

Abstract
The amgRS operon encodes a presumed membrane stress-responsive two-component system linked to intrinsic aminoglycoside resistance in Pseudomonas aeruginosa. Genome sequencing of a lab isolate showing modest pan-aminoglycoside resistance, strain K2979, revealed a number of mutations, including a substitution in amgS that produced an R182C change in the AmgS sensor kinase product of this gene. Introduction of this mutation into an otherwise wild-type strain recapitulated the resistance phenotype, while correcting the mutation in the resistant mutant abrogated the resistant phenotype, confirming that the amgS mutation is responsible for the aminoglycoside resistance of strain K2979. The amgSR182 mutation promoted an AmgR-dependent, 2- to 3-fold increase in expression of the AmgRS target genes htpX and PA5528, mirroring the impact of aminoglycoside exposure of wild-type cells on htpX and PA5528 expression. This suggests that amgSR182 is a gain-of-function mutation that activates AmgS and the AmgRS two-component system in promoting modest resistance to aminoglycosides. Screening of several pan-aminoglycoside-resistant clinical isolates of P. aeruginosa revealed three that showed elevated htpX and PA5528 expression and harbored single amino acid-altering mutations in amgS (V121G or D106N) and no mutations in amgR. Introduction of the amgSV121G mutation into wild-type P. aeruginosa generated a resistance phenotype reminiscent of the amgSR182 mutant and produced a 2- to 3-fold increase in htpX and PA5528 expression, confirming that it, too, is a gain-of-function aminoglycoside resistance-promoting mutation. These results highlight the contribution of amgS mutations and activation of the AmgRS two-component system to acquired aminoglycoside resistance in lab and clinical isolates of P. aeruginosa.
INTRODUCTION
Pseudomonas aeruginosa is a significant opportunistic human pathogen (1, 2) commonly associated with pulmonary infections in patients with cystic fibrosis (CF) (3). Aminoglycosides are an important class of anti-pseudomonal agent often used to treat such infections (4, 5), although their use is associated with resistance development (6, 7). Targeting the 16S rRNA component of the 30S ribosomal subunit, aminoglycosides compromise mRNA translation fidelity, resulting in the production of nonfunctional mistranslated or truncated polypeptides (8). Subsequent cytoplasmic membrane insertion of these aberrant proteins permeabilizes the membrane (9) to allow further aminoglycoside entry and accumulation, ultimately resulting in total inhibition of all cellular ribosomes and cell death (8). Although the exact mechanism(s) by which aminoglycosides achieve their bactericidal effect remains elusive, several recent studies point to the critical involvement of reactive oxygen species (ROS), particularly hydroxyl radicals (10, 11), whose generation is linked to membrane perturbation by aminoglycoside-induced mistranslated products and subsequent activation of an envelope stress response controlled by the CpxRA two-component system (TCS) (12).
Bacterial resistance to aminoglycosides typically results from enzymatic modification of the drug, drug efflux, or target modification (7, 13, 14), with the latter involving mutation of genes for 16S RNA (13, 14) and ribosomal proteins (13, 14) or methylation of 16S RNA by transposon-encoded methyl-transferases (15). 16S rRNA methyl-transferase-mediated aminoglycoside resistance is rarely seen in P. aeruginosa (16), where the most common mechanism of aminoglycoside resistance involves aminoglycoside-modifying enzymes (17) encoded by transmissible genes that are acquired through horizontal gene transfer. This is not, however, the case for CF isolates of P. aeruginosa, where these mechanisms are almost unknown (6) and efflux by the MexXY-OprM multidrug efflux system appears to be the favored aminoglycoside resistance determinant (18–23). P. aeruginosa also possesses and exploits an impressive intrinsic aminoglycoside resistome. Several recent random transposon mutagenesis studies have, for example, identified a large number of genes whose inactivation either enhances (24) or decreases (25–27) aminoglycoside resistance in P. aeruginosa, including several genes involved in lipopolysaccharide (LPS) biosynthesis (26) [LPS is the initial site of aminoglycoside binding during entry into bacterial cells (28, 29)] and energy metabolism (24) [aminoglycoside uptake by bacteria is energy dependent (30)]. Disruption of genes encoding a novel TCS, AmgRS, was also shown to enhance susceptibility to tobramycin (27) and several additional aminoglycosides (26, 31). Intriguingly, a homologue of the EnvZ-OmpR osmoregulatory TCS of Escherichia coli, AmgRS, regulates a number of genes that are more reminiscent of CpxRA targets (27), including a number of proteases (31), and appears to function as part of an envelope stress response to aminoglycoside-induced aberrant polypeptides.
In this report, we identify missense mutations in the sensor component, AmgS, of the AmgRS TCS that are responsible for aminoglycoside resistance in lab and clinical isolates. Dependent on AmgR for resistance, these mutations apparently activate this envelope stress response TCS in P. aeruginosa.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this study are described in Table 1. Bacterial cells were cultured in Luria broth (L broth) and on Luria agar (L agar), with antibiotics as necessary, at 37°C. Plasmid pEX18Tc and its derivatives were maintained or selected in Escherichia coli with 10 μg/ml tetracycline. A tobramycin-resistant derivative of wild-type P. aeruginosa strain K767, K2979, was recovered on L agar containing 1 μg/ml (1× MIC) tobramycin following an 8-day exposure to H2O2 as described previously (32). ΔamgR derivatives of P. aeruginosa were constructed by mobilizing pEX18Tc::ΔamgR (pCG005) into P. aeruginosa from E. coli as described previously (26). Briefly, 700 μl of pCG005-carrying E. coli S17-1 (log phase, cultured at 37°C) was mixed with 300 μl of P. aeruginosa (stationary phase, cultured at 42°C) in a microcentrifuge tube, followed by centrifugation. The resultant cell pellet was then resuspended in 100 μl of L broth and subsequently spotted onto the center of an L agar plate. Following incubation at 37°C for 16 h, bacteria were recovered from the L agar plate in 1 ml of L broth, and P. aeruginosa transconjugants harboring chromosomal inserts of pCG005 were selected on L agar plates containing tetracycline (50 μg/ml) and chloramphenicol (5 μg/ml; to counterselect E. coli S17-1). These were subsequently streaked onto L agar plates containing sucrose (10% [wt/vol]), and sucrose-resistant colonies were screened for chromosomal deletion of amgR using colony PCR with the primers amgRUP-F and amgRDown-R (Table 2) as described elsewhere (26).
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Reference |
|---|---|---|
| E. coli strains | ||
| DH5α | ϕ80d lacZΔM15 endA1 recA1 hsdR17 (rK− mK+) supE44 thi- 1 gyrA96 relA1 F− Δ(lacZYA-argF)U169 | 68 |
| S17-1 | thi pro hsdR recA Tra+ | 69 |
| P. aeruginosa strains | ||
| K767 | PAO1 prototroph (wild-type) | 70 |
| K2979 | Pan-aminoglycoside resistant derivative of K767 | This study |
| K3159 | K767ΔamgR | 26 |
| K3248 | K2979ΔamgR | This study |
| K3249 | K767 derivative carrying the amgSR182C mutation | This study |
| K3250 | K2979 derivative in which the amgSR182C mutation has been restored to wild-type | This study |
| K3260 | K767 derivative carrying the amgSV121G mutation | This study |
| K2153 to K2159, K2161 and K2162 | Pan-aminoglycoside resistant clinical isolates | 20 |
| K3257 | K2161ΔamgR | This study |
| K3198 | K2162ΔamgR | 26 |
| Plasmids | ||
| pEX18Tc | Broad-host-range gene replacement vector; sacB Tcr | 71 |
| pCG005 | pEX18Tc::ΔamgR | 26 |
| pCL4 | pEX18Tc derivative carrying amgSR182C on a 1,801-bp EcoRI-XbaI fragment | This study |
| pCL5 | pEX18Tc derivative carrying amgSWT on a 1,801-bp EcoRI-XbaI fragment | This study |
| pCL6 | pEX18Tc derivative carrying amgSV121G on a 1,801-bp EcoRI-XbaI fragment | This study |
| pCL7 | pEX18Tc derivative carrying amgSD106N on a 1,801-bp EcoRI-XbaI fragment | This study |
Tcr, tetracycline resistant; WT, wild type.
Table 2.
Oligonucleotides used in this study
| Primer | Oligonucleotide sequence (5′→3′) | Reference |
|---|---|---|
| amgRUP-F | GACTGAATTCCTGTAGAAGTCCTGGCGGT | 26 |
| amgRDown-R | GACTCTGCAGCGGCGCTGGAGAAACTGGT | 26 |
| amgS-1801-F | CGCCAGAATTCACGAGGTACACATGCTGACa | This study |
| amgS-1801-R | GCACTGTCTAGATGGCCTTACGGGAAGACCb | This study |
| amgS-V121G-F | CGTCGCAGCTCTGGGGGCGCGCACCGAGCCTCc | This study |
| amgS-V121G-R | GAGGCTCGGTGCGCGCCCCCAGAGCTGCGACGc | This study |
| amgS-D106N-F | GATGGAGCTGGGGCCGAACACCGAGACCCGCCTGc | This study |
| amgS-D106N-R | CAGGCGGGTCTCGGTGTTCGGCCCCAGCTCCATCc | This study |
| amgR-1150-F | GATGCTGTCCATTGATCCAC | This study |
| amgR-1150-R | CGTTCATCAGCAGGTAGACC | This study |
| qPCR-htpX-F | ATCTCCAAGTGGATGGCGA | This study |
| qPCR-htpX-R | CAGCTCTTCGACGGTTTGC | This study |
| qPCR-PA5528-F | ATGCAGCGTGTTCTCAGC | This study |
| qPCR-PA5528-R | CGCTTGGCATTGGCATCCA | This study |
| qPCR-mexX-F | CTATCGGCATCACCAGCG | 37 |
| qPCR-mexX-R | ATCTGGAACAGCACGGTG | 37 |
| qPCR-rpoD-F | ATCCTGCGCAACCAGCAGAA | 37 |
| qPCR-rpoD-R | TCGACATCGCGCGGTTGATT | 37 |
The EcoRI site is underlined.
The XbaI site is underlined.
The mutation site is underlined.
Construction of P. aeruginosa amgS mutants.
To introduce an R182-to-C mutation in amgS (amgSR182C mutation) into P. aeruginosa strain K767, a DNA fragment carrying the mutation was first amplified from the chromosome of strain K2979 and then cloned into the gene replacement vector pEX18Tc. Briefly, a 1,801-bp fragment including ca. 900 bp upstream and downstream of the mutation site within the amgS gene was amplified using PCR. Amplification was achieved using primers amgS-1801-F and amgS-1801-R (Table 2) in a 50-μl reaction mixture containing 10 ng of chromosomal DNA, 1 U of Phusion high-fidelity DNA polymerase (New England BioLabs, Ltd., Pickering, Ontario, Canada), 1× Phusion HF buffer, 5% (vol/vol) dimethyl sulfoxide (DMSO), primers at a 0.6 μM final concentration, and deoxynucleoside triphosphates (dNTPs) at a 0.2 mM final concentration. The mixture was heated for 3 min at 98°C, followed by 35 cycles of 0.5 min at 98°C, 0.5 min at 70.1°C, and 0.9 min at 72°C, before finishing with 10 min at 72°C. The PCR product was subsequently cloned into plasmid pEX18Tc as an EcoRI-XbaI-restricted fragment to yield plasmid pCL4. pCL4 was mobilized into P. aeruginosa strain K767 from E. coli strain S17-1 as described above, and P. aeruginosa transconjugants harboring chromosomal inserts of pCL4 were selected on L agar plates containing tetracycline (50 μg/ml) and chloramphenicol (5 μg/ml; to counterselect E. coli). These transconjugants were then streaked onto L agar plates containing sucrose (10% [wt/vol]), and the resultant sucrose-resistant colonies were screened for the chromosomal amgSR182C mutation by PCR amplification and sequencing of the amgS gene. To construct a derivative of K2979 in which the amgSR182C mutation was corrected, yielding wild-type amgS, the wild-type gene was amplified as described above from strain K767 and cloned into pEX18Tc to yield pCL5. Plasmid pCL5 was mobilized into P. aeruginosa K2979 from E. coli S17-1 as described above, and a derivative of K2979 in which the mutation had been corrected was recovered, again as described above, using sucrose selection, amgS amplification, and sequencing. To introduce an amgSV121G mutation into strain K767, pCL6, a pEX18Tc-based gene replacement vector carrying the desired mutation, was constructed by using a site-directed mutagenesis single-primer PCR protocol with slight modifications (33). Briefly, individual single-primer amplifications were carried out using the mutagenic primers amgS-V121G-F and amgS-V121G-R (Table 2) at a 0.6 μM final concentration in a 50-μl reaction mixture containing 25 ng of pCL5 template, 1 U of Phusion high-fidelity DNA polymerase, 1× Phusion HF buffer, 5% (vol/vol) DMSO, and dNTPs at a 0.2 mM final concentration. The mixtures were heated for 3 min at 98°C, followed by 25 cycles of 0.5 min at 98°C, 0.5 min at 72°C, and 4 min at 72°C, before finishing with 10 min at 72°C. The two PCR products were then combined in equal volumes (giving a total volume of 20 μl) and heated for 5 min at 95°C, followed by a standard slow stepwise cooling to a final temperature of 37°C (33) to promote random reannealing of the denatured plasmid templates and PCR products. Eight units of DpnI (New England BioLabs, Ltd.) was subsequently added to the reannealed templates/products and incubated for 1 h at 37°C to digest the methylated parental plasmid template. Finally, 5 μl of the DpnI-treated material was transformed into E. coli DH5α, plasmids were recovered from transformants, and the amgS gene was sequenced. Plasmid pCL6, in which the amgSV121G mutation was confirmed, was then mobilized into P. aeruginosa K767 from E. coli S17-1 as described above, and a derivative of K767 containing the chromosomal amgSV121G mutation was identified following sucrose selection, amgS amplification, and sequencing. Plasmid pCL7, a pEX18Tc-based gene-replacement vector carrying the amgSD106N mutation, was generated as described above for pCL6 except that the mutagenic primers amgS-D106N-F and amgS-D106N-R (Table 2) were employed.
DNA methods.
Standard protocols were used for restriction endonuclease digestion, ligation, transformation, plasmid isolation, and agarose gel electrophoresis, as described by Sambrook and Russell (34). Plasmid DNAs were also prepared from E. coli using a GeneJET plasmid miniprep kit (Fermentas Canada Inc., Burlington, Ontario, Canada) according to a protocol provided by the manufacturer. Chromosomal DNA of P. aeruginosa was extracted using a DNeasy blood and tissue kit (Qiagen Inc., Mississauga, Ontario, Canada) according to the manufacturer's protocol. DNA fragments used for cloning were extracted from agarose gels using a Wizard SV gel and PCR cleanup system (Fisher Scientific, Ltd., Nepean, Ontario, Canada) and, once cloned, were sequenced to verify that no unintended mutations were introduced during PCR. Competent E. coli cells were prepared as described previously (35). For sequencing, the amgRS genes were individually amplified from the chromosome of P. aeruginosa using the primer pairs amgR-1150-F–amgR-1150-R and amgS-1801-F–amgS-1801-R (Table 2), respectively, using components and conditions as described above for amgS except that a 0.58-min extension time was used for the amgR PCR. Oligonucleotide synthesis was carried out by Integrated DNA Technologies (Coralville, IA), and nucleotide sequencing was carried out by ACGT Corp. (Toronto, Ontario, Canada) using universal and custom primers. Genome sequencing and polymorphism detection, annotation, and validation were carried out as described previously (36).
Quantitative real-time PCR.
Bacterial RNA was isolated, purified, and reverse transcribed into cDNA as described previously (37). The primers used in quantitative real-time PCR (those with a “qPCR” designation [Table 2]) were designed to amplify specific gene fragments with lengths of 99 bp (htpX), 74 bp (PA5528), 142 bp (mexX), or 91 bp (rpoD) and were validated as described elsewhere (the mexX and rpoD primers were validated previously) (37). The amplification efficiencies of the quantitative real-time PCR primer pairs for htpX and PA5528 were determined to be 101.6% (correlation coefficient, r2 = 0.998) and 99.7% (r2 = 0.997), respectively. All quantitative real-time PCR primer pairs used in the present study had a minimum 4-log10 dynamic range. The expression of mexX, htpX, PA5528, and rpoD was assessed by quantitative real-time PCR as described previously using a CFX96 real-time PCR detection system (Bio-Rad) (37). For each gene studied, at least one control reaction with no cDNA template was included in each experiment to check for contamination of the reagent(s) and to identify unintended amplification products (e.g., primer dimers). The levels of expression of the target genes in each strain studied, normalized against that of the reference gene, were calculated using the standard analysis feature of the CFX manager software version 1.6 (Bio-Rad) and are reported here as fold change relative to that in the P. aeruginosa PAO1 wild-type strain K767, unless otherwise specified.
Antibiotic susceptibility assay.
The susceptibility of P. aeruginosa to antimicrobial agents was assessed using the 2-fold serial microtiter broth dilution method described previously (38), with an inoculum of ∼5 × 105 cells per ml. MICs were recorded as the lowest concentration of antibiotic inhibiting visible growth after 18 h of incubation at 37°C.
RESULTS
amgS mutation in pan-aminoglycoside-resistant P. aeruginosa.
In a previous study, extended (8-day) exposure of wild-type P. aeruginosa to peroxide enhanced the recovery of amikacin-resistant mutants that were ultimately revealed to be pan-aminoglycoside resistant (32). Tobramycin-resistant isolates were also recovered but not studied further. One of these, strain K2979, was here assessed for changes in susceptibility to additional aminoglycosides. As seen in Table 3, the mutant showed modest increases in resistance to several aminoglycosides but not to the aminocyclitol spectinomycin. Since spectinomycin is thought to be a substrate for MexXY-OprM—resistance to this agent increases in mutants expressing mexXY (20, 37, 39)—this suggested that aminoglycoside resistance in K2929 was not attributable to MexXY-OprM. Consistent with this, mexXY expression was not elevated in strain K2979 relative to its parent strain (data not shown). To identify the mutation(s) responsible for the modest aminoglycoside resistance of strain K2979 and thus gain some insight into the resistance mechanism, its genome was sequenced. Thirty-seven mutations were identified in the mutant, one of which occurred in the amgS gene [a C-to-T transition at nucleotide 544, resulting in an Arg-to-Cys substitution at amino acid residue 182 (R182C)], encoding the sensor component of the AmgRS TCS (27). As a regulator of a probable envelope stress response, AmgRS contributes to intrinsic aminoglycoside resistance—transposon insertions into amgRS enhances susceptibility to these agents (27). To confirm the contribution of the amgSR182C mutation to aminoglycoside resistance in strain K2979, the mutation was engineered into wild-type P. aeruginosa strain K767, and the impact on the aminoglycoside resistance in the resulting mutant strain, K3249, was assessed. As with strain K2979, strain K3249 showed a modest increase in resistance to all aminoglycosides tested and no change in susceptibility to spectinomycin (Table 3). Similarly, reverting the amgSR182C mutation of strain K2979 to the wild-type sequence in strain K3250 restored aminoglycoside susceptibility to essentially wild-type levels (Table 3). Thus, it is concluded that an amgSR182C mutation promotes modest pan-aminoglycoside resistance in P. aeruginosa.
Table 3.
Effect of amgRS mutations on pan-aminoglycoside resistance in laboratory and clinical P. aeruginosa strains
| Strain | AmgRa | AmgSa | MIC (μg/ml)b |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TOB | GEN | AMI | STR | PAR | NEO | SPC | CAM | TET | ERY | |||
| Lab strainsc | ||||||||||||
| K767 | WT | WT | 1 | 2 | 2 | 16 | 256 | 32 | 1,024 | 64 | 16 | 512 |
| K2979 | WT | R182C | 4 | 4 | 4 | 32 | 512 | 64 | 1,024 | |||
| K3249 | WT | R182C | 2 | 4 | 4 | 32 | 512 | 64 | 1,024 | 64 | 16 | 512 |
| K3250 | WT | WT | 2 | 2 | 2 | 16 | 256 | 32 | 1,024 | |||
| K3159 | − | WT | 0.5 (2)c | 0.5 (4) | 0.25 (8) | 8 (2) | 16 (16) | 4 (8) | 512 (2) | |||
| K3248 | − | R182C | 0.5 (8) | 0.5 (8) | 0.25 (16) | 8 (4) | 16 (32) | 4 (16) | 512 (2) | |||
| K3260 | WT | V121G | 2 | 4 | 4 | 32 | 512 | 64 | 256 | 32 | 8 | 256 |
| Clinical strains | ||||||||||||
| K2161 | WT | D106N | 8 | 32 | 16 | 64 | 2,048 | 512 | 256 | |||
| K3257 | − | D106N | 1 (8) | 2 (16) | 2 (8) | 8 (8) | 64 (32) | 8 (64) | 256 (1) | |||
| K2162 | WT | WT | 64 | 256 | 128 | 256 | >4,096 | 512 | 2,048 | |||
| K3198 | − | WT | 16 (4) | 64 (4) | 32 (4) | 128 (2) | 4,096 (≥2) | 128 (4) | 2,048 (1) | |||
WT, wild type; −, absent.
TOB, tobramycin; GEN, gentamicin; AMI, amikacin; STR, streptomycin; PAR, paromomycin; NEO, neomycin; SPC, spectinomycin; CAM, chloramphenicol; TET, tetracycline; ERY, erythromycin. Values in parentheses are fold reductions in MIC for the amgR deletion mutants relative to their corresponding parental strains.
Strain K767 derivatives.
amgSR182C is a gain-of-function mutation.
AmgRS is proposed to regulate an envelope stress response that responds to membrane perturbation by aminoglycoside-generated mistranslated polypeptides and, as such, is activated by aminoglycosides (27). Two known targets of AmgRS that are upregulated by aminoglycoside exposure and that contribute to intrinsic aminoglycoside resistance in P. aeruginosa are htpX, encoding a cytoplasmic membrane protease, and PA5528, encoding a protein of unknown function (27, 31). We sought to first validate the AmgR-dependent induction of htpX and PA5528 by aminoglycosides in wild-type P. aeruginosa and then assess their AmgR-dependent expression in the amgSR182C mutant strain K2979. Contrary to a previous report (27), neither htpX nor PA5528 was induced by exposure of log-phase cells to tobramycin at 1× MIC (Fig. 1), although increasing tobramycin to 4× MIC did provide for a 2-fold increase in htpX expression (no increase in PA5528 expression was observed) (data not shown). In contrast, the aminoglycosides neomycin, paromomycin, and streptomycin all promoted a 2- to 3-fold induction of htpX at 1× MIC, with neomycin and paromomycin also providing for a 2-fold induction of PA5528 at this level of antimicrobial (Fig. 1). Importantly, and consistent with these genes being AmgRS regulated, the aminoglycoside-promoted induction of htpX and PA5528 was abrogated in the amgR knockout strain K3159 (Fig. 1). Bacteriostatic agents such as chloramphenicol and spectinomycin, which also target the ribosome but do not cause mistranslation or generation of membrane-perturbing aberrant polypeptides, failed to induce htpX or PA5528 (Fig. 1), which is consistent with membrane perturbation being the signal for AmgRS activation. Thus, htpX and PA5528 are markers for AmgRS activation by aminoglycosides, although there is some variability with respect to a given aminoglycoside's ability to activate this system.
Fig 1.

AmgR-dependent induction of htpX and PA5528 by mistranslation-promoting aminoglycosides. Mid- to late-log phase cultures (optical density at 600 nm [OD600] = 0.8 to 0.9) of the P. aeruginosa PAO1 strains K767 (WT) and K3159 (ΔamgR) were exposed to 1× MIC of the indicated antimicrobials (—, no antimicrobial; PAR, paromomycin; NEO, neomycin; STR, streptomycin; TOB, tobramycin; SPC, spectinomycin; CAM, chloramphenicol) for 30 min before harvesting for RNA extraction. The expression of htpX (top) and PA5528 (bottom) was subsequently assessed in these strains using real-time quantitative PCR. Expression was normalized to rpoD and is reported relative to the untreated wild-type strain K767. Values are means ± standard errors of the means (SEMs) (error bars) from at least three independent determinations, each performed in triplicate.
As seen in Fig. 2, expression of these genes was enhanced 2- to 3-fold in the amgSR182C mutant strain K2979 and in the K767 derivative harboring the same mutation (i.e., K3249). Moreover, reversion of the mutation in strain K3250 reversed this increase (Fig. 2) confirming that the amgSR182C mutation was indeed responsible for activating the AmgRS TCS. As with the aminoglycoside-mediated activation of AmgRS, amgSR182C activation of AmgRS in K2979 was nullified in its amgR knockout derivative, where htpX and PA5528 expression was reduced to levels seen in the amgR knockout derivative of K767 (Fig. 2; compare strains K3248 and K3159). These results indicate that amgSR182C is a gain-of-function mutation that mimics the effects of aminoglycosides on the AmgS sensor, promoting AmgR-dependent activation of htpX and PA5528 and, consequently, modest aminoglycoside resistance.
Fig 2.

Effect of amgRS mutations on the expression of known AmgRS-regulated genes in P. aeruginosa. The expression of htpX and PA5528 was assessed in the indicated strains using real-time quantitative PCR. The status of the amgR and amgS gene in each strain is indicated (WT, wild type; −, absent), as is the identity of any mutations. Expression was normalized to rpoD and is reported relative to that of the wild-type P. aeruginosa PAO1 strain K767 (fold-change). Values are means ± standard errors of the means (SEMs) from at least three independent determinations, each performed in triplicate.
Additional amgS gain-of-function mutations in aminoglycoside-resistant clinical isolates of P. aeruginosa.
To assess whether AmgRS, possibly via amgS gain-of-function mutations, contributes to aminoglycoside resistance in clinical P. aeruginosa isolates, several aminoglycoside-resistant CF lung isolates were examined for increased expression of htpX and PA5528, as evidence of AmgRS activation. Nine isolates [including one aminoglycoside-susceptible control, K2155 (20)] were examined, of which three (K2157, K2158, and K2161) showed approximately 2-fold increases in expression of both genes and one (K2162) showed a 2-fold increase in htpX only (Fig. 3). To assess whether this increase in expression and resistance was linked to amgRS, attempts were made to delete the amgR gene in these mutants. This gene had been deleted in K2162 previously (26), but despite numerous attempts with the other three isolates, a knockout was achieved in strain K2161 only. In the case of the K2162 amgR knockout derivative, strain K3198, the impact of losing AmgR on aminoglycoside resistance was modest and comparable to that seen in the K767 amgR knockout derivative, strain K3159, in which AmgRS is not activated by mutation (comparable fold reductions in MICs [Table 3]), and the enhanced htpX expression associated with K2162 was abrogated (Fig. 3). In contrast, loss of amgR in the K2161 derivative, strain K3257, markedly reduced aminoglycoside MICs (Table 3) as well as abrogating the enhanced expression of both htpX and PA5528 (Fig. 3). This suggested that in K2161, and perhaps to a lesser extent in K2162, mutational activation of AmgRS (as reflected by the increased htpX and/or PA5528 expression) contributed to its aminoglycoside-resistant phenotype. To assess this, the amgS gene was amplified from the htpX- and/or PA5528-overexpressing strains K2157, K2158, K2161, and K2162, as well as two control strains that did not show any increases in htpX or PA5528 expression, K2154 and K2155, and sequenced. Ignoring silent mutations, a single missense mutation producing a Val-to-Gly substitution at residue 121 (V121G) of AmgS was identified in strains K2157 and K2158, while a single missense mutation producing an Asp-to-Asn substitution at residue 106 (D106N) of AmgS was identified in strain K2161. Strain K2162, as well as the control strains K2154 and K2155, lacked amino acid-altering mutations in amgS. Subsequent sequencing of the regulator component-encoding gene amgR in all these isolates revealed no mutations, suggesting that in strains K2157, K2158, and K2161, the two aforementioned amgS mutations were responsible for the enhanced htpX and PA5528 expression and, possibly, aminoglycoside resistance of these strains.
Fig 3.
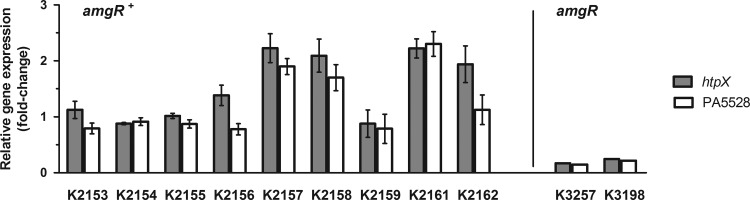
Expression of AmgRS-regulated genes in clinical isolates of P. aeruginosa. The expression of htpX and PA5528 was assessed in the indicated clinical strains using real-time quantitative PCR. The impact of loss of amgR on htpX and PA5528 expression in the K2161 and K2162 derivatives, K3257 and K3198, respectively, is shown at right. Expression was normalized to rpoD and is reported relative to the wild-type P. aeruginosa PAO1 strain K767 (fold-change). Values are means ± standard errors of the means (SEMs) from at least three independent determinations, each performed in triplicate.
To confirm that V121G and D106N were additional amgS gain-of-function mutations contributing to the aminoglycoside resistance of the clinical isolates K2157, K2158, and K2161, we attempted to introduce them into the chromosome of P. aeruginosa wild-type strain K767 and assess their impact on htpX and PA5528 expression and aminoglycoside resistance. The amgSV121G mutation was introduced into K767, yielding strain K3260, in which a 2- to 3-fold increase in expression of htpX and PA5528 was observed (Fig. 2). As expected, strain K3260 also showed a modest increase in resistance to all aminoglycosides tested but not to spectinomycin (Table 3), confirming V121G as a second amgS gain-of-function mutation linked to aminoglycoside resistance in P. aeruginosa, and amgS gain-of-function mutations as contributors to pan-aminoglycoside resistance in clinical isolates. Interestingly, and in contrast to the amgSR182C mutant strain, K3249, where no change was observed, K3260 showed an increase (4-fold) in susceptibility to spectinomycin (Table 3). K3260 but not K3249 was also more susceptible (2-fold) to other ribosome-targeting bacteriostatic agents, including chloramphenicol, tetracycline, and erythromycin (Table 3). Unfortunately, and despite considerable effort, attempts at introducing the amgSD106N mutation into the chromosome of strain K767 were unsuccessful, precluding its study here.
DISCUSSION
The AmgRS TCS of P. aeruginosa is an apparent envelope stress response-regulatory protein pair predicted to respond to membrane perturbation caused by aminoglycoside-generated mistranslated polypeptides and as such plays a role in intrinsic aminoglycoside resistance in this organism (26, 27). Results presented here demonstrate that this TCS also contributes to acquired resistance to these agents, in both lab and clinical isolates, by means of gain-of-function mutations in the AmgS sensor component that activate AmgRS independent of its natural inducer(s). Still, the contribution of these amgS mutations to aminoglycoside resistance is modest (they increase resistance in the lab strain, K767, 2-fold), and, thus, the markedly greater resistance of clinical isolates harboring such mutations (Table 3) likely reflects the presence of additional resistance determinants/mutations in these isolates. In agreement with this, the MexXY-OprM multidrug efflux system has been shown to contribute to the aminoglycoside resistance of clinical isolate K2161 (20), which also harbors the amgSD106N mutation. Given the often-multifactorial nature of resistance in clinical strains, determinants of modest resistance such as amgS gain-of-function mutations can be important contributors.
Consistent with the amgS mutations identified in this study being gain-of-function mutations, they upregulate known AmgRS targets, htpX and PA5528, in an AmgR-dependent manner, reminiscent of the induction of these genes by the aminoglycosides paromomycin and neomycin. Interestingly, only htpX expression is increased in the aminoglycoside-resistant clinical isolate K2162, and while this mutant lacks mutations in amgRS, htpX expression in K2162 is wholly AmgR dependent. This greatly resembles the effect of the aminoglycosides streptomycin (at 1× MIC) and tobramycin (at 4× MIC), which also promote AmgR-dependent htpX expression only. The differential impact of AmgRS activation on the expression of gene targets of this TCS might reflect the strength of the inducing signal, with a stronger signal being needed to upregulate PA5528. Possibly, then, a mutation in K2162 causes an upstream stress/signal that acts on AmgRS to stimulate htpX expression, but this signal is weaker than that provided by an amgS mutation or exposure to paromomycin or neomycin and yet is comparable to the stress/signal provided by the other aminoglycosides. Although paromomycin and neomycin, 4,5-disubstituted aminoglycosides, act on the same site of the ribosome as streptomycin (40) and 4,6-disubstituted aminoglycosides such as tobramycin and gentamicin (41) and like these aminoglycosides cause misreading and production of aberrant polypeptides, there are indications of differences, too. In addition to promoting mistranslation, neomycin and paromomycin also negatively impact assembly of the 30S ribosomal subunit (42), although the 4,6-disubstituted aminoglycosides were not examined in this study and it is unclear if they have ever been assessed in this context. Similarly, switching the 6′ substituent of ring 1 of 4,5- versus 4,6-disubstituted aminoglycosides differentially impacts ribosome inhibitory activity—replacing the 6′ NH2 group with an OH has a major negative impact on ribosome inhibition by the 4,6-disubstituted aminoglycoside kanamycin, while replacing the NH2 of neomycin with OH (resulting in paromomycin) has a minimal impact (43). In any case, there are clearly differences in the AmgRS-responsive stress signals propagated by neomycin and paromomycin compared to the other aminoglycosides. Whether this relates to the degree of stress or differences in stress signals is at present unknown.
A puzzling observation in the present study is the lack of htpX induction by tobramycin at 1× MIC—htpX induction required the use of 4× MIC. Interestingly, and in contrast to the other aminoglycosides and ribosome-targeting agents examined in this study, tobramycin failed to elicit immediate growth inhibition when applied to log-phase P. aeruginosa at 1× MIC—immediate growth inhibition was seen only at 4× MIC for this agent (data not shown). Since by definition 1× MIC tobramycin is sufficient to kill/block growth of P. aeruginosa in a typical susceptibility assay involving diluted overnight cultures (typically 5 × 106 cells/ml), this agent is apparently less effective on denser P. aeruginosa cultures. The apparent lack of perturbation of log-phase cells by 1× MIC tobramycin likely explains the failure of tobramycin to induce htpX at 1× MIC.
As a TCS, the AmgS histidine kinase is expected to sense/bind its cognate ligand/signal, activating its kinase activity to phosphorylate AmgR, which in turn binds to and promotes expression of target genes (e.g., htpX and PA5528). Thus, amgS gain-of-function mutations probably stimulate the kinase activity and AmgR phosphorylation by mimicking the effect of its ligand(s)/signaling molecule(s). Such mutations have been reported for other TCS kinases (44), including EnvZ (45–49), the closest homologue of AmgS and part of the E. coli osmoregulatory TCS, EnvZ-OmpR, which controls expression of the ompC and ompF porin genes (50). These kinases are characterized by an N-terminal periplasmic sensor or input domain flanked by 2 transmembrane domains (51) (Fig. 4) and a C-terminal output module that contains a phosphotransfer/dimerization domain and ATP binding/catalytic domain (52), with the input and output domains being connected by a cytoplasmic linker region that occurs immediately following the second transmembrane domain (47) (Fig. 4). Mutations activating EnvZ (45–47) and other kinases (44, 53, 54) often map to the linker region, a domain called HAMP (because it is present in histidine kinases, adenyl cyclases, methyl-accepting chemotaxis proteins, and phosphatases) that is proposed to transmit signals from the periplasmic input sensor domain to the cytoplasmic output domain (55). Activating mutations in the periplasmic sensor domain in EnvZ have not generally been described, although there is one report of a P148S mutation in the sensor domain of EnvZ in a bile-resistant mutant E. coli strain (56), where bile resistance can be an indicator of increased ompC expression and, therefore, of activation of OmpR. Based on an alignment with EnvZ, the AmgS gain-of-function mutations described in the present study map to the sensor domain (D106N and V121G) and the linker (R182C) (Fig. 4). That the V121G and R182C mutations map to functionally separate regions of AmgS is noteworthy given the differential impact of these mutations on susceptibility to the bacteriostatic agents spectinomycin, chloramphenicol, tetracycline, and erythromycin, which, like aminoglycosides, target the ribosome but, unlike aminoglycosides, do not cause mistranslation. Whether this difference in susceptibility reflects a difference in the region being disrupted (sensor versus linker) or is unique to the V121G mutation is unclear. In any case, it is curious that mutational activation of AmgS would, in some instances at least, specifically compromise resistance to bacteriostatic ribosome-targeting agents while at the same time promoting resistance to bactericidal ribosome-targeting agents like aminoglycosides.
Fig 4.

Sequence alignment of AmgS from P. aeruginosa and its homologue EnvZ from E. coli. Sequences were obtained from the NCBI protein database (NCBI references: AmgS, NP_253886.1; EnvZ, NP_417863.1). A multiple sequence alignment program, T-Coffee (72), was used. Residue numbers (EnvZ annotation; domains based on references 51 and 47) are provided in parentheses. The C-terminal cytoplasmic catalytic domain alignment (EnvZ annotation, residues 237 to 450) is not displayed. The three putative gain-of-function AmgS mutation sites (D106, V121, and R182) are in bold and indicated by asterisks (*). TM, transmembrane domain.
Mutations in envelope stress response TCS sensor kinase genes are not uncommon in bacteria and have been linked to antimicrobial resistance, usually to envelope-targeting agents. For instance, mutations in the cpxA gene encoding the sensor kinase of the CpxRA envelope stress response TCS has been shown to facilitate amikacin (57) and cephalosporin (58) resistance in E. coli and Salmonella enterica serovar Typhimurium, respectively. In addition, a G226D mutation in envZ has also been linked to cephalosporin resistance in S. Typhimurium (58). Similarly, mutations in the vraS (59, 60) and graS (61) sensor kinase genes of the VraRS and GraRS envelope stress response TCSs of Staphylococcus aureus have been shown to promote reduced susceptibility to glycopeptide antibiotics. Mutations in the sensor kinase genes of related envelope-modifying TCSs, including the phoQ and pmrB genes of the PhoPQ and PmrAB TCSs, which are best characterized in Salmonella enterica and P. aeruginosa, also promote antimicrobial resistance. Responsive to Mg2+ and Fe3+, these TCSs control genes linked to LPS modification and promotion of resistance to polycationic antimicrobials, including the polymyxins and antimicrobial peptides that target the cell envelope (62–64). Mutations in the HAMP linker region of PmrB have been found in polymyxin B-resistant lab isolates of Salmonella enterica serovar Typhimurium (65) and colistin-resistant clinical isolates of P. aeruginosa (66), in the latter instance associated with increased expression of the LPS modification arn locus, which is a target for PmrA and a determinant of colistin resistance (63). Mutations in the sensor domain of PmrB have also been reported in polymyxin B (67)- and colistin (66)-resistant clinical isolates of P. aeruginosa, in the latter instance again associated with increased arn expression. Clearly, then, envelope stress responses can promote antimicrobial resistance in P. aeruginosa and other pathogens, and, as such, mutational activation of the regulatory TCSs can be a determinant of resistance in these bacteria.
ACKNOWLEDGMENTS
This work was funded by an operating grant from Cystic Fibrosis Canada (to K.P.). The genome sequencing was supported by the Pathogen Functional Genomics Resource Center (PFGRC) (contract N01-AI15447).
Footnotes
Published ahead of print 4 March 2013
REFERENCES
- 1. Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK. 2008. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect. Control Hosp. Epidemiol. 29:996–1011 [DOI] [PubMed] [Google Scholar]
- 2. Zhanel GG, DeCorby M, Adam H, Mulvey MR, McCracken M, Lagacé-Wiens P, Nichol KA, Wierzbowski A, Baudry PJ, Tailor F, Karlowsky JA, Walkty A, Schweizer F, Johnson J, the Canadian Antimicrobial Resistance Alliance, Hoban DJ. 2010. Prevalence of antimicrobial-resistant pathogens in Canadian hospitals: results of the Canadian Ward Surveillance Study (CANWARD 2008). Antimicrob. Agents Chemother. 54:4684–4693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lyczak JB, Cannon CL, Pier GB. 2002. Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 15:194–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ratjen F, Brockhaus F, Angyalosi G. 2009. Aminoglycoside therapy against Pseudomonas aeruginosa in cystic fibrosis: a review. J. Cyst. Fibros. 8:361–369 [DOI] [PubMed] [Google Scholar]
- 5. Cantón R, Cobos N, De Gracia J, Baquero F, Honorato J, Gartner S, Alvarez A, Salcedo A, Oliver A, García-Quetglas E, the Spanish Consensus Group for Antimicrobial Therapy in the Cystic Fibrosis Patient 2005. Antimicrobial therapy for pulmonary pathogenic colonisation and infection by Pseudomonas aeruginosa in cystic fibrosis patients. Clin. Microbiol. Infect. 11:690–703 [DOI] [PubMed] [Google Scholar]
- 6. Poole K. 2011. Pseudomonas aeruginosa: resistance to the max. Front. Microbiol. 2:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Poole K. 2005. Aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49:479–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davis BD. 1987. Mechanism of bactericidal action of aminoglycosides. Microbiol. Rev. 51:341–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davis BD, Chen LL, Tai PC. 1986. Misread protein creates membrane channels: an essential step in the bactericidal action of aminoglycosides. Proc. Natl. Acad. Sci. U. S. A. 83:6164–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. 2012. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336:315–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810 [DOI] [PubMed] [Google Scholar]
- 12. Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. 2008. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 135:679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shakil S, Khan R, Zarrilli R, Khan A. 2008. Aminoglycosides versus bacteria—a description of the action, resistance mechanism, and nosocomial battleground. J. Biomed. Sci. 15:5–14 [DOI] [PubMed] [Google Scholar]
- 14. Jana S, Deb JK. 2006. Molecular understanding of aminoglycoside action and resistance. Appl. Microbiol. Biotechnol. 70:140–150 [DOI] [PubMed] [Google Scholar]
- 15. Wachino JI, Arakawa Y. 2012. Exogenously acquired 16S rRNA methyltransferases found in aminoglycoside-resistant pathogenic Gram-negative bacteria: an update. Drug Resist. Updates 15:133–148 [DOI] [PubMed] [Google Scholar]
- 16. Yokoyama K, Doi Y, Yamane K, Kurokawa H, Shibata N, Shibayama K, Yagi T, Kato H, Arakawa Y. 2003. Acquisition of 16S rRNA methylase gene in Pseudomonas aeruginosa. Lancet 362:1888–1893 [DOI] [PubMed] [Google Scholar]
- 17. Ramirez MS, Tolmasky ME. 2010. Aminoglycoside modifying enzymes. Drug Resist. Updates 13:151–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Islam S, Jalal S, Wretlind B. 2004. Expression of the MexXY efflux pump in amikacin-resistant isolates of Pseudomonas aeruginosa. Clin. Microbiol. Infect. 10:877–883 [DOI] [PubMed] [Google Scholar]
- 19. Islam S, Oh H, Jalal S, Karpati F, Ciofu O, Høiby N, Wretlind B. 2009. Chromosomal mechanisms of aminoglycoside resistance in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Clin. Microbiol. Infect. 15:60–66 [DOI] [PubMed] [Google Scholar]
- 20. Sobel ML, McKay GA, Poole K. 2003. Contribution of the MexXY multidrug transporter to aminoglycoside resistance in Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 47:3202–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Westbrock-Wadman S, Sherman DR, Hickey MJ, Coulter SN, Zhu YQ, Warrener P, Nguyen LY, Shawar RM, Folger KR, Stover CK. 1999. Characterization of a Pseudomonas aeruginosa efflux pump contributing to aminoglycoside impermeability. Antimicrob. Agents Chemother. 43:2975–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morita Y, Tomida J, Kawamura Y. 2012. MexXY multidrug efflux system of Pseudomonas aeruginosa. Front. Microbiol. 3:408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vettoretti L, Plésiat P, Muller C, El Garch F, Phan G, Attrée I, Ducruix A, Llanes C. 2009. Efflux unbalance in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Antimicrob. Agents Chemother. 53:1987–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El'Garch F, Jeannot K, Hocquet D, Llanes-Barakat C, Plésiat P. 2007. Cumulative effects of several nonenzymatic mechanisms on the resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother. 51:1016–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gallagher LA, Shendure J, Manoil C. 2011. Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. mBio 2:e00315–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krahn T, Gilmour C, Tilak J, Fraud S, Kerr N, Lau CHF, Poole K. 2012. Determinants of intrinsic aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 56:5591–5602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee S, Hinz A, Bauerle E, Angermeyer A, Juhaszova K, Kaneko Y, Singh PK, Manoil C. 2009. Targeting a bacterial stress response to enhance antibiotic action. Proc. Natl. Acad. Sci. U. S. A. 106:14570–14575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hancock RE, Farmer SW, Li ZS, Poole K. 1991. Interaction of aminoglycosides with the outer membranes and purified lipopolysaccharide and OmpF porin of Escherichia coli. Antimicrob. Agents Chemother. 35:1309–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kadurugamuwa JL, Lam JS, Beveridge TJ. 1993. Interaction of gentamicin with the A band and B band lipopolysaccharides of Pseudomonas aeruginosa and its possible lethal effect. Antimicrob. Agents Chemother. 37:715–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Taber HW, Mueller JP, Miller PF, Arrow AS. 1987. Bacterial uptake of aminoglycoside antibiotics. Microbiol. Rev. 51:439–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hinz A, Lee S, Jacoby K, Manoil C. 2011. Membrane proteases and aminoglycoside antibiotic resistance. J. Bacteriol. 193:4790–4797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fraud S, Poole K. 2011. Oxidative stress induction of the mexXY multidrug efflux genes and promotion of aminoglycoside resistance development in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 55:1068–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Edelheit O, Hanukoglu A, Hanukoglu I. 2009. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 9:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 35. Inoue H, Nojima H, Okayama H. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 96:23–28 [DOI] [PubMed] [Google Scholar]
- 36. Boyle-Vavra S, Jones M, Gourley BL, Holmes M, Ruf R, Balsam AR, Boulware DR, Kline S, Jawahir S, DeVries A, Peterson SN, Daum RS. 2011. Comparative genome sequencing of an isogenic pair of USA800 clinical methicillin-resistant Staphylococcus aureus isolates obtained before and after daptomycin treatment failure. Antimicrob. Agents Chemother. 55:2018–2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lau CHF, Fraud S, Jones M, Peterson SN, Poole K. 2012. Reduced expression of the rplU-rpmA ribosomal protein operon in mexXY-expressing pan-aminoglycoside-resistant mutants of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 56:5171–5179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jo JTH, Brinkman FSL, Hancock REW. 2003. Aminoglycoside efflux in Pseudomonas aeruginosa: involvement of novel outer membrane proteins. Antimicrob. Agents Chemother. 47:1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morita Y, Sobel ML, Poole K. 2006. Antibiotic inducibility of the MexXY multidrug efflux system of Pseudomonas aeruginosa: involvement of the antibiotic-inducible PA5471 gene product. J. Bacteriol. 188:1847–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. 2000. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 407:340–348 [DOI] [PubMed] [Google Scholar]
- 41. Francois B, Russell RJ, Murray JB, Aboul-ela F, Masquida B, Vicens Q, Westhof E. 2005. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 33:5677–5690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mehta R, Champney WS. 2002. 30S ribosomal subunit assembly is a target for inhibition by aminoglycosides in Escherichia coli. Antimicrob. Agents Chemother. 46:1546–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Salian S, Matt T, Akbergenov R, Harish S, Meyer M, Duscha S, Shcherbakov D, Bernet BB, Vasella A, Westhof E, Bottger EC. 2012. Structure-activity relationships among the kanamycin aminoglycosides: role of ring I hydroxyl and amino groups. Antimicrob. Agents Chemother. 56:6104–6108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Appleman JA, Stewart V. 2003. Mutational analysis of a conserved signal-transducing element: the HAMP linker of the Escherichia coli nitrate sensor NarX. J. Bacteriol. 185:89–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Giraud A, Arous S, De Paepe M, Gaboriau-Routhiau V, Bambou JC, Rakotobe S, Lindner AB, Taddei F, Cerf-Bensussan N. 2008. Dissecting the genetic components of adaptation of Escherichia coli to the mouse gut. PLoS Genet. 4:e2 doi:10.1371/journal.pgen.0040002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harlocker SL, Rampersaud A, Yang WP, Inouye M. 1993. Phenotypic revertant mutations of a new OmpR2 mutant (V203Q) of Escherichia coli lie in the envZ gene, which encodes the OmpR kinase. J. Bacteriol. 175:1956–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Park H, Inouye M. 1997. Mutational analysis of the linker region of EnvZ, an osmosensor in Escherichia coli. J. Bacteriol. 179:4382–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tokishita S, Kojima A, Mizuno T. 1992. Transmembrane signal transduction and osmoregulation in Escherichia coli: functional importance of the transmembrane regions of membrane-located protein kinase, EnvZ. J. Biochem. 111:707–713 [DOI] [PubMed] [Google Scholar]
- 49. Hsing W, Russo FD, Bernd KK, Silhavy TJ. 1998. Mutations that alter the kinase and phosphatase activities of the two-component sensor EnvZ. J. Bacteriol. 180:4538–4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mizuno T, Mizushima S. 1990. Signal transduction and gene regulation through the phosphorylation of two regulatory components: the molecular basis for the osmotic regulation of the porin genes. Mol. Microbiol. 4:1077–1082 [DOI] [PubMed] [Google Scholar]
- 51. Forst S, Comeau D, Norioka S, Inouye M. 1987. Localization and membrane topology of EnvZ, a protein involved in osmoregulation of OmpF and OmpC in Escherichia coli. J. Biol. Chem. 262:16433–16438 [PubMed] [Google Scholar]
- 52. Park H, Saha SK, Inouye M. 1998. Two-domain reconstitution of a functional protein histidine kinase. Proc. Natl. Acad. Sci. U. S. A. 95:6728–6732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ames P, Parkinson JS. 1988. Transmembrane signaling by bacterial chemoreceptors: E. coli transducers with locked signal output. Cell 55:817–826 [DOI] [PubMed] [Google Scholar]
- 54. del Carmen Burón-Barral M, Gosink KK, Parkinson JS. 2006. Loss- and gain-of-function mutations in the F1-HAMP region of the Escherichia coli aerotaxis transducer Aer. J. Bacteriol. 188:3477–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dunin-Horkawicz S, Lupas AN. 2010. Comprehensive analysis of HAMP domains: implications for transmembrane signal transduction. J. Mol. Biol. 397:1156–1174 [DOI] [PubMed] [Google Scholar]
- 56. Leatham-Jensen MP, Frimodt-Moller J, Adediran J, Mokszycki ME, Banner ME, Caughron JE, Krogfelt KA, Conway T, Cohen PS. 2012. The streptomycin-treated mouse intestine selects Escherichia coli envZ missense mutants that interact with dense and diverse intestinal microbiota. Infect. Immun. 80:1716–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rainwater S, Silverman PM. 1990. The Cpx proteins of Escherichia coli K-12: evidence that cpxA, ecfB, ssd, and eup mutations all identify the same gene. J. Bacteriol. 172:2456–2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sun S, Berg OG, Roth JR, Andersson DI. 2009. Contribution of gene amplification to evolution of increased antibiotic resistance in Salmonella typhimurium. Genetics 182:1183–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kato Y, Suzuki T, Ida T, Maebashi K. 2010. Genetic changes associated with glycopeptide resistance in Staphylococcus aureus: predominance of amino acid substitutions in YvqF/VraSR. J. Antimicrob. Chemother. 65:37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kato Y, Suzuki T, Ida T, Maebashi K, Sakurai M, Shiotani J, Hayashi I. 2008. Microbiological and clinical study of methicillin-resistant Staphylococcus aureus (MRSA) carrying VraS mutation: changes in susceptibility to glycopeptides and clinical significance. Int. J. Antimicrob. Agents 31:64–70 [DOI] [PubMed] [Google Scholar]
- 61. Howden BP, Stinear TP, Allen DL, Johnson PD, Ward PB, Davies JK. 2008. Genomic analysis reveals a point mutation in the two-component sensor gene graS that leads to intermediate vancomycin resistance in clinical Staphylococcus aureus. Antimicrob. Agents Chemother. 52:3755–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Macfarlane EL, Kwasnicka A, Hancock RE. 2000. Role of Pseudomonas aeruginosa PhoP-PhoQ in resistance to antimicrobial cationic peptides and aminoglycosides. Microbiology 146:2543–2554 [DOI] [PubMed] [Google Scholar]
- 63. McPhee JB, Lewenza S, Hancock RE. 2003. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol. Microbiol. 50:205–217 [DOI] [PubMed] [Google Scholar]
- 64. Richards SM, Strandberg KL, Gunn JS. 2010. Salmonella-regulated lipopolysaccharide modifications. Subcell. Biochem. 53:101–122 [DOI] [PubMed] [Google Scholar]
- 65. Sun S, Negrea A, Rhen M, Andersson DI. 2009. Genetic analysis of colistin resistance in Salmonella enterica serovar Typhimurium. Antimicrob. Agents Chemother. 53:2298–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Moskowitz SM, Brannon MK, Dasgupta N, Pier M, Sgambati N, Miller AK, Selgrade SE, Miller SI, Denton M, Conway SP, Johansen HK, Hoiby N. 2012. PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients. Antimicrob. Agents Chemother. 56:1019–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee JY, Lim MH, Heo ST, Ko KS. 2012. Repeated isolation of Pseudomonas aeruginosa isolates resistant to both polymyxins and carbapenems from 1 patient. Diagn. Microbiol. Infect. Dis. 72:267–271 [DOI] [PubMed] [Google Scholar]
- 68. Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. 1992. Short protocols in molecular biology, 2nd ed John Wiley & Sons, Inc., New York, NY [Google Scholar]
- 69. Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Nat. Biotechnol. 1:784–791 [Google Scholar]
- 70. Masuda N, Ohya S. 1992. Cross-resistance to meropenem, cephems, and quinolones in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 36:1847–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86 [DOI] [PubMed] [Google Scholar]
- 72. Notredame C, Higgins DG, Heringa J. 2000. T-coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217 [DOI] [PubMed] [Google Scholar]