC-A | 0.685 | 0.84 | <.0001 | 0.11 | 0.002 | 0.083 | 0.8783 | Equivalent | |
| Dauno + Ara-C | p-Chk2 | Alexa Fluor 647-A | 0.829 | 0.82 | <.0001 | 0.11 | −0.003 | 0.052 | 0.7456 | Equivalent | |
| Etoposide | Cleaved PARP | FITC-A | 0.661 | 1 | <.0001 | 0.02 | 0.014 | 0.041 | 0.0412 | Other | |
| Etoposide | p-Chk2 | Alexa Fluor 647-A | 0.737 | 0.74 | <.0001 | 0.19 | 0.014 | 0.057 | 0.1224 | Equivalent | |
| Signaling | |||||||||||
|
| |||||||||||
| FLT-3 Ligand | p-Akt | Alexa Fluor 647-A | 0.622 | 0.77 | <.0001 | 0.13 | 0.006 | 0.035 | 0.2373 | Equivalent | |
| FLT-3 Ligand | p-ERK | PE-A | 0.651 | 0.72 | <.0001 | 0.16 | 0.004 | 0.049 | 0.5902 | Equivalent | |
| FLT-3 Ligand | p-S6 | Alexa Fluor 488-A | 0.704 | 0.8 | <.0001 | 0.14 | 0.004 | 0.069 | 0.7139 | Equivalent | |
| G-CSF | p-STAT1 | Alexa Fluor 488-A | 0.568 | 0.84 | <.0001 | 0.1 | 0.015 | 0.062 | 0.1119 | Equivalent | |
| G-CSF | p-STAT3 | PE-A | 0.74 | 0.86 | <.0001 | 0.11 | 0.021 | 0.073 | 0.0605 | Equivalent | |
| G-CSF | p-STAT5 | Alexa Fluor 647-A | 0.746 | 0.86 | <.0001 | 0.12 | 0.024 | 0.073 | 0.029 | Other | |
| Hydrogen Peroxide | p-Akt | Alexa Fluor 488-A | 0.681 | 0.83 | <.0001 | 0.12 | −0.013 | 0.054 | 0.1236 | Equivalent | |
| Hydrogen Peroxide | p-PLCγ2 | PE-A | 0.854 | 0.93 | <.0001 | 0.07 | 0.032 | 0.078 | 0.008 | Biased Low | |
| Hydrogen Peroxide | p-SLP-76 | Alexa Fluor 647-A | 0.687 | 0.93 | <.0001 | 0.04 | 0.011 | 0.069 | 0.2816 | Equivalent | |
| IL-27 | p-STAT1 | Alexa Fluor 488-A | 0.827 | 0.9 | <.0001 | 0.07 | −0.001 | 0.06 | 0.8822 | Equivalent | |
| IL-27 | p-STAT3 | PE-A | 0.756 | 0.85 | <.0001 | 0.09 | 0 | 0.061 | 0.9904 | Equivalent | |
| IL-27 | p-STAT5 | Alexa Fluor 647-A | 0.627 | 0.78 | <.0001 | 0.12 | −0.004 | 0.06 | 0.6927 | Equivalent | |
| PMA | p-CREB | PE-A | 0.763 | 0.84 | <.0001 | 0.13 | 0.021 | 0.073 | 0.0663 | Equivalent | |
| PMA | p-ERK | Alexa Fluor 647-A | 0.716 | 0.85 | <.0001 | 0.12 | −0.002 | 0.069 | 0.887 | Equivalent | |
| PMA | p-S6 | Alexa Fluor 488-A | 0.742 | 0.8 | <.0001 | 0.16 | 0.008 | 0.067 | 0.4246 | Equivalent | |
| SCF | p-Akt | Alexa Fluor 647-A | 0.726 | 0.84 | <.0001 | 0.12 | 0.026 | 0.054 | 0.0017 | Other | |
| SCF | p-ERK | PE-A | 0.434 | 0.59 | <.0001 | 0.23 | 0.008 | 0.054 | 0.3389 | Equivalent | |
| SCF | p-S6 | Alexa Fluor 488-A | 0.68 | 0.74 | <.0001 | 0.18 | 0.013 | 0.078 | 0.2677 | Equivalent | |
Uu=Mann-Whitney U comparing modulated and unmodulated wells
P-value of the test that the slope equals 0.
When (BM−PB) and (BM+PB)/2 are not correlated, the difference between the BM and PB mean values is an unbiased estimate of the bias, the difference one would expect on average between BM and PB flow readouts.
When the p-value is marked in dark green, the bias is 0 and BM may be substituted for PB and vice-versa. When the p-value is marked in light green, the substition can be made with the addition (subtraction) of the bias.
Classification of the comparison of BM and PB for each nodemetric following the method of Altman and Bland (1983). Nodemetrics where the BM and PB values are judged equivalent are classified as “Equivalent” and can be used interchangeably on average. Nodemetrics classified as “Other” require further investigation before they can be used interchangeably.
In total, 97% of the 161 node-metrics studied in matched BMMC and PBMC were judged equivalent using either the Bland-Altman method (Table 2, Supplemental Tables S1B, S2B) or Lin’s concordance correlation coefficient (Table 2, Supplemental Tables S1C, S2C). For the majority of signaling node-metrics, the difference between BMMC and PBMC is either not statistically significant at the 5% level or a simple linear transformation equates the two methods on average.
Bland-Altman analysis of the basal signaling as evaluated using the Ua metric across all nodes and all donors revealed equivalence between PBMC and BMMC samples (Table 6) for all nodes except p-Erk-PE and p-S6-Alexa488 in the DMSO vehicle. Visual inspection of the Bland-Altman plots (Supplemental Figure S3) for these two nodes indicate that most donors have high basal levels of expression of p-Erk and p-S6 relative to autofluorescence. With the exception of one datapoint in the p-Erk-PE, the data for both tissue types are tightly clustering around a Ua value between 0.85 and 1.0 and are consistent with each other.
Analysis of modulated signaling measure using Uu across all nodes and all donors revealed equivalence between PBMC and BMMC samples (R2= 0.819, Figure 4).
Figure 4.

Correlation of the SCNP signaling and apoptosis readouts on the Uu metric between BM and PB samples.
Several of the exceptions that displayed poorer linear regression correlations (R2<0.64) belonged to nodes with weak response to modulation (e.g. SCF→p-Erk | Uu). The response for those nodes is consistent between the two tissue types, as can be seen from the Bland-Altman analyses.
Identification and Analysis of Biological Outliers
Two biological outliers (ECOG023 and ECOG025) were identified using methods described above (see Methods, Identification of Outliers) (Figure 5A). Further analysis revealed that biological outliers displayed distinct leukemic cell populations between paired BM and PB samples. Specifically, ECOG023 and ECOG025 BM samples demonstrated a higher proportion of CD34+ cells compared to the paired PB sample (Figure 5B). ECOG023 was collected from a patient with refractory anemia with excess blasts in transformation (t-RAEB). Because CD34+ cells respond to SCF, G-CSF, IL-27 modulators, the marked difference in leukemic cell populations may explain the lower equivalence between PB and BM samples for these two patients.
Figure 5. Process for identification of outlier samples.
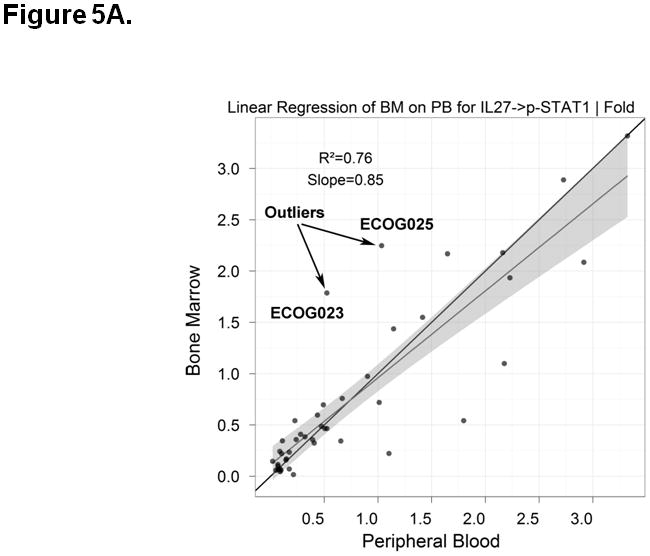
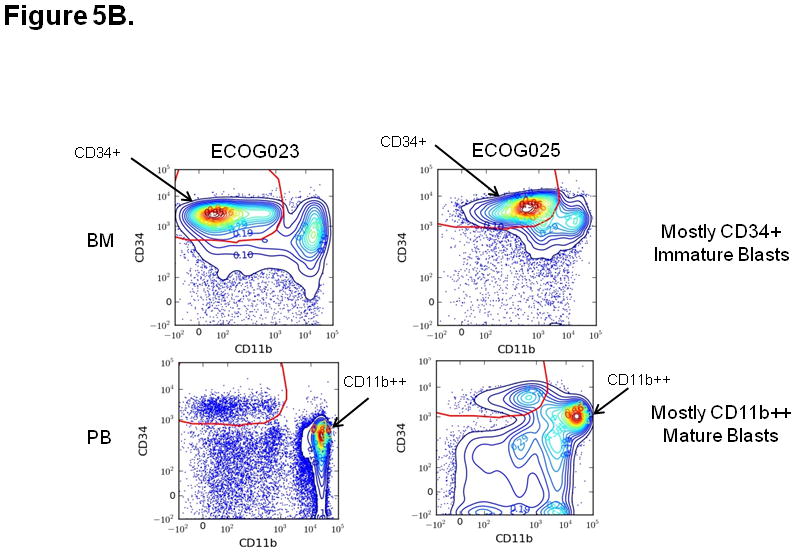
A) Outlier samples were identified by review of linear regression plots of signaling and apoptosis nodes. Results that were outside the 95% confidence interval of the linear regression fit were subject to additional analysis. As an example, the IL-27 -> p-STAT1 | Fold response is higher in BM compared to PB for ECOG023 and ECOG025. B) The gating plots were then visually inspected on the outliers identified from the linear regression plots. Results for CD11b vs. CD34 on both PB and BM for ECOG023 and ECOG025 are shown. In the case of ECOG023 and ECOG025, the BM contains mostly CD34+ immature blasts while the PB contains mostly CD11b++ mature blasts (i.e. “biologic outliers”).
Accounting for Sample Quality Improves the Concordance Between Paired BM and PB Samples
Signaling has been shown to be affected by sample quality (Figure 2). In this study, differences in PB and BM sample quality, as measured by PH, were observed in a few donors (Figure 3). Regression analysis, including PH as a covariate, was performed in order to evaluate the impact of sample quality on concordance between matched PB and BM readouts. The slope for the difference in PH between samples was found to be significant (P < 0.05, slope=0.0) in 68% of Uunode-metrics (13/19) (Table 8). This data shows that sample quality plays a significant role in the concordance between PB and BM sample readouts.
Table 8.
Impact of sample quality on concordance between matched peripheral blood and bone marrow readouts.
| Modulator | Modulator Time | Stain | Color | R2 | R2 when difference in PH is included as a covariate | p (slope=0) for PH1 | N |
|---|---|---|---|---|---|---|---|
| AraC+Daunorubicin | 24 Hrs | cPARP | FITC-A | 0.670 | 0.763 | 0.003 | 31 |
| AraC+Daunorubicin | 24 Hrs | p-Chk2 | Alexa Fluor 647-A | 0.845 | 0.845 | 0.864 | 31 |
| Etoposide | 24 Hrs | cPARP | FITC-A | 0.669 | 0.683 | 0.241 | 35 |
| Etoposide | 24 Hrs | p-Chk2 | Alexa Fluor 647-A | 0.738 | 0.770 | 0.044 | 35 |
| FLT-3 Ligand | 15 min | p-Akt | Alexa Fluor 647-A | 0.612 | 0.656 | 0.041 | 39 |
| FLT-3 Ligand | 15 min | p-ERK | PE-A | 0.644 | 0.671 | 0.094 | 39 |
| FLT-3 Ligand | 15 min | p-S6 | Alexa Fluor 488-A | 0.725 | 0.785 | 0.003 | 39 |
| G-CSF | 15 min | p-STAT1 | Alexa Fluor 488-A | 0.574 | 0.668 | 0.003 | 39 |
| G-CSF | 15 min | p-STAT3 | PE-A | 0.777 | 0.848 | 0.000 | 39 |
| G-CSF | 15 min | p-STAT5 | Alexa Fluor 647-A | 0.777 | 0.844 | 0.000 | 39 |
| Hydrogen Peroxide | 15 min | p-Akt | Alexa Fluor 488-A | 0.727 | 0.729 | 0.661 | 38 |
| Hydrogen Peroxide | 15 min | p-PLCγ2 | PE-A | 0.841 | 0.860 | 0.034 | 38 |
| Hydrogen Peroxide | 15 min | p-SLP76 | Alexa Fluor 647-A | 0.741 | 0.761 | 0.096 | 38 |
| IL-27 | 15 min | p-STAT1 | Alexa Fluor 488-A | 0.857 | 0.880 | 0.012 | 39 |
| IL-27 | 15 min | p-STAT3 | PE-A | 0.808 | 0.826 | 0.066 | 39 |
| IL-27 | 15 min | p-STAT5 | Alexa Fluor 647-A | 0.734 | 0.744 | 0.248 | 39 |
| PMA | 15 min | p-CREB | PE-A | 0.836 | 0.868 | 0.007 | 37 |
| PMA | 15 min | p-ERK | Alexa Fluor 647-A | 0.768 | 0.837 | 0.001 | 37 |
| PMA | 15 min | p-S6 | Alexa Fluor 488-A | 0.806 | 0.842 | 0.009 | 37 |
| SCF | 15 min | p-Akt | Alexa Fluor 647-A | 0.781 | 0.866 | 0.000 | 39 |
| SCF | 15 min | p-ERK | PE-A | 0.397 | 0.445 | 0.087 | 39 |
| SCF | 15 min | p-S6 | Alexa Fluor 488-A | 0.723 | 0.829 | 0.000 | 39 |
The slope for the difference in PH between samples was found to be significant (P < 0.05, slope=0.0)
DISCUSSION
SCNP assay using multiparametric flow cytometry is emerging as a valuable tool to inform on therapy outcomes or relapse risks in AML (22–24). Although SCNP assay has previously been limited to academic settings (16–21), recent studies have shown the potential utility of this assay in clinical settings to inform decision making (22–24). In order to successfully transition this technology from the academic environment to the clinic, instrument, reagent and assay procedure standardization were put in place. Specifically, standard instrument controls (rainbow beads) and cell line controls enabled the assessment of technical variability at the modulation, fixation, staining and acquisition steps in the laboratory work flow thus allowing for the generation of reproducible results across operators, plates and time. These improvements are essential for the development of clinically applicable assays.
In addition, specific metrics were developed to describe and quantify the functional changes observed using the SCNP assay. Classic flow cytometry metrics measure “static” events using surface markers. In contrast, the metrics described in this paper are designed to quantify the biological effect of modulation over time, providing a dynamic and quantitative view of intracellular functional events. These metrics are calculated on a relative basis and compared across patients and tissue types, which underscores the importance of equipment calibration and standardization. Data analysis using a standard set of metrics is a critical component for assay reproducibility and a required step for moving the phosphoflow assay from the laboratory into clinical practice.
Moreover, pre-analytical sample characteristics pose potential barriers to standardization in flow cytometry. In particular, functional assays require that the cells are not only viable but sufficiently “healthy” to signal. Standardization methods based on sample quality could allow for improved control of pre-analytical variables and comparison of samples that were subject to different handling procedures (i.e. cryopreserved vs. fresh).
After the development and implementation of processes to control for technical variability within the assay, the effect of specimen source on proteomic signaling was assessed by comparing signaling readouts for matched viable BMMC and viable PBMC. Because of the way the experiments were designed, the differentiation between technical assay variability and biological variability in leukemic cells from paired BMMC and PBMC specimens was possible.
Overall, strong correlations between the majority of nodes in matched PB and BM samples were observed; correlations were particularly high in signaling nodes which had the higher levels of changes under modulation. The few exceptions that were noted could be classified as either technical or biological “outliers”. Specifically, repeatability analyses identified a low frequency of technical outliers which were attributed to a cytometer clog. The extremely low frequency of technical outliers (2 out of more than 8,000 assays) demonstrated that the instruments performed within expected levels of variance and that the modulation and staining processes were consistent among plates and across days.
In addition, two biological outliers were also identified in which the primary leukemic cell population identified in the BM was almost undetectable in the paired PB samples. This is in alignment with previous observations that a small number of AML patients will not have circulating myeloblasts, and therefore these samples will not be usable for the SCNP assay. It is also of note that overall correlations may be further improved by adjustments to the gating strategy using two methods. First, one could use additional surface markers to better identify the leukemic myeloblasts only. Second, one could utilize subpopulations (i.e. immature and mature myeloblasts) for SCNP signaling readouts within those cell populations only. Despite these future efforts, we have demonstrated that a classifier based on the leukemic blasts as defined in this analysis has been validated and shown repeated accuracy in independent patient sample sets (22,24).
An important observation in this study was the effect of sample quality (pre-analytic variable) on intrinsic cell signaling capability; specifically, controlling for cell health improved correlation between matched PB and BM samples readouts. These results suggest that analysis of sample quality and cell health must be integrated into study design and qualification of samples and used to control/adjust the level of signaling. Currently, two methods have been utilized to account for cell health – gating on live, healthy cells (defined using both Live/Dead Aqua Stain and cleaved PARP) and normalization using a pre-specified adjustment factor for cell health. We have previously reported on the successful application of a normalization adjustment during classifier development (24), which underscores the importance of standardized, pre-specified processes for addressing cell health in signaling assays.
While not the focus of this study, demonstrating a correlation of SCNP readouts between fresh and cryopreserved preparations is essential to applying these proteomic signatures to the clinical setting. Bridging studies which compare the results of SCNP assays between paired fresh and cryopreserved samples have been published (34), and results suggest that SCNP assays developed and validated using cryopreserved samples can be applied to fresh samples and integrated prospectively into frontline clinical trials and clinical practice. Further studies are ongoing.
In summary, our results demonstrate that specimen source (BM or PB) does not significantly affect proteomic signaling in patients with AML and circulating blasts. Therefore, PB myeloblasts can be used as a sample source for SCNP assays to identify functionally distinct leukemic blast cell populations. The ability to use PB as a sample source will greatly improve the utility of these assays, e.g. it can facilitate the analysis of target coverage and off-target activities during preclinical selection of lead molecules and early phase clinical trials and also guide dosing schedules after a drug candidate moves to clinical testing. Moreover, PB can be used as a sample source at time-points when BM aspirates are not clinically justifiable. The development of instrument standards and process controls described here will accelerate the transition of SCNP proteomic assays from the academic laboratory to the clinic.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Footnotes
Conflict of Interest Statement: A. C., D. B. R., P.O., S. P., U. G., and D. C. S. are employees of and holders of equity in Nodality Inc.
References
- 1.Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–62. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 2.Bloomfield CD, Lawrence D, Byrd JC, Carroll A, Pettenati MJ, Tantravahi R, Patil SR, Davey FR, Berg DT, Schiffer CA, et al. Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res. 1998;58:4173–9. [PubMed] [Google Scholar]
- 3.Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, Rees J, Hann I, Stevens R, Burnett A, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood. 1998;92:2322–33. [PubMed] [Google Scholar]
- 4.Byrd JC, Mrozek K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC, Pettenati MJ, Patil SR, Rao KW, Watson MS, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461) Blood. 2002;100:4325–36. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- 5.Advani AS, Rodriguez C, Jin T, Jawde RA, Saber W, Baz R, Kalaycio M, Sobecks R, Sekeres M, Tripp B, et al. Increased C-kit intensity is a poor prognostic factor for progression-free and overall survival in patients with newly diagnosed AML. Leuk Res. 2008;32:913–8. doi: 10.1016/j.leukres.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 6.Boissel N, Leroy H, Brethon B, Philippe N, de Botton S, Auvrignon A, Raffoux E, Leblanc T, Thomas X, Hermine O, et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML) Leukemia. 2006;20:965–70. doi: 10.1038/sj.leu.2404188. [DOI] [PubMed] [Google Scholar]
- 7.Bullinger L, Dohner K, Kranz R, Stirner C, Frohling S, Scholl C, Kim YH, Schlenk RF, Tibshirani R, Dohner H, et al. An FLT3 gene-expression signature predicts clinical outcome in normal karyotype AML. Blood. 2008;111:4490–5. doi: 10.1182/blood-2007-09-115055. [DOI] [PubMed] [Google Scholar]
- 8.Dohner K, Schlenk RF, Habdank M, Scholl C, Rucker FG, Corbacioglu A, Bullinger L, Frohling S, Dohner H. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106:3740–6. doi: 10.1182/blood-2005-05-2164. [DOI] [PubMed] [Google Scholar]
- 9.Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, Habdank M, Spath D, Morgan M, Benner A, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–18. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 10.Schlenk RF, Dohner K. Impact of new prognostic markers in treatment decisions in acute myeloid leukemia. Curr Opin Hematol. 2009;16:98–104. doi: 10.1097/MOH.0b013e3283257adb. [DOI] [PubMed] [Google Scholar]
- 11.Schnittger S, Schoch C, Kern W, Mecucci C, Tschulik C, Martelli MF, Haferlach T, Hiddemann W, Falini B. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood. 2005;106:3733–9. doi: 10.1182/blood-2005-06-2248. [DOI] [PubMed] [Google Scholar]
- 12.Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD, Carroll AJ, Mrozek K, Vardiman JW, George SL, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. 2001;61:7233–9. [PubMed] [Google Scholar]
- 13.Marcucci G. Molecular markers in acute myeloid leukemia. Clin Adv Hematol Oncol. 2009;7:448–51. [PubMed] [Google Scholar]
- 14.Marcucci G, Mrozek K, Bloomfield CD. Molecular heterogeneity and prognostic biomarkers in adults with acute myeloid leukemia and normal cytogenetics. Curr Opin Hematol. 2005;12:68–75. doi: 10.1097/01.moh.0000149608.29685.d1. [DOI] [PubMed] [Google Scholar]
- 15.Becker H, Marcucci G, Maharry K, Radmacher MD, Mrozek K, Margeson D, Whitman SP, Wu YZ, Schwind S, Paschka P, et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:596–604. doi: 10.1200/JCO.2009.25.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud O, Gjertsen BT, Nolan GP. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118:217–28. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 17.Irish JM, Kotecha N, Nolan GP. Mapping normal and cancer cell signalling networks: towards single-cell proteomics. Nat Rev Cancer. 2006;6:146–55. doi: 10.1038/nrc1804. [DOI] [PubMed] [Google Scholar]
- 18.Krutzik PO, Nolan GP. Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nat Methods. 2006;3:361–8. doi: 10.1038/nmeth872. [DOI] [PubMed] [Google Scholar]
- 19.Sachs K, Perez O, Pe’er D, Lauffenburger DA, Nolan GP. Causal protein-signaling networks derived from multiparameter single-cell data. Science. 2005;308:523–9. doi: 10.1126/science.1105809. [DOI] [PubMed] [Google Scholar]
- 20.Perez OD, Nolan GP. Phospho-proteomic immune analysis by flow cytometry: from mechanism to translational medicine at the single-cell level. Immunol Rev. 2006;210:208–28. doi: 10.1111/j.0105-2896.2006.00364.x. [DOI] [PubMed] [Google Scholar]
- 21.Kotecha N, Flores NJ, Irish JM, Simonds EF, Sakai DS, Archambeault S, Diaz-Flores E, Coram M, Shannon KM, Nolan GP, et al. Single-cell profiling identifies aberrant STAT5 activation in myeloid malignancies with specific clinical and biologic correlates. Cancer Cell. 2008;14:335–43. doi: 10.1016/j.ccr.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kornblau SM, Minden MD, Rosen DB, Putta S, Cohen A, Covey T, Spellmeyer DC, Fantl WJ, Gayko U, Cesano A. Dynamic single-cell network profiles in acute myelogenous leukemia are associated with patient response to standard induction therapy. Clin Cancer Res. 2010;16:3721–33. doi: 10.1158/1078-0432.CCR-10-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cesano A, Rosen DB, Putta S, Gayko U, Cripe L, Sun Z, Uno H, Litzow MR, Tallman MS, Paietta E. Specimen source (BM or PB) does not affect proteomic signaling in patients with AML and circulating blasts. Blood (ASH Annual Meeting Abstracts) 2010;116:Abstract 2693. [Google Scholar]
- 24.Lacayo NJCA, Westfall M, Lackey A, Xin X, Gayko U, Putta S, Meshinchi S, Raimondi SC, Alonzo TA, Arceci RJ, et al. Single cell network profiling (SCNP) signatures predict response to induction therapy and relapse risk in pediatric patients with Acute Myeloid Leukemia: Children’s Oncology Group (COG) study POG-9421. Blood (ASH Annual Meeting Abstracts) 2010:116, Abstract 954. [Google Scholar]
- 25.Covey TM, Cesano A. Modulated multiparametric phosphoflow cytometry in hematological malignancies: technology and clinical applications. Best Pract Res Clin Haematol. 2010;23:319–31. doi: 10.1016/j.beha.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Purvis N, Stelzer G. Multi-platform, multi-site instrumentation and reagent standardization. Cytometry. 1998;33:156–65. [PubMed] [Google Scholar]
- 27.Stelzer GT, Goodpasture L. Use of multiparameter flow cytometry and immunophenotyping for the diagnosis and classification of acute myeloid leukemia. In: Stewart CC, Nicholson JKA, editors. Immunophenotyping. Wilmington DE: Wiley-Liss; 2000. pp. 215–238. [Google Scholar]
- 28.Shults KE, Miller DT, Davis BH, Flye L, Hobbs LA, Stelzer GT. A standardized ZAP-70 assay--lessons learned in the trenches. Cytometry B Clin Cytom. 2006;70:276–83. doi: 10.1002/cyto.b.20136. [DOI] [PubMed] [Google Scholar]
- 29.Wang L, Gaigalas AK, Marti G, Abbasi F, Hoffman RA. Toward quantitative fluorescence measurements with multicolor flow cytometry. Cytometry A. 2008;73A:279–88. doi: 10.1002/cyto.a.20507. [DOI] [PubMed] [Google Scholar]
- 30.Putta S, Spellmeyer D, Evensen E, Banville S, Friedland B, Rosen D, Soper D, Purvis N, Covey T, Francis-Lang H, et al. Informatics platform and workflows for robust high throughput single cell network profiling (SCNP) International Society for Advancement of Cytometry Annual Meeting abstract; 2010. [Google Scholar]
- 31.Altman DG, Bland JM. Measurement in Medicine: the Analysis of Method Comparison Studies. The Statistician. 1983:307–317. [Google Scholar]
- 32.Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet. 1986;1:307–10. [PubMed] [Google Scholar]
- 33.Lin LI. A concordance correlation coefficient to evaluate reproducibility. Biometrics. 1989;45:255–68. [PubMed] [Google Scholar]
- 34.Cesano A, Gotlib JR, Lacayo NJ, Putta S, Lackey A, Gayko U, Kornblau SM. Sample Cryopreservation Does Not Affect Functional Read Outs In SCNP Assays: Implications for Biomarker Development. Blood (ASH Annual Meeting Abstracts) 2010;116:4843. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.