

Abstract
A Cationic polymer nanobrush was synthesized, attached to a MALDI target and used for the fractionation of peptides and proteins based on their pI, prior to analysis by MALDI-MS. The cationic polymer nanobrush was synthesized on a gold substrate by AIBN photo-initiated polymerization, using a 30:70 ratio of 2-aminoethyl-methacrylate hydrochloride (AEMA) : N-isopropylacrylamide (NIPAAM). This brush showed selectivity for adsorption of acidic peptides and proteins, and allowed fractionation of simple two-component mixtures to be completed in less than 10 minutes. The brush-adsorbed biomolecules were recovered by treating the nanobrush with ammonium hydroxide, which effectively collapsed the brush thereby releasing the trapped compounds for MALDI MS analysis. These results demonstrate that nanobrush can serve as a convenient platform for rapid fractionation of biomolecules prior to analysis by MALDI-MS.
Keywords: photoinitiation, polymer brush, MALDI-MS, fractionation, polymerization
Introduction
The future of health care is quickly advancing towards the reliance on technology to improve patient care. In order to achieve this goal, it is important to provide fast, robust and efficient methods for the diagnosis and treatment of diseases. In recent years, comparative proteomics has emerged as a useful approach for the study of proteins and their involvement in various diseases.1 Matrix Assisted Laser Desorption Ionization (MALDI) Mass Spectroscopy (MS) is currently one of the two dominant MS-based tools used for analysis of mixtures of peptides and proteins as is typically encountered in the field of proteomics.1 MALDI MS provides exceptional sensitivity and extremely low limits of detection, especially for the analysis of individual or simple mixtures of compounds. However, in the analysis of complex peptide/protein mixtures, there is an apparent inherent decrease in MALDI-MS performance.2,3 It is observed that the MALDI mass spectra of these complex mixtures are biased towards lower molecular weight species at the expense of the higher MW components, and that ion suppression can lead to enhanced signals for basic components at the expense of acidic ones, even to the extent of complete suppression of the acidic species. Consequently, the complexity of a given peptide/protein mixture is typically reduced via one of a number of chromatographic or extraction procedures prior to MALDI-MS analysis.
Chromatographic sample complexity reduction approaches, including liquid chromatography, gel electrophoresis, capillary electrophoresis, etc., clearly offer the highest efficiencies for component separation. However, one common drawback of many of these approaches is that they can be quite time-consuming, often requiring many hours for the separation process to be completed. To address this drawback, a number of methods for rapid fractionation of complex protein mixtures have been developed. Furthermore, such fractionation approaches can be directly coupled to the MALDI target thereby reducing the number of sample manipulation steps, and consequently, reducing the opportunities for sample loss. These on-MALDI-target sample fractionation approaches typically involve direct modification of the target surface to incorporate a selective affinity capture motif, based on either bioselectivity or chemical selectivity.4,5,6 One limitation of these on-MALDI-target fractionation approaches, is the inherent low loading capacities of the modified target surfaces, which may be limited to as little as a monolayer of captured analyte, or even lower. In an effort to address this limitation, while retaining the advantages of the on-MALDI-target approach, our research has focused on the development of functional brush-polymer modified MALDI targets.2 The use of these brush-polymers is expected to increase the peptide/protein loading capacity of the on-MALDI-target affinity capture motif by offering a three-dimensioned volume for uptake of the targeted species.
Currently, polymer nanobrushes have been used to immobilize a variety of proteins, where the efficiency of separation is increased due to the specificity of the brush. Examples of such cases in the literature include: the use of either poly(oligoethylene glycol methacrylate) (POEGMA) or poly(hydroxyethyl methacrylate) (PHEMA) modified with nitrolotiacetic (NTA) to immobilize histidine-tagged proteins selectively and reversibly,7 the use of poly(N-isopropylacryl-amide) (PNIPAAM) and poly(2-vinylpyridine) (P2VP) to achieve temperature dependent switching adsorption behavior of serum albumin,8 poly(methoxyethyl acrylamide) chains were adsorbed on a resin bearing surfaces and used for protein size exclusion studies in Entropic Interaction Chromatography,9 and Bruening’s group used polymer-oxotitanium-modified gold wafer in order to produce a high surface area for enrichment of phosphopeptide from the tryptic digest.10
In this report we describe the use of a cationic copolymer brush of 2-aminoethyl-methacrylate hydrochloride (AEMA) and N-isopropylacrylamide (NIPAAM) synthesized by a grafting-from AIBN type free-radical initiator (Scheme 1) for MALDI target modification. The NIPAAM was integrated into the brush structure to control the hydrophobicity/hydrophilicity of the brush during the transition between swollen and collapsed states. It was shown that the competition between the intramolecular hydrogen bonds of the NIPAAM and the neutral AEMA, and the intermolecular hydrogen bonds of the NIPAAM and the charged AEMA with the aqueous solution dominates the hydrophobicity and hydrophilicity of the mixed polymer brush.11 Using this brush-polymer modified MALDI target, the capture and release of biomolecules is expected to be primarily driven by the pH-adjustable charges on the polymer brush surface and not by the temperature. Specifically, it is expected that at lower pH’s the positively charged polymer brush will adsorb negatively charged peptides/proteins and repel positively charged ones. The adsorbed species can then be released in a separate step by depositing a solution having a high pH, thereby deprotonating the amine functionality, which in turn causes the polymer brush to collapse; this phenomenon was described in a prior publication.2 Both the initially unbound species and the bound and subsequently released species can be analyzed by MALDI-MS of the appropriate solutions. The entire process is extremely simple, robust and fast with the fractionation process typically being completed in less than 10 minutes, and it is these features that make our system desirable. Another advantage is the capability of our brush to be recycled. Since the brush response is based on pH, once the trapped species are released and washed off, the pH manipulation can be reversed, providing a brush ready to be used again.
Scheme 1.
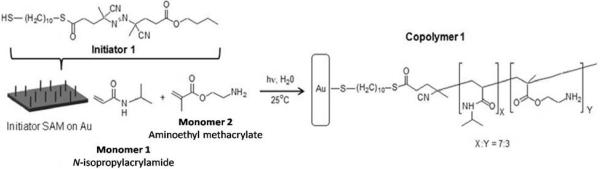
shows the synthesis of nanobrush. Initiator 1 was self-assembled on the gold substrate and the reaction was carried out with a 7:3 NIPAAM:AEMA ratio
Experimental
Materials
All supplies were purchased from Acros, Aldrich, or Fisher Scientific and used as supplied. The peptides tetra-aspartic acid (478.37g/mol, pI 2.8) and bradykinin (1059 g/mol, pI 12), and the small proteins insulin (5.7 kDa, pI 5.3) and lysozyme (14.3 kDa, pI 9.3) were purchased from Sigma Aldrich (St. Louis, MO).
Brush synthesis
Substrate Preparation
The synthesis of initiator 1 has been discussed in previous work.2 A 2 × 2.5 cm gold coated Si wafer was cleaned in Piranha solution for 60 minutes, after which it was rinsed with Milipore-filtered water, followed by tetrahydrofuran (THF) solution. The wafer was then blown dry with liquid nitrogen boil off and then placed in the ozone cleaner for 45 minutes. Next, the wafer was placed in a vial containing a 1 mM THF solution of initiator 1 for 18 hours. Once removed, the wafer was rinsed with THF three times and dried with nitrogen. A Nicolet 670 FTIR spectrometer with a nitrogen cooled MCT-B detector and PIKE grazing angle accessory at 80o grazing angle was used to collect IR spectra of the modified wafers. Self -assembly of a monolayer was confirmed by reflection absorption infrared (RAIR) spectra acquisition
Photopolymerization
AEMA (300 mg) and NIPAAM (700 mg) were dissolved in 5 mL of Millipore-filtered water. The polymer solution and the gold-SAM substrate were then placed in a Schlenk tube, which was degassed by three freeze-thaw cycles, and was then back filled with argon just before being irradiated at 350 nm (~1.6 mW/cm2). Polymerization was accomplished by irradiating the substrate in a Rayonet photochemical reactor (model RMR-600, Southern New England Ultraviolet Co., Branford, CT) for four hours. After irradiation, the wafer was removed from the Schlenk tube. The excess polymer was removed from the wafer surface by rinsing with Milipore-filtered water and the wafer was placed in a fresh Milipore-filtered water bath overnight. The substrate was then removed from the bath, rinsed with fresh Milipore-filtered water and blown dried with liquid nitrogen blow off. RAIR spectra for the brush were recorded, where the NIPAAM carbonyl band was at 1662 cm−1 and the amide band was at 1528 cm−1.12 The presence of AEMA was confirmed with the carbonyl band at 1721 cm−1.13
Fractionation studies
(t-Asp acid and bradykinin)
The polymer brush (30:70 AEMA/NIPAAM copolymer) was initially placed in 100 μL of milliQ water and allowed to stand for 1 hour. Next, a 1 μL aliquot of a peptide mixture (20 pmol tetra-aspartic acid:1 pmol bradykinin) in milliQ water was placed directly onto a conventional MALDI target (control sample) and onto the polymer-modified surface (see Figure 1). After 3 minutes, the residual solution on the polymer-modified surface was removed with a pipet and deposited onto a separate location on a conventional MALDI target. Subsequently, three 1 μL aliquots of milliQ water were deposited onto the peptide coated polymer-modified surface, removed immediately by pipet, and then combined with the originally removed droplet on the MALDI target. Next, 1 μL aliquots of 6 mg/mL α-cyano-4-hydroxycinnamic acid (CHCA) in 0.1% trifluoroacetic acid (TFA) were added to the control sample and to the polymer-fractionated combined droplets on the MALDI target and MALDI mass spectra were acquired from these samples. The peptides that remained bound to the polymer brush were analyzed by first adding 1μL of 10% ammonium hydroxide to the peptide coated polymer brush in order to deprotonate the polymer brush and release the bound peptides. After 3 minutes the 10% ammonium hydroxide droplet was pipetted off and deposited onto a conventional MALDI target. Three additional aliquots of the basic solution were applied to the brush polymer and combined on the conventional MALDI target. Subsequently, 1 μL of the CHCA matrix solution was added to the combined droplets and MALDI mass spectra were acquired. The peptides that remained bound to the polymer brush were also analyzed directly on the polymer brush. Direct analysis was accomplished by placing 1 μL of 6 mg/mL CHCA in 0.1% TFA solution directly on the peptide coated polymer brush. MALDI mass spectra were then acquired directly from the brush polymer surface.
Figure 1.
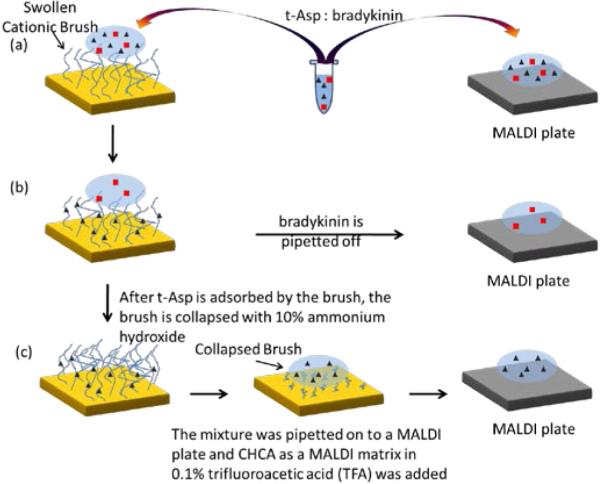
(a) A mixture of t-Asp and bradykinin is placed on swollen brush and a conventional MALDI plate. (b) After 3 min fractionation time, t-Asp is adsorbed by the brush and the eluent containing the unbound bradykinin is pipetted off onto a MALDI plate. (c) The brush is collapsed by treatment with 10% ammonium hydroxide, which expelled the t-Asp that is analyzed on a MALDI plate.
(insulin and lysozyme)
The polymer brush was placed in 100 μL of a 0.100 μM pH 7 ammonium acetate buffer solution and allowed to stand for 1 hour, after which the leftover solution was removed. Next, 1 μL aliquots of a protein mixture (3 pmol insulin: 32 pmol lysozyme in 40:60 acetonitrile: water) were placed directly onto a conventional MALDI target (control sample) and onto a gold coated substrate containing 30:70 AEMA/NIPAAM copolymer. Two different fractionation times of 1 minute and 3 minutes were used in these studies. After the specified time, the residual solution was removed with a pipet and deposited onto a separate location on a conventional MALDI target. Subsequently, three 1 μL aliquots of water were deposited onto the protein coated polymer surface, removed immediately and then combined with the originally removed droplet on the MALDI target. Next, 1 μL aliquots of 8 mg/mL CHCA in 0.1% TFA were added to the control sample and to the polymer-fractionated combined droplets on the MALDI target and MALDI mass spectra were acquired from these samples. To release the polymer bound protein 1 μL of 10% ammonium hydroxide was added to the protein coated polymer, allowed to stand for 3 minutes and then removed by pipet to a conventional MALDI target. Three additional 1 μL droplets of water were applied to the polymer and combined on the conventional MALDI target. Subsequently, 1 μL of the CHCA matrix solution was added to the combined droplets and MALDI mass spectra were acquired.
Mass Spectral Analysis
All mass spectra were acquired using a Bruker Microflex MALDI mass spectrometer having a nitrogen laser operating at 337nm. The laser intensity was kept at 25-35% of full intensity and 100 laser shots were averaged for the peptide spectra. The laser intensity was kept at 75%-80% of full intensity and 500 laser shots were averaged for the protein spectra. The linear mode was used for all mass spectra collection and spectra were calibrated using internal standards. The delay time was 150ns, and ions below mass/charge 400 were deflected.
Results and Discussion
Figure 2A shows the control MALDI mass spectrum of a 1:20 mixture of bradykinin (pI = 12, MW = 1059) and tetra-aspartic acid (t-Asp, pI = 2.8, MW = 478.37g/mol) on a conventional MALDI target. (Note: The series of peaks higher in mass than the protonated t-Asp parent ion at m/z 479 may be attributed to sodium and potassium attached adduct ions.) It is important to note that due to the low ionization efficiency of the t-Asp, a large excess was needed for the control experiment in order to obtain a detectable signal in the mass spectrum in the presence of bradykinin. The peptide mixture was added to a 30:70 AEMA: NIPAAM brush synthesized by the photopolymerization method, and the bound peptides and unbound peptides were separated and analyzed as described in the experimental section. Figure 2B shows the MALDI mass spectrum of the unbound peptides that were washed from the polymer brush surface, while Figure 2C shows the mass spectrum of the peptide that remained bound to the surface.
Figure 2.

(A) MALDI-MS spectra of a 1:20 Bradykinin:tetra-aspartic acid. (B) Shows the spectrum of the peptide that was bound to the surface. (C) Shows the spectrum of the unbound peptide washed off the surface.
Comparison of the MALDI mass spectra reveals a number of important insights. Figure 2A provides a clear example of the impact of the ion suppression effect in the analysis of peptide mixtures, where it is seen that the presence of the strongly basic bradykinin suppresses the ionization of the t-Asp. However, after fractionation, the t-Asp is easily detected in the mass spectrum once it is removed from the brush (Figure 2B). This process occurs by soaking the cationic brush in a basic pH solution to neutralize the amine, thus causing the brush to collapse and release the trapped peptides. The bradykinin on the other hand, remained in the unbound fraction (Figure 2C) as it was repelled by the brush due to like-charges repulsion effect. It is evident from this data that the AEMA:NIPAAM brush can be used to effectively fractionate the peptide mixture by selectively binding the t-Asp and repelling the bradykinin, and this fractionation can substantially overcome the ion suppression effect by isolating the acidic peptide for MALDI analysis. The fractionation process is very efficient and essentially only the bradykinin is detected in the solution initially removed from the polymer surface, while only t-Asp was detected in the bound peptide mass spectrum.
The success in fractionation of peptides led us to explore fractionation of small proteins such as insulin and lysozyme. In the control sample, there was an excess of insulin present; however, its ion signal was greatly suppressed by the presence of lysozyme (Figure 3A). This is another example of the ion suppression effect for proteins. After fractionation for three minutes, the MALDI mass spectrum of the solution containing the unbound protein was shown to contain mostly lysozyme with a very small amount of insulin (Figure 3B). The bound protein was released from the brush in a similar manner as the peptides, and was shown to be only the insulin (Figure 3C). In a second experiment it was found that this protocol can be repeated with the fractionation time reduced to 1 minute (Figures 3D and 3E) with the results being very similar to the three minutes fractionation time. It is worth noting that insulin was again found in the solution containing the unbound proteins, which suggests that the brush was either saturated, or the proteins were not allowed sufficient time to adsorb onto the brush. However, since the mass spectrum for the three minute fractionation was identical to the one minute fractionation, it seems that the brush saturation is the more likely explanation. It is also of interest to note the relative decrease in singly-charged to doubly-charged lysozyme ion signals in the fractionated mass spectra versus the control mass spectrum. This behavior is not entirely understood but may result from substantial reduction of the high concentration insulin species leading to a decrease in competition for matrix protons by the lysozyme molecules.
Figure 3.

(a) MALDI spectra of 2:5 Insulin:Lysozyme calibration protein mixture on a MALDI plate, (b) eluant of 3 minute fractionation study showing Lysozyme fragments, (c) released Insulin 3 minute fractionation study, (d) eluant of 1 minute fractionation study showing Lysozyme fragments, (e) released Insulin after 1 minute fractionation study.
The presence of insulin in the unbound protein droplet suggested that the brush polymer was becoming saturated at a relatively low protein concentration, which is inconsistent with expectations that the brush polymer would have a high capacity for protein uptake. To gain insight into this effect, the polymer brush grafting density was calculated using the following formula,
where σ is the grafting density in polymer chains/nm2, h is the thickness of the film, ρ is the density of the polymer, NA is Avogrado’s number and Mn is the molecular weight of the polymer.14 The grafting density for this polymer with a molecular weight of 1.7 million and an average film thickness of 10 nm is 0.004 chains/nm2, which is significantly less than the grafting density expected for a dense brush structure. This result suggests that our brushes are merely polymer chains spread over the surface of the gold, having more of a ‘mushroom conformation’, than a brush structure. However, since we were still able to obtain selective fractionation of the peptides and proteins, the charged functionality must still be present on the surface of the gold. This conclusion is further confirmed through the measurement of the surface zeta potential of the brush polymer where the isoelectric point is estimated at pH 8.0 (supporting information).15 This data confirms that the brush polymer is positively charged at pH values below 8, consistent with the conditions used for peptide/protein adsorption in these experiments.
Conclusions
In this study a cationic nanobrush was shown to be effective for the fractionation of simple two-component mixtures of peptides and small proteins prior to MALDI-MS analysis. The fractionation process can be accomplished directly on the brush polymer modified MALDI target, with high efficiency and in as little as a few minutes. Work is currently ongoing to demonstrate the efficiency of this technique to improve the protein coverage of digested proteins. Finally, and of particular significance, it is shown that incorporation of this fractionation step in the mixture analysis is particularly valuable to address the ion suppression effect in MALDI. Specifically, the ion signals obtained for acidic peptides/proteins are significantly enhanced after fractionation as a result of the removal of the more efficiently ionized basic peptides/proteins. However, the data obtained in these studies do suggest that the capacity of the brush polymers for peptide/protein adsorption is significantly below what is expected for a dense polymer surface modification. Calculation of the polymer grafting density offers a possible interpretation of this result and suggests that the surface is covered with widely distributed polymer chains rather than a dense polymer brush. Future work will focus on adaptation of synthetic procedures to increase the grafting density of the brush polymer on the MALDI target and on the synthesis of responsive polymer brushes on quartz surfaces for the fractionation of more complex mixture of biomolecules.
Supplementary Material
Acknowledgments
We would like to thank the National Science Foundation (CHE-0719426), and the National Institutes of Health (NIGMS-R15GM083325) for financial support. Thanks to Anton Paar (USA) for the surface zeta potential data.
Footnotes
Supporting Information: Information regarding the measurement of the surface zeta potential is given in the supporting info. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Weston AD, Hood L. Systems Biology, Proteomics, and the Future of Health Care: Toward Predictive, Preventative, and Personalized Medicine. J Proteome Res. 2004;3:179–196. doi: 10.1021/pr0499693. [DOI] [PubMed] [Google Scholar]
- 2.Wong VN, Fernando G, Wagner AR, Zhang J, Kinsel GR, Zaucher S, Dyer DJ. Separation of Peptides with Polyionic Nanosponges for MALDI-MS Analysis. Langmuir. 2009;25:1459–1465. doi: 10.1021/la802723r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hillenkamp F, Peter-Katalinic J. MALDI MS: A Practical Guide to Instrumentation, Methods and Applications. Wiley-VCH Verlag GmbH. 2007 [Google Scholar]
- 4 (a).Lian X, Lubman DM. On-Probe Immunoaffinity Extraction by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. Anal. Chem. 1998;70:498–503. doi: 10.1021/ac9708856. [DOI] [PubMed] [Google Scholar]; (b) Wang H, Tseng K, Lebrilla CB. A General Method for Producing Bioaffinity MALDI Probes. Anal. Chem. 1999;71:2014–2020. doi: 10.1021/ac981347b. [DOI] [PubMed] [Google Scholar]; (c) Bundy JL, Fenselau C. Lectin and Carbohydrate Affinity Capture Surfaces for Mass Spectrometric Analysis of Microorganisms. Anal. Chem. 2001;73:751–757. doi: 10.1021/ac0011639. [DOI] [PubMed] [Google Scholar]; (d) Blackedge JA, Alexander AJ. Polyethylene Membrane as a Sample Support for Direct Matrix-Assisted Laser Desorption/Ionization Mass Spectrometric Analysis of High Mass Proteins. Anal. Chem. 1995;67:843–848. doi: 10.1021/ac00101a009. [DOI] [PubMed] [Google Scholar]; (e) Worrall TA, Cotter RJ, Woods AS. Purification of Contaminated Peptides and Proteins on Synthetic Membrane Surfaces for Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. Anal. Chem. 1998;70:750–756. doi: 10.1021/ac970969e. [DOI] [PubMed] [Google Scholar]; (f) Bai J, Qian MG, Liang X, Lubman DM. Peptide Mapping by CNBr Degradation on a Nitrocellulose Membrane with Analysis by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. Anal. Chem. 1995;67:1705–1710. [Google Scholar]; (g) Berhane BT, Limbach PA. Functional Microfabricated Sample Targets for Matrix-Assisted Laser Desorption/Ionization Spectrometry Analysis of Ribonucleic Acids. Anal. Chem. 2003;75:1997–2003. doi: 10.1021/ac020710i. [DOI] [PubMed] [Google Scholar]
- 5 (a).Brockman AH, Orlando R. Probe-Immobilized Affinity Chromatography/Mass Spectrometry. Anal. Chem. 1995;67:4581–4585. doi: 10.1021/ac00120a024. [DOI] [PubMed] [Google Scholar]; (b) Brockman AH, Orlando R. New immobilization chemistry for probe affinity mass spectrometry. Anal. Chem. 1996;10:1688–1692. doi: 10.1002/(SICI)1097-0231(199610)10:13<1688::AID-RCM717>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; (c) Neuber H, Jacoby E, Bansal S, Iles R, Cowan D, Kicman AT. Enhanced Affinity Capture MALDI-TOF MS: Orientation of an Immunoglobulin G Using Recombinant Protein G. Anal. Chem. 2002;74:3677–3683. doi: 10.1021/ac025558z. [DOI] [PubMed] [Google Scholar]; (d) Warren ME, Brockman AH, Orlando R. On-Probe Solid-Phase Extraction/MALDI-MS Using Ion-Pairing Interactions for the Cleanup of Peptides and Proteins. Anal. Chem. 1998;70:3757–3761. doi: 10.1021/ac980210i. [DOI] [PubMed] [Google Scholar]; (e) Zhang L, Orlando R. Solid-Phase Extraction/MALDI-MS: Extended Ion-Pairing Surfaces for the On-Target Cleanup of Protein Samples. Anal. Chem. 1999;71:4753–4757. doi: 10.1021/ac990328e. [DOI] [PubMed] [Google Scholar]
- 6.Tang N, Tornatore P, Weinberger SR. Current developments in SELDI affinity technology. Mass Spectrometry Reviews. 2004;23:34–44. doi: 10.1002/mas.10066. [DOI] [PubMed] [Google Scholar]
- 7.Gautrot JE, Huck WTS, Welch M, Ramstedt M. Protein-Resistant NTA-Functionalized Polymer Brushes for Selective and Stable Immobilization of Histidine-Tagged Proteins. Appl. Mater. & Inter. 2010;2:193–202. doi: 10.1021/am9006484. [DOI] [PubMed] [Google Scholar]
- 8 (a).Burkert S, Bittrich E, Kuntzsch M, Muller M, Eichhorn K-J, Bellmann C, Uhlmann P, Stamm M. Protein Resistance of PNIPAAm Brushes: Application to Switchable Protein Adsorption. Langmuir. 2010;26:1786–1795. doi: 10.1021/la902505q. [DOI] [PubMed] [Google Scholar]; (b) Morisada S, Namazuda K-I, Suzuki S, Kikuchi N, Kanda H, Hirokawa Y, Nakano Y. Temperature-Swing Adsorption of Proteins in Water Using Cationic Copolymer-Grafted Silica Particles. Ind. Eng. Chem. Res. 2011;50:12358–12365. [Google Scholar]
- 9.Pang P, Koska J, Coad B, Brooks DE, Haynes CA. Entropic interaction chromatography: Separating proteins on the basis of size using end-grafted polymer brushes. Biotech. & Bioeng. 2005;90:1–13. doi: 10.1002/bit.20430. [DOI] [PubMed] [Google Scholar]
- 10.Wang W-H, Palumbo AM, Tan Y-J, Reid GE, Tepe JJ, Bruening ML. Identification of p65-Associated Phosphoproteins by Mass Spectrometry after On-Plate Phosphopeptide Enrichment Using Polymeroxotitanium Films. J. of Prot. Res. 2010;9:3005–3015. doi: 10.1021/pr901200m. [DOI] [PubMed] [Google Scholar]
- 11.Xia F, Feng L, Wang S, Sun T, Song W, Jiang W, Jiang L. Dualresponsive surfaces that switch between superhydrophilicity and superhydrophobicity. Adv. Mater. 2006;18:432–436. [Google Scholar]
- 12 (a).Maeda Y, Higuchi T, Ikeda I. Change in Hydration State during the Coil–Globule Transition of Aqueous Solutions of Poly(N-isopropylacrylamide) as Evidenced by FTIR Spectroscopy. Langmuir. 2000;16:7503–7509. [Google Scholar]; (b) Sun B, Lin Y, Wu P. Structure analysis of poly(N-isopropylacrylamide) sing near-infrared spectroscopy and generalized two-dimensional correlation infrared spectroscopy. Appl. Spectrosc. 2007;61:765–771. doi: 10.1366/000370207781393271. [DOI] [PubMed] [Google Scholar]
- 13.Yang Q, Xu Z-K, Hu M-X, Li J-J, Wu J. Novel Sequence for Generating Glycopolymer Tethered on a Membrane Surface. Langmuir. 2005;21:10717–10723. doi: 10.1021/la051797g. [DOI] [PubMed] [Google Scholar]
- 14.Brittain WJ, Minko S. A Structural Definition of Polymer Brushes. Journal of Polymer Science: Part A: Polymer Chemistry. 2007;45:3505–3512. [Google Scholar]
- 15.SurPASS Prospect Test Report. Anton Paar (USA): [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.