

Abstract
The nucleoprotein (NP) binds the viral RNA genome and associates with the polymerase in a ribonucleoprotein complex (RNP) required for transcription and replication of influenza A virus. NP has no cellular counterpart, and the NP sequence is highly conserved, which led to considering NP a hot target in the search for antivirals. We report here that monomeric nucleoprotein can be inhibited by a small molecule binding in its RNA binding groove, resulting in a novel antiviral against influenza A virus. We identified naproxen, an anti-inflammatory drug that targeted the nucleoprotein to inhibit NP-RNA association required for NP function, by virtual screening. Further docking and molecular dynamics (MD) simulations identified in the RNA groove two NP-naproxen complexes of similar levels of interaction energy. The predicted naproxen binding sites were tested using the Y148A, R152A, R355A, and R361A proteins carrying single-point mutations. Surface plasmon resonance, fluorescence, and other in vitro experiments supported the notion that naproxen binds at a site identified by MD simulations and showed that naproxen competed with RNA binding to wild-type (WT) NP and protected active monomers of the nucleoprotein against proteolytic cleavage. Naproxen protected Madin-Darby canine kidney (MDCK) cells against viral challenges with the H1N1 and H3N2 viral strains and was much more effective than other cyclooxygenase inhibitors in decreasing viral titers of MDCK cells. In a mouse model of intranasal infection, naproxen treatment decreased the viral titers in mice lungs. In conclusion, naproxen is a promising lead compound for novel antivirals against influenza A virus that targets the nucleoprotein in its RNA binding groove.
INTRODUCTION
The propensity of influenza A virus (IAV) to develop resistance to antivirals, as observed in 2009 with oseltamivir (Tamiflu), a neuraminidase inhibitor, calls for the search of new therapeutics. Because of the continuous change in the major viral antigens, vaccine must be renewed each year, and during influenza pandemics, antiviral can provide a first step of protection, at least during the time lapse required for vaccine production. The nucleoprotein (NP) is highly expressed during viral infection and has multiple functions. NP covers the eight single-stranded segments of the genomic RNA and assembles with the three polymerase subunits in a ribonucleoprotein complex (RNP) controlling viral transcription and replication (1). Recent studies unraveled the RNA-free trimeric NP structures of the H1N1 and H5N1 strains of influenza A virus (2–5). NP formed a trimer in the crystal that was stabilized by a swapping loop protruding from one monomer to its neighbor. The overall structure of the nucleoprotein of influenza B virus shared many similarities with its analog of influenza A virus, although NP was tetrameric in the former (6). The oligomerization of NP plays an important role in the maintenance of RNP structure required for function (4, 5, 7–10). Moreover, NP is a highly conserved protein (>90% amino acid sequence identity) among the different viral strains of influenza A virus and has no cellular counterpart. Therefore, the nucleoprotein constitutes a good candidate for inhibition by antiviral compounds (2, 11).
Nucleozin was discovered through high-throughput screening using the readout of a monoclonal antibody directed against NP; it inhibited NP-mediated nuclear transport and protected Madin-Darby canine kidney (MDCK) cells against a viral challenge (12–15). High-order inactive NP oligomers were generated by Gerritz and coworkers by a successful stabilization of the dimeric interface with small ligands possessing antiviral properties (12). The swapping loop, important for the stabilization of the trimeric interface, was considered to be a good target for the design of new NP-directed antivirals based on structural studies (4, 5). Indeed, the salt bridge between E339 and R416 from neighbor monomers required for NP trimerization was targeted by NP inhibitors identified by virtual screening (16). Other compounds targeting cellular proteins involved in the nuclear transport of the RNP or in signaling pathways exploited by influenza A virus also exhibited antiviral effects (17, 18). Novel mercapto triazole derivatives also displayed antiviral properties against influenza NP (19).
In the present work, we used a structure-based approach to identify novel antivirals directed against the nucleoprotein of influenza A virus. NP function requires proper binding of RNA to the protein and the formation of NP-RNA oligomers. Inhibiting these activities is expected to inhibit viral replication. Our strategy relies on the novel hypothesis that an NP inhibitor competing with the viral RNA genome for binding to the nucleoprotein should present antiviral properties. We used a structure-based modeling approach, based on the published X-ray structure of the RNA-free H1N1 NP (5), as a starting point to which unsolved residues in the X-ray structure forming loops were added (20). Using this structure, we defined a site within the RNA groove for the binding of potential NP inhibitors. The choice of this site partly relied on mutation studies that identified conserved residues in the putative RNA groove which are absolute requisite for viral replication (8, 9, 21). Further steps of energy minimization were performed before starting the in silico screening procedure using commercial docking software. The screening identified naproxen (22), a known inhibitor of inducible cyclooxygenase type 2 (COX-2), as a potential antiviral candidate. This paper presents the results of docking experiments, molecular dynamics (MD) simulations, and experimental tests showing that naproxen targets the nucleoprotein and competes with RNA binding to NP. Naproxen has antiviral properties against influenza A virus in MDCK cells and in vivo.
MATERIALS AND METHODS
Molecular modeling.
The virtual screening of small chemical compounds as potential inhibitors of NP-RNA interaction was performed with the computer program Ludi (Discovery Studio; Accelrys). The three-dimensional (3D) crystal structure of the NP monomeric form (Protein Data Bank identification no. [PDB ID] 2IQH [5]), to which we added hydrogen atoms, was used as the target throughout the virtual screening. The 100,335 commercially available compounds from the Sigma-Aldrich database (Sigma catalog 2008–2009) were filtered down by keeping compounds with low molecular mass (≤500 Da) either polar or with a net negative charge. The program OpenBabel was employed to generate 3D structures of the filtered-compound list (23). A sphere of 12-Å radius centered on Y148 was used as the research volume during the virtual screening. One of the best hits was naproxen. The NP-naproxen structure obtained after the virtual screening was further used as the initial structure for MD simulation. The MD simulations of the NP-WT in the presence of naproxen were carried out using the program NAMD (24) with the CHARMM27 force field (25). The system was solvated in TIP3P water molecules (26). The NP-naproxen solute was centered in a cubic cell, with an edge of 113 Å, of pre-equilibrated water. The system was made electrostatically neutral by adding chloride ions at points of minimal electrostatic energy. The system was set to an ionic strength of 0.15 M by adding sodium and chloride ions at random coordinates in solution for a total number of atoms of about 135,500. The van der Waals interactions were smoothly shifted to zero at between 10.0 Å and 12.0 Å. The list of the nonbonded interactions was truncated at 13.5 Å. The electrostatic interactions were calculated with no truncation, using the particle mesh Ewald summation algorithm (27). The lengths of the bonds containing hydrogen atoms were fixed with the SHAKE algorithm (28), and the equations of motion were iterated using a time step of 2 fs in the velocity Verlet integrator. The conjugate gradient energy minimization algorithm was used, while the solute atoms were harmonically restrainted to their initial positions using a force constant of 50.0 kcal/mol/Å2 during 5,000 minimization steps. The system was heated linearly to 300 K over 60 ps. MD simulation in an ensemble with a constant number of atoms at constant pressure and temperature (NPT) was further used to equilibrate the system and for production runs. During the initial 0.2 ns of the equilibration phase, the solute atoms were harmonically restrained with a force constant of 5.0 kcal/mol/Å2, which was gradually reduced to zero during the next 0.3 ns. Five trajectories of 10 ns each were produced for the NP-naproxen complex and analyzed using the package CHARMM (29). In order to extend the space explored by naproxen in the RNA binding site, docking of naproxen on NP was performed using an extended searching box (16 Å by 16 Å by 32 Å) that covered the rigid part of the RNA binding site, using the program AutoDock Vina (30). A total of 4,000 poses were obtained, keeping the best 10 poses of 400 independent docking experiments initiated with different random seed numbers.
Protein expression and purification of wt NP and the Y148A, R355A, and R361A mutants.
Protein expression experiments were performed as described previously (31) with modifications of the purification protocol by affinity chromatography using Ni magnetic beads (PureProteome; Millipore) and performed as recommended by the manufacturer, followed by extensive dialysis using 50 mM NaCl–20 mM Tris buffer at pH = 7.5. After purification, the protein concentration was determined using an extinction coefficient ε of 56,200 M−1 · cm−1 at 280 nm.
Chemicals and antibodies.
For oligonucleotides, biotinylated (5′ end) RNA fragments were purchased from Dharmacon (Thermo-Fisher, France) with high-performance liquid chromatography (HPLC) purification. The following sequences were used: rU25 and Flu1 (5′ UUU GUU ACA CAC ACA CAC GCU GUG 3′). The molecular beacon (Flu2) was purchased from MGW-Biotech (Germany) or Sigma (France) with a Cy3 fluorophore at the 5′ end and a Dabcyl quencher at the 3′ end (5′ AUA UAU AUC GAC AUA GAU AUA UAU 3′). Naproxen sodium (98% purity) was purchased from Sigma (N5160). NP primary monoclonal antibody was purchased from Santa Cruz Biotechnology.
Fluorescence.
The fluorescence measurements were performed on a Jasco fluorimeter equipped with a thermostated cell holder at 20°C. The Cy3-labeled oligonucleotides forming a molecular beacon were excited at 525 nm and the fluorescence detected between 535 and 650 nm. Corresponding excitation spectra were monitored at λem = 580 nm, and λexc varied between 440 and 570 nm. The samples for the fluorescence measurements and dynamic light scattering were prepared in 20 mM Tris buffer at pH = 7.4 with 50 mM NaCl.
SPR experiments.
The binding competition kinetics were performed on a Biacore 3000 apparatus using streptavidin-coated sensor chips (SA; Biacore) prepared as indicated by the manufacturer. Immobilization of the biotinylated oligonucleotide on the streptavidin-coated sensor chip was carried out using phosphate-buffered saline (PBS). The surface plasmon resonance (SPR) kinetic measurements were performed in 20 mM Tris-HCl buffer, containing 300 mM NaCl, at pH = 7.4 and 20°C. The oligonucleotides were denatured at 80°C and renatured slowly at room temperature for 1 h before experiments were performed. The NP protein or its mutants were injected at concentrations of 100 to 500 nM in the presence or absence of 50 nM and 20 μM naproxen (and up to 300 μM naproxen in some experiments). Measurements were conducted at 25°C, and samples were injected at an 25 μl/min flow rate. The direct association of naproxen to NP was performed on CM5 sensor chips with NP bound to the surface via amine coupling as indicated by the manufacturer. The R361A or Y148A proteins were attached to the surface of the same chip at similar resonance units (RUs), and a modified flow cell was used for blank subtraction. The activity of these surface-bound proteins was tested by injecting RNA oligonucleotides.
Cells and virus.
MDCK cells were obtained from the American Type Culture Collection. The influenza viruses A/PR/8/34 (H1N1) and A/Udorn/72 (H3N2) were grown in 8-day-old embryonated hens' eggs, and titers in MDCK cells were determined. A/WSN/33 (H1N1) was produced by reverse genetics: the 12-plasmid reverse genetics system was used as described previously (32) to generate a recombinant virus of the influenza A/WSN/33 strain. Briefly, 293T cells were transfected with eight plasmids encoding the individual vRNA segments of influenza A/WSN/33 (H1N1) virus from a truncated human polymerase I promoter and four expression plasmids encoding the corresponding subunits of the viral polymerase and the nucleoprotein. The generated wild-type (WT) virus was amplified on Madin-Darby canine kidney cells and titrated by plaque assays. MDCK cells were cultured in minimal essential medium (MEM; Sigma) containing 0.2% NaHCO3 (Sigma), MEM amino acids (Gibco), MEM vitamins (Gibco), penicillin-streptomycin-glutamine (PSG), and 5% newborn calf serum (ICN Biochem).
Cell viability assay by MTT assay.
A549 or MDCK cells (3.2 × 105 cells/well) were cultured in 12-well plates at 37°C for 1 day in MEM without fetal calf serum. Serial dilutions of naproxen (1 μM to 10 mM) were added to the cells that were further incubated at 37°C for 24, 48, and 72 h. The same culture conditions were used for the antiviral tests and toxicity measurements. MTT analysis was performed based on the standard method (33). The absorbance was measured at 550 nm, with subtraction of the blank value at 650 nm determined using a plate reader (PerkinElmer). The presence of naproxen in the supernatants did not affect the readings within experimental error. The data at 24 h were best fitted by a dose-response curve in the 1-to-500-μM range. Data at 48 and 72 h were concentration independent. The toxicity data (250 μM to 10 mM) were fitted with a dose-response curve (Origin; v8.5 software) to determine the 50% cytotoxic concentration (CC50). Levels of viability of MDCK cell cultures (3.2 × 105 cells/well) grown in 12-well tissue culture plates were determined by a trypan blue dye exclusion method. After a 24-h period of incubation, the procedure used for the cytotoxicity assay described above was followed. After 48 h, the supernatant was removed, cells were washed and trypsinized, and a trypan blue dye aqueous solution was added to the cell suspension. Viable and dead cells were counted under the phase-contrast microscope. CC50 values were estimated from graphic plots constructed from data of the number of viable cells compared to cell controls in the absence of naproxen.
Antiviral activity of naproxen in MDCK cells.
MDCK cells (0.32 × 106 cells/well) were cultured in 5% CO2 for 24 h to 80% confluence as described above but without fetal calf serum. MDCK cells were infected with A/WSN/33 virus at a multiplicity of infection (MOI) of 10−3 (multicycle growth assay), and naproxen (5 to 500 μM) or vehicle was added to the cells immediately after virus adsorption. Some experiments were also performed at a higher MOI (10−2 or 5) or by replacing Influenza/A/WSN/33 (H1N1) by A/Udorn/72 (H3N2). Viral titers of the clarified cell supernatants were determined by plaque assay using crystal violet readouts at different times as indicated. The experiments were carried out in triplicate and repeated at least twice for confirmation. The 50% effective concentration (EC50) was determined by fitting the data to a dose-response curve (Origin version 8.5). In addition, the same method used to evaluate cell viability as described above was followed to determine the EC50 by MTT tests. The viral RNA content in the cell supernatants purified using Qiagen Minikit (Qiamp) viral RNA was determined using a nanodrop instrument (Thermo-Fisher).
Localization of NP in A549 and MDCK cells infected by influenza A virus in the absence or presence of naproxen as detected by immunofluorescence.
Cells (20,000 cells/P6 well) were grown for 24 h to 80% confluence as detailed above prior to an A/WSN/33 virus challenge (MOI = 1) in the presence or absence of 50 μM naproxen. At the indicated time postinfection (p.i.), cells were treated with 4% paraformaldehyde for 30 min at room temperature and stained with 4′,6′diamidino-2-phenylindole (DAPI). NP locations were revealed using a mouse anti-influenza A virus NP monoclonal antibody and a secondary anti-mouse fluorescein isothiocyanate (FITC)-labeled antibody. The locations of NP and the nuclei were revealed by fluorescence microscopy analyses (Nikon).
Escape mutant production.
MDCK cells (8 × 105 cells/well) were seeded in a 6-well plate and incubated for 24 h. The cells were washed in Eagle minimum essential medium with Earle's balanced salt solution (EMEM)–penicillin-streptomycin (PS)–glutamine (Glu) and infected with A/WSN/33 obtained by reverse genetics with 8 × 103 PFU/well. After 1 h of virus absorption, the cells were washed twice and incubated for 2 days in 4 ml of EMEM-PS-Glu that contained 250 μM naproxen. The supernatant was collected, clarified, and denoted passage 1. The viral titers of the supernatants were determined by plaque assay. The titer obtained with naproxen-treated cells was found to be 2.5 × 10−2 relative to the viral titer obtained with untreated cells. The passage 1 virus was used at a dilution of 10−3 to infect a new cell monolayer, which was further incubated in the presence or absence of 250 μM naproxen. The third, fourth, fifth, and sixth passages were performed with 500 μM naproxen added and by a dilution of 10−3 of the supernatant of the previous passage. The selection was performed using a total of 6 passages with naproxen. The isolated viral particles from the supernatant from the sixth passage were seeded on a petri dish, and 12 clones were selected and amplified during 72 h. The RNAs of 6 of 12 clones were extracted, and the NP segment was amplified by reverse transcription-PCR (RT-PCR) and sequenced (34).
In vivo antiviral efficacy.
Five-week-old female BALB/c mice were housed in the confined facility of the rodent animal center of Jouy en Josas, France, for 1 week before the experiment. On the morning of day 0, mice were fully anesthetized by intraperitoneal (IP) injection of Imalgene 1000/ketamine (Merial, France) and Rompun/xylazine 158 (Alcyon, France) (0.1% ketamine plus 0.06% xylazine; 150 μl for a mouse weighing 20 g) and then infected by intranasal application of 50 μl of virus suspension (25 μl into each nostril) with 2,000 or 50 PFU/ml influenza A/PR8 (H1N1) virus by the intranasal route. They were subsequently treated in the afternoon of day 0 for the first time with naproxen at a dose of 0, 1, 3, 4, or 8 mg dissolved in 50 μl physiological serum (10 mice per group) by intraperitoneal injections or with 0.2 to 3 mg naproxen treatment by the intranasal route. The same treatment was administered the morning of each day postinfection during the experiment up to the day of sacrifice. Under the 2,000-PFU conditions, 94% ± 2% of the infected mice survived and were sacrificed after 7 days; under the 50-PFU conditions, all mice survived and were sacrificed after 14 days. Noninfected mice treated with naproxen or left untreated were used as controls for potential cytotoxic effects. The mouse weights were recorded daily. Mice were sacrificed at the end of the experiment, and their lungs were washed with PBS for collecting epithelial and blood cells (see the supplemental material). The 2,000-PFU dose, corresponding to a ca. 0.4 50% lethal dose (LD50), mimics conditions of human infection without mortality, while the 50-PFU dose mimics mild seasonal infection. Lungs were recovered for subsequent determinations of viral titers. The protocol was performed in agreement with recommendations of the Ethics Committee and French legislation. For statistical analysis, in vivo data are expressed as the median values ± variances of the results of three separate experiments and statistical analyses were performed using paired Student t tests (two tailed; statistical package of Origin software, version 8.5).
RESULTS
In silico screening and MD of the NP-naproxen complex.
The first step of the in silico screening defined the binding site of the inhibitor in the putative RNA binding groove of the nucleoprotein, guided by mutation studies (8, 9, 21). We defined this site as a sphere centered on Y148 with a diameter of 12 Å; Y148 is a residue with proposed interactions with the nucleobases of viral RNA (5). Such an aromatic side chain constitutes a binding platform for a putative NP inhibitor. Y148 is surrounded by the basic residues R361, R355, and R152. Mutation of any of these residues failed to rescue viral replication (21). Y148 is also located in proximity to R150, recently shown to be important for the binding of PB2 polymerase subunit (35). The program (Ludi, Discovery Studio; Accelrys) sorted from the chosen chemical library (100,337 compounds listed in the Sigma-Aldrich 2008–2009 catalog) all compounds that could fit in this selected site. Additional criteria, such as molecular mass, charge, and hydrophobicity, guided the screening and restricted the number of hits. We selected compounds with a molecular mass up to 500 Da that were either neutral or negatively charged and that contained an aromatic scaffold that could stack on Y148. One of the best hits was naproxen. The modeling procedure was further refined by testing the stability of the complex solvated with water molecules by MD. An overall view of the bound naproxen in the RNA binding groove of NP obtained at the end of 10 ns of dynamics analysis is shown in Fig. 1A. An electrostatic potential map projected on the solvent-accessible surface of the protein is presented here. Naproxen was found in a pocket located in the RNA binding groove and defined by two aromatic residues, Y148 and F489. Naproxen made a strong electrostatic interaction with R361 via its carboxylic group (Fig. 1B, green structure). The oxygen of its methoxy group interacted with Q149, while the methyl group was in hydrophobic interaction with F489. The residue Y148 partly stacked on the naphalene core of naproxen, and one of the guanidium groups of R355 also formed cation-π aromatic-ring interactions with naproxen. The structure derived from MD simulations shows that R361, R355, and Y148 are important residues for naproxen binding (Fig. 1B).
Fig 1.
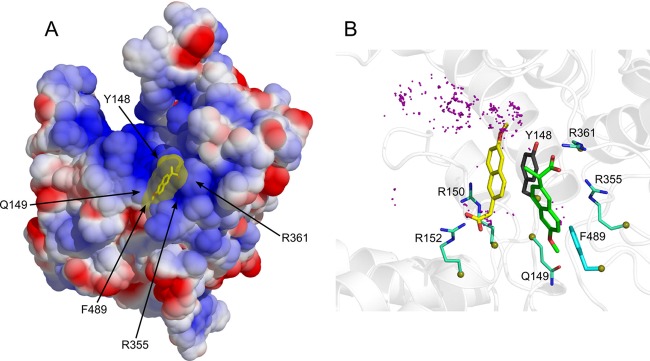
(A) Binding of naproxen to the RNA binding groove of the nucleoprotein from influenza A H1N1 virus based on PDB 2IQH (5). The protein surface is shown according to the electrostatic potential (blue, positive potential arising from the multiple arginine and lysine residues in the RNA binding groove; red, negative potential). This structure of the NP-naproxen complex solvated by water molecules was obtained after 10 ns of MD simulations. Details of the interactions of naproxen with NP inserted in a small hydrophobic cavity defined by Y148 and F489 are shown in panel B. (B) Superposition of the NP-naproxen complexes with the lowest energy. Virtual screening was used to define the possible binding site(s) of naproxen in the RNA binding groove of NP. The most MD-stable structure of the initial naproxen structure (whose carbon atoms are colored in green) is stabilized by hydrophobic interactions with Y148 and F489, a salt bridge with R361, and a water-mediated salt bridge with R355. Y148 stacks on the naphthalene core of naproxen, and the methoxy group of naproxen often forms an H-bond interaction with Q149 (see also Fig. S1 and Movie S6 in the supplemental material). The naproxen was further docked on NP using a searching volume extended to the rigid RNA binding site. The docking poses are represented as purple dots. The naproxen structure (with carbon atoms colored in yellow) with the lowest interaction energy (electrostatic and van der Waals) made a salt bridge with R152, a water-mediated salt bridge with R150, and a π-π interaction with Y148.
Docking of naproxen to NP.
The MD-derived NP-naproxen complexes were initiated from the structure obtained by virtual screening restricted on a sphere of 12-Å radius centered on Y148. To test whether naproxen could fit to an additional site(s) in the RNA groove, the 12-Å radius of the initial sphere was expanded to cover the entire groove on the same face of the protein and included Y148. This docking experiment generated 4,000 poses which were further analyzed after energy minimization (see description of experiments above). The carboxylate carbon atoms of naproxen docking poses are shown as purple spheres in Fig. 1B. The structures with the lowest interaction energy values, generated by MD and docking, are represented in Fig. 1B and 2. The MD simulation allowed naproxen to acquire a more stable configuration by rotating the propanoate group around the boundary that connects it to the naphthalene ring, thus making a salt bridge with R361 and a water-mediated salt bridge with R355, while the hydrophobic interactions with Y148 and F469 and the cation-π interaction with R355 were preserved (Fig. 1B, green [naproxen]; see also Movie S6 and Fig. S1 in the supplemental material). The extended docking revealed an alternative binding mode for naproxen, making a salt bridge with R152 and a water-mediated salt bridge with R150 and a π-π interaction between its naphthalene ring and the phenyl ring of Y148 (Fig. 1B, yellow [naproxen]). The interaction energy of the NP-naproxen complexes was found distributed in two populations (Fig. 2). The NP-naproxen complexes generated during the initial steps of MD simulation had interaction energies distributed around −55 kcal/mol (Fig. 2, basin I, inset A), mainly due to the hydrogen bonds with R361 and the nitrogen backbone atom of Q149. The nearby R361 residue forces the propanoate group to rotate in order to make a salt bridge interaction, with both carboxylate oxygen atoms bonded to it with hydrogen, clustering the NP-naproxen interaction energy around a value of −130 kcal/mol (Fig. 2, basin II, inset B). The extended docking of naproxen generated structures with the interaction energy with NP lying in the same basin (basin II) through the salt bridge with R152 and the electrostatic interaction with R150 (Fig. 2, basin II, inset C). However, the most stable naproxen structures generated by MD and docking had a head-to-tail relative orientation, though keeping the naphthalene core in hydrophobic interaction with Y148 (Fig. 1B, green and yellow naproxen conformations; Fig. 2, insets B and C). In addition, the less favorable binding sites for naproxen were found at the base of loop 1, in agreement with previous results indicating that this loop is at the most positive electrostatic potential (20). Usually, only one arginine contacted the carboxylate and no aromatic residue stacks were on the naphthalene core of naproxen in these poses.
Fig 2.

Interaction energy of the NP-naproxen complexes: the energy corresponding to interaction between naproxen and NP during the MD simulations revealed a bimodal behavior. A salt bridge between the carboxylate of naproxen and R361 shifted the naproxen from basin I (Eb1 = −55 ± 13 kcal/mol [inset A]) to basin II (Eb2 = −130 ± 14 kcal/mol [inset B]). The docking of naproxen in the extended RNA binding site generated structures with minimum interaction energy (see Fig. 1B) lying in basin II (inset C).
Experimentally, R355A, R361A, and R152A NP mutants were expressed to discriminate between the two structures (Fig. 1B). We also generated the Y148A mutant, expected to reduce the interactions between naproxen and NP in both structures.
Evidence for naproxen competing with RNA for binding to NP and for the formation of a naproxen-NP complex from surface plasmon resonance (SPR) experiments.
Naproxen is expected to compete with RNA for binding to NP according to the choice of the naproxen binding site in the RNA groove of the protein. This competition can be observed by SPR using immobilized RNA on the sensor surface and adding NP to form the NP-RNA complex in the absence or presence of naproxen. In this experiment, the competition of naproxen with RNA is deduced from the decrease of the NP-RNA complex formation upon coaddition of naproxen with NP on immobilized RNA. An alternative to this indirect method involves a surface-bound NP protein and monitoring the NP-naproxen complex by direct injection of various naproxen concentrations. SPR signals are proportional to the molecular mass of the added analytes and the respective molecular masses of NP and naproxen are 57 kDa and 252 Da. Therefore, we expect a good signal-to-noise ratio from the indirect competition experiment in which NP binds to a surface-bound RNA in the presence or absence of naproxen. In contrast, naproxen binding to surface-bound NP should give a SPR signal of only a few RU. Both experimental approaches are presented in Fig. 3. The nucleoprotein tightly binds to RNA, with dissociation constant (Kd) values ranging between 14 and 40 nM in the trimeric and monomeric forms, respectively (4, 5, 7, 31). Therefore, a NP concentration of 300 nM was sufficient for full binding to a biotinylated RNA probe attached to a streptavidin-coated sensor chip (1/1 complex). Addition of increasing naproxen concentrations to NP decreased the signal due to the NP-RNA complex. A plateau was obtained at 70% inhibition, and the 50% inhibition (IC50) value for naproxen was (1.5 ± 0.5) μM (Fig. 3A and D). Repeating these experiments at various NP concentrations in the range 100 to 500 nM and using the Cheng-Prusoff equation, the inhibitory constant Ki = IC50/(1 + [NP]/Kd) was found to be Ki = 0.20 ± 0.05 μM. The SPR experiments were also performed with the R361A, R355A, R152A, and Y148A mutants in the presence or absence of naproxen. The R361A and Y148A mutations were expected to reduce naproxen binding by disruption of the strong electrostatic interactions between naproxen and R361 and hydrophobic interaction with Y148, respectively, according to the MD results (Fig. 2, inset B). Figure 3B shows that naproxen (5 to 20 μM) could no longer compete with RNA binding to R361A (20), in contrast with naproxen inhibiting WT NP-RNA interactions (Fig. 3A). Naproxen was also unable to compete with RNA binding to Y148A (see Fig. S2 in the supplemental material) and had strongly reduced affinity to the R355A mutant (IC50 ≫ 20 μM; see Fig. S2 in the supplemental material). In contrast, naproxen binding was unaffected by the R152A mutation and IC50 = 1.7 ± 0.2 μM, which was similar to the value determined for naproxen-WT NP interactions (Fig. 3B). These data clearly supported the binding mode of naproxen to NP deduced from MD simulations.
Fig 3.

Naproxen binding to NP in competition with RNA shown by surface plasmon resonance experiments. (A) Comparison of the NP-RNA complexes formed in the absence (solid black line) and in the presence of 10 μM naproxen (dashed blue line). In this experiment, the RNA fragment was attached via its biotin 5′ extremity to surface-bound streptavidin. (B) Same experiment as described for panel A performed with the R361A mutant (solid black and dashed red lines), showing that naproxen could not compete with RNA binding to this mutant; therefore, R361 is required for the competitive binding of naproxen to NP. In contrast, naproxen competed with RNA binding to R152A (full dark greens and dotted orange lines), showing that naproxen binding is not extensively affected by mutation of this residue. (C) Binding of naproxen (1 μM) to surface-bound NP (blue trace, left scale). Note the absence of binding of naproxen (1 μM) to R361A (red trace, left scale). Both surface-bound proteins remained active and could bind RNA (dashed blue and red lines, right scale [NP and R361A, respectively]). (D) Dose dependence of naproxen binding to NP (300 nM) in competition with RNA. The maximum RU (RUmax) and RU are the signals observed with NP in the absence and presence of naproxen, respectively. Data are the means of the results of three experiments with standard deviations and could be fitted by a simple binding isotherm, yielding IC50 = 1.5 ± 0.5 μM.
Direct measurements of naproxen binding to NP required immobilizing the protein to the sensor chip, and Fig. 3C shows that the WT NP and R361A proteins remained active and could effectively bind RNA despite their immobilization on the surface. Addition of 10 μM naproxen induced the formation of a NP-naproxen complex displaying a 5-RU signal but no signal due to naproxen binding to R361A. Indeed, R361 was in a strong interaction with naproxen (Fig. 2, inset B). Naproxen largely inhibited RNA binding to R355A, in agreement with the expected interactions of naproxen and R355. We could not detect naproxen binding to R416A, which had strongly reduced affinity for RNA (data not shown) (9, 31, 36).
Competition with respect to naproxen for RNA binding to the nucleoprotein evidenced by the fluorescence change of a RNA beacon.
We designed a beacon, named Flu2, to exploit the selectivity of NP for single-stranded RNA in preference to double-stranded RNA (37). Flu2 beacon was constituted by a 9-bp AU-rich stem substituted at its 5′ end with a Cy3 chromophore and with a Dabcyl quencher at its 3′ end (cartoon, Fig. 4A). In the absence of NP, Flu2 is base paired and its fluorescence quenched by the proximity of the Dabcyl quencher to the Cy3 chromophore. Since NP has much greater affinity for linear, single-stranded RNA than for duplex RNA, NP binding disrupted the AU base pairs of Flu2. As a result of Flu2 hairpin melting, the Cy3-Dabcyl quencher distance increased, resulting in a fluorescence enhancement due to the binding of NP. Panels B and C in Fig. 4 show typical fluorescence enhancements observed upon NP binding to Flu2 that leveled off at saturating NP concentrations at a ratio of about 1 NP per RNA. The enhanced fluorescence of the NP-RNA complex decreased upon addition of naproxen, with a concomitant release of the free beacon, resulting in a clear fluorescence decrease (Fig. 4D). As found in the SPR experiments, the competition of naproxen with RNA binding to NP was not total. The data could be fitted by a simple inhibition mechanism, Ki = 0.3 ± 0.1 μM, using Km = 61 ± 15 nM. By replacing the WT protein with an R361A mutant, the addition of naproxen could not compete with RNA binding to NP.
Fig 4.

Fluorescence assay of naproxen binding to NP. (A) Schematic principle of the assay using a fluorescent RNA hairpin labeled at its 5′ end with Cy3 fluorescent dye and at its 3′ end with a quencher (called an RNA beacon). (B) Fluorescence spectrum of the free beacon and in the presence of increasing NP concentrations. (C) Binding of NP to the RNA beacon monitored at 560 nm. In the free beacon, the proximity of the 3′ and 5′ ends in the paired hairpin quenched the Cy3 fluorescence due to the proximity of the quencher. Because NP preferentially binds to linear single-stranded RNA (52), NP binding to the RNA hairpin dissociates its stem (consecutive fluorescence enhancements are shown in panels B and C). These curves allow a quantitative determination of the Ki of NP for RNA. (D) Naproxen binding to NP competes with the dissociation of the hairpin stem; this is not the case in the R361A mutant. In this experiment, naproxen was added to highly fluorescent RNA-bound NP complex. Naproxen competed with RNA binding to NP and released free RNA beacon, which folded back to form the initial paired hairpin; thus, the fluorescence level decreased (black squares). Under the same conditions, the R361A mutant formed a weaker complex with the RNA beacon, as reflected by the lower initial fluorescence intensity, in agreement with previous results (20). Naproxen binding to R361A was impeded by this mutation, in agreement with the lack of fluorescence change seen upon addition of naproxen (blue circles). The values shown in panels C and D represent the results of 1 of 2 experiments with standard errors (SE).
Stabilization of NP against proteolytic cleavage by naproxen binding.
The monomeric nucleoprotein contains flexible loops and linkers that could be prone to cleavage by protease. Indeed, freshly purified NP migrates on an SDS-PAGE gel according to its molecular mass of 57 kDa; NP undergoes cleavage upon storage at 4°C for 1 month or at room temperature for a week (see Fig. S3A and B in the supplemental material). The cleaved fragments have molecular masses of 45 and 9 kDa, as determined by electrospray mass spectrometry. The Western blot analysis performed using an anti-His antibody of the C-terminally labeled NP confirmed the C-terminal fragment proteolytic cleavage upon storage at room temperature, in agreement with the mass spectrometry results. In contrast with the cleavage of free NP, the monomeric protein is stabilized against cleavage by addition of naproxen (see Fig. S3B in the supplemental material). These data suggest that naproxen protects and stabilizes the C-terminal part of NP, in agreement with F489 being part of the naproxen binding site.
EC50, CC50, and SI determinations from cellular assays.
Naproxen (1 to 500 μM) was nontoxic to A549 lung epithelial cells and Madin-Darby canine kidney (MDCK) cells as tested by the MTT assay 48 or 72 h after administration of naproxen (Fig. 5A and Table 1). The data obtained after incubation of 24 h with high naproxen concentrations even showed somewhat increased metabolism of the cells. In a second set of experiments, we used an MDCK cell-based influenza A virus infection assay performed under the same culture conditions as described above in which naproxen was found to be nontoxic to (uninfected) MDCK cells to determine the antiviral activity of naproxen using a multiplicity of infection (MOI) of 10−3 H1N1 A/WSN/33 virus. The cells were treated with 1 to 500 μM naproxen immediately postinfection, and the absorbance values measured by the MTT test presented a typical dose-response curve reaching an asymptotic optical density (OD) value similar to that observed in noninfected controls, in contrast with the marked viability loss of IAV-infected untreated cells (Fig. 5B, right scale, and Table 1). The infectious titers of these cell supernatants treated with naproxen were determined by a plaque assay. The decrease of viral titers as a function of the naproxen concentration was fitted by a dose-response curve, yielding the naproxen concentration required to reduce the viral titer to 50% of that seen with the infected untreated control (EC50 = 11 ± 4 μM) at 48 h p.i. (Table 1). Under these conditions, the amount of RNA found in the cell supernatants decreased with the naproxen concentration (EC50 = 16 ± 5 μM) (Table 1). To quantify the kinetics of the antiviral effect, the viral titers of the cell supernatants in the presence of naproxen (0 to 500 μM) added to the cells immediately after infection were determined at time points 24, 48, and 72 h postinfection (Fig. 5C [means of the results of 4 experiments]). Addition of naproxen to the virus-challenged cells was found to be protective for the MDCK cells, with a mean EC50 value of 16 ± 5 μM. We observed a slight increase of the viral titers at low naproxen concentrations after 24 h which paralleled the higher cell viability deduced from the MTT tests (Fig. 5A and C). We attribute this titer increase to the larger number of cells in each well at low naproxen concentrations at 24 h p.i.
Fig 5.

Effects of naproxen on cell viability and viral titers in the absence or presence of influenza A virus. (A) MTT tests. Data represent levels of viability (± SE) of noninfected A549 cells (black squares and blue triangles) and MDCK cells (brown triangles and green open circles) in the presence of various concentrations of naproxen added to the cell supernatants at t = 0 after incubation for 24 or 48 h (or 72 h; see Table 1). (B) Left scale (blue squares), ratio (R) of titers of treated cells (t) to titers of untreated infected cells (t0), R = t/t0; right scale (open black circles), results (± SE) of MTT viability tests of MDCK cells infected with IAV at MOI = 10−3 in the presence or absence of different naproxen concentrations at 48 h postinfection (p.i.). The experiments whose results are shown in panels A to D were performed under the same culture conditions. (C) Kinetics of viral titers of MDCK cells infected with influenza A/WSN/33 virus at MOI = 10−3. A large decrease of the titers was observed in the presence of increasing naproxen concentrations indicated as follows: 15 μM, blue circles; 50 μM, green triangles; 100 μM, gray triangles; 250 μM, brown diamonds; 500 μM, red triangles (compared to the infected and nontreated controls for [naproxen] = 0 [black squares] at 24, 48, and 72 h postinfection). The values shown in panel C are the means of the results of 4 experiments with standard deviations. (D) Toxicity assays for CC50 of naproxen determinations (see also Table 1). Left scale (gray circles), MTT tests; right scale (brown open squares), trypan blue test. Data represent results ± SE. The lines in panels B and D correspond to the fit of the data to a dose-response curve. (E) Similar levels of protection by naproxen treatment of MDCK cells challenged by two viral strains of influenza A virus, WSN/33 (H1N1) and UDORN/72 (H3N2), 24 h p.i. at MOI = 10−2.
Table 1.
Effective naproxen concentration for reduction of MDCK cell viral titersa
| [Naproxen] (μM) | Method | Time p.i. (h) | MOI | EC50 (μM ± SD) | CC50 (μM ± SD) | SI |
|---|---|---|---|---|---|---|
| 0–10 | MTT (n = 3) | 24 | 1,119 ± 320 | |||
| 48 | 1,435 ± 550 | |||||
| 72 | 2,167 ± 600 | |||||
| 0–10 | Trypan blue (n = 2) | 48 | 1,700 ± 900 | |||
| Mean | 1,605 ± 328 | |||||
| 0–500 | MTT (n = 3) | 48 | 10−3 WSN/33 | 15 ± 5 | ||
| 0–500 | Viral titers (n = 4) | 24 | 10−3 WSN/33 | 20 ± 7 | ||
| 48 | 11 ± 4 | |||||
| 72 | 18 ± 5 | |||||
| 0–500 | Viral RNA (n = 2) | 48 | 10−3 WSN/33 | 16 ± 5 | ||
| Mean | 16 ± 5 | 100 ± 30 | ||||
| 0–500 | Viral titers (n = 2) | 48 | 10−2 WSN/33 | 25 ± 7 |
Data in columns 5, 6, and 7 represent EC50 (cellular toxicity of naproxen on uninfected MDCK cells under the same cell growth conditions), CC50, and selectivity index (SI [CC50/EC50]) values, respectively.
An estimate of the 50% cytotoxic concentration (CC50 = 1.44 ± 0.55 mM) of naproxen was obtained from the plot of the cell viability as a function of the naproxen concentration (millimolar range) (Fig. 5D; 48 h p.i.). Cell counting using the trypan blue assay yielded a similar value: CC50 = 1.7 ± 0.9 mM. Therefore, the selectivity index (SI) for naproxen, defined as SI = CC50/EC50, was found to be SI = 100 ± 30 (Table 1). Figure 5E compares the reduction of the titer of cells challenged with either WSN (H1N1) or Udorn (H3N2) virus at MOI = 10−2 and treated with 100 and 500 μM naproxen to the titer of controls. Naproxen reduced the titer of both viral strains, being as efficient with H1N1 as with H3N2.
We used an anti-NP monoclonal antibody to test if naproxen blocked the transport from the cytoplasm to the nucleus as reported for another NP inhibitor (14) (see Fig. S4 in the supplemental material): the cellular localization of NP remained the same with or without naproxen at early times (3 h) postinfection, a time found to restrict the localization of NP to cell nuclei. Likewise, no significant change of NP distribution induced by the presence of naproxen could be detected at a later time postinfection (24 h p.i.).
Attempted selection of resistance mutation.
An influenza A/WSN/33 virus obtained by reverse genetics was used to infect cells with 8 × 103 PFU/well in the presence or absence of 250 μM naproxen (see Materials and Methods). The cell supernatants were used to infect a new monolayer, and the procedure was repeated for six passages. The naproxen concentration was increased to 500 μM for the third to sixth passages. After the sixth passage, six clones were isolated and the NP fragment was amplified. Sequencing revealed that in the six selected clones, NP did not carry a detectable mutation, suggesting that no escape mutants could still be selected.
In vivo effects of naproxen on mice inoculated with influenza A/PR8 virus.
Six-week-old BALB/c female mice were inoculated via the intranasal route with 2,000 PFU or 50 PFU of influenza A/PR8 (H1N1) virus, corresponding to 0.4 and 0.01 LD50, respectively (see Fig. S5 in the supplemental material). The mice were treated or not with naproxen 4 h after infection and each day thereafter, either by intraperitoneal (IP) injection or by intranasal treatment, until the end of the experiments. Treated and noninfected mice with matched naproxen doses were used for toxicity tests. Naproxen was nontoxic at up to 8 mg (4 mg administered twice daily; protocol guided by published data on 50% naproxen being metabolized in 24 h [38]). Alternatively, naproxen was intranasally administered at lower doses of 0.2 to 3 mg/day without weight loss in noninfected mice. At days 5 to 7 postinfection (at 2,000 PFU), the mice that reached a critical weight loss (and all remaining mice at the end of the experiments) were sacrificed. Most of the mice (94% ± 2%) survived the 2,000-PFU viral challenge, while all of them survived the 50-PFU challenge, and infection was resolved. Mice lungs were dissected for subsequent virus titer determinations. Panels A and B in Fig. 6 show that administration of naproxen at doses of 1, 3, and 4 to 8 mg by IP injection shifted the onset of weight loss compared to the infected untreated controls and lowered the maximal weight loss. The uninfected mice at the highest doses showed a low loss of weight during the first 3 to 4 days of treatment that progressively resolved, indicative of a transient weak toxicity, followed by weight normalization. The decrease of the viral titer in mice lungs following treatment was dependent on the naproxen dose, the EC50 being about 0.8 mg/mice/day or 40 mg/kg of body weight/day (Fig. 6C and D). Given that the LD50 of naproxen is 1,234 mg/kg for mice (39), the therapeutic index (TI) can be calculated to be TI = 1,234/40 = 31. Many blood cells were present in the bronchoalveolar fluids of infected and untreated mice compared to healthy controls, while naproxen treatment at 3 mg/day markedly reduced the number of red blood cells (see Fig. S6 in the supplemental material). Figure 7 shows that mice challenged with a low viral load (50 PFU) were totally protected from weight loss with 2 mg/day naproxen administered to mice by the intranasal route or with 0.2 mg/day oseltamivir (data not shown) by IP injections. Under these conditions, all mice survived the viral challenge.
Fig 6.

In vivo protection by naproxen treatment of mice infected with influenza A/PR8 virus loads of 2,000 PFU. (A and B) Weight change of infected mice with or without naproxen treatment administered via IP injection at the specified doses (in mg/mouse/day) to mice I-0, I-1, I-3, I-4, and I-8 (0, 1, 3, 4, and 8 mg naproxen, respectively) and the matched uninfected controls (NI-0, NI-1, NI-3, NI-4, and NI-8, respectively) at the same naproxen concentration. Each panel represents a single experiment of n = 3 experiments performed. A total of 7 to 10 mice per naproxen concentration were used in each experiment. The data are expressed as median values of the weight change in percent ± variance. Statistics analysis with the paired t test revealed meaningful differences (P being below 0.05) between I-0 and I-1 or I-3 or I-4 or I-8 and between I-1 and I-3. (B) No statistical differences between the data for I-4 and I-8 were seen. (C) Decrease of the viral titers in mice lungs as a function of the naproxen dose; statistically significant differences were found between the lung viral titer with no naproxen treatment and each of the naproxen concentrations, namely, I-0 versus I-1 (P < 0.01) and I-0 versus I-3 and I-0 versus I-8 (P < 0.005, respectively). Under these conditions, the decrease of the viral titer seen after administration of 0.2 mg oseltamivir was ca. 100-fold compared to 42-fold with 2 doses of 4 mg naproxen. (D) Comparison of the weight loss decrease and the viral titer decrease values as a function of the naproxen concentration; the fits determined by a dose-response model yielded quite similar values of EC50 = 1.6 ± 0.5 and 0.85 ± 0.42 mg/day, respectively. In this figure and Fig. 7, the data are expressed as the medians and variances of the weight changes at day i = 100 × (w0 − wi)/w0, where wi and w0 are the weights at day i and 0 postinfection calculated for each condition for infected and uninfected mice at each naproxen dose.
Fig 7.

In vivo protection by naproxen treatment of mice infected with influenza A/PR8 virus loads of 50 PFU. (A) Weight change of mice infected with 50 PFU IAV and its uninfected control. (B) Same experiment as described for panel A, with mice treated by intranasal administration of naproxen at a dose of 2 mg/day. Note that both naproxen and oseltamivir (not shown) completely inhibited the weight loss in the infected untreated animals. There were statistical differences between the untreated and treated mice (I-0 and I-2 [A and B]) and the infected and noninfected untreated mice (NI-0 and I-0 [A]).
DISCUSSION
Naproxen binds to the targeted site in the RNA groove of NP.
In this work, we aimed at inhibiting the binding of the nucleoprotein to RNA for interference with NP function. To achieve this goal, part of the RNA binding groove was blocked by naproxen, a compound identified by virtual screening which binds within the RNA binding groove of the nucleoprotein (Fig. 1A). After MD simulations, naproxen was found in a binding pocket defined by two aromatic residues, Y148 and F489. The naproxen naphtalene core was stabilized by π-stacking interactions with Y148, F489 formed hydrophobic interactions with the methoxy moiety of naproxen, and strong electrostatic interactions between the carboxylate of naproxen and the guanidium of R361 stabilized naproxen in its binding site (Fig. 1B). Docking experiments performed by greatly extending the naproxen binding space identified an alternative binding site in which the interactions of naproxen with NP involved Y148, R150, and R152 (Fig. 1B and Fig. 2). In each of the two NP-naproxen complexes, the naproxen binding mode shared some of the features of naproxen bound to COX-2, including a strong carboxylate-arginine interaction and hydrophobic contact of the methoxy group with an aromatic residue; however, naproxen was less buried in NP than in COX-2 (40).
Direct evidence for naproxen binding to the nucleoprotein was given by the specific signal of the naproxen-NP complex using NP attached to the surface (Fig. 3C). SPR also showed that naproxen binding to NP was competitive with RNA and disrupted the RNA-NP complex (Fig. 3A and D). Quantitative analysis of the IC50 values as a function of the NP concentration yielded Ki = 0.20 ± 0.05 μM and Ki = 0.30 ± 0.05 μM in SPR and fluorescence assays, respectively. Knowledge of the putative naproxen binding site(s) enabled identification of the interactions of naproxen with NP and gave us the possibility to perturb them by point mutagenesis. Therefore, naproxen binding to the mutants R361A, R355A, R152A, and Y148A was compared with its binding to WT NP. The specific signal of naproxen binding to NP was not observed by replacing NP with R361A, Y148A, or R355A. As expected from the naproxen binding to NP deduced from MD, naproxen could not compete with RNA binding to R361A and Y148A; naproxen hardly competed with RNA binding to R355A (Fig. 3B and C and Fig. 4; see also Fig. S2 in the supplemental material). However, naproxen could displace about 70% to 80% of the NP-RNA complex. Higher concentrations may be required for full competition, suggesting an alternative/additional mode(s) of binding of naproxen to NP. These experimental SPR and fluorescence data were consistent with the MD study that identified the naproxen binding shown in Fig. 1 and 2 as the preferred naproxen binding site. At high naproxen concentrations, an alternative site is likely to be occupied. Such a site was also defined by docking experiments shown in Fig. 1 and 2. Naproxen binding was observed at a 1:1 stoichiometric naproxen/NP ratio and was specific to NP purified as monomers (31). We did not observe a disruption of RNA-free trimers formed by either Y148A or R355A, suggesting that naproxen preferentially binds to NP monomer (20, 31). This hypothesis is consistent on the one hand with the F489 C-terminal residue being located in the naproxen binding site in NP monomer and on the other hand with residues 486 to 490 proposed to inhibit NP oligomerization (10). In agreement with naproxen binding involving interactions with the F489 C-terminal residue (Fig. 2), NP was stabilized against proteolytic cleavage of its C terminus by addition of naproxen (see Fig. S3 in the supplemental material). Close to the naproxen binding site, a few residues can be phosphorylated: S478 and S486 (41) are located on the same linker as F489; and S165 (42, 43) is close to the naproxen binding site (Fig. 1 and 2). This suggests that the negative charge of naproxen is a sort of mimic of NP phosphorylation that may stabilize monomers of NP (42, 43). In agreement with this last hypothesis and with the relationships found between nuclear export and NP oligomerization (44), inhibition of NP by naproxen results in antiviral effects without modifying the NP location in the nucleus. All together, the results determined in this work show that targeting the RNA binding groove of NP provides a way to inhibit and possibly sequestrate active NP monomers, as shown here with naproxen (31). Shen and coworkers have shown that direct targeting of the highly conserved E339-R416 salt bridge by small compounds successfully impeded the NP-NP interactions and stabilized NP monomers (16). Thus, there are at least two sites where drugs can inhibit the NP-NP or NP-RNA interactions and stabilize a monomer, suggesting cross talk between different parts of the protein (20). These drugs lead to antiviral activity, emphasizing the critical role of the nucleoprotein in the viral cycle (12, 14, 16).
Antiviral and anti-inflammatory effects of naproxen in IAV-infected cells.
Cyclooxygenase type 2 (COX-2) is a component of the arachidonic acid cascade thought to be involved in inflammatory and immune responses; naproxen is a COX-2 inhibitor used to treat these symptoms in humans (40). Influenza A viruses induce high levels of proinflammatory cytokines in macrophages and alveolar epithelial cells (45, 46), and COX-2 plays a regulatory role in induction of H5N1-mediated proinflammatory responses in vitro (47). Such cytokine deregulation was proposed to be a major contributor to the pathogenesis of H5N1 disease in humans (48). Coadministration of the COX-2 inhibitors mesalamine and celecoxib with an antiviral (zanamivir) significantly improved survival in mice challenged with H5N1 infection compared with infected mice treated with the antiviral alone (49). Recently, a selective inhibitor of COX-2, nimesulide, was shown to suppress H9N2 replication in human macrophages but was less potent for the H1N1 strain (50). The EC50 value of nimesulide required to reduce the viral titer of infected macrophages with H5N1 virus (MOI = 2) to 50% was 170 μM, with a toxicity CC50 > 100 μM (50). In this work, the mean EC50 value for naproxen was 16 ± 5 μM at MOI = 10−3, and administration of concentrations of 100 μM and 500 μM to MDCK-infected cells with influenza A/WSN/33 virus reduced the infectious titers to 10% ± 3% and 0.3% ± 0.1% (Fig. 5B and C) whereas at MOI = 5, naproxen (100 μM) reduced the titers to 7% relative to control levels (data not shown). Naproxen cellular toxicity was observed in the millimolar range, yielding a CC50 value of 1.6 ± 0.3 mM and a selectivity index (SI) EC50/CC50 = 100 ± 30. Thus, the naproxen antiviral effect was significantly more potent than those of nimesulide and the other COX-2 selective inhibitors tested. Naproxen presents a reasonable selectivity index compared to other antivirals that present a wide distribution, with SI = 1,562 and 3,623 (nucleozin derivatives [14]), SI = 13 and >1,000 (compound 3 and peptide 2 [16]), SI = 17 (pyrazole derivatives [34]), and SI = 38 (isoquercitin [51]). Despite a high pressure selection exerted by 500 μM naproxen, the virus had no mutation on the NP after six passages and more passages may be required to generate escape mutants. In vivo experiments using intranasal infection with Influenza/A/PR8 virus in a mice model yielded protection similar to that observed in cells when the mice were treated by IP administration of naproxen: the infectious viral titer of mice lungs treated twice daily with 4 mg naproxen was reduced to 4% relative to the infected control level (Fig. 6C). The in vivo therapeutic index (TI) calculated from the EC50 determined from the lung viral titer-naproxen dose response of EC50 = 40 mg/kg and from the in vivo toxicity index of 1,234 mg/kg is TI = 31. Besides its antiviral effect, naproxen reduced lung bleeding (see Fig. 6 in the supplemental material). Although naproxen is less effective than nucleozin and its analogs in inhibiting viral replication (the smallest values for antiviral EC50 against H1N1 A/WSN/33 obtained in three studies were 0.7 μM for nucleozin and 0.07 μM for compound 5 [12, 14, 15]), and our results are potentially very significant on the basis of the following considerations.
Naproxen constitutes a lead compound to be improved by drug design; its main binding site is known and was shown by the use of site-directed mutants. Docking experiments identified residues involved in additional binding modes of naproxen.
Naproxen is equally effective against the viral variants H1N1 and H3N2.
Importantly, we demonstrated that blockade of the RNA binding groove of NP in its monomeric form results in antiviral effects, presumably through sequestration of active NP monomers. We did not find an inhibitory effect of naproxen on trimeric NP mutants for RNA binding. This result is in line with our previous reports that provided evidence for a monomeric RNA-free NP (20, 31, 43). Our studies showed that RNA-free monomeric nucleoprotein is a target for inhibition of influenza A virus which paves the way for further insight on NP function.
In conclusion, in the era of dual pharmacology, where the efficacy of a drug is improved by reaching more than one target while being nontoxic, naproxen has the advantage of inhibiting both COX-2 and the nucleoprotein of influenza A virus. Naproxen is more potent than COX-2 inhibitors such as nemesulide against influenza A virus challenge. Importantly, naproxen is a safe, potential pharmaceutical readily available for cases of resistance to antiviral and if an IAV infection pandemic emerges.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the French agency for research (ANR) for funding (Flunucleovir; ANR-2010-Blanc-1307-01 to A.S.-S.), by a grant of the Programme de Recherche Influenza A (H1N1), coordinated by the Institut de Microbiologie et Maladies Infectieuses of the Institut National de la Santé et de la Recherche Médicale, INSERM, France, to B.D. This work was granted access to the HPC resources of IDRIS made by GENCI (Grand Equipement National de Calcul Intensif) under grants 2010-99636 and 2011-076378 attributed to A.S.-S. and B.T.
We thank Sabine Riffault for helpful discussions, Stéphane Biacchesi for his help with fluorescence microscopy measurements, and Bruno Da Costa for his help with NP amplification and sequencing. All the people in charge of the animal care facilities at INRA are gratefully acknowledged for their help. We thank Olivier Bakowiez for preliminary experiments. We are grateful to the structural biology facility (UMS 3033/US001) of the Institut Européen de Chimie et Biologie (Pessac, France) for access to the Biacore 3000 instrument.
Footnotes
Published ahead of print 4 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02335-12.
REFERENCES
- 1. Ruigrok RW, Crepin T, Kolakofsky D. 2011. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr. Opin. Microbiol. 14:504–510 [DOI] [PubMed] [Google Scholar]
- 2. Das K, Aramini JM, Ma LC, Krug RM, Arnold E. 2010. Structures of influenza A proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol. 17:530–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krug RM, Aramini JM. 2009. Emerging antiviral targets for influenza A virus. Trends Pharmacol. Sci. 30:269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ng AK, Zhang H, Tan K, Li Z, Liu JH, Chan PK, Li SM, Chan WY, Au SW, Joachimiak A, Walz T, Wang JH, Shaw PC. 2008. Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. FASEB J. 22:3638–3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ye Q, Krug RM, Tao YJ. 2006. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444:1078–1082 [DOI] [PubMed] [Google Scholar]
- 6. Ng AK, Lam MK, Zhang H, Liu J, Au SW, Chan PK, Wang J, Shaw PC. 2012. Structural basis for RNA binding and homo-oligomer formation by influenza B virus nucleoprotein. J. Virol. 86:6758–6767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chan WH, Ng AK, Robb NC, Lam MK, Chan PK, Au SW, Wang JH, Fodor E, Shaw PC. 2010. Functional analysis of the influenza virus H5N1 nucleoprotein tail loop reveals amino acids that are crucial for oligomerization and ribonucleoprotein activities. J. Virol. 84:7337–7345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elton D, Medcalf E, Bishop K, Digard P. 1999. Oligomerization of the influenza virus nucleoprotein: identification of positive and negative sequence elements. Virology 260:190–200 [DOI] [PubMed] [Google Scholar]
- 9. Mena I, Jambrina E, Albo C, Perales B, Ortin J, Arrese M, Vallejo D, Portela A. 1999. Mutational analysis of influenza A virus nucleoprotein: identification of mutations that affect RNA replication. J. Virol. 73:1186–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ng AK, Wang JH, Shaw PC. 2009. Structure and sequence analysis of influenza A virus nucleoprotein. Sci. China C Life Sci. 52:439–449 [DOI] [PubMed] [Google Scholar]
- 11. Das K. 2012. Antivirals targeting influenza A virus. J. Med. Chem. 55:6263–6277 [DOI] [PubMed] [Google Scholar]
- 12. Gerritz SW, Cianci C, Kim S, Pearce BC, Deminie C, Discotto L, McAuliffe B, Minassian BF, Shi S, Zhu S, Zhai W, Pendri A, Li G, Poss MA, Edavettal S, McDonnell PA, Lewis HA, Maskos K, Mortl M, Kiefersauer R, Steinbacher S, Baldwin ET, Metzler W, Bryson J, Healy MD, Philip T, Zoeckler M, Schartman R, Sinz M, Leyva-Grado VH, Hoffmann HH, Langley DR, Meanwell NA, Krystal M. 2011. Inhibition of influenza virus replication via small molecules that induce the formation of higher-order nucleoprotein oligomers. Proc. Natl. Acad. Sci. U. S. A. 108:15366–15371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hagiwara KKY, Ueda A, Yamada K, Goto H, Watanabe T, Nakata T, Osada H, Aida Y. 2010. Discovery of novel antiviral agents directed against the influenza A virus nucleoprotein using photo-cross-linked chemical arrays. Biochem. Biophys. Res. Commun. 394:721–727 [DOI] [PubMed] [Google Scholar]
- 14. Kao RY, Yang D, Lau LS, Tsui WH, Hu L, Dai J, Chan MP, Chan CM, Wang P, Zheng BJ, Sun J, Huang JD, Madar J, Chen G, Chen H, Guan Y, Yuen KY. 2010. Identification of influenza A nucleoprotein as an antiviral target. Nat. Biotechnol. 28:600–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Su CY, Cheng TJ, Lin MI, Wang SY, Huang WI, Lin-Chu SY, Chen YH, Wu CY, Lai MM, Cheng WC, Wu YT, Tsai MD, Cheng YS, Wong CH. 2010. High-throughput identification of compounds targeting influenza RNA-dependent RNA polymerase activity. Proc. Natl. Acad. Sci. U. S. A. 107:19151–19156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shen YF, Chen YH, Chu SY, Lin MI, Hsu HT, Wu PY, Wu CJ, Liu HW, Lin FY, Lin G, Hsu PH, Yang AS, Cheng YS, Wu YT, Wong CH, Tsai MD. 2011. E339.R416 salt bridge of nucleoprotein as a feasible target for influenza virus inhibitors. Proc. Natl. Acad. Sci. U. S. A. 108:16515–16520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haidari M, Zhang W, Ganjehei L, Ali M, Chen Z. 2011. Inhibition of MLC phosphorylation restricts replication of influenza virusto—a mechanism of action for anti-influenza agents. PLoS One 6:e21444 doi:10.1371/journal.pone.0021444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pinto R, Herold S, Cakarova L, Hoegner K, Lohmeyer J, Planz O, Pleschka S. 2011. Inhibition of Influenza virus-induced NF-kappaB and RAF/MEK/ERK activation can reduce both virus titers and cytokine expression simultaneously in vitro and in vivo. Antiviral Res. 92:45–56 [DOI] [PubMed] [Google Scholar]
- 19. Fedichev P, Timakhov R, Pyrkov T, Getmantsev E, Vinnik A. 2011. Structure-based drug design of a new chemical class of small molecules active against influenza A nucleoprotein in vitro and in vivo. PLoS Curr. 3:RRN1253 doi:10.1371/currents.RRN1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tarus B, Chevalier C, Richard CA, Delmas B, Di Primo C, Slama-Schwok A. 2012. Molecular dynamics studies of the nucleoprotein of influenza a virus: role of the protein flexibility in RNA binding. PLoS One 7:e30038 doi:10.1371/journal.pone.0030038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Z, Watanabe T, Hatta M, Watanabe S, Nanbo A, Ozawa M, Kakugawa S, Shimojima M, Yamada S, Neumann G, Kawaoka Y. 2009. Mutational analysis of conserved amino acids in the influenza A virus nucleoprotein. J. Virol. 83:4153–4162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Slama-Schwok AT B, Bertrand H, Quideau S, Delmas B. 2012. Compositions antivirales dirigees contre la nucleoproteine des virus influenza. International patent PCT/EP2012/001720 deposited 21 April 2012
- 23. O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. 2011. Open Babel: an open chemical toolbox. J. Cheminform. 3:33 doi:10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. MacKerell AD, Jr, Banavali N, Foloppe N. 2000. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 56:257–265 [DOI] [PubMed] [Google Scholar]
- 26. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935 [Google Scholar]
- 27. Darden T, York DM, Pedersen L. 1993. Particle mesh Ewald: an N×log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092 [Google Scholar]
- 28. Ryckaert JP, Ciccotti G, Berendsen HJC. 1977. Numerical-integration of Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comp. Phys. 23:327–341 [Google Scholar]
- 29. Brooks BR, Brooks CL, III, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. 2009. CHARMM: the biomolecular simulation program. J. Comp. Chem. 30:1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trott O, Olson AJ. 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31:455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tarus B, Bakowiez O, Chenavas S, Duchemin L, Estrozi LF, Bourdieu C, Lejal N, Bernard J, Moudjou M, Chevalier C, Delmas B, Ruigrok RW, Di Primo C, Slama-Schwok A. 2012. Oligomerization paths of the nucleoprotein of influenza A virus. Biochimie 94:776–785 [DOI] [PubMed] [Google Scholar]
- 32. Fodor E, Devenish L, Engelhardt OG, Palese P, Brownlee GG, Garcia-Sastre A. 1999. Rescue of influenza A virus from recombinant DNA. J. Virol. 73:9679–9682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sieuwerts AM, Klijn JG, Peters HA, Foekens JA. 1995. The MTT tetrazolium salt assay scrutinized: how to use this assay reliably to measure metabolic activity of cell cultures in vitro for the assessment of growth characteristics, IC50-values and cell survival. Eur. J. Clin. Chem. Clin. Biochem. 33:813–823 [DOI] [PubMed] [Google Scholar]
- 34. Shih SR, Chu TY, Reddy GR, Tseng SN, Chen HL, Tang WF, Wu MS, Yeh JY, Chao YS, Hsu JT, Hsieh HP, Horng JT. 2010. Pyrazole compound BPR1P0034 with potent and selective anti-influenza virus activity. J. Biomed. Sci. 17:13 doi:10.1186/1423-0127-17-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ng AK, Chan WH, Choi ST, Lam MK, Lau KF, Chan PK, Au SW, Fodor E, Shaw PC. 2012. Influenza polymerase activity correlates with the strength of interaction between nucleoprotein and PB2 through the host-specific residue K/E627. PLoS One 7:e36415 doi:10.1371/journal.pone.0036415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Elton D, Medcalf L, Bishop K, Harrison D, Digard P. 1999. Identification of amino acid residues of influenza virus nucleoprotein essential for RNA binding. J. Virol. 73:7357–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baudin F, Bach C, Cusack S, Ruigrok RW. 1994. Structure of influenza virus RNP. I. Influenza virus nucleoprotein melts secondary structure in panhandle RNA and exposes the bases to the solvent. EMBO J. 13:3158–3165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jung J, Park M, Park HJ, Shim SB, Cho YH, Kim J, Lee HS, Ryu DH, Choi D, Hwang GS. 2011. (1)H NMR-based metabolic profiling of naproxen-induced toxicity in rats. Toxicol. Lett. 200:1–7 [DOI] [PubMed] [Google Scholar]
- 39. Roszkowski AP, Rooks WH, II, Tomolonis AJ, Miller LM. 1971. Anti-inflammatory and analgetic properties of d-2-(6′-methoxy-2′-naphthyl)-propionic acid (naproxen). J. Pharmacol. Exp. Ther. 179:114–123 [PubMed] [Google Scholar]
- 40. Duggan KC, Walters MJ, Musee J, Harp JM, Kiefer JR, Oates JA, Marnett LJ. 2010. Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J. Biol. Chem. 285:34950–34959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Poole E, Elton D, Medcalf L, Digard P. 2004. Functional domains of the influenza A virus PB2 protein: identification of NP- and PB1-binding sites. Virology 321:120–133 [DOI] [PubMed] [Google Scholar]
- 42. Hutchinson EC, Denham EM, Thomas B, Trudgian DC, Hester SS, Ridlova G, York A, Turrell L, Fodor E. 2012. Mapping the phosphoproteome of influenza A and B viruses by mass spectrometry. PLoS Pathog. 8:e1002993 doi:10.1371/journal.ppat.1002993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chenavas S, Estrozi LF, Slama-Schwok A, Delmas B, Di Primo C, Baudin F, Li X, Crepin T, Ruigrok RWH. Monomeric nucleoprotein of Influenza A virus. PLoS Pathog., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yu M, Liu X, Cao S, Zhao Z, Zhang K, Xie Q, Chen C, Gao S, Bi Y, Sun L, Ye X, Gao GF, Liu W. 2012. Identification and characterization of three novel nuclear export signals in the influenza A virus nucleoprotein. J. Virol. 86:4970–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Simmons C, Farrar J. 2008. Insights into inflammation and influenza. N. Engl. J. Med. 359:1621–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zeng H, Goldsmith C, Thawatsupha P, Chittaganpitch M, Waicharoen S, Zaki S, Tumpey TM, Katz JM. 2007. Highly pathogenic avian influenza H5N1 viruses elicit an attenuated type i interferon response in polarized human bronchial epithelial cells. J. Virol. 81:12439–12449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee SM, Cheung CY, Nicholls JM, Hui KP, Leung CY, Uiprasertkul M, Tipoe GL, Lau YL, Poon LL, Ip NY, Guan Y, Peiris JS. 2008. Hyperinduction of cyclooxygenase-2-mediated proinflammatory cascade: a mechanism for the pathogenesis of avian influenza H5N1 infection. J. Infect. Dis. 198:525–535 [DOI] [PubMed] [Google Scholar]
- 48. Peiris JS, Cheung CY, Leung CY, Nicholls JM. 2009. Innate immune responses to influenza A H5N1: friend or foe? Trends Immunol. 30:574–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zheng BJ, Chan KW, Lin YP, Zhao GY, Chan C, Zhang HJ, Chen HL, Wong SS, Lau SK, Woo PC, Chan KH, Jin DY, Yuen KY. 2008. Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc. Natl. Acad. Sci. U. S. A. 105:8091–8096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee SM, Gai WW, Cheung TK, Peiris JS. 2011. Antiviral activity of a selective COX-2 inhibitor NS-398 on avian influenza H5N1 infection. Influenza Other Respi. Viruses 5(Suppl 1):230–232 [PubMed] [Google Scholar]
- 51. Thapa M, Kim Y, Desper J, Chang KO, Hua DH. 2012. Synthesis and antiviral activity of substituted quercetins. Bioorg. Med. Chem. Lett. 22:353–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ruigrok RW, Baudin F. 1995. Structure of influenza virus ribonucleoprotein particles. II. Purified RNA-free influenza virus ribonucleoprotein forms structures that are indistinguishable from the intact influenza virus ribonucleoprotein particles. J. Gen. Virol. 76(Pt 4):1009–1014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.