

Abstract
Background
D-Serine, an endogenous co-agonist of the N-methyl-D-aspartate (NMDA) receptor, is synthesized from L-serine by serine racemase (SRR). Given the role of D-serine in both neurodevelopment and the pathophysiology of schizophrenia, we examined whether neonatal disruption of D-serine synthesis by SRR inhibition could induce behavioral abnormalities relevant to schizophrenia, in later life.
Methodology/Principal Findings
Neonatal mice (7–9 days) were injected with vehicle or phenazine methosulfate (Met-Phen: 3 mg/kg/day), an SRR inhibitor. Behavioral evaluations, such as spontaneous locomotion, novel object recognition test (NORT), and prepulse inhibition (PPI) were performed at juvenile (5–6 weeks old) and adult (10–12 weeks old) stages. In addition, we tested the effects of D-serine on PPI deficits in adult mice after neonatal Met-Phen exposure. Finally, we assessed whether D-serine could prevent the onset of schizophrenia-like behavior in these mice. Neonatal Met-Phen treatment reduced D-serine levels in the brain, 24 hours after the final dose. Additionally, this treatment caused behavioral abnormalities relevant to prodromal symptoms in juveniles and to schizophrenia in adults. A single dose of D-serine improved PPI deficits in adult mice. Interestingly, chronic administration of D-serine (900 mg/kg/day from P35 to P70) significantly prevented the onset of PPI deficits after neonatal Met-Phen exposure.
Conclusions/Significance
This study shows that disruption of D-serine synthesis during developmental stages leads to behavioral abnormalities relevant to prodromal symptoms and schizophrenia, in later life. Furthermore, early pharmacological intervention with D-serine may prevent the onset of psychosis in adult.
Introduction
Accumulating evidence suggests that hypofunction of glutamatergic neurotransmission via the N-methyl-D-aspartate (NMDA) receptor plays a crucial role in the pathophysiology of schizophrenia [1]–[6]. D-Serine, an endogenous co-agonist of the NMDA receptor is synthesized from L-serine by serine racemase (SRR) [7]–[9]. Recent studies using Srr knock-out (KO) mice show that levels of D-serine in the forebrain of Srr-KO mice are 80–90% lower than in wild-type (WT) mice [10]–[13], implying that D-serine production in the forebrain is largely dependent on SRR activity. D-Serine also plays a crucial role in neurotransmission via the NMDA receptor, throughout development and into adulthood [14]–[16].
Several studies have highlighted evidence suggesting that disturbed NMDA receptor neurotransmission due to decreased D-serine levels, is a causative factor in the pathophysiology of schizophrenia [3], [5], [6], [17]–[19]. These findings include firstly, lower levels of D-serine in the blood, cerebrospinal fluid, and postmortem brain tissue from patients with schizophrenia, relative to normal controls [20]–[24]. Secondly, treatment with D-serine is beneficial for reducing positive, negative and cognitive symptoms in patients with schizophrenia [25], [26]. A recent meta-analysis supports findings that D-serine is effective in the treatment of schizophrenia [27]. Thirdly, mRNA expression and activity of D-amino acid oxidase (DAAO), which metabolizes D-serine, is increased in the postmortem brains of schizophrenic patients [28], [29]. Fourthly, the G72 gene at chromosome 13q is significantly associated with schizophrenia [30], [31]. This gene has been designated a DAAO activator, since the G72 protein interacts physically with DAAO [30]. A recent meta-analysis provided evidence of highly significant association between nucleotide variations in the G72/G30 region and schizophrenia [32]. In addition, multiple epidemiological surveys support the neurodevelopmental hypothesis for the pathogenesis of schizophrenia [33]–[35]. Taken together, these findings point to the possibility that hypofunction of the NMDA receptor, resulting from reduced D-serine levels during gestation could interfere with normal fetal brain neurodevelopment and that these deficits are causative to the onset of schizophrenia in adulthood.
Phenazine methosulfate (Met-Phen) is an inhibitor (IC50 = 3.0 µM) of SRR [36]. At present, there are no reports of using Met-Phen for in vivo manipulation of SRR in brain. Furthermore, the effect of Met-Phen on brain levels of D-serine relative to other amino acids is currently unknown. This study was, therefore, undertaken to examine whether neonatal Met-Phen exposure in mice could lead to behavioral phenotypes similar to those seen in juvenile and adult human schizophrenia. Auditory sensory gating prepulse inhibition (PPI) deficits were used as an animal model of schizophrenia. We also assessed effects of D-serine on behavioral abnormalities in adult mice after neonatal Met-Phen exposure. Finally, we examined whether D-serine could prevent the onset of schizophrenia-like behavior in adult mice after neonatal Met-Phen exposure.
Materials and Methods
Animals
DDY mice (Japan SLC Inc., Shizuoka, Japan) were mated at age 10 weeks and the offspring were used for these experiments. Male and female neonatal mice (10 days old, weight 0.4–0.6 g), male juvenile mice (5–6 weeks old, weight 24–30 g) and male adult mice (10–12 weeks old, weight 34–44 g) bred in our laboratory were used for experiments. Animals were housed under controlled temperature and 12 h light/dark cycles (lights on between 07∶00–19∶00), with ad libitum food and water. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, USA. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Chiba University (Permit Number: #22–98). For the measurement of amino acids, mice were sacrificed under CO2, and all efforts were made to minimize suffering.
Neonatal Administration of Met-Phen
On postnatal day 7, baby mice were divided randomly into control (saline treated) or phenazine methosulfate treated groups (Met-Phen, #P9625, Sigma-Aldrich, St Louis, MO) [36]. From P7 to P9, the pups were injected intraperitoneally (i.p.) with Met-Phen (3.0 mg/kg/day) or saline (1.0 ml/kg/day). In a preliminary experiment, we examined the effects of Met-Phen on brain levels of D-serine. The dose (3.0 mg/kg/day for 3 days) of Met-Phen established in this experiment was used for this study. Male mice were separated from their mothers after 3 weeks and mice were caged in separate groups, depending on treatment.
Measurement of Amino Acids in the Brain
At postnatal (P10), juvenile (P35–P42), and adult (P70–P84) stages, mice were sacrificed, and their brains excised for measurement of amino acids. The cerebellum, frontal cortex, hippocampus and striatum were quickly dissected from whole brain after decapitation. The dissected tissues were weighed and stored at −80°C until assayed. Measurements of D- and L- serine, glutamate, glutamine and glycine levels were carried out using a column-switching high performance liquid chromatography (HPLC) system (Shimadzu Corporation, Kyoto, Japan), as described previously [37]–[39].
Locomotor Activity in Mice
Both horizontal and rearing activity were monitored by an infrared ray passive sensor system (SCANET-SV10, Melquest Ltd, Toyama, Japan), and activity was integrated every 10 minutes, as previously reported [40]–[42]. Individual mice were placed in activity chambers and allowed 2 hours of free exploration as spontaneous activity.
Novel Object Recognition Test (NORT)
The NORT was performed as previously reported [43]–[45]. Before testing, mice were habituated in the box for 3 days. During a training session, two objects (differing in shape and color but of similar size) were placed in the box 35.5 cm apart (symmetrically), and each animal was allowed to explore in the box for 5 minutes. The animals were considered to be exploring the object when the head of the animal was both facing and within 2.54 cm of the object or when any part of the body, except for the tail was touching the object. The time that mice spent exploring each object was recorded. After training, mice were immediately returned to their home cages, and the box and objects were cleaned with 75% ethanol, to avoid any possible instinctive odorant cues. Retention tests were carried out at one-day intervals, following the respective training. During the retention test, each mouse was reintroduced into their original test box, and one of the training objects was replaced by a novel object. The mice were then allowed to explore freely for 5 minutes, and the time spent exploring each object was recorded. Throughout the experiments, the objects were counter-balanced, in terms of their physical complexity and emotional neutrality. A preference index, that is, the ratio of time spent exploring either of the two objects (training session) or the novel object (retention test session) over the total time spent exploring both objects, was used.
Acoustic Startle and Prepulse Inhibition (PPI) Test
The mice were tested for their acoustic startle responses in a startle chamber (SR-LAB, San Diego Instruments, San Diego, CA), using a standard method described previously [41], [46]. Test sessions started after an initial 10-minute acclimation period in the chamber. The mice were subjected to one of six trials: (1) pulse alone, as a 40 ms broadband burst; a pulse (40 ms broadband burst) preceded by 100 ms with a 20 ms prepulse that was (2) 4 dB, (3) 8 dB, (4) 12 dB, or (5) 16 dB over background (65 dB); and (6) background only (no stimulus). Prepulse inhibition (PPI) was expressed as the percentage decrease in amplitude of startle reactivity, caused by a prepulse (% PPI).
To examine effects of D-serine on PPI deficits in adult mice after neonatal Met-Phen exposure, D-serine (900 mg/kg, Sigma-Aldrich, St. Louis, MO) and saline (10 ml/kg) were administered i.p. The drugs and vehicles were injected i.p., 30 minutes before the start of testing.
Prevention of Met-Phen-induced PPI deficits by D-serine
To examine whether D-serine could prevent the onset of PPI deficits in adult mice after neonatal Met-Phen exposure, D-serine (900 mg/kg/day for 35 days) or vehicle (saline; 10 ml/kg/day for 35 days) were administered i.p. from P35 to P70; this period is thought to represent pre-adolescence to adulthood. One week (P77) after the last dose of D-serine, PPI testing was performed, as described.
Statistical Analysis
All data are shown as mean ± standard error of the mean (S.E.M.). The results of D-serine levels, locomotion, NORT and social interaction were analyzed by Student’s t-test. PPI data were analyzed by multivariate analysis of variance (MANOVA). Where relevant at individual dB levels, the data were compared by Student’s t-test or one-way ANOVA, followed LSD test. Significance for results was set at p<0.05.
Results
Effects of Neonatal Met-Phen Treatment on Levels of Amino Acids in the Brain
Treatment with Met-Phen (3.0 mg/kg/day for 3 days from P7–P9) significantly decreased the levels of D-serine in the frontal cortex (t = 2.131, p<0.05) and cerebellum (t = 2.736, p<0.01) at P10 ( Table 1 ), suggesting that Met-Phen affects the synthesis of D-serine. Treatment with Met-Phen also significantly decreased levels of L-serine in the frontal cortex, but not cerebellum. Moreover, the levels of glutamate and glutamine in the frontal cortex and cerebellum of Met-Phen treated mice were significantly higher than those of saline-treated mice ( Table 1 ). In contrast, levels of glycine remained the same ( Table 1 ).
Table 1. Levels of amino acids in the frontal cortex and cerebellum 24 hours after the last administration of Met-Phen.
| Glutamate | Glutamine | Glycine | L-Serine | D-Serine | |
| Frontal cortex | |||||
| Control | 4.729±0.294 | 3.649±0.373 | 1.892±0.442 | 0.746±0.036 | 0.150±0.005 |
| Met-Phen | 5.857±0.314 (124%)* | 4.371±0.141 (120%)* | 1.463±0.369 (77.3%) | 0.585±0.019 (78.4%)*** | 0.130±0.004 (86.7%)* |
| Cerebellum | |||||
| Control | 3.509±0.220 | 7.118±0.298 | 2.602±0.153 | 0.848±0.034 | 0.154±0.006 |
| Met-Phen | 4.375±0.221 (125%)* | 9.200±0.423 (129%)** | 2.294±0.153 (88.2%) | 0.827±0.039 (97.5%) | 0.125±0.006 (81.2%)* |
Data (nmol/mg tissue) are expressed as the mean ± SEM (Control: n = 9, Met-Phen: n = 21).
Parenthesis is the percentage of control values.
p<0.05,
p<0.01,
p<0.001 compared to control group (Student’s t test).
Amino acids in the frontal cortex and hippocampus of 4 week old mice did not differ between the two groups, although levels of D-serine and L-serine in the cerebellum of Met-Phen treated mice were significantly higher than those of controls (Table S1). In adult mice (10 weeks old), there were no changes in the levels of amino acids in the frontal cortex and striatum between the two groups. Furthermore, levels of all amino acids in the hippocampus of Met-Phen treated mice were significantly lower than those of controls (Table S2).
Effects of Neonatal Met-Phen Treatment on Locomotor Activity
In the open field test, spontaneous locomotion, such as horizontal activity and rearing activity, was measured at juvenile and adult stages. Horizontal activity was unchanged between the groups at both stages ( Figure 1A and 1B ). However, rearing activity in Met-Phen treated juvenile mice was significantly (t = −5.742, p<0.001) higher than that of the saline treated group ( Figure 1C ), suggesting that Met-Phen treated mice show stereotypy at the juvenile stage. In adult mice, no differences were detected between the treatment groups in rearing activity ( Figure 1D ).
Figure 1. Spontaneous locomotion after neonatal Met-Phen treatment.

Saline (1.0 ml/kg/day) or Met-Phen (3.0 mg/kg/day) was administered i.p. from P7 to P9. Horizontal activity and rearing activity were performed at juvenile (5–6 weeks old) and adult stages (10–12 weeks old). Data represent the mean ± S.E.M. (n = 9 mice for control group, n = 16 for Met-Phen group). ***P<0.001 compared with saline treated group.
Effects of Neonatal Met-Phen Treatment on Cognition
Using NORT, we measured cognition at juvenile and adult stages. Treatment with Met-Phen (3.0 mg/kg/day for 3 days from P7–P9) caused significant cognitive deficits in juvenile (t = 4.719, p<0.001) and adult mice (t = 3.458, p<0.01) ( Figure 2A and 2B ). These results imply that neonatal Met-Phen exposure induces cognitive deficits in juvenile and adult mice.
Figure 2. Cognition after neonatal Met-Phen treatment.
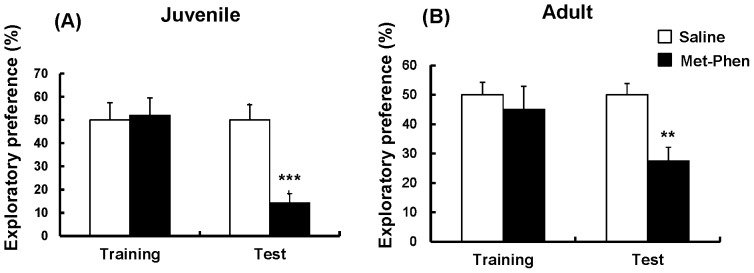
Saline (1.0 ml/kg/day) or Met-Phen (3.0 mg/kg/day) was administered i.p. from P7 to P9. NORT was performed at juvenile (5–6 weeks old) and adult stages (10–12 weeks old). Data represent the mean ± S.E.M. (n = 8–11 mice for control group, n = 11 or 12 for Met-Phen group). *P<0.05, **P<0.01, *** P<0.001 compared with saline treated group.
Effects of Neonatal Met-Phen Treatment on PPI
Deficits in sensorimotor gating are a well characterized endophenotype of schizophrenia. Figure 3 shows PPI data for juvenile and adult mice. MANOVA analysis of all data from test and control juvenile mice revealed no differences, indicating that treatment with Met-Phen (3.0 mg/kg/day for 3 days from P7–P9) did not cause PPI deficits in juvenile animals ( Figure 3A ). In contrast, MANOVA analysis of adult data revealed significant differences. Subsequent analysis showed that treatment with Met-Phen (3.0 mg/kg/day for 3 days from P7–P9) caused significant PPI deficits in three groups (69, 73, and 77 dB) ( Figure 3B ). No differences in startle response were found between the groups (data not shown).
Figure 3. Auditory sensory gating PPI deficits after neonatal Met-Phen treatment.
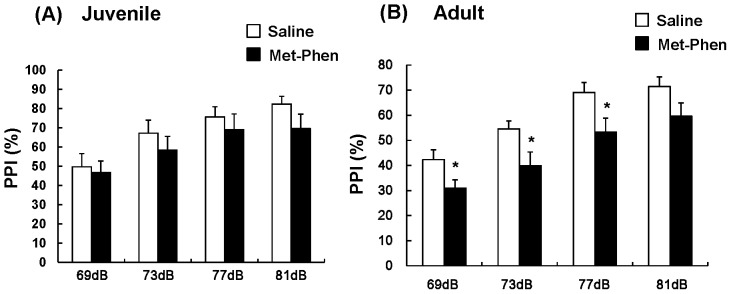
Saline (1.0 ml/kg/day) or Met-Phen (3.0 mg/kg/day) was administered i.p. from P7 to P9. Auditory sensory gating PPI test was performed at juvenile (5–6 weeks old) and adult stages (10–12 weeks old). Data represent the mean ± S.E.M. (n = 11 mice for control group, n = 12 or 13 for Met-Phen group). *P<0.05 compared with saline treated group.
Effects of D-serine on PPI Deficits in Adult Mice after Neonatal Met-Phen Exposure
We examined the effects of D-serine on PPI deficits in adult mice after neonatal Met-Phen exposure. D-Serine (900 mg/kg) or a vehicle was administered i.p., 30 minutes before PPI testing. MANOVA analysis revealed a significant effect for D-serine. Additional analysis showed that a single dose of D-serine significantly improved neonatal Met-Phen induced PPI deficits in all tested groups (69, 73, 77, and 81 dB) ( Figure 4 ).
Figure 4. Effects of D-serine on PPI deficits at adult after neonatal Met-Phen treatment.
Met-Phen (3.0 mg/kg/day) was administered i.p. from P7 to P9. PPI test was performed at adult (10–12 weeks old). D-Serine (900 mg/kg, i.p.) or vehicle (saline; 10 ml/kg) was administered 30 minutes before PPI test. Data represent the mean ± S.E.M. (n = 7 mice for control group, n = 12 for D-serine group). *P<0.05, **P<0.01 compared with Met-Phen+vehicle treated group.
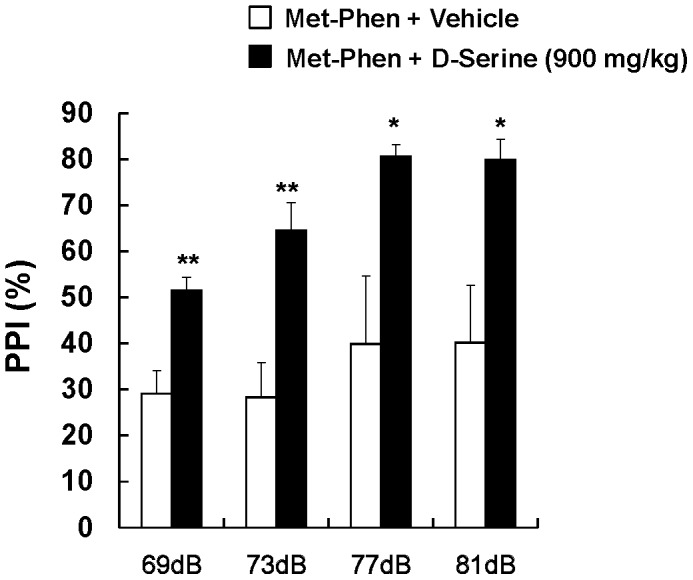
D-serine Prevents PPI Deficits in Adult Mice after Neonatal Met-Phen Exposure
We examined whether D-serine was capable of preventing schizophrenia-like behavioral abnormalities in adult mice, after neonatal Met-Phen exposure. From P35 to P70, D-serine (900 mg/kg/day) or a vehicle (10 ml/kg/day) was chronically administered to mice with neonatal Met-Phen exposure. To exclude the acute effects of D-serine, PPI testing was performed one week after the last dose of D-serine or vehicle. As with clozapine, MANOVA analysis revealed a significant effect for D-serine. Subsequent analysis confirmed that chronic administration of D-serine significantly improved PPI deficits seen in neonatal Met-Phen exposure, in all the tested groups (69, 73, 77, and 81 dB) ( Figure 5 ).
Figure 5. Preventive effects of D-serine on PPI deficits at adult after neonatal Met-Phen treatment.

Met-Phen (3.0 mg/kg/day) was administered i.p. from P7 to P9. PPI test was performed at adult (11 weeks old). D-Serine (900 mg/kg/day, i.p.) or vehicle (saline; 10 ml/kg/day, i.p.) was administered chronically from P35 (5 weeks old) to P70 (10 weeks old). PPI test was performed 1 week (P77) after the final administration of D-serine. Data represent the mean ± S.E.M. (n = 5 mice for control group, n = 5 for D-serine group). *P<0.05, **P<0.01, ***P<0.001 compared with Met-Phen+vehicle treated group.
Discussion
In this study, we found that neonatal administration of Met-Phen, an SSR inhibitor, caused behavioral abnormalities, such as increased rearing, and cognitive impairment in juvenile mice and cognitive impairment, and auditory sensory gating PPI deficits in adult mice. Furthermore, PPI deficits in adult animals with neonatal Met-Phen exposure were improved by a single dose of D-serine, indicating that D-serine could possess antipsychotic activity in this model. In this study, we found that the absolute values of PPI differed slightly between experiments ( Figures 3 , 4 , and 5 ), suggesting a degree of variance between experiments. Moreover, we observed increased rearing activity in juvenile mice after neonatal Met-Phen exposure. This behavior may be due to alterations in amino acids levels within the cerebellum, since NMDA receptors in this region play a role in motor function. Considering the crucial role of D-serine in brain development, it is likely that disruption of NMDA receptor neurotransmission, by decreased D-serine levels during this period, may contribute to the later life behavioral abnormalities seen after neonatal Met-Phen exposure. Further detailed studies on how postnatal Met-Phen treatment induces schizophrenia-like behavioral abnormalities in adulthood are needed.
At birth, substantial amounts of D-serine were observed in all brain regions, including the cerebellum [16]. Levels of D-serine in the forebrain increased dramatically up to P21, and then remained roughly constant, whereas in the cerebellum, D-serine increased up to P7 and then declined dramatically to trace levels between P12 to P18 [16]. In this study, Met-Phen was administered from P7 to P9, since this period showed high levels of D-serine and SSR in all brain regions, including the forebrain and cerebellum [16]. Within 24 hours of the last dose of Met-Phen, levels of D-serine in mouse brains were significantly lower than those of the control group, indicating that Met-Phen inhibited SRR activity in the brain. Furthermore, there were alternations in the levels of glutamine and glutamate in the brains of Met-Phen treated mice, suggesting disruption of the glutamine-glutamate cycle in these animals. In the juvenile stage, Met-Phen treated mice showed increased levels of L-and D-serine in the cerebellum compared with controls. In adults, all amino acids were significantly lower in the hippocampus of Met-Phen treated mice relative to control mice, although no changes were observed in the frontal cortex and striatum. It is likely, that disrupting D-serine synthesis during development contributes to long-lasting changes in the synthesis and metabolism of amino acids associated with NMDA receptor function in the brain, in later life. These Met-Phen induced alterations to NMDA receptor neurotransmission persist through adulthood, even though Met-Phen is washed out from the adult brain.
In the NORT, the exploratory preference (14.2% in juveniles and 27.5% in adults) in mice after neonatal Met-Phen exposure was significantly lower than that of control mice, suggesting that the behavior of Met-Phen-treated mice may not be due to memory impairment. Reports show that repeated administration of phencyclidine (PCP) causes social interaction deficits in animals [47], [48]. In addition, negative symptoms such as social withdrawal are related to cognitive deficits in patients with schizophrenia [49]. Considering these findings, it is likely that our model of Met-Phen-treated cognitive deficits using the NORT, may show negative symptoms such as social withdrawal, which as stated before, are related to cognitive deficits [50]. Interestingly, we found that in the PCP-induced NORT model, D-serine could attenuate PCP-induced cognitive deficits in mice [51].
PPI deficits are generally recognized as an animal model of schizophrenia [52], as these deficits are observed in patients with psychiatric diseases, including schizophrenia [52]. In this study, we found that neonatal Met-Phen administration causes PPI deficits in adult, but not juvenile stages, suggestive of maturation-dependent auditory sensory gating deficits. In addition, adult mice with neonatal Met-Phen exposure, showed behavioral abnormalities, such as cognitive impairment, social interaction deficits and sensory gating PPI deficits, all of which are relevant to schizophrenia. We were able to improve these PPI deficits in adult mice with a single dose of D-serine. From the perspective of NMDA receptor related, neurodevelopmental processes playing a critical role in both normal brain development and schizophrenia, it is plausible that adult mice neonatally exposed to Met-Phen, may constitute an animal model of schizophrenia. A further detailed study will obviously be needed to confirm this hypothesis.
Accumulating evidence suggests that patients with schizophrenia show nonpsychotic and nonspecific prodromal symptoms such as depression, social withdrawal, and cognitive impairment, for several years preceding the onset of frank psychosis [53]–[58]. A recent meta-analysis of 27 studies showed that the average rate of transition to full psychosis among such patients is 22% within the first year and 36% within three years [57]. Therefore, providing early intervention at the prodromal phase of schizophrenia and related psychosis is one of the most important and challenging tasks in psychiatry [58]. In this study, we found that neonatal Met-Phen treatment induced behavioral abnormalities, such as stereotypy, cognitive impairment and social withdrawal in juveniles, suggesting that these mice may show prodromal, or at risk of psychosis symptoms. Interestingly, we found that chronic administration of D-serine from pre-adolescent to adult stages prevented the onset of PPI deficits in adult mice. A recent report on two pilot studies using prodromal subjects showed that glycine, an endogenous agonist of the NMDA receptor, reduced symptoms with promising effect sizes [59]. This would imply that targeting the glycine agonist sites of the NMDA receptor could provide promising therapy for the prodromal phase of psychotic disorders. This makes D-serine an attractive potential drug for early intervention in the onset of schizophrenia, since D-serine is effective in treating several symptoms of schizophrenia [25]–[27], [60].
Before D-serine could be considered as a therapeutic agent, it would be necessary to solve the problem of bioavailability. Orally administered D-serine is heavily metabolized by D-amino acid oxidase (DAAO) in peripheral organs, diminishing its bioavailability in human subjects. Administering higher doses could potentially lead to nephrotoxicity, although no significant adverse events have as yet been observed at doses of up to 4 g/day [19]. Therapeutic levels of D-serine at lower doses could be achieved if D-serine were co-administered with DAAO inhibitors [17], [46]. This combination could represent a new pharmacological intervention for the onset of schizophrenia and related psychosis.
Finally, as with studies of this nature, there are some limitations to this report. The main limitation was the use of Met-Phen, particularly since the precise pharmacology of Met-Phen as an SRR inhibitor has not been fully elucidated. Although several SSR inhibitors have been reported, there is no data on the selectivity of these inhibitors [9]. Therefore, further studies using selective SSR inhibitors or SSR gene deletion during the neonatal period are needed. It is also reported that phenazine (Phen) is an inactive inhibitor at SSR [36]. Thus, these experiments could be strengthened if Phen were used as a negative control for this study. Another limitation was the noted changes on brain amino acid levels, after neonatal Met-Phen exposure. In this study, we found alternations in brain levels of other amino acids including glutamate, glutamine, glycine and L-serine, all of which could affect NMDA receptor neurotransmission [5], [38], [39], [61], [62]. It is reported that the glutamine-glutamate cycle and L-serine-glycine pathway, play a role in NMDA receptor neurotransmission in the brain [5], [39], [61], [62]. Given the key role of the NMDA receptor in the pathophysiology of schizophrenia [1]–[6], [63], our model represents a new tool for evaluating the contribution of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Accumulating evidence suggests that the potent antioxidant glutathione plays a role in the modulation of redox-sensitive sites on the NMDA receptor and in the pathophysiology of schizophrenia [64]–[70]. Since glutathione is synthesized in the body from L-glutamate, glycine and L-cysteine, it would also be of great interest to measure the brain glutathione levels of animals in this model.
In conclusion, our results suggest that neonatal Met-Phen exposure causes behavioral abnormalities, relevant to prodromal symptoms and schizophrenia during juvenile and adult stages, respectively. Interestingly, chronic administration of D-serine from juvenile to adult stages may prevent the onset of schizophrenia-like symptoms in adults after neonatal Met-Phen exposure, indicating that D-serine may serve as an early intervention for psychosis. It is therefore likely that neonatal Met-Phen treatment may constitute a new neurodevelopmental animal model of schizophrenia, based on the NMDA receptor hypofunction hypothesis.
Supporting Information
Effect of neonatal Met-Phen treatment on levels of amino acids in the brain at juvenile.
(XLSX)
Effect of neonatal Met-Phen treatment on levels of amino acids in the brain at adult.
(XLSX)
Funding Statement
This study was supported by a Grant-in-Aid from the Minister of Education, Culture, Sports, Science, and Technology of Japan (to KH), and a Grant-in-Aid for Scientific Research on Innovative Areas of the Ministry of Education, Culture, Sports, Science and Technology, Japan (to KH). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Javitt DC, Zukin SR (1991) Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 148: 1301–1308. [DOI] [PubMed] [Google Scholar]
- 2. Krystal JH, D’Souza DC, Petrakis IL, Belger A, Berman RM, et al. (1999) NMDA agonists and antagonists as probes of glutamatergic dysfunction and pharmacotherapies in neuropsychiatric disorders. Harv Rev Psychiatry 7: 125–143. [PubMed] [Google Scholar]
- 3. Coyle JT, Tsai G (2004) The NMDA receptor glycine modulatory site: a therapeutic target for improving cognition and reducing negative symptoms in schizophrenia. Psychopharmacology (Berl) 174: 32–38. [DOI] [PubMed] [Google Scholar]
- 4. Javitt DC, Coyle JT (2004) Decoding schizophrenia. Sci Am 290: 48–55. [DOI] [PubMed] [Google Scholar]
- 5. Hashimoto K, Shimizu E, Iyo M (2005) Dysfunction of glia-neuron communication in pathophysiology of schizophrenia. Curr Psychiatry Rev 1: 151–163. [Google Scholar]
- 6. Hashimoto K (2006) The NMDA receptor hypofunction hypothesis for schizophrenia and glycine modulatory sites on the NMDA receptors as potential therapeutic drugs. Clin Psychopharmacol Neurosci 4: 3–10. [Google Scholar]
- 7. Wolosker H, Sheth KN, Takahashi M, Mothet JP, Brady RO Jr, et al. (1999) Purification of serine racemase: biosynthesis of the neuromodulator D-serine. Proc Natl Acad Sci USA 96: 721–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wolosker H, Blackshaw S, Snyder SH (1999) Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc Natl Acad Sci USA 96: 13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wolosker H, Mori H (2012) Serine racemase: an unconventional enzyme for an unconventional transmitter. Amino Acids 43: 1895–1904. [DOI] [PubMed] [Google Scholar]
- 10. Inoue R, Hashimoto K, Harai T, Mori H (2008) NMDA- and β-amyloid1–42-induced neurotoxicity is attenuated in serine racemase knock-out mice. J Neurosci 28: 14486–14491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Basu AC, Tsai GE, Ma CL, Ehmsen JT, Mustafa AK, et al. (2009) Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol Psychiatry 14: 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horio M, Kohno M, Fujita Y, Ishima T, Inoue R, et al. (2011) Levels of D-serine in the brain and peripheral organs of serine racemase (Srr) knock-out mice. Neurochem Int 59: 853–859. [DOI] [PubMed] [Google Scholar]
- 13.Horio M, Mori H, Hashimoto K (in press) Is D-cycloserine a pro-drug for D-serine in the brain? Biol Psychiatry 2012 Aug 7. [Epub ahead of print]. [DOI] [PubMed]
- 14. Hashimoto A, Nishikawa T, Oka T, Takahashi K (1993) Endogenous D-serine in rat brain: N-methyl-D-aspartate receptor-related distribution and aging. J Neurochem 60: 783–786. [DOI] [PubMed] [Google Scholar]
- 15. Schell MJ, Brady RO Jr, Molliver ME, Snyder SH (1997) D-Serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptor. J Neurosci 17: 1604–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang LZ, Zhu XZ (2003) Spatiotemporal relationships among D-serine, serine racemase, and D-amino acid oxidase during mouse postnatal development. Acta Pharmacol Sci 24: 965–974. [PubMed] [Google Scholar]
- 17. Ferraris DV, Tsukamoto T (2011) Recent advances in the discovery of D-amino acid oxidase inhibitors and their therapeutic utility in schizophrenia. Curr Pharm Des 17: 103–111. [DOI] [PubMed] [Google Scholar]
- 18. Labrie V, Wong AH, Roder JC (2012) Contributions of the D-serine pathway to schizophrenia. Neuropharmacology 62: 1484–1503. [DOI] [PubMed] [Google Scholar]
- 19. Javitt DC, Zukin SR, Heresco-Levy U, Umbricht D (2012) Has an angel shown the way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophr Bull 38: 958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hashimoto K, Fukushima T, Shimizu E, Komatsu N, Watanabe H, et al. (2003) Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry 60: 572–576. [DOI] [PubMed] [Google Scholar]
- 21. Yamada K, Ohnishi T, Hashimoto K, Ohba H, Iwayama-Shigeno Y, et al. (2005) Identification of multiple serine racemase (SRR) mRNA isoforms and genetic analyses of SRR and DAO in schizophrenia and D-serine levels. Biol Psychiatry 57: 1493–1503. [DOI] [PubMed] [Google Scholar]
- 22. Hashimoto K, Engberg G, Shimizu E, Nordin C, Lindström LH, et al. (2005) Reduced D-serine to total serine ratio in the cerebrospinal fluid of drug naive schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry 29: 767–769. [DOI] [PubMed] [Google Scholar]
- 23. Bendikov I, Nadri C, Amar S, Panizzutti R, De Miranda J, et al. (2007) A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophr Res 90: 41–51. [DOI] [PubMed] [Google Scholar]
- 24. Calcia MA, Madeira C, Alheira FV, Silva TC, Tannos FM, et al. (2012) Plasma levels of D-serine in Brazillian individuals with schizophrenia. Schizophr Res 142: 93–87. [DOI] [PubMed] [Google Scholar]
- 25. Tsai G, Yang P, Chung LC, Lange L, Coyle JT (1998) D-serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry 44: 1081–1089. [DOI] [PubMed] [Google Scholar]
- 26. Heresco-Levy U, Javitt DC, Ebstein R, Vass A, Lichtenberg P, et al. (2005) D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol Psychiatry 57: 577–585. [DOI] [PubMed] [Google Scholar]
- 27. Tsai GE, Lin PY (2010) Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia. A critical review and meta-analysis. Curr Pham Des 16: 522–537. [DOI] [PubMed] [Google Scholar]
- 28. Verrall L, Walker A, Rawlings N, Benzel I, Kew JNC, et al. (2007) D-Amino acid oxidase and serine racemase in human brain: normal distribution and altered expression in schizophrenia. Eur J Neurosci 26: 1657–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Madeira C, Freitas ME, Vargas-Lopes C, Wolosker H, Panizzutti R (2008) Increased brain D-amino acid oxidase (DAAO) activity in schizophrenia. Schizophr Res 101: 76–83. [DOI] [PubMed] [Google Scholar]
- 30. Chumakov I, Blumenfeld M, Guerassimenko O, Cavarec L, Palicio M, et al. (2002) Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc Natl Acad Sci USA 99: 13675–13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kvajo M, Dhilla A, Swor DE, Karayiorgou M, Gogos JA (2008) Evidence implicating the candidate schizophrenia/bipolar disorder susceptibility gene G72 in mitochondrial function. Mol Psychiatry 13: 685–696. [DOI] [PubMed] [Google Scholar]
- 32. Detetra-Wadleigh SD, McMahon FJ (2006) G72/G30 in schizophrenia and bipolar disorder: review and meta-analysis. Biol Psychiatry 60: 106–114. [DOI] [PubMed] [Google Scholar]
- 33. Weinberger DR (1987) Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry 44: 660–669. [DOI] [PubMed] [Google Scholar]
- 34. Wyatt RJ (1996) Neurodevelopmental abnormalities and schizophrenia. A family affair. Arch Gen Psychiatry 53: 11–15. [DOI] [PubMed] [Google Scholar]
- 35. McGrath JJ, Féron FP, Burne TH, Mackay-Sim A, Eyles DW (2003) The neurodevelopmental hypothesis of schizophrenia: a review of recent developments. Ann Med 35: 86–93. [DOI] [PubMed] [Google Scholar]
- 36. Kim PM, Aizawa H, Kim PS, Huang AS, Wickramasinghe SR, et al. (2005) Serine racemase: activation by glutamate neurotransmission via glutamate receptor interacting protein and mediation of neuronal migration. Proc Natl Acad Sci USA 102: 2105–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fukushima T, Kawai J, Imai K, Toyo’oka T (2004) Simultaneous determination of D- and L-serine in rat brain microdialysis sample using a column-switching HPLC with fluorimetric detection. Biomed Chromatogr 18: 813–819. [DOI] [PubMed] [Google Scholar]
- 38. Hashimoto K, Engberg G, Shimizu E, Nordin C, Lindstrom LH, et al. (2005) Elevated glutamine/glutamate ratio in cerebrospinal fluid of first episode and drug naive schizophrenic patients. BMC Psychiatry 5: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hashimoto K, Sawa A, Iyo M (2007) Increased levels of glutamate in brains from patients with mood disorders. Biol. Psychiatry 62: 1310–1316. [DOI] [PubMed] [Google Scholar]
- 40. Ozawa K, Hashimoto K, Kishimoto T, Shimizu E, Ishikura H, et al. (2006) Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry 59: 546–554. [DOI] [PubMed] [Google Scholar]
- 41. Zhang L, Shirayama Y, Iyo M, Hashimoto K (2007) Minocycline attenuates hyperlocomotion and prepulse inhibition deficits in mice after administration of the NMDA receptor antagonist dizocilpine. Neuropsychopharmacology 32: 2004–2010. [DOI] [PubMed] [Google Scholar]
- 42. Hagiwara H, Iyo M, Hashimoto K (2009) Mithramycin protects dopaminergic neurotoxicity in mouse brain after administration of methamphetamine. Brain Res 1301: 189–196. [DOI] [PubMed] [Google Scholar]
- 43. Hashimoto K, Fujita Y, Shimizu E, Iyo M (2005) Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of clozapine, but not haloperidol. Eur J Pharmacol 519: 114–117. [DOI] [PubMed] [Google Scholar]
- 44. Hashimoto K, Fujita Y, Iyo M (2007) Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of fluvoxamine: Role of sigma-1 receptors. Neuropsychopharmacology 32: 514–521. [DOI] [PubMed] [Google Scholar]
- 45. Hashimoto K, Ishima T, Fujita Y, Matsuo M, Kobashi T, et al. (2008) Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of the novel selective α7 nicotinic receptor agonist SSR180711. Biol Psychiatry 63: 92–97. [DOI] [PubMed] [Google Scholar]
- 46. Hashimoto K, Fujita Y, Horio M, Kunitachi S, Iyo M, et al. (2009) Co-administration of a D-amino acid oxidase inhibitor potentiates the efficacy of D-serine in attenuating prepulse inhibition deficits after administration of dizocilpine. Biol Psychiatry 65: 1103–1106. [DOI] [PubMed] [Google Scholar]
- 47. Sams-Dodd F (1998) Effects of continuous D-amphetamine and phencyclidine administration on social behaviour, stereotyped behaviour, and locomotor activity in rats. Neuropsychopharmacology 19: 18–25. [DOI] [PubMed] [Google Scholar]
- 48. Mandillo S, Rinaldi A, Oliverio A, Mele A (2003) Repeated administration of phencyclidine, amphetamine and MK-801 selectively impairs spatial learning in mice: a possible model of psychotomimetic drug-induced cognitive deficits. Behav Pharmacol 14: 533–544. [DOI] [PubMed] [Google Scholar]
- 49. Zakzanis KK (1998) Neuropsychological correlates of positive vs. negative schizophrenic symptomatology. Schizophr Res 29: 227–233. [DOI] [PubMed] [Google Scholar]
- 50. Hashimoto K, Fujita Y, Shimizu E, Iyo M (2005) Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of clozapine, but not haloperidol. Eur J Pharmacol 519: 114–117. [DOI] [PubMed] [Google Scholar]
- 51. Hashimoto K, Fujita Y, Ishima T, Chaki S, Iyo M (2008) Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of glycine transporter-1 inhibitor NFPS and D-serine. Eur Neuropsychopharmacol 18: 414–421. [DOI] [PubMed] [Google Scholar]
- 52. Braff DL, Geyer MA (1990) Sensorimotor gating and schizophrenia. Human and animal model studies. Arch Gen Psychiatry 47: 181–188. [DOI] [PubMed] [Google Scholar]
- 53. Lieberman JA, Perkins D, Belger A, Chakos M, Jarskog F, et al. (2001) The early stages of schizophrenia: speculations on pathogenesis, pathophysiology, and therapeutic approaches. Biol Psychiatry 50: 884–897. [DOI] [PubMed] [Google Scholar]
- 54. Lencz T, Smith CW, Auther A, Correll CU, Cornblatt B (2004) Nonspecific and attenuated negative symptoms in patients at clinical high-risk for schizophrenia. Schizophr Res 68: 37–48. [DOI] [PubMed] [Google Scholar]
- 55. Carrión RE, Goldberg TE, McLaughlin D, Auther AM, Correll CU, et al. (2011) Impact of neurocognition on social and role functioning in individuals at clinical high risk for psychosis. Am J Psychiatry 168: 806–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McGorry PD, Nelson B, Amminger GP, Bechdolf A, Francey SM, et al. (2009) Intervention in individuals at ultra-high risk for psychosis: a review and future directions. J Clin Psychiatry 70: 1206–1212. [DOI] [PubMed] [Google Scholar]
- 57. Fusar-Poli P, Deste G, Smieskova R, Barlati S, Yung AR, et al. (2012) Cognitive functioning in prodromal psychosis: a meta-analysis. Arch Gen Psychiatry 69: 562–571. [DOI] [PubMed] [Google Scholar]
- 58. Sabbag R, Levin R, Edelman S, Heresco-Levy U (2011) Preventive pharmacological treatment – An evolving new concept in schizophrenia. Isr J Psychiatry Relat Sci 48: 82–90. [PubMed] [Google Scholar]
- 59.Woods SW, Walsh BC, Hawkins KA, Miller TJ, Saksa JR, et al. (in press) Glycine treatment of the risk syndrome for psychosis: Report of two pilots studies. Eur Neuropsychopharmacol 2012 Oct 19. doi:pii: S0924–977X(12)00272–6. 10.1016/j.euroneuro.2012.09.008. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 60. Kantrowitz JT, Malhotra AK, Cornblatt B, Silipo G, Balla A, et al. (2010) High dose of D-serine in the treatment of schizophrenia. Schizophr Res 121: 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hashimoto K (2009) Emerging role of glutamate in the pathophysiology of major depressive disorder. Brain Res Rev 61: 105–123. [DOI] [PubMed] [Google Scholar]
- 62. Maekawa T, Ohnishi T, Hashimoto K, Watanabe A, Iwayama Y, et al. (2010) Analyses of mechanism for mouse strain-dependent prepulse inhibition point to a role of Shmt1 (SHMT1) in mice and schizophrenia. J Neurochem 115: 1374–1385. [DOI] [PubMed] [Google Scholar]
- 63.Hashimoto K, Malchow B, Falkai P, Schmitt P (in press) Glutamate modulators as potential therapeutic drugs in schizophrenia and affective disorders. Eur Arch Psychiatry Clin Neurosci 2013 Mar 1. [Epub ahead of print]. [DOI] [PubMed]
- 64. Sucher NJ, Lipton SA (1991) Redox modulatory site of the NMDA receptor-channel complex: Regulation by oxidized glutathione. J Neurosci Res 30: 582–591. [DOI] [PubMed] [Google Scholar]
- 65. Kohr G, Eckardt S, Luddens H, Monyer H, Seeburg PH (1994) NMDA receptor channels: Subunit-specific potentiation by reducing agents. Neuron 12: 1031–1040. [DOI] [PubMed] [Google Scholar]
- 66. Varga V, Jenei Z, Janaky R, Saransaari P, Oja SS (1997) Glutathione is an endogenous ligand of rat brain N-methyl-D-aspartate (NMDA) and 2-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors. Neurochem Res 22: 1165–1171. [DOI] [PubMed] [Google Scholar]
- 67. Matsuzawa D, Obata T, Shirayama Y, Nonaka H, Kanazawa Y, et al. (2008) Negative correlation between brain glutathione levels and negative symptoms in schizophrenia: a 3T 1H-MRS study. PLoS ONE 3: e1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hashimoto K (2008) Correspondence: Regarding “N-Acetyl cysteine as a glutathione precursor for schizophrenia – a double-blind, randomized, placebo-controlled trial”. Biol Psychiatry 64: e1. [DOI] [PubMed] [Google Scholar]
- 69. Matsuzawa D, Hashimoto K (2011) Magnetic resonance spectroscopy study of antioxidant defense system in schizophrenia. Antioxi Redox Sig15: 2057–2065. [DOI] [PubMed] [Google Scholar]
- 70. Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M (2009) Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol 19: 220–230. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of neonatal Met-Phen treatment on levels of amino acids in the brain at juvenile.
(XLSX)
Effect of neonatal Met-Phen treatment on levels of amino acids in the brain at adult.
(XLSX)