

Abstract
Objective
The death rate of mature hepatocytes is chronically increased in various liver diseases, triggering responses that prevent liver atrophy, but often cause fibrosis. Mice with targeted disruption of Inhibitor kappa beta kinase (Ikk) in hepatocytes (HEP) provide a model to investigate this process because inhibiting Ikk-NFκB signaling in hepatocytes increases their apoptosis.
Methods
Cell proliferation, apoptosis, progenitors, fibrosis, and production of Hedgehog (Hh) ligands (progenitor and myofibroblast growth factors) were compared in ΔHEP and control mice before and after feeding methionine choline-deficient ethionine-supplemented (MCDE) diets. Ikkβ was deleted from primary hepatocytes to determine effects on Hh ligand production; Hh signaling was inhibited directly in progenitors to determine effects on viability. Liver sections from patients were examined to assess relationships between hepatocyte production of Hh ligands, accumulation of myofibroblastic cells, and liver fibrosis.
Results
Disrupting the Ikk-NFkB pathway in hepatocytes inhibited their proliferation but induced their production of Hh ligands. The latter provided viability signals for progenitors and myofibroblasts, enhancing accumulation of these cell types, and causing fibrogenesis. Findings in the mouse models were recapitulated in diseased human livers.
Conclusion
Dying mature hepatocytes produce Hh ligands which promote the compensatory outgrowth of progenitors and myofibroblasts. These results help to explain why diseases that chronically increase hepatocyte death promote cirrhosis.
Keywords: Hedgehog, Progenitors, Ikkβ, nonalcoholic steatohepatitis (NASH)
Introduction
Repetitive exposure to various insults chronically increases the death rate of mature hepatocytes and triggers reparative responses to reconstitute damaged hepatic epithelium. Chronic liver injury is often accompanied by progressive liver fibrosis [1]. However, it is not fully understood why situations that cause chronic, but relatively incremental, increases in the rate of mature hepatocyte death typically result in some degree of hepatic architectural distortion and tend to promote liver fibrosis, when such outcomes rarely complicate recovery from acute injuries that abruptly cause massive hepatocyte loss [2].
In order to investigate regenerative mechanisms associated with chronically increased rates of hepatocyte death, we took advantage of mice with targeted disruption of the kinase that promotes nuclear accumulation of nuclear factor κB (NFκB), a transcription factor that controls expression of genes that encode hepatocyte viability factors. NFκB is held in the cytoplasm in its inactive form by a specific inhibitor, inhibitor kB (IκB). The IκB kinase (Ikk) complex phosphorylates IκB, allowing NFκB to translocate into the nucleus [3]. Ikk is comprised of 3 subunits, Ikkα, Ikkβ and Ikkγ. Hepatocyte specific-deletion of Ikkβ inhibits nuclear localization of NFkB, reduces expression of hepatocyte viability factors, and increases hepatocyte susceptibility to apoptosis [4, 5, 6].
The present study evaluates the hypothesis that hepatocytes that are prone to apoptosis have an impaired proliferative capacity, but elaborate paracrine signals that trigger compensatory growth of progenitors which ultimately regenerate the hepatic parenchyma. Further, we theorize that this process also promotes hepatic fibrogenesis. Hedgehog ligands activate a morphogenic signaling pathway that orchestrates tissue construction during development and adult wound healing. Because Hedgehog pathway activity is low in healthy livers but high in various types of diseased livers, and Hedgehog signaling promotes the growth of liver progenitors and myofibroblasts [7, 8], our aims were to determine if, and how, disrupting NFkB-survival signals in hepatocytes activates the Hh pathway in other liver cell types that are involved in fibrogenic repair of damaged livers. The results confirm our hypothesis and demonstrate an unsuspected mechanism that couples hepatocyte death to regenerative strategies that result in fibrogenic repair of liver damage.
Material and Methods
Experimental Animals
IkkβF/F and IkkβΔHep mice were generous gifts from Dr. Michael Karin (University of California San Diego). IkkβF/F mice were bred with Alb-Cre mice to generate mice in which all Albumin-expressing cells (e.g., hepatocytes) and their progeny are Ikkβ-deficient (referred to as IkkβΔHep), All mice were housed in a facility with a 12-h light/dark cycle and allowed free access to food and water. Adult mice were used between 5 and 8 months of age. To induce oxidative liver injury, inhibit replication of mature hepatocytes, and activate HSC and liver progenitor populations, IkkβF/F (n=7) and IkkβΔHep mice (n = 9) were fed methionine/choline-deficient diet supplemented with 0.15% ethionine (MCDE). Surviving mice were sacrificed after being fed MCDE diets for 1 week (IkkF/F mice n = 7; Ikk ΔHep n=5). Chow-fed IkkβF/F (n=6) and IkkβΔHep mice (n = 6) were also sacrificed at the same time point.
Animal care and surgical procedures were approved by the Duke University Medical Center Institutional Animal Care and Use Committee as set forth in the “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health.
Human Subjects
Anomymized liver biopsy-proven non-alcoholic steatohepatitis (NASH) (n=5)and NASH-related cirrhosis (n=6) were obtained from the Duke University, Division of Gastroenterology and Department of Pathology. The control liver tissues were obtained from the Duke University School of Medicine Tissue Bank Shared Resource and studied in accordance with NIH and Institutional guidelines for human subject research.
Liver Histology and Immunohistochemistry
Specimens fixed in formalin and embedded in paraffin were cut into 4μm sections, dewaxed, hydrated, subsequently incubated for 10 min in 3 % hydrogen peroxide to block endogenous peroxidase. Antigen retrieval was performed by heating in 10mM sodium citrate buffer (pH 6.0) for 10 min or incubation with 0.25% pepsin for 10 min. Sections were blocked in Dako protein block (X9090;Dako) for 30 min and incubated with primary antibodies, active capase 3 (9661, 1: 250; Cell Signaling), Ki-67(NCL-Ki67, novocastra, 1:1000; ), AE1/AE3 (18-0132,1:500; invitrogen), A6 (a gift from Dr. Valenia Factor, National Institutes of Health, Bethesda, MD), NFkBp65 (ab7970 1;5000: Abcam), Gli2(26056 1:4500 Dako), and Shh (1843-1, 1:7500;Epitomics) at 4°C overnight. Other sections also incubated at 4°C overnight in nonimmune sera. Polymer-HRP anti-rabbit (K4003; Dako), anti-mouse (K4001;Dako) or MACH3 mouse AP polymer kit (MP530, Biocare medical) was used as secondary antibody. DAB reagent was employed in the detection procedure. Omitting primary antibodies from the reactions eliminated staining, demonstrating staining specificity.
Sirius red staining, AE1/AE3 and A6 immunohistochemical staining were assessed by morphometry (MetaView software, Universal Imaging Corp, Downington, PA). To quantify Sirius red staining, ten randomly chosen 20X fields/section were evaluated for each mouse. The quantification of AE1/AE3 and A6 staining was evaluated by choosing randomly eight portal tract (PT) excluding the major bile duct, at 40X fields/section for each mouse. After excluding the major bile duct in each portal tract (PT) from consideration, cells staining positively for Ki67, p65 (cytosol or nuclear) or Gli2 (a Hh-regulated transcription factor) in 8 PT/slide were counted at 40X magnification. PT selected for analysis contained a portal vein that ranged from 120–180μm. The average number of Ki67 or Gli2-positive ductular cells was obtained by dividing the total number of positive cells by the total number of portal tracts. Ki-67, p65 or Gli2-positive hepatocytes were quantified by counting total number of Ki-67, p-65 or Gli(+) hepatocytes/field and dividing by the total number of hepatocytes/field.
RNA and protein analysis
Total RNA and protein were extracted from freeze-clamped liver samples that had been stored at −80°C. A detailed protocol and antibodies used are listed in Supplemental Materials and Methods. The sequences of primers for mouse are summarized in Supplemental table 1.
Hepatic hydroxyproline assay
Hydroxyproline content in whole liver specimens was quantified colorimetrically as described in Supplemental Materials and Methods.
Isolation and Study of Primary Liver Cell from Mice
Primary hepatocytes were obtained as described [6, 9]. To isolate primary liver progenitors, we performed a two-step collagenase perfusion according to the protocol of Selgen et al [10], and collected the nonparenchymal cell (NPC) fraction that contained progenitors using gradient centrifugation. Isolated cells were incubated with anti-Sca (stem cell antigen)-1 FITC-conjugated antibody (130-092-529 MACs, Miltenyi Biotec.), followed by anti-FITC microbeads. After incubation, cells were positively selected using MACS sort [11, 12]. As determined by Trypan-blue exclusion, cell viability was > 90% in all experiments.
To evaluate apoptotic activity, hepatocytes and progenitors were cultured for 24 hours and then cytocentrifuged to slides and examined for annexin V (ab14196, Abcam). For double immunofluorescent staining, cytospinned cells were fixed, permeabilized, and processed for immunostaining with primary antibody annexin V and albumin (MAB1455; 1:100; R&D Systems). Alexa Fluor 568 and Alexa Fluor 488 (Molecular Probes) were used as secondary antibodies. DAPI counterstaining was employed to demonstrate nuclei. Hepatocytes were also labeled with anti-mouse CD26 (559653 BD Bioscience) and Annexin V (559933 BD Bioscience) or matched isotype control antisera and analyzed using the FACS VantageSe (Beckton Dickinson).
In separate experiments other freshly isolated progenitors were seeded in 96-well tissue culture plates (2000cells/well) and grown in IMDM supplemented with 10% FBS. After 72 hours, the medium was replaced with 1% FBS IMDM; the next morning cells were treated with either cyclopamine (3μM, Toronto Research Chemicals, Inc) or tomatidine (3μM, Calbiochem) for 24 hours. Control wells were treated with an equal volume of vehicle (DMAO) for the same treatment period. At the end of the treatment period, cell numbers were determined by Cell Counting Kit-8 (CCK-8, Dojindo, Maryland). A calibration curve was prepared using wells containing a fixed numbers of viable cells. A FLUOstar OPTIMA micro-plate reader (BMG Labtech, Durham, NC) was used for absorbance measurements.
Primary hepatocytes isolated from WT and IkkβF/F mice (n = 5/group) were cultured in the presence of adenoviral vectors carrying green fluorescent protein (AdGFP) or Cre recombinanse (AdCre) (MOI 25). Cultures were analyzed at 24 and 48 hours.
Statistical Analysis
Results are expressed as mean ± SEM. Significance was established using the student’s t-test and analysis of variance when appropriate. Differences were considered significant when p < 0.05.
Results
IkkβΔHep mice have more apoptotic hepatocytes and increased liver progenitors
Although livers of IkkβΔHep mice appeared grossly normal at baseline (Supple Fig. 1), they had increased caspase 3 activity by immunohistochemistry (IHC) (Fig. 1A,B) and western blot analysis (Fig. 1C). IHC of primary hepatocytes that were double-stained for annexin V, a marker of cells undergoing apoptosis, and albumin, a marker for hepatocytes, confirmed that many albumin-positive hepatocytes from IkkβΔHep mice were annexin V-positive, whereas albumin-positive hepatocytes from IkkβF/F mice were generally negative for annexin V (Fig. 1D). FACS analysis of primary hepatocytes which were double-stained for annexin V and CD26, another marker for mature hepatocytes, verified that more hepatocytes from IkkβΔHep mice were positive for annexin V (Fig. 1E),. Hence, hepatocyte- specific deletion of Ikkβ increased baseline hepatocyte apoptotic activity.
Figure 1. Increased hepatocyte apoptosis in IkkβΔHep mice.
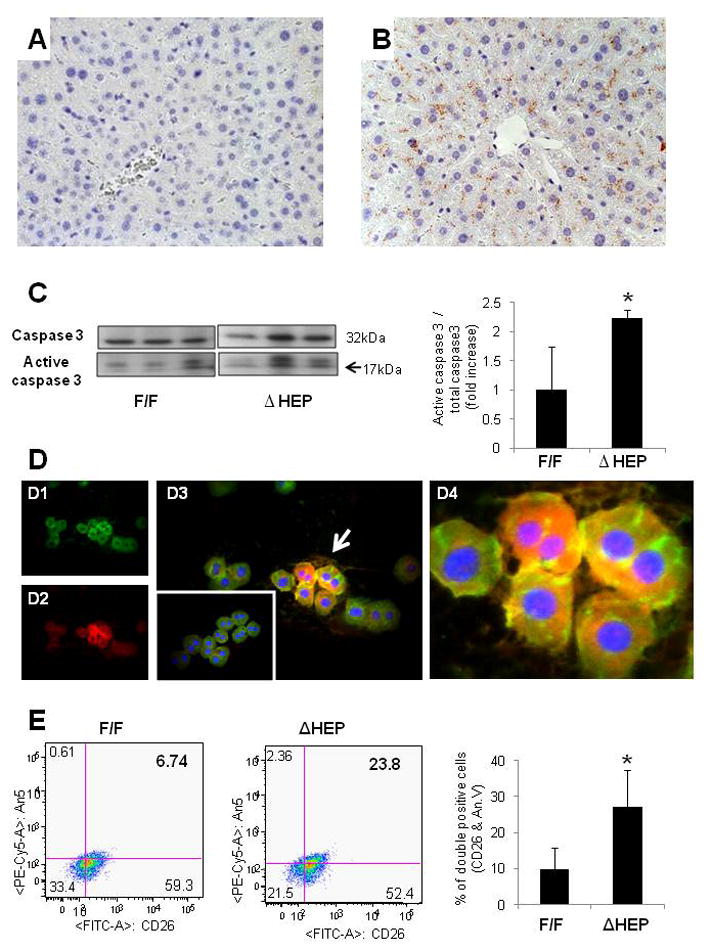
(A–B) Liver sections stained to demonstrate activated caspase 3 from representative IkkβF/F (A), and IkkβΔHep (B) mice (X40). (C) Western blot analysis for caspase 3 in whole liver extracts from IkkβF/F and IkkβΔHep mice (n = 3 mice/group). Densitometric data are displayed as mean ± SD (*P < 0.05 vs IkkβF/F group). (D1–4) Double immunoflorescent staining for albumin (green) and annexin V (red) in representative primary hepatocyte cytospins from IkkβF/F and IkkβΔHep mice: Albumin (green, D1), Annexin V (red, D2) and merged image of Albumin and Annexin V (yellow, D3) in hepatocytes from IkkβΔHep mice. Arrows indicate the albumin and annexin V- double positive (yellow) hepatocytes which are magnified in d4. The inserted image in D3 shows the merged images of representative Albumin/Annexin V-stained hepatocytes from IkkβF/F mice (X40). (E) Representative FACS analysis of primary hepatocytes from IkkbF/F and Ikkβ ΔHep mice. Cells were incubated with antibodies to CD26 and Annexin V. (F) Graphic summary of FACS data (n = 4 mice/group). Results are expressed as mean ± SD (*P < 0.05 vs IkkβF/F group).
To determine if this led to expansion of hepatocyte progenitor populations, livers from IkkβΔHep mice and IkkβF/F controls were stained for AE1/AE3 and A6, two different markers of liver progenitors. Compared to livers of IkkβF/F mice, livers of IkkβΔHep mice contained more AE1/AE3-positive cells, and more A6-positive cells at baseline (Fig. 2A–C). Hence, hepatocyte-specfic deletion of Ikkβ expanded hepatic progenitor populations in seemingly-healthy IkkβΔHep mice.
Figure 2. Increased hepatic progenitor populations in IkkβΔHep mice before and after hepatic damage.
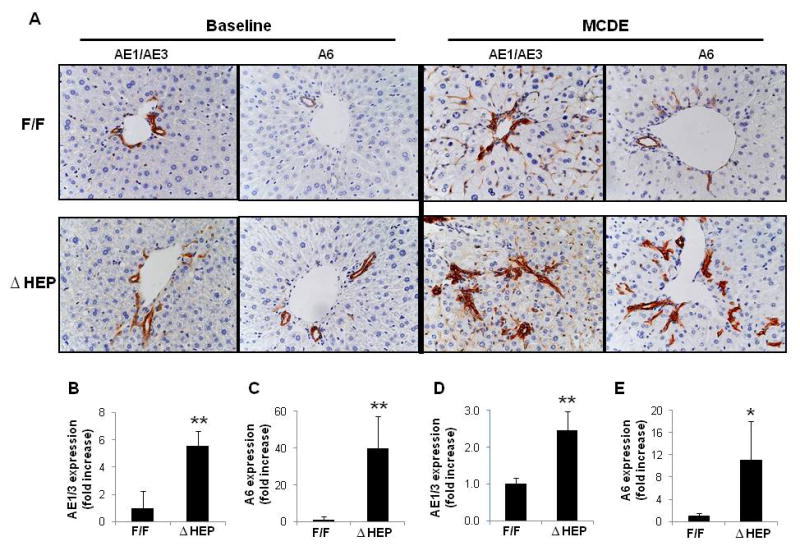
(A) Immunohistochemical staining for progenitor markers, AE1/AE3 and A6 in liver sections from representative IkkβF/F and IkkβΔHep mice before (baseline) and after MCDE diets for 1week (X40). Quantitative AE1/AE3 (B, D) or A6 (C, E) immunohistochemistry data from IkkβF/F and IkkβΔHep mice before (B; AE1/AE3, C: A6) and after injury (D; AE1/AE3, E: A6). AE1/AE3 or A6-positive cells were measured by morphometry in 8 portal triads (PT)/section. Mean ± SD are graphed (*P<0.05, **P<0.005 vs IkkβF/F group).
Decreased nuclear localization of NFkBp65 and Ki67 expression in IkkβΔHep hepatocytes after liver injury
To investigate the effects of superimposed liver injury on IkkβF/F and IkkβΔHep mice, we fed mice with methionine choline-deficient, ethionine-supplemented (MCDE) diets for 1week. This is known to induce liver injury and compensatory proliferation of liver progenitors in normal rodents [13]. Only 50 % of IkkβΔHep mice survived during MCDE treatment. Survivors also demonstrated less hepatomegaly than IkkβF/F mice (LW/BW ratio 5.09±0.41% vs 7.22±1.02%, p<0.05).
Hepatic injury generally activates the Ikk complex. This allows cytosolic NFkB to translocate into the nuclear compartment, eventually exerting viability functions in hepatocytes. Because IkkβΔHep mice have been reported to exhibit impaired NFkB activation, we compared the nuclear localization of NFkB in IkkβF/F and IkkβΔHep mice after MCDE treatment. As assessed by IHC, IkkβF/F and IkkβΔHep mice exhibited dramatic differences in NFκB localization in response to liver injury. Strong nuclear localization of NFkB p65 was observed in MCDE-fed IkkβF/F mice, whereas relatively few hepatocyte nuclei contained NFκB p65 in MCDE-fed IkkβΔHep mice (Fig. 3A,B). NFκBp65 was localized predominately in the cytosol of both hepatocytic and ductular cells in IkkβΔHep mice (Fig. 3A,B and Supplemental Fig 2). Therefore, after liver injury IkkβΔHep mice had more NFκB p65 in hepatocyte cytosol and less NFκB p65 in hepatocyte nuclei than IkkβF/F mice.
Figure 3. Reduced nuclear localization of NFκBp65 and decreased hepatoctye Ki67 expression in IkkβΔHep mice after hepatic injury.
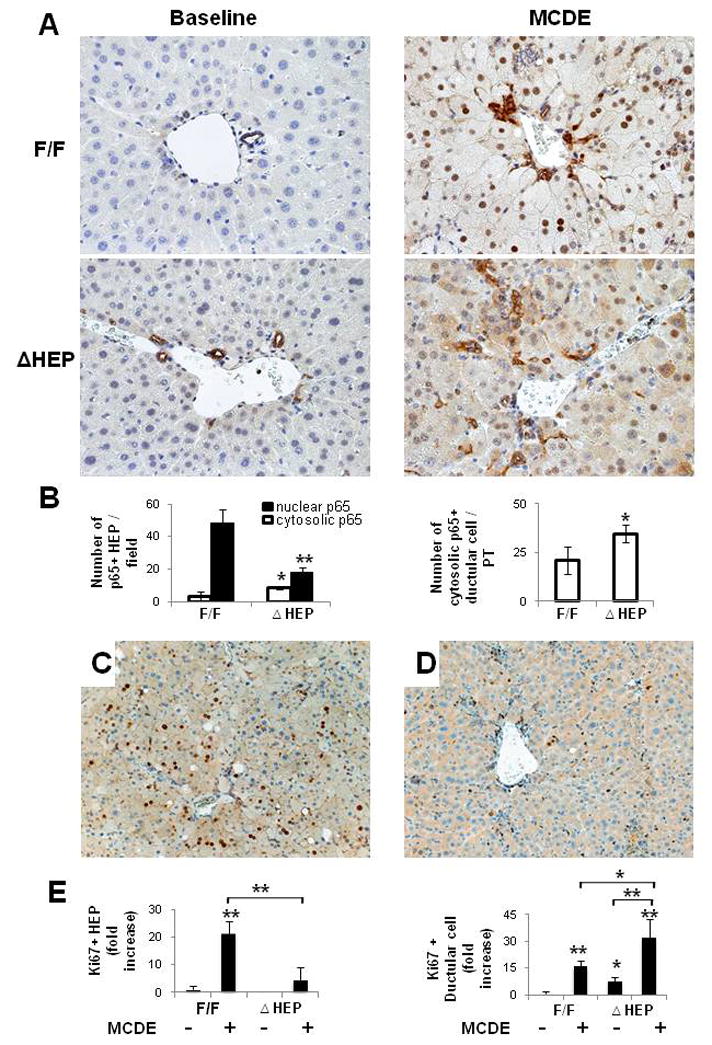
(A) Immunohistochemical staining for NFκB p65 in liver from representative IkkβF/F and IkkβΔHep mice before (baseline) and after MCDE diets for 1week (X40). (B) Quantitative NFkB p65 immunohistochemistry data from MCDE-fed mice (n = 4 mice/group). The numbers of hepatocytic cells (HEP) or ductular cells with nuclear or cytosolic NFκB p65(+) were counted in 8 portal triads (PT)/section. Results are expressed as numbers of NFκB p65(+) cells per PT and graphed as mean ± SD results (*P<0.05, **P<0.005 vs IkkβF/F MCDE-fed group). (C–D) Immunohistochemical staining with Ki67 (cell proliferation marker) in liver sections from representative IkkβF/F (D) and IkkβΔHep (E) mice after MCDE diet-induced liver injury. (E) Quantitative Ki67 immunohistochemistry data from all mice (n = 4 mice/group). The numbers of Ki67 (+) hepatocytic cells (HEP) and ductular cells were counted in 8 PT/section. Ki67 (+) HEP were quantified by counting total number of Ki 67(+) HEP/field and dividing by the total number of hepatocyes/field. Ki67(+) ductular cells were quantified by dividing the total number of positive cells by the total number of PT. Mean ± SD results are graphed (*P<0.05, **P<0.005 vs IkkβF/F chow-fed control group).
To determine if these changes in NFκB localization were associated with differences in hepatocyte proliferative responses, liver sections from MCDE-fed IkkβF/F mice and IkkβΔHep mice were stained for Ki 67, a marker of S phase. The injured livers of IkkβF/F mice contained 20 fold more Ki 67-positive hepatocytic cells than those of IkkβΔHep mice (Fig. 3C,E). Interestingly, although IkkβΔHep mice had reduced hepatocyte replication, these mice demonstrated greater proliferation of ductular cells than IkkβF/F mice, both at baseline and after liver injury (Fig. 3D,E). Because the ductular cell population is enriched with liver progenitors [14], these findings suggested that hepatocyte-specific deletion of Ikkβ promoted proliferation of liver progenitor populations.
Increased progenitors in IkkβΔHep mice during liver damage
To investigate this possibility further, livers from MCDE diet-fed IkkβF/F mice and IkkβΔHep mice were stained for the progenitor markers, AE1/3 and A6. Liver injury induced greater accumulation of AE1/AE3 (+) cells in IkkβΔHep mice than in IkkβF/F mice (Fig. 2A,D). Similarly, numbers of A6 (+) progenitors were also greater in IkkβΔHep mice than in IkkβF/F mice (Fig. 2A,E). Therefore, hepatocyte-specific deletion of Ikkβ promoted compensatory expansion of progenitor populations after liver injury.
Increased hedgehog signaling in IkkβΔHep mice promotes progenitor viability
Evidence that mature hepatocytes from IkkβΔHep mice were prone to apoptosis, but that progenitors accumulated in IkkβΔHep livers, suggested that hepatic progenitors might have survival mechanisms that were distinct from those that operate in mature hepatocytes. Since activation of the Hedgehog (Hh) pathway is known to regulate the viability of many types of progenitors, including fetal liver progenitors and ductular-type progenitors in adult livers [8], we next evaluated Hh pathway activity in IkkβΔHep mice.
First we clarified which types of liver cells were capable of Hh signaling by isolating progenitor populations and mature hepatocytes from healthy IkkβΔHep mice and IkkβF/F controls. In both cell types, we compared expression of the mutant Ikkβ allele, albumin, and cre recombinase, and correlated those results with expression of Gli2 protein, a marker of Hh signaling. Results in progenitors from IkkβF/F mice were compared to findings in progenitors from IkkβΔHep mice. Although progenitor populations from IkkβF/F mice and IkkβΔHep mice all expressed albumin mRNA, only the IkkβΔHep progenitors expressed cre recombinase and the mutant Ikkβ allele. Similarly, expression of Cre and the mutant Ikkβ allele were noted only in mature hepatocytes from IkkβΔHep mice (Fig. 4A).
Figure 4. Cell type-specific differences in survival factor signaling in progenitors and mature hepatocytes from IkkβΔHep mice, as well as IkkβΔHep livers.
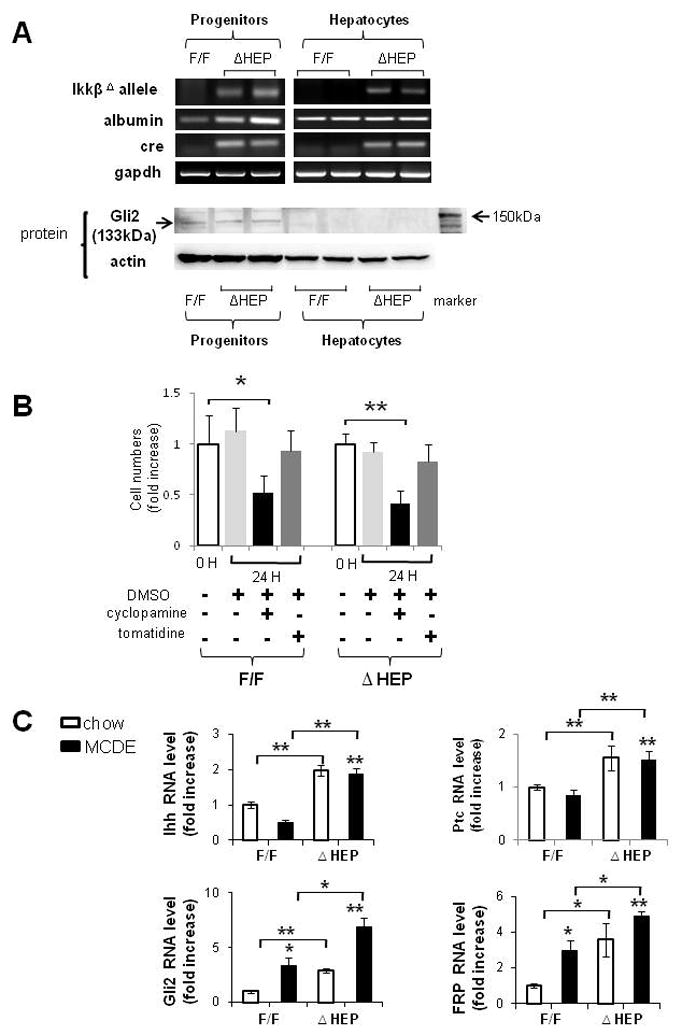
(A) Primary hepatocytes and liver progenitors were harvested from IkkβF/F mice and IkkβΔHep mice (n = 15 mice/group). Due to the relatively low numbers of progenitors in adult livers, progenitor cells were pooled from 2–3 mice/group for RNA and protein extraction. QRT-PCR analysis of RNA expression of the mutant Ikkβ allele, albumin, cre recombinase and gapdh in representative mice. Western blot analysis of Gli2 (133kDa) and β actin expression in the same mice. (B)Primary progenitors from both groups were then cultured with cyclopamine (3μM) or tomatidine (3μM) for 24 h. Control wells were treated with an equal volume of vehicle (DMSO) for the same treatment period. At the end of the treatment period, growth (cell number) was assessed using the CCK-8 assay. Each experiment was replicated three times. Mean ± SD results are graphed (*P<0.05, **P<0.005 vs respective 0 hour control group). (C) QRT-PCR analysis of liver RNA from chow-fed IkkβF/F and IkkβΔHep mice (open bars), and MCDE diet-fed IkkβF/F and IkkβΔHep mice (closed bars) for Indian hedgehog (Ihh), Patched (Ptc), Gli2 and Frizzled related peptide (Frp)1 (n = 4 mice/group/treatment). Mean ± SD results are graphed (*P<0.05, **P<0.005 vs IkkβF/F chow-fed control group).
Despite these engineered-differences in Ikkβ expression, immunohistochemistry demonstrated that NFkB p65 remained mostly cytosolic in ductular-type progenitors during liver injury in both IkkβF/F mice and IkkβΔHep mice (Supple Fig 2). This finding suggested that liver progenitors may not require nuclear localization of NFkBp65 to survive liver injury. In contrast, progenitor populations from both IkkβF/F mice and IkkβΔHep mice expressed Gli2 proteins, whereas mature hepatocytes from neither strain expressed Gli2 (Fig. 4A). Thus, the hepatic progenitor populations were capable of activating Hh signaling and retained this capability even when their NFκB pathway was disrupted.
To more directly investigate the role of Hh signaling in maintaining progenitor viability during liver injury, we isolated progenitor populations from MCDE-fed IkkβF/F mice and IkkβΔHep, placed them in culture, and treated the cultures with vehicle, cyclopamine, a specific pharmacologic inhibitor of the Hh pathway[15], or tomatidine, an inactive cyclopamine analog. Effects on cell numbers were quantified. Neither vehicle, nor tomatidine, altered the number of IkkbF/F or IkkβΔHep progenitors. In contrast, cyclopamine significantly reduced progenitor numbers in both groups, decreasing cell counts by more than 50% within 24 hours (Fig. 4B). Hence, Hh signaling permitted the survival of liver progenitor populations during chronic liver injury, including the IkkβΔHep progenitors that had a defective NFκB pathway.
Increased expression of Hh ligands and target genes in livers of IkkβΔHep mice
Because progenitor populations were increased in the livers of IkkβΔHep mice, both at baseline and during superimposed injury, we next asked if the Hh pathway was generally activated in mice with disruption of Ikk-NFkB signaling in hepatocytes. Compared to IkkβF/F mice, IkkβΔHep mice had greater mRNA expression of the Hh ligand, Indian Hh (Ihh), and several Hh target genes, including the Hh receptor, Patched (Ptc), Gli-2, a Hh-regulated transcription factor, and frizzled related peptide (Frp)-1, a Hh-target gene and soluble inhibitor of canonical Wnt signaling, both before and after liver injury (Fig. 4C).
Disrupting the Ikk-NFkB pathway in hepatocytes triggers their production of Hh ligands
Since differences in Hh pathway activation between IkkβΔHep mice and IkkβF/F mice accompany targeted deletion of Ikkβ in mature hepatocytes, we isolated primary hepatocytes from IkkβF/F mice and wild type (WT) mice and treated the cells with adenoviral vectors carrying either Cre recombinase (AdCre) or green fluorescent protein (AdGFP) to determine if acutely deleting Ikkβ function influenced hepatocyte production of Hh ligands (Fig. 5). Incubation with AdGFP efficiently transduced virtually all of the hepatocytes and did not reduce cell viability or effect Hh ligand expression. Similarly, treating WT hepatocytes with AdCre had no effect on either of these parameters. In contrast, AdCre treatment significantly reduced viability and increased mRNA expression of Ihh ligand in hepatocytes from IkkβF/F mice (Fig. 5A,B). Western blot analysis of protein lysates from IkkβF/F hepatocytes that were treated with AdGFP or AdCre demonstrated that acute AdCre-mediated disruption of Ikkβ led to caspase 3 activation and increased cellular production of both Shh and Ihh proteins (Fig. 5C,D). However, AdCre-treated hepatocytes did not exhibit increased expression of Hh target genes, such as Ptc or Gli2 (data not shown). Therefore, interrupting Ikkβ function in isolated hepatocytes reduced their viability and induced them to produce Hh ligands that function as viability factors for liver progenitors.
Figure 5. Deleting Ikkb in hepatocytes promotes production of Hh ligands.
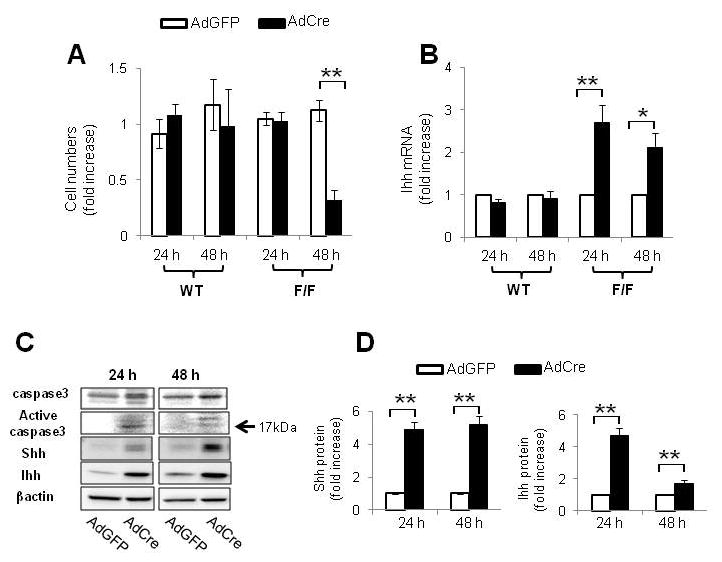
(A) Primary hepatocytes were isolated from WT and IkkβF/F mice (n = 5/group) and cultured in the presence of adenoviral vectors carrying green fluorescent protein (AdGFP) or Cre recombinanse (AdCre). Cultures were analyzed at 24 and 48 hours. In each experiment, quintuplicate wells were assayed. Each experiment was replicated three times. (A) Cell numbers were measured by CCK8 assay and (B) Indian hedgehog ligand (Ihh) expression was evaluated by QRT PCR analysis. Mean ± SD results are graphed (*P<0.05, **P<0.005 vs AdGFP-treated cells in Ikkβ F/F group). (C) Representative Western blot analysis of caspase 3, Shh and Ihh ligands in primary hepatocytes isolated from IkkβF/F mice and treated with either AdGFP or AdCre for 24 or 48 h. (D) Cumulative densitometric analysis of Shh and Ihh Western Blots Results are displayed as mean ± SD (**P<0.005 vs AdGFP-treated cells in Ikkβ F/F group ).
Increased production of Hh ligands causes hepatic accumulation of Hh-responsive cells
Subsequent experiments used IHC for the Hh-regulated transcription factor, Gli2, to identify which types of liver cells were Hh-responsive in intact animals. Although few Gli2(+) cells were evident in the uninjured livers of either IkkβF/F mice or IkkβΔHep mice, quantitative analysis demonstrated that IkkβΔHep had more Gli(+)-hepatocytic cells and Gli(+)-ductular cells than IkkβF/F mice at baseline. Numbers of Gli2(+) cells increased in both groups after MCDE diet treatment, but the greatest numbers of these Hh-responsive cells accumulated in IkkβΔHep livers (Fig. 6A–C). After only one week of diet-induced liver injury, the net numbers of Hh-responsive cells were about three-fold greater in IkkβΔHep mice than in IkkβF/F mice.
Figure 6. Increased accumulation of Hh-responsive cells in Ikk ΔHep mice after liver injury.
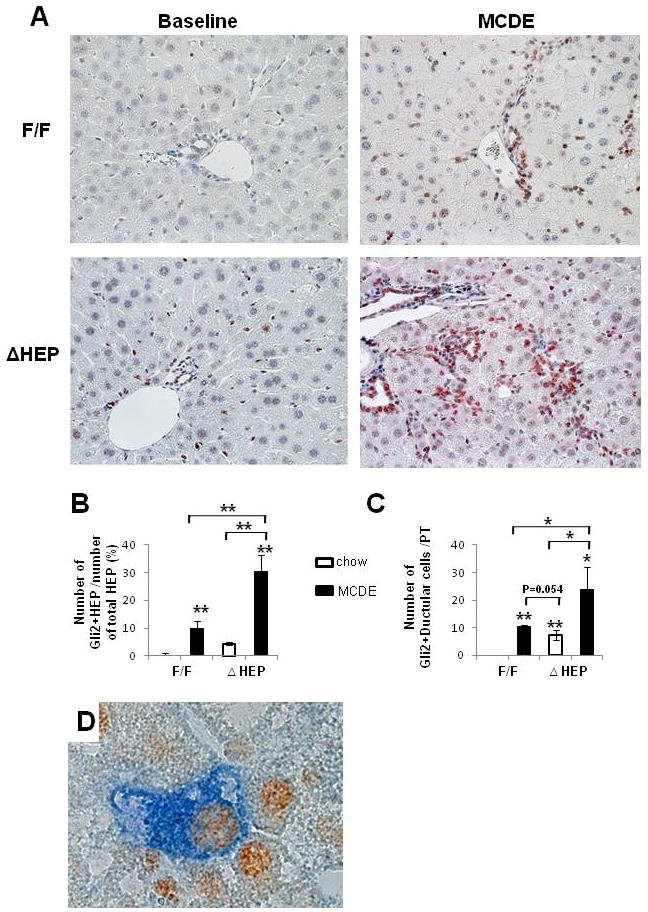
(A) lmmunohistochemical staining for Gli2 in representative IkkβF/F and IkkβΔHep mice before (baseline) and after MCDE diets for 1week (X40). (B–C) Quantitative Gli2 immunohistochemistry data from all mice (n = 4 mice/group). Gli2 (+) hepatocytic cells (HEP) or ductular cells were counted in 8 (X40 magnification) fields that contained portal triads (PT). (B) Gli2 (+) hepatocytic cells were expressed as % of Gli2 (+) hepatocyte nuclei/total HEP. (C) Gli2 (+) ductular cells were quantified by dividing the total number of positive cells by the total number of PT. Mean ± SD results are graphed (*P<0.05, **P<0.005 vs IkkβF/F chow-fed control group). (D) Double immunohistochemical staining with Gli2 (brown) and Pan CK (blue) in Ikkβ ΔHep after MCDE treatment (original magnification X63).
Evidence that neither mature hepatocytes from IkkβΔHep mice nor hepatocytes from IkkβΔHep mice expressed Gli2 at baseline, and that targeted disruption of Ikkβ function in cultured hepatocytes failed to activate expression of Hh-regulated genes, suggested that the Gli2-positive hepatocytic cells in injured livers might have been derived from Hh-responsive progenitors that survived liver injury and then partially differentiated to replace dead hepatocytes. This concept was evaluated by double immunostaining liver sections for Gli2 and pancytokeratin, a marker of immature hepatocytes [16]. Co-localization of Gli2 and pancytokeratin was demonstrated in hepatocytic cells, particularly peri-portally (Fig. 6D), supporting the possibility that Gli2-expressing hepatocytic cells in IkkβΔHep mice were immediate progeny of Gli2(+) progenitors that survived liver injury.
Enhanced fibrogenesis and liver fibrosis in IkkβΔHep mice, and increased hepatocyte expression of Shh in patients with liver fibrosis
Hepatocytes in healthy livers lack a basement membrane and hence, are immediately adjacent to hepatic stellate cells (HSC) that reside in the space of Disse. Cells that produce Hh ligands release these factors in membranous particles, and this process increases during apoptosis [17]. Thus, HSC are proximal targets of Hh ligands that are released from hepatocytes in injured livers. This may promote fibrogenesis because Hh ligands activate quiescent HSC to become myofibroblastic (MF) [18] and promote the viability and proliferation of MF-HSC [19]. To assess this issue, we performed Western blot analysis to compare expression of α–SMA, a marker of MF-HSC, in IkkβF/F mice and IkkβΔHep mice. Levels of α-SMA protein were significantly greater in the IkkβΔHep group, both before and after MCDE diet treatment (Fig. 7A). Sirius red staining demonstrated that IkkβΔHep mice also had more peri-cellular and sinusoidal deposition of collagen fibrils than IkkβF/F mice at both time points (Fig. 7B). Biochemical determination of hepatic hydroxyproline content, a quantitative measure of liver fibrosis, confirmed that IkkβΔHep mice had significantly more liver fibrosis than IkkβF/F mice (Fig. 7C). Therefore, activation of the Hh pathway promoted accumulation of myofibroblastic cells and increased fibrogenesis in mice with chronically increased hepatocyte apoptosis.
Figure 7. Enhanced fibrogenesis and liver fibrosis in IkkβΔHep mice.
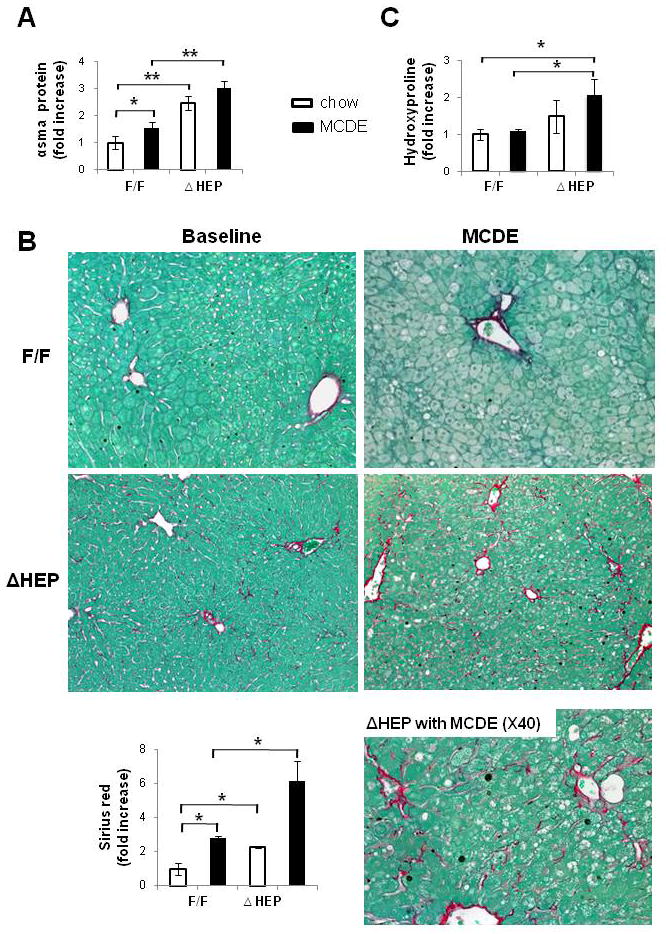
(A)α-smooth muscle actin (α-sma) expression was assessed in whole liver proteins from IkkβF/F and IkkβΔHep mice (n = 3/group/treatment) using Western blot analysis. (B) Sirius red staining in liver sections from representative mice (X20 or X40). Graph demonstrates mean ± SD morphometric data from all mice (n = 4/group/greatment). (C) Hepatic hydroxyproline content in all mice (n = 4 mice/group). All results are displayed as mean ± SD (*P<0.05, **P<0.005 vs IkkβF/F chow-fed control group).
To determine if a similar process might occur in humans, we performed immunostaining for Sonic hedgehog (Shh) and α–SMA on liver biopsy samples from 12 patients with early (stage F2) or advanced (stage F4) fibrosis due to chronic nonalcoholic steatohepatitis (NASH). In parallel, similar staining was done on healthy control liver tissue. NASH livers were selected for scrutiny because levels of hepatic apoptotic activity correlate with the severity of liver fibrosis in this disease [20].
No Shh (+) hepatocytes were identified in healthy control livers (data not shown). In NASH livers, hepatocytes that stained for Shh were variably detected, with greater numbers of more strongly-stained hepatocytes occurring in livers with more fibrosis (Fig. 8A–C, Supplemental Fig 3A–B). Fibrotic NASH livers typically demonstrated foci of hepatocytes that were surrounded by stromal cells that expressed the myofibroblast marker, alpha smooth muscle actin (α-sma). Double-immunostaining for Shh (brown) and α-sma (blue) confirmed that α-sma(+) myofbroblastic cells tended to localize near regions of Shh-expressing hepatocytes within the hepatic parenchyma (Fig. 8D–E). Thus, in NASH, a type of human liver disease in which there is increased hepatocyte apoptosis, increased hepatocyte expression of Hh ligands occurs, and this is accompanied by evidence of HSC activation and increased fibrogenesis.
Figure 8. Increased expression of Hh ligand by hepatocytes in NASH patients with fibrosis.
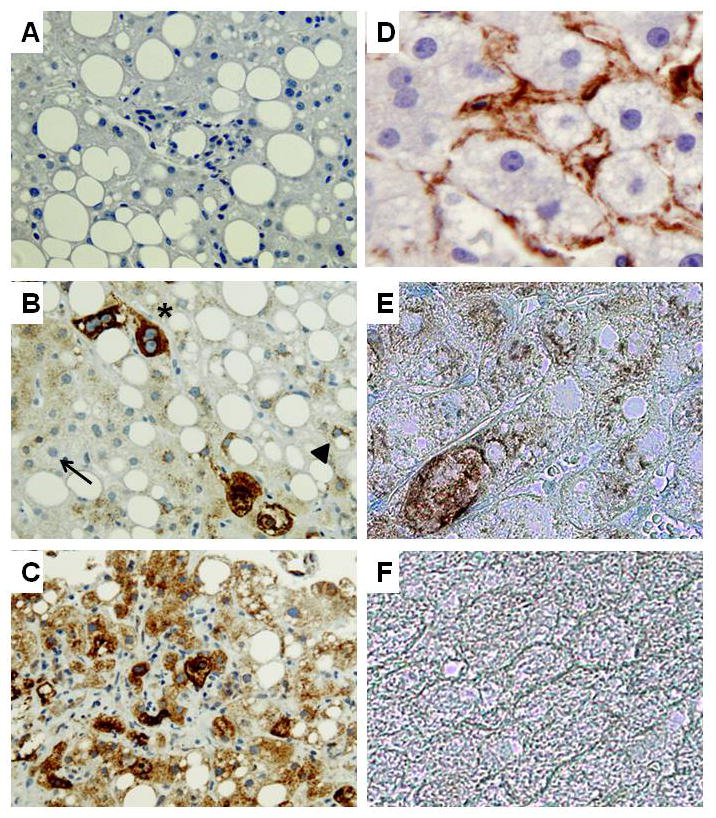
Immunohistochemistry was used to assess Shh and αSMA expression in healthy control livers (n = 3) and in livers from patients with NASH with early (F2) fibrosis (n = 6) or advanced (F4) fibrosis (n = 6). Representative photomicrographs are shown. A) Negative control for Shh immunostaining in which the primary anti-Shh antibody was eliminated and section was exposed to secondary antibody only. B) NASH liver with early fibrosis shows hepatocytes that express no Shh (thin arrow), low levels of Shh (arrow head), or higher levels of Shh (*) C) NASH liver with advanced fibrosis shows many hepatocytes that are strongly stained for Shh. D) Single immunostaining for a-SMA (brown) in NASH liver with early fibrosis. E) Double staining for a-SMA (blue) and Shh (brown) in NASH liver with early fibrosis. F) Double staining for a-SMA and Shh in healthy control liver (A–C:X40, D–F: X100).
Discussion
Our results in IkkβΔHep mice, a murine model of increased hepatocyte death, demonstrate that optimal viability and proliferative capacities of mature hepatocytes are heavily dependent upon an intact NFkB pathway, whereas Hh signaling in liver progenitors provides an alternative survival mechanism that permits them to survive and proliferate even when NFkB nuclear localization is impaired. Moreover, we discovered that when the Ikkβ-NFkB pathway is disrupted in mature hepatocytes, they produce Hh ligands. This enriches the hepatic microenvironment with factors that enhance the growth of Ikkβ-deficient progenitor cells. As these progenitors mature, they eventually lose the ability to transduce Hh-initiated signals, and become more reliant on NFkB-regulated viability factors. Thus, their vulnerability to apoptosis increases because Ikkβ-NFkB signaling has been disrupted, and this perpetuates the increased rate of mature hepatocyte apoptosis. The latter maintains heightened production of Hh ligands and stimulates the continued outgrowth of Hh-responsive nonparenchymal cells that rely on Hh signaling to survive, including myofibroblasts. Progressive hepatic accumulation of such cells promotes fibrogenic repair, helping to explain why liver fibrosis is increased in IkkβΔHep mice.
Increased hepatocyte production of Hh ligands and accumulation of Hh target cells (i.e., myofibroblastic cells) were also demonstrated in patients with NASH. Increased rates of hepatocyte apoptosis are known to occur in this common type of chronic human liver disease. Moreover, hepatic apoptotic activity predicts the severity of liver fibrosis in NASH, and is significantly greater in patients with advanced fibrosis than in patients with early fibrosis [20]. MF-HSC play major roles in excessive matrix accumulation during many types of chronic liver disease, including NASH. Earlier work showed that phagocytosis of apoptotic bodies activates Q-HSC to become MF-HSC [21]. Therefore, the early stages of fibrosis in NASH are characterized by deposition of fibrous matrix around hepatocytes and along sinusoids [22].
The new evidence that injured hepatocytes in NASH produce Shh ligands provides another mechanism that helps to explain the pattern of liver fibrosis that occurs in NASH because HSC are capable of responding to Hh ligands. In healthy livers, Q-HSC produce relatively large amounts of (Hh interacting protein) Hip, a potent Hh ligand antagonist. However, HSC expression of Hip is abolished by liver injury (or culture), permitting Hh-dependent induction of myofibroblastic (MF) genes and enhancing the viability and proliferative activity of MF-HSC [18, 19]. MF-HSC themselves produce and release Hh ligands[19], further enriching the hepatic microenvironment with these factors.
Mature Hh ligands diffuse away from the cells that produce them, and variably modulate gene expression in distant Hh-responsive cells. Target cell effects depend upon the concentration and duration of Hh ligand exposure [23]. Membranous particles containing biologically-active Hh ligands have been purified from the blood and bile of experimental animals with liver injury [17], suggesting routes by which Hh ligands generated by apoptotic hepatocytes and MF-HSC might be disseminated both extra-hepatically and to other parts of the liver lobule. Hepatic progenitors that reside in the Canals of Hering and bile ductular cells in portal tracts are Hh-responsive, and Hh ligands released from MF-HSC have been shown to enhance the viability and proliferative activity of both cell types [24]. This promotes the ductular response that often accompanies chronic liver injury [24, 25]. Hh ligands also stimulate immature ductular cells to undergo epithelial-to-mesenchymal transition [26], a process that may furher contribute to liver fibrosis [26]. Indeed, QRT-PCR analysis of liver RNA from IkkβΔHep mice demonstrated increased expression of various EMT-related factors (Supple Fig 4). Hence, evidence that dying hepatocytes release morphogens that trigger hepatic accumulation of cell types that mediate fibrogenic repair suggests a unifying mechanism that helps to explain the strong association between chronic liver injury and fibrogenesis. This discovery identifies novel diagnostic and therapeutic targets for further research that is relevant to various types of chronic liver injury.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Summary Box.
What is already known about this subject
Disruption of the Ikkβ kinase, and other factors that inhibit nuclear localization of the pro-survival transcription factor NF-kB, promote the death of mature hepatocytes.
The death rate of hepatocytes is increased chronically in many types of chronic hepatitis, including NonAlcoholic Steatohepatitis (NASH)
Chronic efforts to regenerate dead hepatocytes are often associated with liver fibrosis.
Chronic regenerative responses also increase the risk of liver cancer.
What are the new findings
Dying hepatocytes generate Hedgehog (Hh) ligands, but are not Hh-responsive themselves.
Hh ligands are viability factors for liver progenitors, and thus, promote hepatic accumulation of cells that might contribute to cancer formation.
Hh ligands also promote hepatic accumulation of myofibroblasts, and thus, increase fibrogenesis..
How might it impact on clinical practice in the foreseeable future?
Noninvasive tests that detect/quantify Hh pathway activity might provide novel biomarkers of fibrogenesis and cancer risk in NASH.
Hh inhibitors might reduce development of cirrhosis and liver cancer by removing viability signals for myofibroblasts and immature liver cells.
Acknowledgments
We like to thank Dr. Michael Karin (Uni. of California, San Diego) for providing the IkkF/F and Ikk ΔHEP mice and helpful discussion.
Funding
This work was supported by NIH grant 5R01-DK077794 to AMD.
Abbreviations
- Hh
Hedgehog
- Ikk
Inhibitor kappa beta kinase
- NFkB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- Ihh
Indian hedgehog
- Shh
Sonic hedgehog
- Ptc
Patched
- Gli
Glioblastoma
- FRP
Frizzled-related peptide
- BMP
Bone morphogenic protein
- MMP
Metalloproteinase
- EMT
Epithelial-to- mesenchymal transition
Footnotes
Disclosures
All authors declare that they have no conflict of interests or financial interests
References
- 1.Henderson NC, Forbes SJ. Hepatic fibrogenesis: from within and outwith. Toxicology. 2008;254:130–5. doi: 10.1016/j.tox.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 2.Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60–6. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 3.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 4.Cosulich SC, James NH, Needham MR, et al. A dominant negative form of IKK2 prevents suppression of apoptosis by the peroxisome proliferator nafenopin. Carcinogenesis. 2000;21:1757–60. doi: 10.1093/carcin/21.9.1757. [DOI] [PubMed] [Google Scholar]
- 5.Li ZW, Chu W, Hu Y, et al. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–45. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwabe RF, Bennett BL, Manning AM, et al. Differential role of I kappa B kinase 1 and 2 in primary rat hepatocytes. Hepatology. 2001;33:81–90. doi: 10.1053/jhep.2001.20799. [DOI] [PubMed] [Google Scholar]
- 7.Sicklick JK, Li YX, Choi SS, et al. Role for hedgehog signaling in hepatic stellate cell activation and viability. Lab Invest. 2005;85:1368–80. doi: 10.1038/labinvest.3700349. [DOI] [PubMed] [Google Scholar]
- 8.Sicklick JK, Li YX, Melhem A, et al. Hedgehog signaling maintains resident hepatic progenitors throughout life. Am J Physiol Gastrointest Liver Physiol. 2006;290:G859–70. doi: 10.1152/ajpgi.00456.2005. [DOI] [PubMed] [Google Scholar]
- 9.Maeda S, Chang L, Li ZW, et al. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19:725–37. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 10.Seglen PO. Hepatocyte suspensions and cultures as tools in experimental carcinogenesis. J Toxicol Environ Health. 1979;5:551–60. doi: 10.1080/15287397909529766. [DOI] [PubMed] [Google Scholar]
- 11.Petersen BE, Grossbard B, Hatch H, et al. Mouse A6-positive hepatic oval cells also express several hematopoietic stem cell markers. Hepatology. 2003;37:632–40. doi: 10.1053/jhep.2003.50104. [DOI] [PubMed] [Google Scholar]
- 12.Deng J, Steindler DA, Laywell ED, et al. Neural trans-differentiation potential of hepatic oval cells in the neonatal mouse brain. Exp Neurol. 2003;182:373–82. doi: 10.1016/s0014-4886(03)00058-x. [DOI] [PubMed] [Google Scholar]
- 13.Hixson DC, Allison JP. Monoclonal antibodies recognizing oval cells induced in the liver of rats by N-2-fluorenylacetamide or ethionine in a choline-deficient diet. Cancer Res. 1985;45:3750–60. [PubMed] [Google Scholar]
- 14.Roskams T, Yang SQ, Koteish A, et al. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am J Pathol. 2003;163:1301–11. doi: 10.1016/S0002-9440(10)63489-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454–72. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 16.Omenetti A, Popov Y, Jung Y, et al. The hedgehog pathway regulates remodelling responses to biliary obstruction in rats. Gut. 2008;57:1275–82. doi: 10.1136/gut.2008.148619. [DOI] [PubMed] [Google Scholar]
- 17.Witek RP, Yang L, Liu R, et al. Liver cell-derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology. 2009;136:320–30 e2. doi: 10.1053/j.gastro.2008.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi SS, Omenetti A, Witek RP, et al. Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2009 doi: 10.1152/ajpgi.00292.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L, Wang Y, Mao H, et al. Sonic hedgehog is an autocrine viability factor for myofibroblastic hepatic stellate cells. J Hepatol. 2008;48:98–106. doi: 10.1016/j.jhep.2007.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feldstein AE, Wieckowska A, Lopez AR, et al. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology. 2009;50:1072–8. doi: 10.1002/hep.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Canbay A, Feldstein AE, Higuchi H, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. 2003;38:1188–98. doi: 10.1053/jhep.2003.50472. [DOI] [PubMed] [Google Scholar]
- 22.Brunt EM. Pathology of nonalcoholic steatohepatitis. Hepatol Res. 2005;33:68–71. doi: 10.1016/j.hepres.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Jeong J, McMahon AP. Cholesterol modification of Hedgehog family proteins. J Clin Invest. 2002;110:591–6. doi: 10.1172/JCI16506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Omenetti A, Yang L, Li YX, et al. Hedgehog-mediated mesenchymal-epithelial interactions modulate hepatic response to bile duct ligation. Lab Invest. 2007;87:499–514. doi: 10.1038/labinvest.3700537. [DOI] [PubMed] [Google Scholar]
- 25.Richardson MM, Jonsson JR, Powell EE, et al. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology. 2007;133:80–90. doi: 10.1053/j.gastro.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 26.Omenetti A, Porrello A, Jung Y, et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J Clin Invest. 2008;118:3331–42. doi: 10.1172/JCI35875. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.