

Abstract
Resolving single fluorescent molecules in the presence of high fluorophore concentrations remains a challenge in single-molecule biophysics that limits our understanding of weak molecular interactions. Total internal reflection fluorescence (TIRF) imaging, the workhorse of single-molecule fluorescence microscopy, enables experiments at concentrations up to about 100 nM, but many biological interactions have considerably weaker affinities, and thus require at least one species to be at micromolar or higher concentration. Current alternatives to TIRF often require three-dimensional confinement, and thus can be problematic for extended substrates, such as cytoskeletal filaments. To address this challenge, we have demonstrated and applied two new single-molecule fluorescence microscopy techniques, linear zero-mode waveguides (ZMWs) and convex lens induced confinement (CLIC), for imaging the processive motion of molecular motors myosin V and VI along actin filaments. Both technologies will allow imaging in the presence of higher fluorophore concentrations than TIRF microscopy. They will enable new biophysical measurements of a wide range of processive molecular motors that move along filamentous tracks, such as other myosins, dynein, and kinesin. A particularly salient application of these technologies will be to examine chemomechanical coupling by directly imaging fluorescent nucleotide molecules interacting with processive motors as they traverse their actin or microtubule tracks.
OCIS codes: (000.1430) Biology and medicine, (170.2520) Fluorescence microscopy, (180.25200) Fluorescence microscopy
1. Introduction
Reducing background fluorescence from fluorophores in solution presents a challenge to many single-molecule fluorescence experiments. For single-molecule fluorescence imaging of surface-immobilized species, total internal reflection fluorescence (TIRF) microscopy is usually used to interrogate a thin layer of molecules close to the surface in order to suppress background fluorescence from molecules freely diffusing in solution. By coupling to fluorophores using an evanescent field, TIRF restricts the excitation volume to a layer of molecules within a few hundred nanometers above the coverslip surface, enabling individual fluorophores attached to the surface to be resolved in the presence of freely diffusing fluorophores at concentrations up to approximately 100 nM [1,2], many times higher than is possible using epifluorescent excitation. However, 100 nM is still well below the affinity of many biologically relevant associations. Furthermore, the kinetics of binding events at such low concentrations are often problematically slow. Consequently, the development and application of single-molecule imaging techniques that probe low affinity interactions inaccessible to TIRF microscopy remains an outstanding challenge to experimental biophysics.
Recently, a few alternatives to TIRF have begun to increase the concentration limit at which single fluorescent molecules can be observed. For instance, circular zero-mode waveguides (ZMWs) constrain fluorescent excitation in sub-micron scale chambers, as described in more detail below [3]. Alternatively, fluorescent substrates can be constrained in lipid vesicles, which can either be trapped or surface-tethered, or in trapped droplets suspended in oil [4–6]. All of these approaches allow imaging at up to micromolar concentrations by limiting the excitation volume in all three dimensions, which works very well for studying substrates whose dimensions are all sub-micron and that do not undergo micron-scale motions. However, for studying extended substrates or for substrates that transit distances of more than a few hundred nanometers, including cytoskeletal motors moving along actin or microtubule tracks, three-dimensional confinement is not appropriate. Furthermore, lipid vesicles or trapped droplets do not generally allow exchange of buffer within the confined volume, which is problematic for studying enzymes, since the reactants may quickly become consumed. Although nanovesicles can be adapted to allow exchange of small molecules through nanopores with the buffer in which the vesicles are immersed, this is not useful in the case that small molecule reactants, such as ATP, are the fluorescent substrates to be imaged [7]. Another alternative strategy for increasing the concentration limit that avoids three-dimensional confinement, PhotoActivation, Diffusion, and Excitation (PhADE), was recently demonstrated [8]. This method of activating fluorophores near the surface via TIRF and then waiting for fluorescent species not bound to molecules of interest to diffuse away is likely to prove useful in many contexts; however, it does require the use of specialized photoactivatable fluorophores and its time resolution is limited by the time required for both the activation and diffusion.
The association of processive cytoskeletal molecular motors with fluorescently labeled nucleotide molecules is an important class of experiments that remains inaccessible to standard microscopy techniques. Most dimeric processive motors, including myosins V and VI, kinesin, and, to some extent, dynein, appear to coordinate their stepwise motion with kinetic rates that vary depending on which motor “head” is in the leading vs. trailing position [9–14]. Since their kinetic cycles are highly coupled to nucleotide binding, hydrolysis, and release, open questions on the underlying mechanochemical coupling and coordination mechanisms could be directly addressed by visualizing single molecules of fluorescently labeled ATP binding to and releasing from single processive motors as they advance along their tracks. However, the fluorescent ATP concentrations accessible to single-molecule TIRF microscopy are prohibitive in such an experiment, since the motors would move extremely slowly, and might exhibit off-pathway kinetics, and the alternatives involving nano-confinement are prohibitive for observing multiple steps along their extended tracks. A few experiments have successfully visualized the association of fluorescent nucleotides with myosin II [15–18] or myosin V [1,10,19], but with one exception [10] (see below) these experiments required very low concentrations of nucleotide (up to 100 nM) and/or fractional labeling of nucleotides (10% fluorescent). Because of these limitations, these experiments were able to detect ATP turnover by myosin motors and correlate that with a myosin power-stroke but, for the most part, were not able to detect coordination between motor domains. Thus far, myosin V is the only processive motor for which detection of fluorescent nucleotides has been used to directly measure intra-molecular coordination, and doing so required the use of a custom-prepared fluorescent nucleotide analog with the unusual property of brightening on binding to the motor [10]; while this custom fluorescent nucleotide minimally perturbed myosin V, its kinetics were altered somewhat, and such effects can be very enzyme-specific, meaning that this specific approach will not necessarily be applicable to other myosins, or to kinesin or dynein. New and accessible technology that enables imaging single molecules at higher concentrations, while also allowing flexibility in the choice of a fluorescent nucleotide analog, would therefore be a powerful addition to the single-molecule microscopy arsenal. There are several molecular motors such as myosin VI and dynein, for which mechanisms of coordination remain controversial or insufficiently characterized [11,13,14,20–23], and for which direct detection of coordination, for instance through imaging of fluorescent ATP binding and release, has not yet been possible. These are especially important systems for applying such a technology.
In this work, we demonstrate the feasibility of two new technologies for imaging processive myosins with enhanced background suppression: linear zero-mode waveguides (ZMWs) and convex lens induced confinement (CLIC). While these two techniques have been used in the past, here we describe significant alterations and advances that were essential for applying these technologies to cytoskeletal motors moving along tracks. This work demonstrates that both of these techniques can be used with myosin motors moving along actin filaments and can be extended to a broad range of applications, such as imaging fluorescent nucleotides associating with actin or microtubule motors or examining polymerization dynamics of actin filaments or microtubules. Both of these techniques function by decreasing the observation volume in which fluorophores are excited in one or two, but importantly not all three, dimensions. Linear ZMWs decrease the observation volume primarily by confining the excitation to a thin layer close to the entrance of the waveguide (at least an order of magnitude thinner than TIRF) and by physically confining the sample in a narrow strip, reducing the excitation volume by a few fold in one of the lateral dimensions as well (Fig. 1(a) ). The CLIC imaging chamber, whose thickness varies continuously from 2 nm to several microns, decreases the observation volume by physically confining the fluorescent molecules within a thin layer (Fig. 1(b)).
Fig. 1.
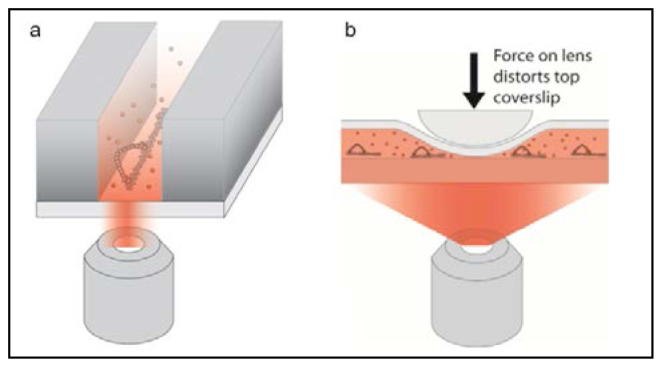
Schematics of new microscopy methods demonstrated in this work that suppress background fluorescence. (a) Myosin walking on actin in a linear ZMW (not to scale). Fluorescent ATP molecules (red dots) are only excited when they are located very near the entrance to the ZMW. Since the ZMW slits are narrower than the wavelength of light, the excitation volume is confined both vertically and in the direction in which the slits are narrowest. (b) Myosin walking in the CLIC device (not to scale). Near the contact region, the thin CLIC imaging chamber confines the excited molecules to a thin layer.
A zero-mode waveguide (ZMW) consists of an aluminum film deposited on a fused silica surface and then etched using lithography to create small holes (waveguides) in the film that are narrower than the wavelength of light [3]. When light enters these waveguides from the fused silica side, the sub-wavelength gaps prevent the light from propagating, creating an evanescent field inside the waveguide whose intensity falls off much more quickly with depth than TIRF illumination. Thus, ZMWs can be used to resolve single molecules at a much higher concentration of fluorophores since fewer background molecules are excited by the very shallow evanescent field. To this end, cylindrical zero-mode waveguides have been used to image DNA polymerase synthesizing individual fluorescent nucleotides into DNA at fluorophore concentrations up to 10 µM and tRNAs transiting on translating ribosomes at micromolar concentrations [3,24,25]. While this technology has had several important applications, the cylindrical geometry severely restricts the kind of experiments for which it is useful. For example, this geometry is prohibitive for processive cytoskeletal motors, since the diameter of the cylinders (around 100 nm) would only allow the insertion of filaments that would be much too short to detect more than a very small number of steps. Here, we have developed a new kind of ZMW with a very different geometry: waveguides that are narrow in one dimension but microns long (Fig. 1(a)). Using polarized light, we demonstrate that, with these new ZMWs, the excitation can still be confined near the surface. We show that actin can be introduced into the linear ZMWs by polymerization inside the waveguides, and that myosin V can move along these actin filaments.
Convex lens induced confinement (CLIC) has previously been reported as a method to reject background fluorescence and to image single molecules under confinement [26]. In its published implementation, the CLIC imaging chamber was formed between the curved surface of a convex lens and a coverslip, placed in direct contact. The CLIC imaging chamber confined molecules to a thin film whose thickness was defined by the local chamber height. This height was limited by surface roughness (~2 nm) at the contact point, and increased gradually over several fields of view (e.g., by tens of nanometers over hundreds of microns close to the contact point). Using this approach, individual protein molecules and fluorophores have been resolved in the presence of background concentrations up to 2 µM. Here, we demonstrate a new implementation of CLIC imaging, and use it to show for the first time that CLIC is compatible with biological activity. This new version of CLIC uses a standard flow chamber (two planar coverslips separated by double stick tape). This flow chamber is subsequently deformed by a lens that presses downward on the top surface until it comes into contact with the bottom coverslip (Fig. 1(b)). This implementation makes CLIC easier to use than the previous design: it consists of disposable parts, eliminates the need for cleaning the lens between measurements, facilitates serial sample replacement and imaging, reduces sample evaporation, and requires a smaller sample volume. These changes dramatically improve the accessibility of CLIC, since it can now be easily combined with very standard biophysical assays performed using flow cells. Here, we use flow cell CLIC to demonstrate the first biological application of this technology, showing that the processive motion of myosin VI can be imaged in the CLIC device at coverslip-coverslip separations as small as 60 nm, corresponding to five-fold enhanced background rejection in comparison to TIRF.
2. Methods
2.1 Preparation of myosin and actin proteins
Myosin V with a C-terminal YFP tag was produced using the baculovirus expression system and FLAG affinity purified as previously described [27]. Briefly, SF9 cells were infected simultaneously with one virus for producing both myosin V and calmodulin and another virus for producing essential light chain (ELC). The myosin V construct included the chicken myosin V sequence up through residue 1099, followed by a YFP tag, with an N-terminal FLAG tag for purification. Myosin was purified using FLAG affinity chromatography. After purification, protein was dialyzed into buffer containing 50% glycerol for long-term storage at −20°C. Immediately before performing experiments, myosin V was fluorescently labeled by diluting to ~10 nM (in assay buffer, described below, with the addition of 100 µg/mL calmodulin), mixing with 10-fold molar excess of CF594 fluorescently labeled anti-GFP antibody (Sigma), which cross-reacts with YFP, and incubating for at least half an hour to allow binding. The fluorescently-labeled antibody was used instead of imaging YFP directly because of the antibody’s compatibility with our existing excitation laser and because YFP is much dimmer and less photostable than organic fluorophores.
Myosin VI was expressed, purified, and fluorescently labeled as previously described [28]. Briefly, porcine myosin VI through residue 991, fused with a C-terminal dimerization domain (GCN4), HaloTag, and Flag purification tag, was cloned into pBiEx-1 and transfected into SF9 cells, Flag affinity purified, and labeled with TMR-HaloTag ligand (Promega). Purified proteins were stored at 4°C on ice for no more than one week.
Actin was prepared from chicken breast by a method previously described [29] and stored in its filamentous (F) form. Biotinylated actin filaments were prepared by polymerizing at 10 µM concentration actin that had been labeled with biotin as previously described [23]. For use as seeds for polymerization inside linear ZMWs, these filaments were stabilized with equimolar amounts of phalloidin. To prepare for inserting in the waveguides, the biotinylated filaments were diluted to a concentration of 200 nM and sheared by sonicating in a microcentrifuge tube in a bath sonicator for three minutes immediately before using them in the ZMW assays described below. For use in CLIC experiments, the actin was polymerized with 33% biotinylated actin and 67% unlabeled actin, and after polymerization, the F-actin was labeled with an equimolar concentration of Alexa 633-labeled phalloidin (Invitrogen).
For polymerizing in linear ZMWs, globular (G) actin was freshly prepared by dialyzing F-actin into G buffer (2 mM TRIS, pH 8, 0.2 mM CaCl2, 0.5 mM DTT) overnight. Here, we did not include ATP, which is normally in canonical G buffer, to prevent contamination with dark ATP, in anticipation of future experiments with fluorescently labeled nucleotide. The dialyzed solution was centrifuged in a TLA 100 rotor (Beckman) at 95k RPM to remove filaments that did not fully depolymerize. G actin was stored on ice for no more than a few days.
2.2 Computer simulations of excitation intensity inside linear ZMWs
Finite-element time-domain simulations of excitation light intensity at the entrance of a linear ZMW were performed with the electric field vector of the excitation light either parallel or perpendicular to the long dimension of the waveguide. Because the simulations benefited from symmetry, they were performed in 2D. All simulations were performed by the COMSOL Multiphysics package (Burlington, MA).
2.3 Imaging excitation polarization dependent background fluorescence of free fluorophores in linear ZMWs
Average fluorescence intensity from 1 µM of freely diffusing Cy3-ATP (prepared in imaging buffer, described below) was observed as a function of the polarization angle of excitation light by a 532 nm laser (Coherent). The light introduced was linearly polarized, and the polarization was rotated using a half-wave plate. Imaging was conducted on a home-built inverted epifluorescence microscope using a 100x 1.4 NA oil immersion objective (Zeiss). Excitation light was filtered out using a notch filter (Semrock) and imaged onto an EMCCD camera (Andor iXon).
2.4 Polymerization of actin in linear zero-mode waveguides
In accord with our need for a new ZMW geometry, linear ZMW chips, whose bottom surfaces were functionalized with biotin [30], were custom-made for us by Pacific Biosciences. The chips were mounted by gluing them under a 3 mm diameter hole in a glass microscope slide, so that the walls of the hole in the slide formed a small well over the waveguide chip that could hold about 10 μL of buffer. In order to introduce actin into these linear ZMW chips, the following new methodology was developed for polymerizing actin inside the waveguides.
To prevent nonspecific sticking, linear ZMWs were blocked with 1 mg/mL BSA in assay buffer (25 mM imidazole-HCl (pH 7.4), 25 mM KCl, 1 mM EGTA and 10 mM DTT) for five minutes. Blocking buffer was removed, and 1 mg/mL streptavidin (Invitrogen) was allowed to bind to the biotinylated surface for 5 minutes. The ZMW chamber was rinsed three successive times with assay buffer plus BSA, this was removed, and sheared actin filaments were added to the chip and allowed to bind for 5 minutes (Fig. 2(a) ). Excess actin filaments that did not bind to the waveguides were removed by three successive washes with assay buffer plus BSA. G actin was freshly diluted into assay buffer (whose salts induce polymerization) at a concentration of approximately 3 μM and added to the chip. The G actin was allowed to polymerize off the seed filaments already attached to the bottom of the waveguide for 20 minutes (Fig. 2(b)). Note that polymerization occurred successfully despite not adding ATP to the polymerization buffer. The ZMW chip was rinsed with assay buffer plus BSA three times to remove excess G actin or filaments that polymerized but were not attached to the surface. Biotinylated phalloidin at a concentration of 1 μM in assay buffer was added to the chip to attach the nascent filaments to the bottom of the waveguides; it was allowed to incubate for 5 minutes (Fig. 2(c)). The biotin phalloidin solution was then removed and replaced with imaging buffer, as described below.
Fig. 2.
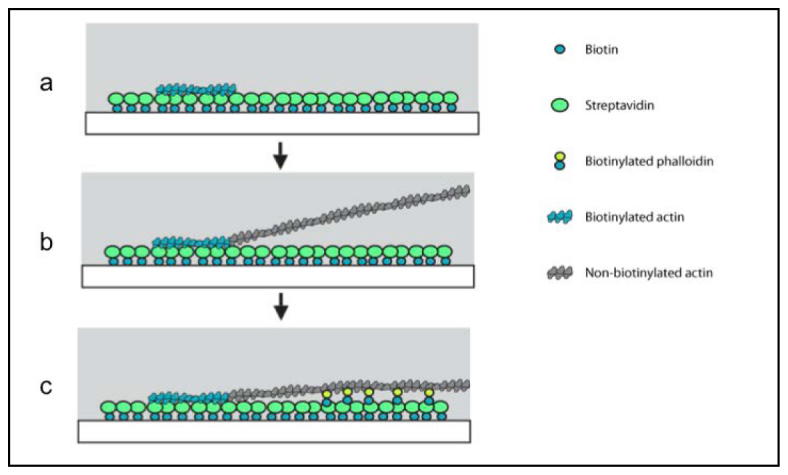
Strategy for polymerizing actin in a linear ZMW (not to scale). Each panel shows a side view of the waveguide. Gray and white boxes indicate the aluminum film and glass coverslip, respectively. (a) Glass at the bottom of the waveguide is selectively functionalized with biotin, which is then covered with a layer of streptavidin. Next, short biotinylated actin filament “seeds” are sparsely added to the waveguide and bind to the bottom surface. (b) Non-biotinylated G-actin is added to the waveguide under salt conditions that allow polymerization, and polymerization occurs off of the seed filaments. The nascent filaments are not attached to the surface during the polymerization step. (c) Biotinylated phalloidin is added to the slide, pulling down the nascent filaments and attaching them to the bottom surface of the waveguide.
2.5 Imaging myosin in linear zero-mode waveguides
After polymerizing actin and attaching it to the surface, as described above, imaging buffer that included fluorescently labeled myosin V and 50 µM ATP was added to the waveguide chip. The imaging buffer consisted of assay buffer (above) with the addition of an oxygen scavenging system consisting of 0.5% glucose, 0.2 mg/mL glucose oxidase (from Aspergillus niger, Calbiochem), and 36 μg/mL catalase (from Aspergillus niger, Calbiochem); 1.2 mM Trolox for improved photostability; and 0.05% Triton X-100 to prevent nonspecific sticking.
For imaging, an Axiovert (Zeiss) with home-built epifluorescence path was used. A 532 nm laser (Coherent) was focused in the center of the back focal plane of a 100x Zeiss TIRF objective. The laser beam was linearly plane wave polarized, and the polarization was rotated with a half-wave plate so that the electric field vector pointed along the waveguides, which was the condition that minimized leakage of incident light into the waveguide (Fig. 4). Imaging was conducted on an EMCCD camera (Andor iXon).
2.6 Convex lens induced confinement (CLIC) with flow cells
This work introduces a new implementation of CLIC imaging using flow cells that are standard in many microscopy labs, an important addition as CLIC is consequently now compatible with existing assays and materials and straightforward to implement. Two initially planar sheets of glass comprise the top and bottom surfaces of the imaging chamber (#0 glass coverslip from Ted Pella, # 1.5 glass coverslips from VWR, respectively). In the work described here, these coverslips were cleaned using a 1:2 solution of 30% hydrogen peroxide to 95-98% sulfuric acid, at room temperature for 30 minutes (piranha solution). We have also cleaned coverslips for these experiments using successive sonication washes with Alconox detergent dissolved in water; acetone; ethanol; 1 M KOH; and ultra-pure water; with several washes with ultrapure water between each sonication step. The latter presents a less harsh though more time-consuming approach. Flow chambers were constructed by adhering two coverslips together with double-sided tape (Scotch brand Permanent Double Sided tape).
To prepare the sample, the flow cell was first coated non-specifically with biotinylated-BSA (Sigma) by incubating at a concentration of 1 mg/mL in assay buffer (described above) for 2 minutes. The following solutions were then added to the chamber and allowed to incubate for 2 minutes each: 30 µL of assay buffer plus 1 mg/mL BSA; then 10 µL of 1 mg/mL streptavidin (Invitrogen) in assay buffer. The chamber was rinsed with 30 µL of assay buffer plus BSA. Biotinylated actin filaments were diluted in assay buffer to 200 nM (monomer concentration), and 10 µL were added to the chamber and incubated for two minutes. The actin was rinsed out with 30 µL of assay buffer plus BSA, and finally 10 µL of imaging buffer containing myosin VI and 50 µM ATP was added to the chamber. Imaging buffer was the same as that described above for ZMWs, except that it did not contain Triton-X 100, and additionally contained an ATP regeneration system consisting of 1 mM phosphocreatine and 100 μg/mL creatine phosphokinase (Calbiochem), as well as a spectrally separate, non-reactive fluorophore (Alexa 488, 50 nM), whose fluorescence served as an in situ diagnostic of the chamber height.
To form the imaging chamber, the flow cell CLIC device (Fig. 3 ) was assembled as in Fig. 3(a). The interface between the pushing lens and the top coverslip was coated with index-matching immersion oil (Cargille HF), to prevent reflections from surfaces other than the two coverslip-buffer interfaces. The mechanical clamp was used to press the lens downward on the top coverslip (thin and flexible), which caused the top coverslip to bow downward until it made contact with the bottom coverslip (thicker and less flexible) at a single point. The device function and mechanics are shown in Fig. 3.
Fig. 3.
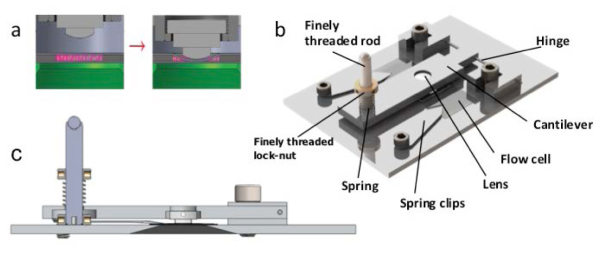
Flow-cell CLIC apparatus. (a) Side view of the flow-cell CLIC apparatus (not to scale). The lens (Thorlabs LA4966) is initially raised for sample insertion (left) and subsequently lowered for imaging (right), decreasing the volume of excited fluorophores (pink) in the imaging region. Excitation light entering from below is shown in green. (b) Top-view of the CLIC device (to scale). A cantilever controls the lens positioning. It contains a clearance hole for a finely threaded rod (Thorlabs F25US200). A spring (Small Parts CSXX-0160-05) is mounted around the rod and directly above the cantilever. A finely threaded lock-nut (Thorlabs LN25100) is mounted on the rod directly above the spring. The downward rotation of the lock-nut presses against the spring and consequently lowers the lens. The spring resists the downward motion of the lock-nut with approximately the same force (~1 N) as is required to deform the coverslip in this geometry. The initial flow chamber (~8 mm wide, ~100 µm thick) is confined vertically by the top and bottom coverslip surfaces and laterally by the edges of double-sided tape. The cantilever is mounted, using custom clamps, on a thin aluminum hinge aligned so that the lens surface is in contact with the top of the flow cell when the cantilever is horizontal. The flow cell is held in place by custom spring clips. (c) Side view of the CLIC device showing the imaging chamber (to scale). The dark gray trapezoid depicts a hole in the plate that gives clearance for the objective.
Molecules were imaged in the deformed chamber using an inverted fluorescence microscope (Nikon TI-E with 1.5x added magnification; 60x oil immersion objective; Andor 897 camera). The microscopy was performed in 4 sequential steps: first, the central region of the imaging chamber was imaged to determine the intensity profile of freely diffusing fluorophores. The center contact point was identified by the minimum dye fluorescent intensity. Next, the surface-immobilized actin filaments were imaged, and then movies were taken of the myosin proteins at several chamber locations (and therefore gap heights). Finally, interferometry of the imaging chamber was performed to spatially resolve its height profile. In each step, a 5x5 grid of high-resolution images (91 × 91 µm2), spanning a total area of 278 × 291 µm2, was acquired by translating the sample relative to the objective using a programmed microscope stage (Biopoint2, Ludl).
The free dye was imaged using a 488 nm laser (Sapphire 100 mW, Coherent), 10 mW illumination, 1 frame per location, and a 100 ms exposure time. The actin filaments were imaged using a 640 nm laser (OBIS 100 mW, Coherent), 7 mW illumination, 1 frame per location, and 100 ms exposure time. The myosin proteins were imaged using a 561 nm laser (Sapphire 100 mW, Coherent), 20 mW illumination, 100 frames per location, and a 500 ms exposure time. Interferometry of the chamber height profile was performed by directly imaging the 488 nm laser profile, using 0.1 mW illumination, 1 frame per location, and a 30 ms exposure time. The emission filters were in place for fluorescence imaging and removed for interferometry [26].
2.7 Characterization of CLIC imaging chamber height
The dye fluorescence intensity and chamber interferometry (corresponding to Newton’s rings) were analyzed to characterize the chamber heights at the locations of the myosin motors. Newton’s rings are separated by 184 nm, taking the refractive index of solution and probe wavelength to be 1.33 and 488 nm respectively. To calculate the chamber height, the normalized dye fluorescence (proportional to the local chamber height) was fit to a sixth-order polynomial. The proportionality constant between the chamber height and the dye fluorescence was calculated using the location of the first interference ring. The re-scaled fit of the dye fluorescence was used as an input to a direct least-squares fit of the measured interference profile (using the 488 nm probe). In estimating the error on the determination of the height of the CLIC chamber, it was assumed that the dominant source of error was the proportionality constant between the dye fluorescence and the chamber height. Since the constant was calculated using the location of the first interference fringe, the error was estimated by varying the constant such that the first contour of the fitted chamber height profile lay either just outside or just inside the edges of the first minimum ring on the measured interferometry image.
3. Results
3.1 Linear zero-mode waveguides can reduce background fluorescence when the light polarization is parallel to the slits
The new linear, rather than cylindrical, geometry was essential to our application of linear ZMWs to experiments with myosin. To test whether this new geometry could be used to restrict excitation of fluorophores to a layer very close to the surface of the waveguide and significantly shallower than TIRF, we conducted finite element simulations of excitation light entering the linear ZMW, with polarization both parallel and perpendicular to the slits (Figs. 4(a) , 4(b), and 4(c)). We found that when the electric field vector of the excitation light was perpendicular to the slits, the light propagated through the slit and deeply into solution (Fig. 4(a)), which would cause a high background fluorescence from excitation of fluorophores in solution. However, when the electric field was directed parallel to the slits, excitation fell off quickly with depth and was confined within a few nanometers of the entrance to the linear ZMW; the intensity was decreased by 2-3 orders of magnitude by the top of the 120 nm thick aluminum film (Fig. 4(b)). This matched well with previous experiments with rectangular nanoslits, which were comparable in width to our linear ZMWs, although these nanoslits were much shorter [31].
Fig. 4.
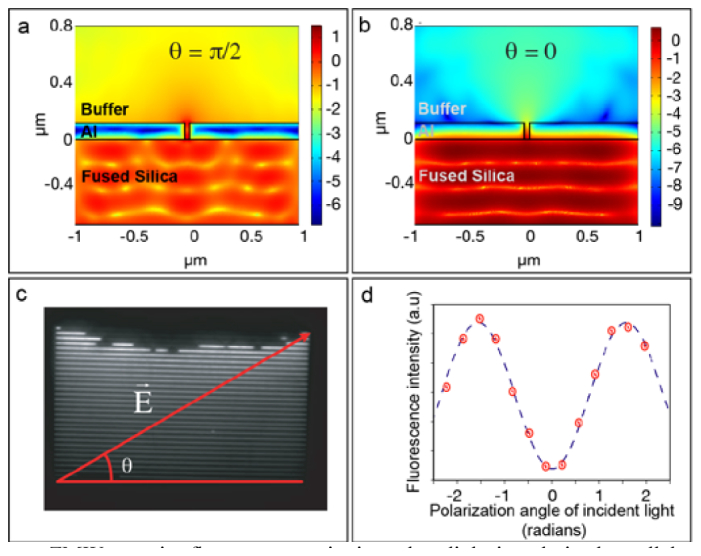
Linear ZMWs restrict fluorescent excitation when light is polarized parallel to the slits. (a) 2D finite-element time-domain simulations of excitation light intensity with polarization perpendicular to the long dimension of the waveguide. Simulations were performed for a 50 nm linear ZMW and the intensity is plotted on a log scale. Under this condition, a significant amount of excitation light propagates up into solution. (b) Simulations analogous to panel (a), but with excitation light polarization parallel to the long dimension of the waveguide (condition that minimizes background fluorescence). Under this condition, intensity has decreased by about 3 orders of magnitude over the 120 nm depth of the linear ZMW. Note that the log intensity scale varies between panels (a) and (b). (c) Image of nanofabricated linear ZMW, imaged with a 20x objective and a white light source. Linear ZMWs range from ~50-250 nm in width. The linear ZMW at the top of the image include manufacturing defects of unknown cause. The definition of the polarization angle for panels (a) and (b) is drawn on top of the image. (d) Average fluorescence intensity (red dots) due to the diffuse background of 1 µM free Cy3-ATP as a function of the excitation polarization angle with fit to theory (blue line). When the polarization is parallel to the longest dimension of the ZMW, the background fluorescence is minimized.
To verify this excitation confinement experimentally, we imaged freely diffusing fluorescent dye (Cy3-ATP) inside linear ZMWs at a concentration of 1 µM while varying the polarization (Fig. 4(d)). As expected from the simulations, polarization perpendicular to the slits caused a considerable amount of background. This background was significantly reduced when the polarization was parallel to the slits.
3.2 Actin filaments can be introduced into linear ZMWs by polymerizing inside the waveguides
A major challenge to the use of linear ZMWs for myosin motility experiments was loading actin filaments into the bottom of the waveguides. We tried two methods of doing so: by specifically attaching actin filaments to the bottom of the slits and by developing a new strategy to polymerize actin filaments inside the waveguides.
Loading already-polymerized actin filaments into the narrow waveguide slits was likely to prove challenging since long actin filaments rotationally diffuse slowly enough that they are rarely oriented properly to allow diffusion to the bottom of the waveguide. Indeed, we found that when we blocked the aluminum surface and preferentially functionalized the bottom of the waveguides with biotin [30], actin filaments that were long enough for myosin to take more than a few steps were only rarely attached to the bottom of the waveguides when they were introduced by diffusion. We attempted to increase the loading of actin filaments to the bottom of the slits by using increasing concentrations of actin, by waiting longer amount of time for them to attach, and by shearing the actin to produce shorter filaments, but we were not able to reproducibly introduce actin filaments longer than a few hundred nanometers into the linear ZMWs with these approaches.
We then took an alternative approach of loading actin by polymerizing the filaments inside the linear ZMWs, as described in detail in the methods section. This new strategy for attaching actin to a coverslip surface was much more successful. We verified that actin was present by adding fluorescently labeled myosin, and looking for processive myosin runs that would indicate the presence of actin filaments, as described below (Fig. 5 ).
Fig. 5.
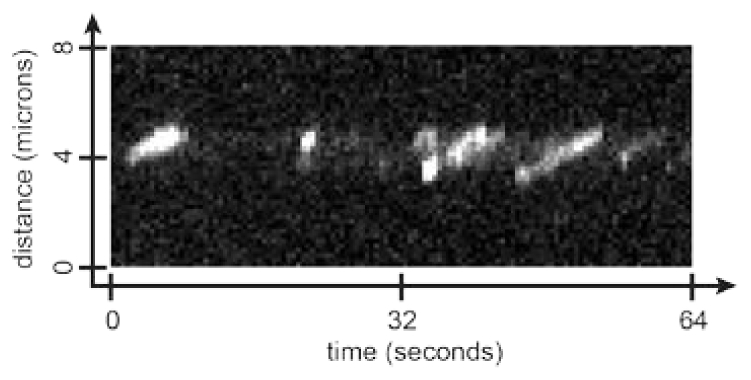
Example kymograph of myosin V molecules walking along actin in a linear ZMW. Note that in some of the myosin runs shown in this kymograph, such as the one beginning around 5 seconds and the one beginning around 35 seconds, two myosin molecules are walking along the actin filament very close together.
3.3 Myosin moves along actin filaments inside linear ZMWs
To observe myosin motility on polymerized actin filaments, we added fluorescently labeled myosin V and ATP to the ZMWs. We were able to see myosin V runs of >1 micron, as shown in the kymograph in Fig. 5, indicating that long actin filaments had been polymerized inside the linear ZMWs. Since myosin V steps are ~36 nm in length, this is long enough to allow at least 30 steps along actin inside the linear ZMWs.
3.4 Myosin walks in the flow-cell CLIC device under significant confinement
The CLIC imaging chamber confines molecules to a thin film to suppress background fluorescence. When the imposed confinement is on the same order as the molecular size, the confinement can alter molecular behavior. This was an important potential hurdle to overcome with CLIC, as it had not previously been demonstrated that the mechanical confinement introduced by the CLIC device was compatible with biological activity. In this work, we demonstrate the first biological application of CLIC by characterizing the processive movement of myosin along actin filaments at various degrees of confinement. This was an important step toward applying CLIC imaging to studying interactions of myosin with fluorescently labeled ATP at high concentrations. This characterization provides not only a technical measure of the potential improvement in imaging quality, but also insight into how confinement or crowding affects molecular interactions in this system and others.
The applied confinement, corresponding to the chamber height, is shown in Fig. 6(a) . The contours of the least-squares fit to the interference pattern and dye fluorescence are superimposed on the data (see Methods Sect. 2.5).
Fig. 6.

Processive myosin movement is observed under confinement in the flow-cell CLIC device. (a) Interferometry of and least-squares fit to the CLIC imaging chamber height profile. The first two labeled interference minima (blue rings) correspond to heights of 184 nm and 368 nm respectively. (b) Two example kymographs of myosin proteins imaged in the chamber at heights of ~80 nm. (c) Distribution of the number of myosins exhibiting processive motion as a function of chamber height, normalized by area. Error bars represent the expected standard deviation assuming Poissonian counting statistics. (d) Scatter plot of processive motor speed vs. chamber height. The speed shows little correlation with chamber height, indicating that confinement does not significantly perturb the motor behavior at heights where motors continue to walk.
The spatial distribution of actin filaments within the chamber was uniform, which was expected since the filaments were inserted before the chamber was compressed. The trajectories of individual myosin proteins at different locations in the chamber were imaged (e.g. Figure 6(b)). Figure 6(c) shows the area density at which we observed processive myosins as a function of the chamber height. Processive myosin movement was not observed in chambers with heights less than 58 nm ( + 6 nm/-12 nm; see Methods Sect. 2.7 for error analysis); instead, these motor proteins were either diffusing or immobile. Using the trajectories of motors that did undergo processive movement, the speed was calculated at different locations in the chamber. Little correlation was seen between velocity and chamber height, indicating that, if motile, the myosin movement is largely unperturbed by the confinement of the CLIC device (Fig. 6(d)). The chamber height measured by interferometry is the coverslip-coverslip separation, which exceeds the effective chamber height experienced by the proteins (e.g. due to surface-blocking agents, see Discussion).
4. Discussion and conclusions
Both new technologies that we demonstrate, linear ZMWs and flow cell CLIC, show promise for single-molecule imaging of fluorescent molecules at higher concentrations than can be accessed via TIRF microscopy. This work represents and important advance, since, unlike other alternatives to TIRF, such as cylindrical ZMWs or nanoconfinement within vesicles or droplets, both of our techniques allow imaging of fluorescent molecules interacting with geometrically extended substrates. We have demonstrated here that processive molecular motors can be imaged with both novel technologies, under conditions that suppress background fluorescence in comparison to TIRF imaging and that do not alter the motor behavior. This is an important first step toward further biological applications.
Cylindrical ZMWs have already proven useful for observing single fluorophores at concentrations up to 10 µM [3], approximately 100-fold higher than TIRF, and the new geometry we present here will dramatically expand the types of experiments that can be used with this technology, allowing not only a fluorescence signal but also the location and movement of that fluorescent label (in one dimension) to be observed simultaneously. Though linear ZMWs may produce somewhat higher levels of background than have been demonstrated with circular ZMWs, due to their larger excitation volume in the linear dimension, they are still likely to allow single-molecule imaging with ~30-fold higher concentrations than TIRF.
Using flow cell CLIC imaging, we have demonstrated the first biological application of CLIC, showing that myosin motors can walk along actin filaments down to a chamber confinement as narrow as 60 nm. In the initial demonstration of CLIC imaging, at confinements narrower than about 300 nm, CLIC imaging was shown to improve the signal-to-background in comparison to TIRF imaging [26]. Therefore, we expect that, at the confinements demonstrated in this study, implementing CLIC imaging will suppress the background from fluorophores in solution by at least five-fold, making single-molecule imaging possible at fluorophore concentrations at least five times higher than with TIRF. In fact, it may be that even further improvements are possible, since the BSA coating used to block both surfaces in this study may substantially increase the effective confinement experienced by molecules in the imaging chamber. While these concentration improvements are not as dramatic as those possible with ZMWs, they are significant, and CLIC has the advantage of the ease with which it may be implemented.
Thus, both technologies will have an important place, with ZMWs possibly more applicable to systems where large increases in concentration are needed, and CLIC useful when more modest concentration increases are sufficient but a simpler experimental setup is desirable. Though not employed in this study, the CLIC geometry has an additional imaging characteristic that is advantageous for some experiments: it extends typical diffusion-limited observation periods of small proteins from ~10 microseconds for ZMWs (or ~100 microseconds for TIRF) to ~seconds for CLIC imaging. These improvements to both suppressing background fluorescence and extending observation periods make a wide range of new biophysical measurements possible.
In conclusion, we have demonstrated two new microscopy technologies that will facilitate a wide range of new single-molecule studies of low affinity molecular interactions and dynamics, particularly with substrates where one dimension extends beyond 100 nm. We have already described one of the most obvious applications: looking at fluorescent nucleotide binding and releasing as processive motors move along filaments. There are many systems for which such an experiment will be informative, including the processive myosins, kinesins, and dynein. Another important application is in examining polymerization of biological filaments, such as actin. For instance, it has been suggested that actin may be able to undergo dynamic instability [32], analogous to that of microtubules. Although there is some evidence from experiments using TIRF that dynamics that are more transient and smaller in scale may occur in actin compared to those in microtubules [33,34], dynamic instability of actin has not been clearly observed experimentally. Imaging polymerization at higher concentrations of fluorescently labeled monomers will facilitate direct measurements of the polymerization and depolymerization kinetics of actin and of other biological filaments, such as microtubules or the many recently-discovered bacterial actin homologues [35].
Statement of competing financial interests
JK is a full-time employee at Pacific Biosciences, a company commercializing single-molecule, real-time nucleic acid sequencing technologies.
Acknowledgments
MWE and JAS were supported by NIH Grant GM33289. MWE thanks the Gerald J. Lieberman Fellowship (Stanford University) for support. SRL thanks the Mary Feiser Fellowship (Harvard University) for support during early development of this work. SRL thanks NSERC EQPEQ 422513-12 and NSERC RGPIN 418915-12 for financial support of the presented CLIC microscopy (performed at McGill University). CM and JSL thank NSERC’s Cellular Dynamics of Molecular Complexes CREATE Program for fellowship support. AEC was supported by NSF Grant CHE-0910824 and by the Harvard Materials Research Science and Engineering Center (MRSEC) under grant DMR-0820484. The authors thank Daniel Berard for assistance with flow cell preparation and for preparing the device schematic in Fig. 3 and Blair Jia for experimental assistance with flow cell preparation in the Leslie laboratory.
References and links
- 1.Komori T., Nishikawa S., Ariga T., Iwane A. H., Yanagida T., “Simultaneous measurement of nucleotide occupancy and mechanical displacement in myosin-V, a processive molecular motor,” Biophys. J. 96(1), L04–L06 (2009). 10.1016/j.bpj.2008.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adachi K., Oiwa K., Nishizaka T., Furuike S., Noji H., Itoh H., Yoshida M., Kinosita K., Jr, “Coupling of rotation and catalysis in F(1)-ATPase revealed by single-molecule imaging and manipulation,” Cell 130(2), 309–321 (2007). 10.1016/j.cell.2007.05.020 [DOI] [PubMed] [Google Scholar]
- 3.Levene M. J., Korlach J., Turner S. W., Foquet M., Craighead H. G., Webb W. W., “Zero-mode waveguides for single-molecule analysis at high concentrations,” Science 299(5607), 682–686 (2003). 10.1126/science.1079700 [DOI] [PubMed] [Google Scholar]
- 4.Boukobza E., Sonnenfeld A., Haran G., “Immobilization in Surface-Tethered Lipid Vesicles as a New Tool for Single Biomolecule Spectroscopy,” J. Phys. Chem. B 105(48), 12165–12170 (2001). 10.1021/jp012016x [DOI] [Google Scholar]
- 5.Chiu D. T., Lorenz R. M., Jeffries G. D. M., “Droplets for ultrasmall-volume analysis,” Anal. Chem. 81(13), 5111–5118 (2009). 10.1021/ac900306q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benítez J. J., Keller A. M., Chen P., “Nanovesicle trapping for studying weak protein interactions by single-molecule FRET,” Methods Enzymol. 472, 41–60 (2010). 10.1016/S0076-6879(10)72016-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okumus B., Arslan S., Fengler S. M., Myong S., Ha T., “Single molecule nanocontainers made porous using a bacterial toxin,” J. Am. Chem. Soc. 131(41), 14844–14849 (2009). 10.1021/ja9042356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loveland A. B., Habuchi S., Walter J. C., van Oijen A. M., “A general approach to break the concentration barrier in single-molecule imaging,” Nat. Methods 9(10), 987–992 (2012). 10.1038/nmeth.2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rief M., Rock R. S., Mehta A. D., Mooseker M. S., Cheney R. E., Spudich J. A., “Myosin-V stepping kinetics: a molecular model for processivity,” Proc. Natl. Acad. Sci. U.S.A. 97(17), 9482–9486 (2000). 10.1073/pnas.97.17.9482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakamoto T., Webb M. R., Forgacs E., White H. D., Sellers J. R., “Direct observation of the mechanochemical coupling in myosin Va during processive movement,” Nature 455(7209), 128–132 (2008). 10.1038/nature07188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De La Cruz E. M., Ostap E. M., Sweeney H. L., “Kinetic mechanism and regulation of myosin VI,” J. Biol. Chem. 276(34), 32373–32381 (2001). 10.1074/jbc.M104136200 [DOI] [PubMed] [Google Scholar]
- 12.Block S. M., “Kinesin motor mechanics: binding, stepping, tracking, gating, and limping,” Biophys. J. 92(9), 2986–2995 (2007). 10.1529/biophysj.106.100677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiu W., Derr N. D., Goodman B. S., Villa E., Wu D., Shih W., Reck-Peterson S. L., “Dynein achieves processive motion using both stochastic and coordinated stepping,” Nat. Struct. Mol. Biol. 19(2), 193–200 (2012). 10.1038/nsmb.2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeWitt M. A., Chang A. Y., Combs P. A., Yildiz A., “Cytoplasmic dynein moves through uncoordinated stepping of the AAA+ ring domains,” Science 335(6065), 221–225 (2012). 10.1126/science.1215804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Funatsu T., Harada Y., Higuchi H., Tokunaga M., Saito K., Ishii Y., Vale R. D., Yanagida T., “Imaging and nano-manipulation of single biomolecules,” Biophys. Chem. 68(1-3), 63–72 (1997). 10.1016/S0301-4622(97)00008-2 [DOI] [PubMed] [Google Scholar]
- 16.Ishijima A., Kojima H., Funatsu T., Tokunaga M., Higuchi H., Tanaka H., Yanagida T., “Simultaneous observation of individual ATPase and mechanical events by a single myosin molecule during interaction with actin,” Cell 92(2), 161–171 (1998). 10.1016/S0092-8674(00)80911-3 [DOI] [PubMed] [Google Scholar]
- 17.Tokunaga M., Kitamura K., Saito K., Iwane A. H., Yanagida T., “Single molecule imaging of fluorophores and enzymatic reactions achieved by objective-type total internal reflection fluorescence microscopy,” Biochem. Biophys. Res. Commun. 235(1), 47–53 (1997). 10.1006/bbrc.1997.6732 [DOI] [PubMed] [Google Scholar]
- 18.Oiwa K., Eccleston J. F., Anson M., Kikumoto M., Davis C. T., Reid G. P., Ferenczi M. A., Corrie J. E., Yamada A., Nakayama H., Trentham D. R., “Comparative single-molecule and ensemble myosin enzymology: sulfoindocyanine ATP and ADP derivatives,” Biophys. J. 78(6), 3048–3071 (2000). 10.1016/S0006-3495(00)76843-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Komori T., Nishikawa S., Ariga T., Iwane A. H., Yanagida T., “Measurement system for simultaneous observation of myosin V chemical and mechanical events,” Biosystems 93(1-2), 48–57 (2008). 10.1016/j.biosystems.2008.04.004 [DOI] [PubMed] [Google Scholar]
- 20.Elting M. W., Bryant Z., Liao J. C., Spudich J. A., “Detailed tuning of structure and intramolecular communication are dispensable for processive motion of myosin VI,” Biophys. J. 100(2), 430–439 (2011). 10.1016/j.bpj.2010.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sweeney H. L., Park H., Zong A. B., Yang Z., Selvin P. R., Rosenfeld S. S., “How myosin VI coordinates its heads during processive movement,” EMBO J. 26(11), 2682–2692 (2007). 10.1038/sj.emboj.7601720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oguchi Y., Mikhailenko S. V., Ohki T., Olivares A. O., De La Cruz E. M., Ishiwata S., “Load-dependent ADP binding to myosins V and VI: implications for subunit coordination and function,” Proc. Natl. Acad. Sci. U.S.A. 105(22), 7714–7719 (2008). 10.1073/pnas.0800564105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunn A. R., Chuan P., Bryant Z., Spudich J. A., “Contribution of the myosin VI tail domain to processive stepping and intramolecular tension sensing,” Proc. Natl. Acad. Sci. U.S.A. 107(17), 7746–7750 (2010). 10.1073/pnas.1002430107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B., Bibillo A., Bjornson K., Chaudhuri B., Christians F., Cicero R., Clark S., Dalal R., Dewinter A., Dixon J., Foquet M., Gaertner A., Hardenbol P., Heiner C., Hester K., Holden D., Kearns G., Kong X., Kuse R., Lacroix Y., Lin S., Lundquist P., Ma C., Marks P., Maxham M., Murphy D., Park I., Pham T., Phillips M., Roy J., Sebra R., Shen G., Sorenson J., Tomaney A., Travers K., Trulson M., Vieceli J., Wegener J., Wu D., Yang A., Zaccarin D., Zhao P., Zhong F., Korlach J., Turner S., “Real-time DNA sequencing from single polymerase molecules,” Science 323(5910), 133–138 (2009). 10.1126/science.1162986 [DOI] [PubMed] [Google Scholar]
- 25.Uemura S., Aitken C. E., Korlach J., Flusberg B. A., Turner S. W., Puglisi J. D., “Real-time tRNA transit on single translating ribosomes at codon resolution,” Nature 464(7291), 1012–1017 (2010). 10.1038/nature08925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leslie S. R., Fields A. P., Cohen A. E., “Convex lens-induced confinement for imaging single molecules,” Anal. Chem. 82(14), 6224–6229 (2010). 10.1021/ac101041s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Churchman L. S., Okten Z., Rock R. S., Dawson J. F., Spudich J. A., “Single molecule high-resolution colocalization of Cy3 and Cy5 attached to macromolecules measures intramolecular distances through time,” Proc. Natl. Acad. Sci. U.S.A. 102(5), 1419–1423 (2005). 10.1073/pnas.0409487102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liao J.-C., Elting M. W., Delp S. L., Spudich J. A., Bryant Z., “Engineered myosin VI motors reveal minimal structural determinants of directionality and processivity,” J. Mol. Biol. 392(4), 862–867 (2009). 10.1016/j.jmb.2009.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pardee J. D., Spudich J. A., “Purification of muscle actin,” Methods Cell Biol. 24, 271–289 (1982). 10.1016/S0091-679X(08)60661-5 [DOI] [PubMed] [Google Scholar]
- 30.Korlach J., Marks P. J., Cicero R. L., Gray J. J., Murphy D. L., Roitman D. B., Pham T. T., Otto G. A., Foquet M., Turner S. W., “Selective aluminum passivation for targeted immobilization of single DNA polymerase molecules in zero-mode waveguide nanostructures,” Proc. Natl. Acad. Sci. U.S.A. 105(4), 1176–1181 (2008). 10.1073/pnas.0710982105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wenger J., Lenne P.-F., Popov E., Rigneault H., Dintinger J., Ebbesen T., “Single molecule fluorescence in rectangular nano-apertures,” Opt. Express 13(18), 7035–7044 (2005). 10.1364/OPEX.13.007035 [DOI] [PubMed] [Google Scholar]
- 32.Theriot J. A., Mitchison T. J., “The nucleation-release model of actin filament dynamics in cell motility,” Trends Cell Biol. 2(8), 219–222 (1992). 10.1016/0962-8924(92)90298-2 [DOI] [PubMed] [Google Scholar]
- 33.Littlefield R., Fowler V. M., “A minor actin catastrophe,” Nat. Cell Biol. 4(9), E209–E211 (2002). 10.1038/ncb0902-e209 [DOI] [PubMed] [Google Scholar]
- 34.Fujiwara I., Takahashi S., Tadakuma H., Funatsu T., Ishiwata S., “Microscopic analysis of polymerization dynamics with individual actin filaments,” Nat. Cell Biol. 4(9), 666–673 (2002). 10.1038/ncb841 [DOI] [PubMed] [Google Scholar]
- 35.Carballido-López R., “The bacterial actin-like cytoskeleton,” Microbiol. Mol. Biol. Rev. 70(4), 888–909 (2006). 10.1128/MMBR.00014-06 [DOI] [PMC free article] [PubMed] [Google Scholar]