

Summary
Strong epidemiologic evidence indicates that tobacco smoke influences frequency and severity of respiratory infections. Previously, we have shown that infection with respiratory syncytial virus upregulates expression of neurotrophic factors and receptors in the lungs, but the effect of tobacco exposure on neurotrophins is unknown. Therefore, we first sought to determine the expression of neurotrophic pathways in lungs of rats chronically exposed to nicotine, and then we studied the interactions between pollution and infection by inoculating virus after nicotine exposure. Expression of the neurotrophins nerve growth factor (NGF) and brain-derived neurotrophic factor, of their high-affinity tyrosine kinase receptors (trkA and trkB, respectively), and of the low-affinity receptor p75NTR was measured in the lungs of nicotine-exposed rats both at the mRNA level by reverse-transcription polymerase chain reaction and at the protein level by enzyme-linked immunoassay. Nicotine increased NGF expression both at the mRNA and protein level and also created a receptor imbalance deriving from increased expression of the pro-inflammatory p75NTR receptor without any concomitant change in the high-affinity trkA receptor. Viral infection after chronic nicotine exposure exerted an additive effect on NGF expression, and resulted in exaggerated neurogenic airway inflammation that was abolished by selective inhibition. In conclusion, nicotine levels comparable to those found in smokers are per se able to upregulate the expression of critical neurotrophic molecules in the respiratory tract, and combination of an acute infection following chronic nicotine exposure produces more severe neurotrophic dysregulation and neurogenic-mediated inflammation compared to either infection or nicotine alone.
Keywords: asthma, airway inflammation, bronchiolitis, nerve growth factor, tobacco smoke
INTRODUCTION
Strong epidemiologic evidence indicates that exposure to tobacco smoke increases frequency and severity of respiratory tract infections. In particular, several studies1–5 have found a significant association between parental smoking and the rate of hospitalization for respiratory syncytial virus (RSV) bronchiolitis in infants and children. Vice versa, viral infections may amplify and prolong the adverse effects of pollution, and recent data suggest that although tobacco smoking remains the major cause of chronic obstructive pulmonary disease, RSV infection may sustain airway inflammation in these patients leading to accelerated and progressive loss of lung function.6
Despite the large body of evidence underscoring the importance of interactions between tobacco smoke and viral respiratory infections, the pathophysiologic mechanisms involved remain largely unclear and controversial. Surprisingly, previous studies have reported a decrease in lung inflammation and mucus production when rodents exposed to side-stream cigarette smoke or to nicotine were infected with RSV7 or influenza virus.8 Although this finding is consistent with the hypothesis that certain components of tobacco smoke (specifically nicotine) might have anti-inflammatory properties,9,10 it does not fit well with the epidemiologic link between tobacco smoke exposure and virus-induced airway inflammation and hyperreactivity in humans.
Previous studies in animal models have shown that RSV infection upregulates the expression of neurotrophic factors and receptors in the lungs.11,12 The increased neurotrophic activity leads to pre-synaptic and post-synaptic modifications responsible for the inflammation and hyperreactivity that develop in virus-infected airways when airborne irritants stimulate intra- and subepithelial nociceptive nerves.13–15 The same pathophysiologic mechanisms appear to be implicated in human disease, as in recent clinical studies we have shown increased expression of neurotrophic factors and receptors in the airways of RSV-infected infants.16 Another environmental agent capable of modulating the activity of sensory neurons and contribute to the process of neurogenic inflammation is nicotine, the major tobacco alkaloid present in both the gaseous and particulate phase of cigarette smoke,17–19 but its effect on neurotrophins expression has not been investigated.
Therefore, in the present study, we first analyzed the expression of neurotrophic pathways in lungs chronically exposed to nicotine, which was infused into pathogen-free rats using micro-osmotic pumps implanted subcutaneously. Expression of nerve growth factor (NGF) and related neurotrophic factors and receptors was measured in the lungs of exposed rats both at the mRNA level by reverse-transcription polymerase chain reaction (RT-PCR) and at the protein level by enzyme-linked immunoassay. In the second part of this study, we explored the interactions between pollution and infection by inoculating RSV in rats previously exposed to nicotine for 28 days. In these rats we also measured neurogenic-mediated airway inflammation and assessed the pathophysiologic role played by the NGF pathway using a specific blocking antibody.
MATERIALS AND METHODS
Animals
We used pathogen-free Fischer 344 (F-344) rats purchased from Charles River Breeding Laboratories (Raleigh, NC). All rats were housed in polycarbonate cages isolated by polyester filter covers and placed on racks that provided positive individual ventilation with class-100 air to each cage at the rate of 1 cage change of air/min (Maxi-Miser, Thoren Caging System, Hazleton, PA). We used separate rooms for housing infected and pathogen-free rats, both of which were serviced by specifically trained husbandry technicians. All manipulations were conducted inside class-100 laminar flow hoods. Bedding, water, and food were autoclaved before use and unpacked only under laminar flow. Cages and water bottles were run through a tunnel washer after every use and disinfected with both chemicals and heat. The institutional Animal Use Committees approved all experimental procedures followed in this study.
Nicotine Exposure
Rats were anesthetized with an intramuscular injection of ketamine-xylazine (14 mg/kg of body weight) and topical 1% lidocaine injected subcutaneously at the implantation site. An osmotic mini-infusion pump (Alzet-Durect, Cupertino, CA) was placed subcutaneously under sterile conditions, and the incision was closed with VetBond tissue adhesive (3M, St. Paul, MN). All rats tolerated the procedure well and resumed their normal behavior within 2 hr.
Each mini-pump was filled with a solution of nicotine bitartrate (Sigma–Aldrich, St. Louis, MO) dissolved in 0.9% NaCl at a concentration of 33 mg/ml and infused at a rate of 60 μl/day to deliver a dose of 6 mg/kg/day,20 which results in plasma levels similar to those detected in humans who smoke or use transdermal nicotine patches.21 Control rats were infused with the vehicle used to dissolve the nicotine using the same delivery system, rate, and duration. Groups of six rats each were infused with either nicotine or vehicle for 14 or 28 days.
Two pregnant rats were implanted with a subcutaneous osmotic pump on the second day of their pregnancy. The pumps were set to deliver 6 mg/kg/day either nicotine or vehicle until delivery. At delivery (i.e., 19 days after pump implant), all newborns from each mother (n = 9 rats from the mother exposed to nicotine and n = 8 rats from the mother exposed to vehicle) were sacrificed and their lungs were harvested.
RT-PCR
The frozen specimens were placed in disruption buffer and homogenized using a conventional rotor-stator homogenizer (Brinkmann Instruments, Westbury, NY) for 45–60 sec. Total RNA was extracted using RNEasy Midi-Kits (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s specifications. These RNA samples were added to a 50 μl master mix consisting of 400 μM each of deoxynucleotide triphosphates (dNTPs), 10U of RNAse inhibitor, 2 μl of enzyme mix containing TaqDNA Polymerase (one-step RT-PCR; Promega, Madison, WI) and 50 pmol each of primers flanking the nucleotide sequence for NGF, brain-derived neurotrophic factor (BDNF), p75NTR, trkA, trkB, and the housekeeping gene β-actin.22 The lungs of rats infected with RSV and their pathogen-free controls were also tested for viral transcripts. The same master mix without the RNA sample was used as a negative control. The primers pairs were designed on the basis of previously published protocols and were used to differentiate cDNA-generated PCR products from genomic DNA contamination. The specific primers sequences (sense and anti-sense) are illustrated in Table 1. Amplification was performed using a Gene-Amp PCR System 9600 thermal cycler (Perkin-Elmer, Waltham, MA). The process was started with an initial step of 50°C for 30 min then 95°C for 15 min followed by 25–35 cycles with a denaturing step followed by an annealing step, an extension step, and then one final extension step at 68–72°C for 10 min. All programs included a 4°C hold step at the end. Amplified PCR products were size-fractionated by electrophoresis through a 2% agarose gel and stained with ethidium bromide. The gels were then photographed using an imaging system (FOTO/Analyst Luminary Workstation, Fotodyne, Hartland, WI). The intensity of DNA bands was analyzed by computerized densitometry (TotalLab TL-101 Image Analysis Software) and expressed as the ratio of the densitometric score measured for each target gene normalized by the β-actin control.
TABLE 1.
Primers for RT-PCR Amplification
| Target | Sense amino acids | Anti-sense amino acids | Product size bp | Accession number | GenInfo ID |
|---|---|---|---|---|---|
| NGF | 100–105 | 225–230 | 395 | M36589 | 310738 |
| BDNF | 129–134 | 230–236 | 293 | M61178 | 24225 |
| p75NTR | 60–65 | 274–279 | 663 | M012601 | 24596 |
| trkA | 138–145 | 354–360 | 690 | M021589 | 59109 |
| trkB | 42–49 | 251–260 | 664 | M55291 | 25054 |
| RSV | 71–75 | 201–205 | 409 | DQ7805631 | 11259 |
| β-Actin | 216–223 | 369–375 | 285 | C005111.2 | 81822 |
NGF, nerve growth factor, BDNF, brain-derived neurotrophic factor; trk, tyrosine kinase; RSV, respiratory syncytial virus.
NGF Immunoassay
NGF protein levels in the lungs were measured with a commercial kit (Promega) using the antibody sandwich technique. In brief, lung tissue samples (100 mg) were homogenized in 5 volumes of lysis buffer containing Tris–HCl (20 mM), NaCl (150 mM), NP-40 (1%), glycerol (10%), phenylmethylsulfonyl fluoride (1 mM), aprotinin (10 μg/ml), leupeptin (1 μg/ml), and sodium vanadate (0.5 mM). The supernatants of homogenized tissue samples were incubated for 18 hr at 4°C in 96-well plates coated with 100 μl of anti-NGF polyclonal antibody (1.5 μg/ml in 100mMcarbonate coating buffer; pH 9.7) to bind NGF from the homogenates. After washing, a specific rat monoclonal antibody was applied (0.6 μg/ml in buffer, 100 μl/well) and incubated for 18 hr at 4°C to bind the captured NGF. The plates were again thoroughly washed and horseradish peroxidase-conjugated antibody to rat IgG was added to each well and incubated for 3 hr at room temperature to detect the amount of specifically bound monoclonal antibody. After final washing to remove unbound antibody conjugate, the chromogenic substrate was added and the color change generated by the reaction was read at a wavelength of 450 nm. Test samples and NGF standards (100 μl/well) were measured in duplicate. Using this assay, NGF can be quantified with a lower detection limit of 15.6 pg/ml and <2% cross-reactivity with other neurotrophic factors.
RSV Preparation and Inoculation23–25
HEp-2 cells were grown in Eagle’s minimum essential medium (MEM) supplemented with 10% fetal bovine serum (GIBCO-BRL, Grand Island, NY). Confluent monolayers of HEp-2 cells were infected with 0.1 plaque-forming units of human RSV strain ALong and the infection was allowed to proceed at 37°C in 5% CO2 atmosphere until more than 75% of the cells exhibited cytopathic effect.25 Cell debris was removed by centrifugation at 9,500g for 20 min in a centrifuge refrigerated at 4°C. Aliquots of the virus stock were snap-frozen in liquid nitrogen and stored at −70°C. Prior to inoculation, the viral stock was titrated and diluted as needed for a final titer of 5 × 104 TCID50 (50% tissue culture infective dose) in 0.1 ml. Supernatants and cell lysates from virus-free flasks of HEp-2 cells in MEM were harvested, centrifuged, and aliquoted following the same protocol to obtain the virus-free medium used as a sham infection control.
Rats were anesthetized with pentobarbital sodium (50 mg/kg i.p.) at the end of a 28-day infusion of nicotine or its vehicle and inoculated with a volume of 0.4 ml/kg body weight of RSV suspension or virus-free medium through an endotracheal cannula. Groups of six rats each inoculated with virus or medium were sacrificed 5 days after inoculation.
Neurogenic Inflammation25–27
Five days after inoculation with RSV or virus-free medium, rats were anesthetized with pentobarbital sodium (50 mg/kg i.p.) and the femoral vein was exposed. The rats were injected with Evans blue dye (30 mg/kg i.v. over 5 sec) to measure the extravasation of albumin from airway blood vessels.25 Immediately after the injection of the tracer, all rats received a 2-min intravenous infusion of 75 μg/kg of capsaicin (8-methyl-N-vanillyl-6-nonenamide; Sigma-Aldrich, St. Louis, MO) dissolved in a vehicle having a final concentration of 0.75% ethanol, 0.375% Tween-80, and 0.85% NaCl in aqueous solution. Capsaicin is a neurotoxin that binds the transient receptor potential cation channel, subfamily V, member 1 (TRPV1) highly expressed on C-type afferent neurons, thereby releasing the neurotransmitters stored in their terminal varicosities.28 All chemicals were delivered in a volume of 1 ml/kg of body weight.
Five minutes after the injection of the tracer, the chest was opened and a 22-gauge cannula was inserted into the ascending aorta through the left ventricle. After incision of the left atrium, the circulation was perfused for 2 min with PBS using a syringe pump set at the rate of 50 ml/min. The airways were dissected, blotted, weighed, and incubated in 1ml of formamide (Sigma) at 50°C for 18 hr to extract the extravasated Evans blue dye. The extravasation of Evans blue-labeled albumin was quantified by measuring the optical density of the formamide extracts at a wavelength of 620 nm. Evans blue extravasation expressed in nanograms per milligram of wet-tissue weight was interpolated from a standard curve of Evans blue concentrations (0.5–10 μg/ml).
NGF Inhibition
As neurotrophic factors are highly conserved in different species,29 we blocked endogenous NGF activity using a polyclonal rabbit anti-mouse antibody bioactivity: 1:10,000 dilution blocks NGF-induced growth of PC-12 cells; Sigma). Anti-NGF (1:2,000 dilution, 4 ml/kg) or control antibody (rabbit purified IgG, 1:2,000 dilution, 4 ml/kg; Sigma) was injected subcutaneously in groups of five rats 60 min before each experimental intervention, that is, 60 min before starting the infusion of nicotine or its vehicle, 60 min before inoculation of RSV or virus-free medium, and 60 min before the infusion of capsaicin (n = 5 rats per group). A single dose of anti-NGF antibody has been shown to inhibit sympathetic innervation in a fetal lamb model for at least 55 days.30 Timing and doses of anti-NGF treatment were chosen on the basis of previous studies that confirmed the efficacy of this antibody in blocking NGF biological effects in our rat model of RSV infection and compared its activity to chemical inhibition of the NGF signaling cascade by K-252a.11,12
Statistical Analysis
Data are expressed as mean ± SEM. Two-factor ANOVA was used to analyze differences between rats exposed to nicotine or vehicle and to RSV or virus-free medium.31 Multiple comparisons between means were performed with the Fisher Protected Least Significant Difference test.32 Statistical analysis was performed using the software StatView version 5.0.1 (SAS Institute, Cary, NC) for Windows. Differences having a P-value <0.05 were considered significant.
RESULTS
Nicotine
Figure 1 shows a representative RT-PCR analysis of mRNA transcripts for NGF (top panel) and its p75NTR (mid panel) and trkA (bottom panel) receptors in lung tissues from a pathogen-free rat exposed to nicotine for 28 days using a subcutaneous osmotic mini-infusion pump (right bands) versus its control exposed to vehicle with the same technique (left bands).
Fig. 1.

RT-PCR analysis of mRNA transcripts for NGF (top panel) and its p75NTR (mid panel) and trkA (bottom panel) receptors in lung tissues from a pathogen-free rat exposed to nicotine for 28 days using a subcutaneous osmotic mini-infusion pump (right bands) versus its control exposed to vehicle with the same technique (left bands). The representative electrophoresis on ethidium bromide-stained agarose gel shows increased NGF and p75NTR expression without change in trkA.
RT-PCR analysis (Fig. 2, left) of lung tissues from rats exposed to nicotine for 14 days revealed a significant increase in NGF mRNA expression compared to controls infused with vehicle (n = 6 rats per group; P<0.05), and a similar increase was measured after 28 days of nicotine exposure (n = 6 rats per group; P<0.01). Immunoassay analysis (Fig. 2, right) confirmed that NGF protein concentration was significantly increased in the lungs of nicotine-exposed rats compared with controls both after 14 days (P<0.01) or 28 days (P<0.05) of exposure.
Fig. 2.
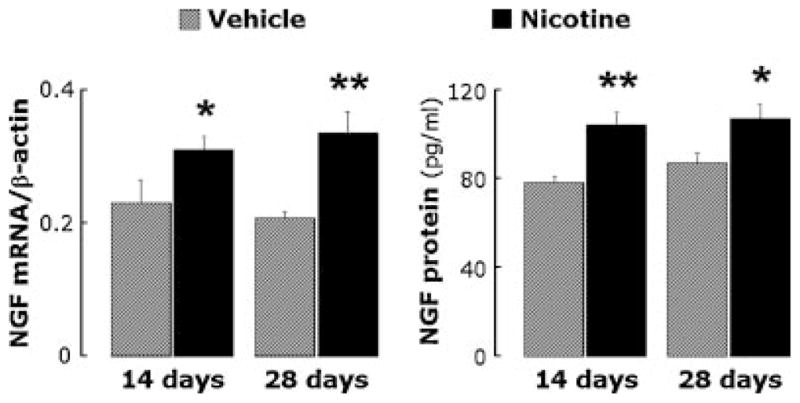
RT-PCR (left panel) and immunoassay (right panel) analysis of lung tissues from rats exposed to nicotine for 14 or 28 days. NGF expression at the mRNA and protein level increased in nicotine-exposed rats at both time points. Data are expressed as mean ± SEM, n = 6 rats per group. *P<0.05; **P<0.01, significantly different from vehicle controls.
RT-PCR analysis was also used to measure expression of the NGF receptors p75NTR and trkA in lung homogenates. Rats exposed to nicotine had increased p75NTR receptor mRNA expression both at 14 days (P<0.05; Fig. 3, left) and at 28 days (P<0.01, Fig. 3, right), whereas there was no change in trkA mRNA levels after a 14-day (P = 0.8) or 28-day exposure (P = 0.7). Consequently, the p75NTR/trkA ratio in lung tissues was markedly increased after exposure to nicotine.
Fig. 3.
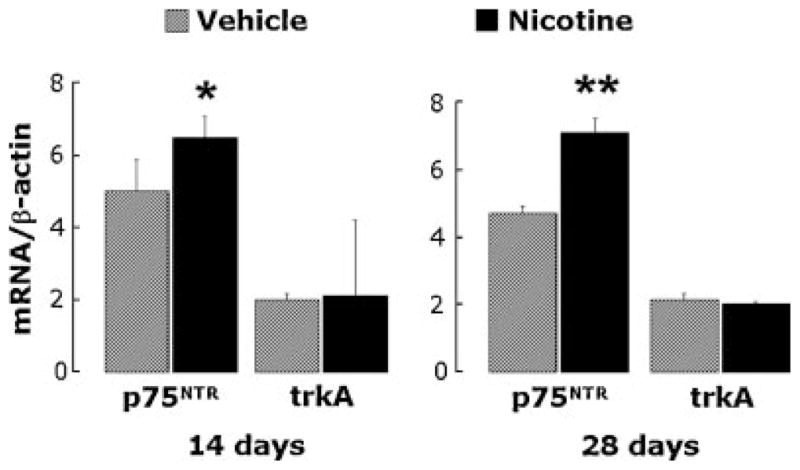
RT-PCR analysis of NGF receptors mRNA expression in lung tissues from rats exposed to nicotine for 14 (left panel) or 28 (right panel) days. Expression of the low-affinity pan-neurotrophin p75NTR receptor increased in nicotine-exposed rats at both time points, whereas the specific high-affinity trkA receptor did not change. Data are expressed as mean ± SEM, n = 6 rats per group. *P<0.05; **P<0.01, significantly different from vehicle controls.
As for the induction of NGF, most of the changes in the expression of neurotrophic receptors occurred within the first 2 weeks of exposure to nicotine, and persisted with only minor changes over the following 2 weeks of exposure. In contrast, we found no significant change in the mRNA expression of the BDNF (1.55 ± 0.09 vs. 1.52 ± 0.06; P = 0.8) or of its specific high-affinity receptor trkB (0.42 ± 0.16 vs. 0.33 ± 0.09; P = 0.7) in the same lung tissues exposed for 28 days, and therefore we elected to focus our attention on NGF and its receptors.
The lungs of newborn rats born from a mother exposed to nicotine for 19 days during pregnancy (i.e., from pregnancy day 2 to birth; n = 9 rats) did not show upregulation of NGF (Fig. 4, left) or its receptors (Fig. 4, right) in lung tissues, and actually we found a significant decrease in NGF and trkA transcripts compared to the lungs of newborns born from a mother infused for 19 days with vehicle (n = 8 rats; P<0.01).
Fig. 4.

RT-PCR analysis of lung tissues from newborn rats exposed to nicotine in utero for 19 days (i.e., from day 2 of pregnancy to birth). NGF and trkA mRNA expression decreased in nicotine-exposed newborns, whereas the p75NTR receptor did not change. Data are expressed as mean ± SEM, n = 8–9 rats per group. **P<0.01, significantly different from vehicle controls.
Nicotine and RSV
The lungs of rats exposed to nicotine for 28 days and then inoculated with virus-free medium (Fig. 5) expressed a significantly higher level of NGF protein compared to rats inoculated with virus-free medium after a 28-day infusion of vehicle (n = 6 rats per group; P<0.05). Five days of RSV infection after a 28-day infusion of vehicle produced a similar increase in NGF (n = 6 rats; P<0.05). However, 5 days infection with RSV following a 28-day exposure to nicotine had an additive effect on NGF protein expression compared to the effects of either nicotine alone (n = 6 rats; P<0.001) or RSV alone (P<0.001). As a result, the combination of chronic exposure to nicotine and acute viral infection approximately doubled NGF expression in lung tissues.
Fig. 5.

NGF protein expression in the lungs of rats infused with nicotine or its vehicle for 28 days and then inoculated with RSV or virus-free medium 5 days before sacrifice. Acute RSV infection following chronic exposure to nicotine had additive effect on NGF protein expression compared to the effects of either nicotine alone or RSV alone. Data are expressed as mean ± SEM, n = 6 rats per group. *P<0.05; ***P<0.001, significantly different from controls infused with nicotine vehicle and inoculated with virus-free medium.
Although there was a trend for increased expression of both p75NTR receptors (0.418 ± 0.173 vs. 0.266 ± 0.014; P = 0.4) and trkA receptors (0.068 ± 0.033 vs. 0.048 ± 0.017; P = 0.6) in rats infected with RSV after nicotine exposure compared to rats infected after vehicle infusion, these differences were not statistically significant.
Pharmacological stimulation of nociceptive nerve fibers with capsaicin (Fig. 6) increased by 82% the extravasation of Evans blue-labeled albumin in the airways of rats inoculated with medium after a 28-day infusion of nicotine compared to control rats inoculated with virus-free medium after a 28-day infusion of vehicle (n = 5 rats per group; P<0.05). Instead, capsaicin increased by 207% the extravasation of Evans blue in the airways of rats infected for 5 days with RSV after a 28-day infusion of vehicle (n = 5 rats; P<0.0001). The same infection after a 28-day exposure to nicotine increased neurogenic extravasation by 342% (n = 5 rats; P<0.0001), which was significantly larger than the effect of either nicotine alone (P<0.0001) or RSV alone (P<0.001) and was consistent with their additive effect on NGF expression.
Fig. 6.
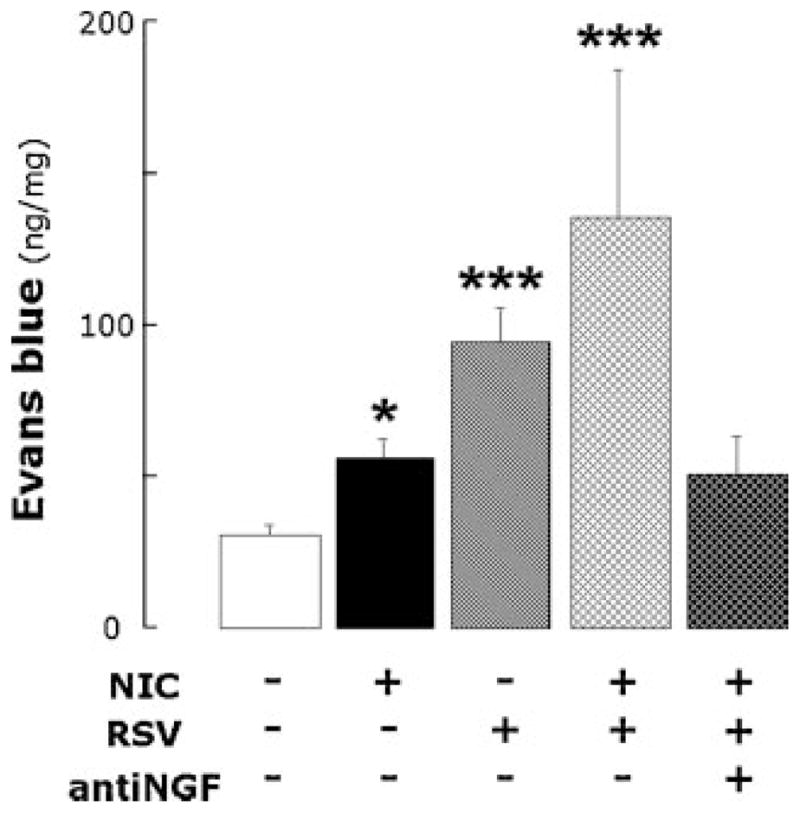
Neurogenic inflammation in the airways of rats infused with nicotine or its vehicle for 28 days and then inoculated with RSV or virus-free medium 5 days before sacrifice. Acute RSV infection following chronic exposure to nicotine had additive effect on capsaicin-induced extravasation of Evans blue-labeled albumin compared to the effects of either nicotine alone or RSV alone, and selective NGF inhibition abolished the combined inflammatory effect. Data are expressed as mean ± SEM, n = 5 rats per group. *P<0.05; ***P<0.001, significantly different from controls infused with nicotine vehicle and inoculated with virus-free medium.
Inhibition of NGF activity with a specific blocking antibody (Fig. 6) reduced Evans blue extravasation by 63% (n = 5 rats; P<0.0001). After NGF blockade, Evans blue extravasation in the airways of nicotine-exposed rats infected with RSV was not significantly different from pathogen-free controls infused with vehicle (P = 0.07). The anti-NGF antibody had no inhibitory effect on capsaicin-induced Evans blue extravasation in the airways of rats inoculated with virus-free medium after a 28-day infusion of vehicle (56.54 ± 4.49 ng/mg; n = 5 rats; P = 0.4), and therefore we can exclude that the antibody reduced capsaicin effect per se.
DISCUSSION
In this study we use a rat model of chronic nicotine exposure to explore environmentally driven changes in the expression of neurotrophic factors and receptors in the respiratory tract. This exposure results in plasma levels similar to those detected in humans who smoke or use nicotine transdermal patches.21 Our data obtained in this model show that nicotine significantly increases expression of the prototypical neurotrophic factor NGF in the lungs, both at the mRNA and protein level. We believe this is the first study to show that expression of growth factors critical for the development of airway innervation can be modified by tobacco smoking, either alone or in combination with other environmental agents.
A potentially more important neurotrophic effect of nicotine is the receptor imbalance deriving from increased low-affinity pan-neurotrophin p75NTR receptor expression without any concomitant change in the specific high-affinity trkA receptor. This imbalance in neurotrophic receptors expression has important pathophysiologic implications given the central role played by p75NTR in the mechanisms of neurogenic-mediated airway inflammation and hyperreactivity33 and the antagonistic nature of the biological effects mediated by the two receptor subtypes.34 Both NGF and p75NTR upregulation develop within the first 2 weeks of exposure to nicotine, and persist without major change when the exposure is prolonged for 2 additional weeks.
Chronic exposure of pregnant rats to nicotine has the opposite effect on the expression of neurotrophic pathways in the lungs of the offspring. This remarkable difference between pre- and post-natal effects is surprising, as it is known that nicotine passes through the placental barrier and that fetuses of smoking women are exposed to nicotine levels as high as those found in active smokers.35 We speculate that nicotine pharmacokinetic and/or pharmacodynamic profiles in fetal lung tissues may differ from adult ones, or that transcriptional or post-transcriptional regulation of neurotrophin genes expression may undergo substantial qualitative changes during development.
Although this mechanism may protect the fetus from neurotrophic and neurogenic-mediated inflammatory activity, it also has important implications concerning the physiologic development and differentiation of the fetal brain structures controlling ventilatory drive and arousal and the peripheral neural networks of the respiratory tract, which are both critically dependent on neurotrophic modulation. Further studies are needed to verify whether this pre-natal disruption of neurotrophic pathways contributes to the strong epidemiologic associations between maternal smoking during pregnancy and childhood respiratory pathology involving abnormal ventilatory control (e.g., apnea and sudden infant death) or small airways structure and function (e.g., asthma).36
We also found several differences between nicotine exposure and viral infection in terms of their individual influence on neurotrophin expression in the respiratory tract. Differently from what we have shown in our previous work with RSV,11,16 nicotine did not increase significantly the expression of BDNF or its high-affinity receptor trkB, and the nicotine-induced upregulation of NGF and its receptors was not dependent on post-natal age, that is, similar effects were measured in weanling and adult rats (data not shown). For these reasons, we focused our subsequent studies on the modulation of the NGF pathway in adult airways.
The most important finding of our study is perhaps that acute RSV infection and chronic nicotine exposure exert comparable and additive effects on NGF expression, doubling the concentration of this critical neurotrophin in lung tissues. A similar interaction was observed in our measurements of neurogenic airway edema, although in these experiments the effect of RSV alone was 2.5-fold that of nicotine alone. The additive nature of this interaction indicates that separate mechanisms are involved, and is not consistent with an interference of nicotine with viral replication or infectivity. Overall, the findings of this study suggest that the final expression of critical growth factors in the lungs derives from combinations of a multiplicity of biological and chemical environmental factors establishing complex interactions with the individual genetic background. Thus, the influence of tobacco smoking on airway modeling and remodeling can amplify and be amplified by concomitant exposures to other environmental agents, for example, common respiratory viruses.
Very little is known about the interactions at the subcellular level between nicotine and airway nerves. Although airway nerves do not express nicotinic acetyl-choline receptors, these receptors are expressed by epithelial cells at all levels of the intrapulmonary airways and in close proximity to nociceptive C-fibers,37 which are believed to be the major sensory fibers mediating the effects of nicotine associated with tobacco smoke.38 This arrangement suggests that chemical substances released locally by epithelial cells expressing nicotinic receptors may play a role in the stimulation and modulation of pulmonary C-fiber afferents. Based on our present data and on the recent finding that bronchial epithelial cells are a primary source ofNGF,39 we propose that NGF is indeed the chemical signal coordinating neuro–epithelial interactions in nicotine-exposed airways (Fig. 7).
Fig. 7.
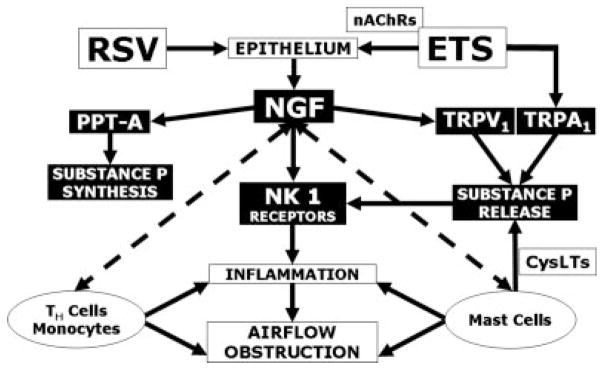
Pathways involved in the neuro-inflammatory and neuro-immunomodulatory effects of RSV infection and environmental tobacco smoke (ETS) exposure. RSV11,16 and the nicotinic fraction of ETS (via activation of epithelial nicotinic cholinergic receptors, nAChRs37) have additive stimulating effects on NGF synthesis from the bronchial epithelium. NGF increases pre-synaptically the expression of the preprotachykinin A (PPT-A) gene encoding substance P,45 and post-synaptically the expression of the high-affinity substance P receptor neurokinin 1 (NK1).11 Furthermore, NGF-trkA binding amplifies neurotransmitter release from nerve terminals by increasing expression and function of the vanilloid receptor TRPV1.43 ETS also contains α, β-unsaturated aldehydes stimulating the other excitatory ion channel TRPA1 co-expressed with TRPV1 on nociceptive nerves.40 The resulting neurogenic-mediated inflammatory response is amplified by the NGF-driven differentiation and activation of mast cells (via release of cysteinyl leukotrienes, CysLTs)46 and the recruitment and activation of CD4+T-helper lymphocytes and monocytes.47
Despite nicotine per se does not evoke neurogenic inflammation,40 our present data indicate that because of its effects on the NGF pathway this molecule augments significantly TRPV1-mediated inflammatory responses during viral respiratory infections. In addition to increasing indirectly the effects of TRPV1 agonists with its nicotinic fraction, tobacco smoke also contains α,β-unsaturated aldehydes (crotonaldehyde and acrolein) that can trigger neurogenic inflammation via direct activation of another TRP channel co-expressed with TRPV1 on vagal nociceptive neurons, the TRPA1.40 Taken together, these studies suggest that a number of chemicals inhaled during tobacco smoking have an extremely complex and profound influence on neural functions in the respiratory tract, and that immunologic or chemical inhibition of specific TRP channels and/or neurotrophic pathways may have important therapeutic potential for highly prevalent respiratory disorders like asthma in smokers and in patients heavily exposed to second-hand smoke. Interestingly, this patient population is frequently resistant to traditional anti-inflammatory strategies like inhaled corticosteroids,41 and the mechanism of this resistance could be explained by the activation of steroid-insensitive inflammatory systems like the neurotrophic pathways.12
Neurotrophins modulate physiologically the development and functional activities of peripheral neurons in a number of ways that collectively define “neuronal plasticity,” and their overexpression in pathologic conditions can replicate the molecular mechanisms of inflammatory hyperalgesia.42 This process results in increased expression of genes encoding pro-inflammatory neuropeptides like substance P, not only in the nociceptive unmyelinated C-fibers that physiologically contain these peptides, but also in myelinated A-fibers that usually do not express tachykinins and therefore switch to a non-physiologic nociceptive phenotype. Furthermore, NGF-trkA binding amplifies neurotransmitter release from nerve terminals by increasing expression and function of the vanilloid receptor TRPV1 (the capsaicin receptor). This mechanism has been shown to be primarily mediated by increased PI3 kinase-dependent phosphorylation of a single tyrosine residue of TRPV1, which promotes the trafficking of these channels to the cell surface and their insertion into the membrane by regulated exocytosis.43 Finally, NGF and its receptors are expressed by several non-neuronal cell types including epithelial, inflammatory and immunocompetent cells, thereby functioning as a potent and eclectic neuro-immunomodulator that activates and is activated by a variety of other inflammatory pathways.
The clinical relevance of our findings is underscored by recent studies in patients with asthma showing high serum levels of NGF that correlate with disease severity and IgE titers, thus suggesting an important pathogenetic role of neurotrophins in allergic respiratory disorders probably mediated through the p75NTR receptor activation.33 This receptor is a member of the tumor necrosis factor receptor superfamily and binds at low-affinity all neurotrophins, but can also bind at high-affinity the NGF precursor (pro-NGF) and regulate the affinity of trkA for its cognate ligand. Our previous clinical studies have implicated similar mechanisms in the inflammatory process evoked by common respiratory viruses like RSV,16 which fits well with the prominent role of these pathogens as triggers of bronchospastic exacerbations. The data reported in the present study provide a plausible mechanism for the increase morbidity deriving from viral infections in children and adults exposed to tobacco smoke.
It should be noted that tobacco smoking has also been associated with a lower incidence of several inflammatory diseases,9 and specific immunomodulatory, anti-inflammatory, and neuroprotective properties have been attributed to nicotine.10 In particular, tobacco smoke has been found to inhibit the production of TH1-type cytokines and tissue inflammation in RSV-infected rat lungs.7 It is therefore conceivable that concomitant anti-inflammatory effects exerted by nicotine alone determined the lower neurogenic inflammatory activity compared to RSV alone, despite the similar degree of NGF upregulation found in our study. As NGF drives airway inflammation and hyperreactivity and contributes to the pathogenesis of asthma,44 the results of the present study suggest that nicotine has a complex influence on airway inflammation, inhibiting certain pathways while potentiating others.
We conclude that nicotine levels comparable to those found in smokers are per se able to upregulate the expression of NGF and its low-affinity pro-inflammatory p75NTR receptor in the lung tissues of rats. More importantly, the combination of nicotine exposure and RSV infection produces more severe dysregulation of NGF expression compared to either infection or nicotine alone. This predisposes the respiratory tract to exaggerated neurogenic-mediated inflammatory responses, amplified by sequential or combined exposures to airborne irritants, which can be prevented by selective inhibition of NGF activity (Fig. 7). The neurotrophic effects of nicotine may contribute to the increased frequency and severity of respiratory infections in individuals exposed to tobacco smoke and may represent a novel target for the therapy of highly prevalent diseases like asthma.
Acknowledgments
We thank Edward Mager (University of Miami), Debbie Piktel, Lennie Samsell, and Cheryl Walton (WVU Pediatric Research Institute) for their technical help with PCR and ELISA analysis. This research was supported in part by grants from the National Institutes of Health (NHLBI HL-61007 and NICHD NCS-07-11) to Dr. Giovanni Piedimonte.
Footnotes
Some of the findings reported in this paper were presented at the 2008 International Conference of the American Thoracic Society in Toronto, Canada.
References
- 1.Bradley JP, Bacharier LB, Bonfiglio J, Schechtman KB, Strunk R, Storch G, Castro M. Severity of respiratory syncytial virus bronchiolitis is affected by cigarette smoke exposure and atopy. Pediatrics. 2005;115:e7–e14. doi: 10.1542/peds.2004-0059. [DOI] [PubMed] [Google Scholar]
- 2.Carbonell-Estrany X, Quero J IRIS Study Group. Hospitalization rates for respiratory syncytial virus (RSV) infection in premature infants born during two consecutive seasons. Pediatr Infect Dis J. 2001;20:874–879. doi: 10.1097/00006454-200109000-00010. [DOI] [PubMed] [Google Scholar]
- 3.Gurkan F, Kiral A, Dagli E, Karakoc F. The effect of passive smoking on the development of respiratory syncytial virus bronchiolitis. Eur J Epidemiol. 2000;16:465–468. doi: 10.1023/a:1007658411953. [DOI] [PubMed] [Google Scholar]
- 4.McConnochie KM, Roghmann KJ. Parental smoking, presence of older siblings, and family history of asthma increase risk of bronchiolitis. Am J Dis Child. 1986;140:806–812. doi: 10.1001/archpedi.1986.02140220088039. [DOI] [PubMed] [Google Scholar]
- 5.Simoes EA. Environmental and demographic risk factors for respiratory syncytial virus lower respiratory tract disease. J Pediatr. 2003;143:S118–S126. doi: 10.1067/s0022-3476(03)00511-0. [DOI] [PubMed] [Google Scholar]
- 6.Wilkinson TM, Donaldson GC, Johnston SL, Openshaw PJ, Wedzicha JA. Respiratory syncytial virus, airway inflammation, and FEV1 decline in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:871–876. doi: 10.1164/rccm.200509-1489OC. [DOI] [PubMed] [Google Scholar]
- 7.Phaybouth V, Wang SZ, Hutt JA, McDonald JD, Harrod KS, Barrett EG. Cigarette smoke suppresses Th1 cytokine production and increases RSV expression in a neonatal model. Am J Physiol Lung Cell Mol Physiol. 2006;290:L222–L231. doi: 10.1152/ajplung.00148.2005. [DOI] [PubMed] [Google Scholar]
- 8.Razani-Boroujerdi S, Singh SP, Knall C, Hahn FF, Pena-Philippides JC, Kalra R, Langley RJ, Sopori ML. Chronic nicotine inhibits inflammation and promotes influenza infection. Cell Immunol. 2004;230:1–9. doi: 10.1016/j.cellimm.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol. 2002;2:372–377. doi: 10.1038/nri803. [DOI] [PubMed] [Google Scholar]
- 10.Sopori ML, Kozak W, Savage SM, Geng Y, Soszynski D, Kluger MJ, Perryman EK, Snow GE. Effect of nicotine on the immune system: possible regulation of immune responses by central and peripheral mechanisms. Psychoneuroendocrinology. 1998;23:189–204. doi: 10.1016/s0306-4530(97)00076-0. [DOI] [PubMed] [Google Scholar]
- 11.Hu C, Wedde-Beer K, Auais A, Rodriguez MM, Piedimonte G. Nerve growth factor and nerve growth factor receptors in respiratory syncytial virus-infected lungs. Am J Physiol Lung Cell Mol Physiol. 2002;283:L494–L502. doi: 10.1152/ajplung.00414.2001. [DOI] [PubMed] [Google Scholar]
- 12.Mohtasham L, Auais A, Piedimonte G. Nerve growth factor mediates steroid-resistant inflammation in respiratory syncytial virus infection. Pediatr Pulmonol. 2007;42:496–504. doi: 10.1002/ppul.20607. [DOI] [PubMed] [Google Scholar]
- 13.Piedimonte G. Virus induction of neurogenic inflammation and airway responsiveness. In: Johnston S, O’Byrne P, editors. Exacerbations of asthma. Boca Raton, FL: Taylor and Francis; 2006. pp. 143–166. [Google Scholar]
- 14.Piedimonte G, Perez MK. Role of early-life environmental influences in the development of asthma: how painful is it when you catch a bad cold too early? J Asthma. 2008;45:25–28. doi: 10.1080/02770900802569991. [DOI] [PubMed] [Google Scholar]
- 15.Zeldin DC, Eggleston P, Chapman M, Piedimonte G, Renz H, Peden D. How exposures to biologics influence the induction and incidence of asthma. Environ Health Perspect. 2006;114:620– 626. doi: 10.1289/ehp.8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tortorolo L, Langer A, Polidori G, Vento G, Stampachiacchere B, Aloe L, Piedimonte G. Neurotrophin overexpression in lower airways of infants with respiratory syncytial virus infection. Am J Respir Crit Care Med. 2005;172:233–237. doi: 10.1164/rccm.200412-1693OC. [DOI] [PubMed] [Google Scholar]
- 17.Bayer C, Black M. Thermal desorption/gas chromatography/mass spectrometric analysis of volatile organic compounds in the offices of smokers and nonsmokers. Biomed Environ Mass Spectrom. 1987;14:363–367. doi: 10.1002/bms.1200140803. [DOI] [PubMed] [Google Scholar]
- 18.Guerin M, Jenkins R, Tomkins B, Eisenberg M, editors. The chemistry of environmental tobacco smoke: composition and measurement. Chelsea: Lewis Publishers; 1992. [Google Scholar]
- 19.Chuang J, Kuhlman M. Evaluation of methods for simultaneous collection and determination of nicotine and polynuclear aromatic hydrocarbons in indoor air. Environ Sci Technol. 1990;24:661–665. [Google Scholar]
- 20.Fewell JE, Smith FG. Perinatal nicotine exposure impairs ability of newborn rats to autoresuscitate from apnea during hypoxia. J Appl Physiol. 1998;85:2066–2074. doi: 10.1152/jappl.1998.85.6.2066. [DOI] [PubMed] [Google Scholar]
- 21.Murrin LC, Ferrer JR, Zeng WY, Haley NJ. Nicotine administration to rats: methodological considerations. Life Sci. 1987;40:1699–1708. doi: 10.1016/0024-3205(87)90020-8. [DOI] [PubMed] [Google Scholar]
- 22.Piedimonte G. Pathophysiological mechanisms for the respiratory syncytial virus-reactive airway disease link. Respir Res. 2002;3:S21–S25. doi: 10.1186/rr185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King KA, Hu C, Rodriguez MM, Romaguera R, Jiang X, Piedimonte G. Exaggerated neurogenic inflammation and substance P receptor upregulation in RSV-infected weanling rats. Am J Respir Cell Mol Biol. 2001;24:101–107. doi: 10.1165/ajrcmb.24.2.4264. [DOI] [PubMed] [Google Scholar]
- 24.Piedimonte G, King KA, Holmgren NL, Bertrand PJ, Rodriguez MM, Hirsch RL. A humanized monoclonal antibody against respiratory syncytial virus (palivizumab) inhibits RSV-induced neurogenic-mediated inflammation in rat airways. Pediatr Res. 2000;47:351–356. doi: 10.1203/00006450-200003000-00011. [DOI] [PubMed] [Google Scholar]
- 25.Piedimonte G, Rodriguez MM, King KA, McLean S, Jiang X. Respiratory syncytial virus upregulates expression of the substance P receptor in rat lungs. Am J Physiol Lung Cell Mol Physiol. 1999;277:L831–L840. doi: 10.1152/ajplung.1999.277.4.L831. [DOI] [PubMed] [Google Scholar]
- 26.Piedimonte G. Neural mechanisms of respiratory syncytial virus-induced inflammation and prevention of respiratory syncytial virus sequelae. Am J Respir Crit Care Med. 2001;163:S18– S21. doi: 10.1164/ajrccm.163.supplement_1.2011113. [DOI] [PubMed] [Google Scholar]
- 27.Saria A, Lundberg JM, Skofitsch G, Lembeck F. Vascular protein leakage in various tissues induced by substance P, capsaicin, bradykinin, serotonin, histamine, and by antigen challenge. Naunyn Schmiedebergs Arch Pharmacol. 1983;324:212– 218. doi: 10.1007/BF00503897. [DOI] [PubMed] [Google Scholar]
- 28.Holzer P. Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacol Rev. 1991;43:143–201. [PubMed] [Google Scholar]
- 29.Rubin JS, Bradshaw R. Isolation and partial amino acid sequence analysis of nerve growth factor from guinea pig prostate. J Neurosci Res. 1981;6:451–464. doi: 10.1002/jnr.490060403. [DOI] [PubMed] [Google Scholar]
- 30.Schuijers JA, Walker DW, Browne CA, Thorburn GD. Peripheral and brain tissue catecholamine content in intact and anti-NGF-treated fetal sheep. Am J Physiol Regul Integr Comp Physiol. 1987;252:R7–R12. doi: 10.1152/ajpregu.1987.252.1.R7. [DOI] [PubMed] [Google Scholar]
- 31.Zar JH. Biostatistical analysis. Englewood Cliffs, NJ: Prentice-Hall, Inc; 1984. [Google Scholar]
- 32.Wallenstein S, Zucker CL, Fleiss JL. Some statistical methods useful in circulation research. Circ Res. 1980;47:1–9. doi: 10.1161/01.res.47.1.1. [DOI] [PubMed] [Google Scholar]
- 33.Kerzel S, Path G, Nockher WA, Quarcoo D, Raap U, Groneberg DA, Dinh QT, Fischer A, Braun A, Renz H. Pan-neurotrophin receptor p75 contributes to neuronal hyperreactivity and airway inflammation in a murine model of experimental asthma. Am J Respir Cell Mol Biol. 2003;28:170–178. doi: 10.1165/rcmb.4811. [DOI] [PubMed] [Google Scholar]
- 34.Kernie SG, Parada LF. The molecular basis for understanding neurotrophins and their relevance to neurologic disease. Arch Neurol. 2000;57:654–657. doi: 10.1001/archneur.57.5.654. [DOI] [PubMed] [Google Scholar]
- 35.Foundas M, Hawkrigg NC, Smith SM, Devadason SG, Le Souef PN. Urinary cotinine levels in early pregnancy. Aust N Z J Obstet Gynaecol. 1997;37:383–386. doi: 10.1111/j.1479-828x.1997.tb02443.x. [DOI] [PubMed] [Google Scholar]
- 36.Landau LI. Smoking: from womb to tomb. Paediatr Respir Rev. 2008;9:1–2. doi: 10.1016/j.prrv.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 37.Dehkordi O, Rose JE, Balan KV, Kc P, Millis RM, Jayam-Trouth A. Neuroanatomical relationships of substance P-immunoreactive intrapulmonary C-fibers and nicotinic cholinergic receptors. J Neurosci Res. 2009;87:1670–1678. doi: 10.1002/jnr.21967. [DOI] [PubMed] [Google Scholar]
- 38.Xu J, Yang W, Zhang G, Gu Q, Lee LY. Calcium transient evoked by nicotine in isolated rat vagal pulmonary sensory neurons. Am J Physiol Lung Cell Mol Physiol. 2007;292:L54–L61. doi: 10.1152/ajplung.00182.2006. [DOI] [PubMed] [Google Scholar]
- 39.Othumpangat S, Gibson L, Samsell L, Piedimonte G. Respiratory syncytial virus (RSV) modulates NGF and neurotrophin receptor expression in human tracheal and bronchial cells. Am J Respir Crit Care Med. 2008;177:A932. [Google Scholar]
- 40.Andrè E, Campi B, Materazzi S, Trevisani M, Amadesi S, Massi D, Creminon C, Vaksman N, Nassini R, Civelli M, Baraldi PG, Poole DP, Bunnett NW, Geppetti P, Patacchini R. Cigarette smoke-induced neurogenic inflammation is mediated by alpha, beta-unsaturated aldehydes and the TRPA1 receptor in rodents. J Clin Invest. 2008;118:2574– 2582. doi: 10.1172/JCI34886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomson NC, Chaudhuri R, Livingston E. Asthma and cigarette smoking. Eur Respir J. 2004;24:822–833. doi: 10.1183/09031936.04.00039004. [DOI] [PubMed] [Google Scholar]
- 42.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1768. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X, Huang J, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 2005;24:4211–4223. doi: 10.1038/sj.emboj.7600893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nockher WA, Renz H. Neurotrophins and asthma: novel insight into neuroimmune interaction. J Allergy Clin Immunol. 2006;117:67–71. doi: 10.1016/j.jaci.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 45.Lindsay RM, Harmar AJ. Nerve growth factor regulates expression of neuropeptide genes in adult sensory neurons. Nature. 1989;337:362–364. doi: 10.1038/337362a0. [DOI] [PubMed] [Google Scholar]
- 46.Wedde-Beer K, Hu C, Rodriguez MM, Piedimonte G. Leukotrienes mediate neurogenic inflammation in lungs of young rats infected with respiratory syncytial virus. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1143–L1150. doi: 10.1152/ajplung.00323.2001. [DOI] [PubMed] [Google Scholar]
- 47.Auais A, Adkins B, Napchan G, Piedimonte G. Immunomodulatory effects of sensory nerves during respiratory syncytial virus infection in rats. Am J Physiol Lung Cell Mol Physiol. 2003;285:L105–L113. doi: 10.1152/ajplung.00004.2003. [DOI] [PubMed] [Google Scholar]