

Abstract
We report that GRL-0519, a novel nonpeptidic human immunodeficiency virus type 1 (HIV-1) protease inhibitor (PI) containing tris-tetrahydrofuranylurethane (tris-THF) and a sulfonamide isostere, is highly potent against laboratory HIV-1 strains and primary clinical isolates (50% effective concentration [EC50], 0.0005 to 0.0007 μM) with minimal cytotoxicity (50% cytotoxic concentration [CC50], 44.6 μM). GRL-0519 blocked the infectivity and replication of HIV-1NL4-3 variants selected by up to a 5 μM concentration of ritonavir, lopinavir, or atazanavir (EC50, 0.0028 to 0.0033 μM). GRL-0519 was also potent against multi-PI-resistant clinical HIV-1 variants isolated from patients who no longer responded to existing antiviral regimens after long-term antiretroviral therapy, highly darunavir (DRV)-resistant variants, and HIV-2ROD. The development of resistance against GRL-0519 was substantially delayed compared to other PIs, including amprenavir (APV) and DRV. The effects of nonspecific binding of human serum proteins on GRL-0519's antiviral activity were insignificant. Our analysis of the crystal structures of GRL-0519 (3OK9) and DRV (2IEN) with protease suggested that the tris-THF moiety, compared to the bis-THF moiety present in DRV, has greater water-mediated polar interactions with key active-site residues of protease and that the tris-THF moiety and paramethoxy group effectively fill the S2 and S2′ binding pockets, respectively, of the protease. The present data demonstrate that GRL-0519 has highly favorable features as a potential therapeutic agent for treating patients infected with wild-type and/or multi-PI-resistant variants and that the tris-THF moiety is critical for strong binding of GRL-0519 to the HIV protease substrate binding site and appears to be responsible for its favorable antiretroviral characteristics.
INTRODUCTION
Combination antiretroviral therapy (cART) has had a major impact on the AIDS epidemic in industrially advanced nations. Recent analyses have revealed that mortality rates for human immunodeficiency virus type 1 (HIV-1)-infected persons have become close to those of the general population (1–4). However, eradication of HIV-1 does not appear to be currently possible, in part due to the viral reservoirs remaining in blood and infected tissues. Moreover, we have encountered a number of challenges in bringing the optimal benefits of the currently available therapeutics for AIDS and HIV-1 infection to individuals receiving cART (5–7). They include (i) drug-related toxicities, (ii) inability to fully restore normal immunologic functions once individuals developed AIDS, (iii) development of various cancers as a consequence of survival prolongation, (iv) flaring up of inflammation in individuals receiving cART or immune reconstruction syndrome (IRS), and (v) increased cost of antiviral therapy. Such limitations and flaws of cART are exacerbated by the development of drug-resistant HIV-1 variants (8–12), although the recent first-line cART with boosted protease inhibitor (PI)-based regimens has made the development of HIV-1 resistance less likely over an extended period of time (13).
Successful antiviral drugs, in theory, produce their virus-specific effects by interacting with viral receptors, virus-encoded enzymes, viral structural components, viral genes, or their transcripts without disturbing cellular metabolism or function. However, at present, no antiretroviral drugs or agents are likely to be completely specific for HIV-1 or to be devoid of toxicity or side effects in the therapy of AIDS. This is a critical issue, because patients with AIDS and its related diseases will have to receive antiretroviral therapy for a long time, perhaps for the rest of their lives. Thus, the identification of new classes of antiretroviral drugs that have a unique mechanism(s) of action and produce no or minimal side effects remains an important therapeutic objective.
We have been focusing on the design and synthesis of nonpeptidyl PIs that are potent against HIV-1 variants resistant to the currently approved PIs. One such anti-HIV-1 agent, darunavir (DRV) (Fig. 1), containing a structure-based designed privileged nonpeptidic P2 ligand, 3(R),3a(S),6a(R)-bis-tetrahydrofuranylurethane (bis-THF) (14–16), has been approved as a first-line therapeutic agent for the treatment of individuals who are infected with HIV-1. In the present work, we examined and characterized the nonpeptidic HIV-1 protease inhibitors GRL-0519 (17) and its stereoisomer, GRL-0529, both of which contain the tris-THF moiety and a sulfonamide isostere. We found that GRL-0519 exerts highly potent activity against a wide spectrum of laboratory HIV-1 strains and primary clinical isolates, including multi-PI-resistant variants with minimal cytotoxicity. In addition, GRL-0519 was active against HIV-2ROD, as well as HIV-1 isolates examined. We also selected HIV-1 variants with GRL-0519 by propagating a laboratory wild-type HIV-1NL4-3 in MT-4 cells in the presence of increasing concentrations of GRL-0519 and determined the amino acid substitutions that emerged under the pressure of GRL-0519 in the protease-encoding region. In addition, we evaluated the effects of nonspecific binding of physiological human serum proteins on GRL-0519's anti-HIV-1 activity. We further analyzed the previously published crystal structure of GRL-0519 with protease (Protein Data Bank [PDB] ID, 3OK9) to gain a better understanding of the present antiviral data. The crystal structure analyses indicated that GRL-0519 has strong polar interactions with key residues in the active site of the protease. GRL-0519 also has several water-mediated polar interactions and tight van der Waals interactions with protease residues, suggesting that GRL-0519 binds very tightly in the active site of the protease.
Fig 1.

Structures of GRL-0519, GRL-0529, amprenavir, and darunavir. M.W., molecular weight.
MATERIALS AND METHODS
Cells and viruses.
MT-2 and MT-4 cells were grown in RPMI 1640-based culture medium supplemented with 10% fetal calf serum (FCS) (JRH Biosciences, Lenexa, MD), 50 units/ml penicillin, and 100 μg/ml kanamycin. The following HIV strains were employed for the drug susceptibility assay (see below): HIV-1LAI, HIV-1NL4-3, HIV-2ROD, HIV-1ERS104pre (18), clinical HIV-1 strains isolated from drug-naive patients with AIDS, and six HIV-1 clinical strains that were originally isolated from patients with AIDS who had received 9 to 11 anti-HIV-1 drugs over the past 32 to 83 months and that were genotypically and phenotypically characterized as multi-PI-resistant HIV-1 variants (19, 20). All primary HIV-1 strains were passaged once or twice in 3-day-old phytohemagglutinin-activated peripheral blood mononuclear cells (PHA-PBM), and the virus-containing culture supernatants were stored at −80°C until they were used as sources of infectious virions.
Antiviral agents and human serum proteins.
Roche Products Ltd. (Welwyn Garden City, United Kingdom) and Abbott Laboratories (Abbott Park, IL) kindly provided saquinavir (SQV) and ritonavir (RTV), respectively. Amprenavir (APV) was a courtesy gift from GlaxoSmithKline, Research Triangle Park, NC. Lopinavir (LPV) was kindly provided by Japan Energy Inc., Tokyo, Japan. Atazanavir (ATV) was a contribution from Bristol Myers Squibb (New York, NY). Darunavir (DRV) was synthesized as previously described (21). Human serum albumin (HSA) and α1-acid glycoprotein (AAG) were purchased from Sigma-Aldrich (St. Louis, MO).
Drug susceptibility assay.
The susceptibility of HIV-1LAI or HIV-2ROD to various drugs was determined as previously described with minor modifications. Briefly, MT-2 cells (104/ml) were exposed to 100 50% tissue culture infectious doses (TCID50) of HIV-1LAI or HIV-2ROD in the presence or absence of various concentrations of drugs in 96-well microculture plates and incubated at 37°C for 7 days. After incubation, 100 μl of the medium was removed from each well and 3-(4,5-dimetylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (10 μl, 7.5 mg/ml in phosphate-buffered saline) was added to each well in the plate, followed by incubation at 37°C for 2 h. After incubation to dissolve the formazan crystals, 100 μl of acidified isopropanol containing 4% (vol/vol) Triton X-100 was added to each well, and the optical density was measured in a kinetic microplate reader (Vmax; Molecular Devices, Sunnyvale, CA). All assays were performed in duplicate or triplicate. In some experiments, MT-2 cells were chosen as target cells in the MTT assay, since these cells undergo greater HIV-1-elicited cytopathic effects than MT-4 cells. To determine the sensitivity of primary HIV-1 isolates to drugs, PHA-PBM (106/ml) were exposed to 50 TCID50 of each primary HIV-1 isolate and cultured in the presence or absence of various concentrations of drugs in 10-fold serial dilutions in 96-well microculture plates. In determining the drug susceptibilities of certain laboratory HIV-1 strains, MT-4 cells were employed as target cells as previously described, with minor modifications. In brief, MT-4 cells (105/ml) were exposed to 100 TCID50 of drug-resistant HIV-1 strains in the presence or absence of various concentrations of drugs and incubated at 37°C. On day 7 of culture, the supernatants were harvested and the amounts of p24 (capsid [CA]) Gag protein were determined by using a fully automated chemiluminescent enzyme immunoassay system (Lumipulse F; Fujirebio Inc., Tokyo, Japan) (22, 23). Drug concentrations that suppressed the production of p24 Gag protein by 50% (50% effective concentration [EC50]) were determined by comparison with the p24 production level in a drug-free control cell culture. All assays were performed in duplicate or triplicate. PHA-PBM were derived from a single donor in each independent experiment. Thus, to obtain the data, three different healthy donors were recruited. For determining the antiretroviral activity and cytotoxicity of a drug, we used the same cells and cultured them for the same 7 days. The MTT assay was employed for HIV-1LAI and HIV-2ROD, while the p24 assay was employed for clinical HIV-1 isolates and drug-resistant HIV-1 strains.
Creation of PI-resistant HIV-1 variants in vitro.
MT-4 cells (105/ml) were exposed to HIV-1NL4-3 (500 TCID50) and cultured in the presence of various PIs at an initial concentration equal to its EC50. Viral replication was monitored by the determination of the amount of p24 Gag produced by MT-4 cells. The culture supernatants were harvested on day 7 and were used to infect fresh MT-4 cells for the next round of culture in the presence of increasing concentrations of each drug. When the virus began to propagate in the presence of the drug, the drug concentration was increased generally 2- to 3-fold. Proviral DNA samples obtained from the lysates of infected cells were subjected to nucleotide sequencing. This drug selection procedure was carried out until the drug concentration reached 5 μM, as previously described (24–26). In the experiments for selecting drug-resistant variants, MT-4 cells were also exploited as target cells, since HIV-1 in general replicates at higher levels in MT-4 cells than in MT-2 cells, as described above.
Determination of nucleotide sequences.
Molecular cloning and determination of the nucleotide sequences of HIV-1 strains passaged in the presence of anti-HIV-1 agents were performed as previously described (24). In brief, high-molecular-weight DNA was extracted from HIV-1-infected MT-4 cells by using the InstaGene Matrix (Bio-Rad Laboratories, Hercules, CA) and was subjected to molecular cloning, followed by sequence determination. The primers used for the first round of PCR with the entire Gag- and protease-encoding regions of the HIV-1 genome were LTR F1 (5′-GAT GCT ACA TAT AAG CAG CTG C-3′) and PR12 (5′-CTC GTG ACA AAT TTC TAC TAA TGC-3′). The first-round PCR mixture consisted of 1 μl of proviral DNA solution, 10 μl of Premix Taq (Ex Taq version; TaKaRa Bio Inc., Otsu, Japan), and 10 pmol of each of the first PCR primers in a total volume of 20 μl. The PCR conditions used were an initial 3 min at 95°C, followed by 35 cycles of 40 s at 95°C, 20 s at 55°C, and 2 min at 72°C, with a final 10 min of extension at 72°C. The first-round PCR products (1 μl) were used directly in the second round of PCR with primers LTR F2 (5′-GAG ACT CTG GTA ACT AGA GAT C-3′) and KSMA2.1 (5′-CCA TCC CGG GCT TTA ATT TTA CTG GTA C-3′) under the following PCR conditions: an initial 3 min at 95°C, followed by 35 cycles of 30 s at 95°C, 20 s at 55°C, and 2 min at 72°C, with a final 10 min of extension at 72°C (a stick diagram of HIV-1 genome PCR amplification for sequence analysis is shown in Fig. S1 in the supplemental material). The second-round PCR products were purified with spin columns (MicroSpin S-400 HR columns; Amersham Biosciences Corp., Piscataway, NJ), cloned directly, and subjected to sequencing with a model 3130 automated DNA sequencer (Applied Biosystems, Foster City, CA).
Determination of replication kinetics of GRL-0519-resistant HIV-1NL4-3 variants and wild-type HIV-1NL4-3.
The GRL-0519-resistant variant at passage 37 was propagated in fresh MT-4 cells without GRL-0519 for 7 days, and aliquoted HIV-1519RP37 viral stocks were stored at −80°C until use. MT-4 cells (3.2 × 105) were exposed to the HIV-1519RP37 or wild-type HIV-1NL4-3 preparation containing 10 ng/ml p24 in 6-well culture plates for 3 h, and the newly infected MT-4 cells were washed with fresh medium and divided into 4 fractions, each cultured with or without GRL-0519 (final concentration of MT-4 cells, 104/ml; drug concentrations, 0, 0.005, 0.01, and 0.015 μΜ). The amounts of p24 were measured every 2 days for up to 7 days.
Generation of recombinant HIV-1 clones.
To generate HIV-1 clones carrying the desired amino acid substitutions, site-directed mutagenesis was performed with a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA), and the amino acid substitution-containing genomic fragments were introduced into pHIV-1NL4-3Sma. Determination of the nucleotide sequences of the plasmids confirmed that each clone had the desired amino acid substitution but no unintended amino acid substitutions. Each recombinant plasmid was transfected into COS7 cells with Lipofectamine LTX transfection reagent (Invitrogen, Carlsbad, CA), and the infectious virions thus made were harvested for 72 h after transfection and stored at −80°C until use.
Structural analysis of interactions of GRL-0519 and DRV with protease.
The crystal structures of HIV-1 protease complexed with GRL-0519 or DRV were obtained from the protein data bank (PDB ID, 3OK9 and 2IEN, respectively). The inhibitor conformation with the higher occupancy in the crystal structure was considered for analysis. Bond orders were properly assigned to the inhibitor molecules. Hydrogens were added to all the heavy atoms, and their positions were optimized in an OPLS2005 force field (27) with constraints on heavy atom positions. A cutoff distance of 3.0 Å between a polar hydrogen and oxygen or nitrogen was used to determine the presence of hydrogen bonds. The structures were analyzed using Maestro version 9.3 (Schrödinger, LLC, New York, NY, 2012).
RESULTS
Antiviral activities of GRL-0519 and -0529 against HIV-1LAI and HIV-2ROD and their cytotoxicities.
We first examined the antiviral potencies of GRL-0519 and -0529 against a variety of HIV-1 isolates. GRL-0529 showed only moderate anti-HIV-1 activity against a laboratory wild-type HIV-1 strain, HIV-1LAI, and an HIV-2 strain, HIV-2ROD, with EC50s of 0.33 and 0.40 μM, respectively (Table 1). Conversely, GRL-0519 was extremely potent against HIV-1LAI, with an EC50 of 0.0007 μM compared to other clinically available Food and Drug Administration (FDA)-approved PIs examined, including DRV (Table 1), as assessed with the MTT assay using MT-2 target cells, while its cytotoxicity was evident only at high concentrations (50% cytotoxic concentration [CC50], 44.6 μM) and the selectivity index proved to be highly favorable at 63,714 (Table 1). GRL-0519 was also very potent against HIV-2ROD, with an EC50 of 0.0004 μM (Table 1).
Table 1.
Antiviral activities of GRL-0519 and -0529 against HIV-1LAI or HIV-2ROD
| Compound | EC50 (μM)a |
CC50 (μM) (±SD) | Selectivity indexb | |
|---|---|---|---|---|
| HIV-1LAI | HIV-2ROD | |||
| GRL-0519 | 0.0007 ± 0.0005 | 0.0004 ± 0.0002 | 44.6 ± 3.5 | 63,714 |
| GRL-0529 | 0.33 ± 0.04 | 0.40 ± 0.06 | 38.7 ± 3.7 | 118 |
| SQV | 0.026 ± 0.006 | 0.003 ± 0.001 | 19.8 ± 2.9 | 773 |
| APV | 0.033 ± 0.002 | 0.37 ± 0.11 | 84.7 ± 5.4 | 2,590 |
| ATV | 0.0048 ± 0.0001 | 0.0077 ± 0.0006 | 27.6 ± 2.7 | 5,520 |
| DRV | 0.0042 ± 0.0006 | 0.0088 ± 0.0004 | 152.7 ± 10.1 | 36,357 |
MT-2 cells (104/ml) were exposed to 100 TCID50 of HIV-1LAI or HIV-2ROD and cultured in the presence of various concentrations of each PI, and the EC50 values were determined using the MTT assay. All assays were conducted in duplicate, and the data shown represent mean value (±1 standard deviation [SD]) derived from the results of three independent experiments.
Each selectivity index denotes a ratio of CC50 to EC50 against HIV-1LAI.
GRL-0519 is potent against various PI-selected laboratory HIV-1 variants.
We also examined GRL-0519 against an array of HIV-1NL4-3 variants, which had been selected by propagating HIV-1NL4-3 in the presence of increasing concentrations (up to 5 μM) of each of 4 FDA-approved PIs (RTV, APV, ATV, and LPV) in MT-4 cells (24). Such variants had acquired various PI resistance-associated amino acid substitutions in the protease-encoding region of the viral genome (Table 2, note a). Each variant was highly resistant to the PI by which the variant was selected and showed significant resistance, with an EC50 of >1 μM. GRL-0519 was highly active against all the variants (except HIV-1APVR5μM), with EC50s of 2.8 to 3.3 nM (differences were 6- to 7-fold greater compared to those against HIV-1NL4-3) (Table 2).
Table 2.
Antiviral activities of GRL-0519 and -0529 against laboratory PI-resistant HIV-1 variants
| Virusa | EC50 (nM)b |
||||||
|---|---|---|---|---|---|---|---|
| GRL-0519 | GRL-0529 | APV | ATV | LPV | TPV | DRV | |
| HIV-1NL4-3 | 0.5 ± 0.1 | 356.3 ± 33.8 | 24.9 ± 0.1 | 4.2 ± 1.1 | 37.8 ± 4.5 | 362.9 ± 104.4 | 3.9 ± 0.6 |
| HIV-1RTVR5μM | 2.8 ± 0.4 (6) | 504.3 ± 26.6 (1) | 532.9 ± 25.9 (21) | 36.4 ± 3.0 (9) | 501.2 ± 44.9 (13) | 402.1 ± 56.1 (1) | 29.1 ± 2.1 (7) |
| HIV-1APVR5μM | 38.0 ± 0.9 (76) | >1,000 (>3) | >1,000 (>40) | 371.0 ± 7.8 (88) | >1,000 (>26) | >1,000 (>3) | 368.5 ± 32.4 (94) |
| HIV-1ATVR5μM | 3.3 ± 1.6 (7) | 376.9 ± 164.8 (1) | 390.0 ± 11.1 (16) | >1,000 (>238) | >1,000 (>26) | >1,000 (>3) | 28.1 ± 6.4 (7) |
| HIV-1LPVR5μM | 3.2 ± 0.4 (7) | >1,000 (>3) | 420.1 ± 61.9 (17) | 35.1 ± 4.7 (8) | >1,000 (>26) | >1,000 (>3) | 32.9 ± 1.3 (8) |
The amino acid substitutions identified in the protease-encoding region compared to the wild-type HIV-1NL4-3 include the following: M46I, V82F, and I84V in HIV-1RTVR5μM; L10F, V32I, M46I, I54 M, A71V, and I84V in HIV-1APVR5μM; L23I, E34Q, K43I, M46I, I50L, G51A, L63P, A71V, V82A, and T91A in HIV-1ATVR5μM; and L10F, M46I, I54V, and V82A in HIV-1LPVR5μM.
The EC50 values were determined by using MT-4 cells as target cells. MT-4 cells (105/ml) were exposed to 100 TCID50 of each HIV-1 strain, and the inhibition of p24 Gag protein production by each drug was used as an endpoint. The numbers in parentheses represent the fold changes of EC50s for each isolate compared to the EC50s for HIV-1NL4-3. All assays were conducted in duplicate or triplicate, and the data shown represent mean values (±1 SD) derived from the results of two or three independent experiments.
GRL-0519 exerts potent activity against highly PI-resistant clinical HIV-1 isolates.
In our previous work, we isolated highly multi-PI-resistant primary HIV-1 strains, HIV-1MDR/B, HIV-1MDR/C, HIV-1MDR/G, HIV-1MDR/TM, HIV-1MDR/MM, and HIV-1MDR/JSL, from patients with AIDS who had failed then-existing anti-HIV regimens after receiving 9 to 11 anti-HIV-1 drugs over 32 to 83 months (19, 20). These primary strains contained 9 to 14 amino acid substitutions in the protease-encoding region, which have reportedly been associated with HIV-1 resistance against various PIs (Table 3, note a). The potency of APV, ATV, and LPV against such clinical multidrug-resistant HIV-1 strains was significantly compromised, as examined in PHA-PBM as target cells using p24 production inhibition as an endpoint (Table 3). However, GRL-0519 exerted quite potent antiviral activity, and its EC50s against those clinical variants were quite low: 0.8 to 3.4 nM (Table 3). The antiviral activity of GRL-0519 proved to be most potent against the six multidrug-resistant clinical HIV-1 variants examined compared to the four FDA-approved PIs (APV, ATV, LPV, and DRV). We also examined the antiviral activity of GRL-0519 against highly DRV-resistant variants (28). These variants were created using the mixture of 8 highly multi-PI-resistant clinical isolates as a starting HIV-1 source and selected with increasing concentrations of DRV. GRL-0519 maintained its activity against highly DRV-resistant MDR mixtures (EC50, 5.6 to 30.0 nM), being more potent than DRV by 7.8- to 8.5-fold (Table 3). Overall, GRL-0519 exerted stronger antiviral activity against various wild-type HIV-1 strains, drug-resistant variants, and HIV-2 strains than DRV by 5- to 22-fold. Furthermore, GRL-0519 was more potent than APV by 118- to 925-fold against HIV-2ROD and drug-resistant variants (Tables 1 to 3).
Table 3.
Antiviral activities of GRL-0519 and -0529 against multidrug-resistant clinical isolates and highly DRV-resistant MDR mixtures in PHA-PBM
| Virusa | EC50 (nM)b |
|||||
|---|---|---|---|---|---|---|
| GRL-0519 | GRL-0529 | APV | ATV | LPV | DRV | |
| HIV-1WT/ERS104pre | 0.6 ± 0.2 | 347.4 ± 27.3 | 33.8 ± 5.1 | 2.7 ± 0.6 | 31.4 ± 4.2 | 3.9 ± 0.6 |
| HIV-1MDR/B (X4) | 3.4 ± 0.5 (6) | 611.8 ± 72.8 (2) | 459.4 ± 99.2 (14) | 469.7 ± 7.4 (174) | > 1,000 (>32) | 27.8 ± 5.9 (7) |
| HIV-1MDR/C (X4) | 0.8 ± 0.2 (1) | 514.4 ± 130.6 (1) | 346.1 ± 55.2 (10) | 38.8 ± 2.8 (14) | 436.5 ± 3.5 (14) | 10.3 ± 2.4 (3) |
| HIV-1MDR/G (X4) | 2.6 ± 1.3 (4) | 655.8 ± 292.9 (2) | 462.6 ± 64.5 (14) | 19.4 ± 7.5 (7) | 181.3 ± 23.0 (6) | 27.8 ± 5.2 (7) |
| HIV-1MDR/TM (X4) | 2.1 ± 0.3 (4) | 530.0 ± 74.7 (2) | 476.4 ± 8.1 (14) | 74.5 ± 2.6 (28) | 422.9 ± 82.0 (13) | 30.0 ± 1.0 (8) |
| HIV-1MDR/MM (R5) | 2.5 ± 0.5 (4) | 787.4 ± 251.4 (2) | 338.9 ± 15.5 (10) | 204.8 ± 23.5 (76) | 622.5 ± 82.0 (20) | 13.3 ± 6.2 (3) |
| HIV-1MDR/JSL (R5) | 2.5 ± 0.2 (4) | > 1,000 (>3) | 436.3 ± 90.4 (13) | 211.3 ± 98.6 (78) | > 1,000 (>32) | 22.1 ± 9.3 (6) |
| HIV-1DRVR10P | 5.6 ± 0.4 (9) | > 1,000 (>3) | > 1,000 (>32) | 322.9 ± 10.4 (77) | > 1,000 (>32) | 43.4 ± 13.4 (11) |
| HIV-1DRVR20P | 30.0 ± 9.8 (50) | > 1,000 (>3) | > 1,000 (>32) | > 1,000 (>370) | > 1,000 (>32) | 255.2 ± 4.0 (64) |
The amino acid substitutions identified in the protease-encoding region compared to the consensus type B sequence cited from the Los Alamos database include the following: L63P in HIV-1ERS104pre; L10I, K14R, L33I, M36I, M46I, F53I, K55R, I62V, L63P, A71V, G73S, V82A, L90 M, and I93L in HIV-1MDR/B; L10I, I15V, K20R, L24I, M36I, M46L, I54V, I62V, L63P, K70Q,V82A, and L89 M in HIV-1MDR/C; L10I, V11I, T12E, I15V, L19I, R41K, M46L, L63P, A71T, V82A, and L90 M in HIV-1MDR/G; L10I, K14R, R41K, M46L, I54V, L63P, A71V, V82A, L90 M, and I93L in HIV-1MDR/TM; L10I, K43T, M46L, I54V, L63P, A71V, V82A, L90 M, and Q92K in HIV-1MDR/MM; L10I, L24I, I33F, E35D, M36I, N37S, M46L, I54V, R57K, I62V, L63P, A71V, G73S, and V82A in HIV-1MDR/JSL; L10I, I15V, K20R, L24I, V32I, M36I, M46L, I54V, I62V, L63P, K70Q, V82A, and L88 M in HIV-1DRVR10P; and L10I, I15V, K20R, L24I, V32I, M36I, M46L, L63P, A71T, V82A, and L88 M in HIV-1DRVR20P. HIV-1ERS104pre served as a source of wild-type HIV-1. DRV-resistant HIV-1 variants (HIV-1DRVR10P and HIV-1DRVR20P) were selected in vitro by propagating a mixture of eight HIV-1MDR isolates in the presence of increasing concentrations of DRV in MT-4 cells. Six of the eight isolates were the same as those described above. Amino acid substitutions identified in proteases of the other two isolates compared to the consensus type B sequence cited from the Los Alamos database include the following: L10I, I15V, E35D, N37E, K45R, I54V, L63P, A71V, V82T, L90 M, I93L, and C95F in HIV-1MDR/A; L10R, N37D, M46I, I62V, L63P, A71V, G73S, V74I, V82T, L90 M, and I93L in HIV-1MDR/SS.
The EC50 values were determined by using PHA-PBM as target cells, and the inhibition of p24 Gag protein production by each drug was used as an endpoint. The numbers in parentheses represent the fold changes of EC50s for each isolate compared to the EC50s for HIV-1ERS104pre. All assays were conducted in duplicate or triplicate, and the data shown represent mean values (±1 SD) derived from the results of three independent experiments. PHA-PBM were derived from a single donor in each independent experiment.
Effects of human serum proteins on the antiretroviral activity of GRL-0519.
The binding of human serum proteins to a drug is an important determinant of its pharmacological activity in vivo, because overly tight binding may result in reducing interactions between the drug and its target (29). We thus determined the effects of the binding of HSA and AAG on GRL-0519's antiretroviral activity in vitro. Physiologically normal concentrations of HSA (40 mg/ml) and AAG (10 μM) were used to evaluate their binding effects on GRL-0519's activity against a wild-type clinical isolate, HIV-1ERS104pre. All four FDA-approved drugs substantially maintained their activity in the presence of HSA, with reduction of activity by up to 5-fold relative to the activity in the absence of additional HSA (Table 4). The activities of those PIs were reduced in the presence of AAG by 7- to 12-fold (Table 4). However, the binding effects of both HSA and AAG on GRL-0519's activity were insignificant, only a 3- to 4-fold difference. Of note, GRL-0519's EC50s with HSA or AAG (1.9 to 2.4 nM) significantly exceeded those of the four FDA-approved PIs (Table 4).
Table 4.
Nonspecific binding effects of human serum proteins on GRL-0519's antiviral activity
| Compound | EC50 (nM)a |
||
|---|---|---|---|
| None | HSA | AAG | |
| GRL-0519 | 0.6 ± 0.2 | 1.9 ± 1.1 (3) | 2.4 ± 0.3 (4) |
| GRL-0529 | 347.4 ± 27.3 | 351.5 ± 22.7 (2) | 649.0 ± 48.9 (2) |
| APV | 31.5 ± 5.5 | 30.5 ± 2.6 (1) | 286.3 ± 14.8 (9) |
| ATV | 2.7 ± 0.6 | 14.6 ± 1.2 (5) | 32.7 ± 15.9 (12) |
| LPV | 31.4 ± 4.2 | 32.5 ± 2.7 (1) | 232.1 ± 15.2 (7) |
| DRV | 3.9 ± 1.0 | 7.4 ± 1.2 (2) | 33.8 ± 3.5 (9) |
HSA (40 mg/ml) or AAG (10 μM) was used to evaluate the binding effects of human serum proteins on GRL-519 and -529 antiviral activities. The EC50 values against HIV-1ERS104pre with or without HSA or AAG were determined by p24 assay using PHA-PBM as target cells, and the inhibition of p24 Gag production by each drug was used as an endpoint. The numbers in parentheses represent the fold changes of the EC50 values compared to the values without HSA or AAG. The data shown represent mean values derived from the results of two or three independent experiments. PBM were derived from a single donor in each independent experiment.
In vitro selection of HIV-1 variants resistant to GRL-0519.
We next attempted to select HIV-1 variants resistant to GRL-0519 by propagating a laboratory HIV-1 strain, HIV-1NL4-3, in MT-4 cells in the presence of increasing concentrations of GRL-0519 as previously described (24). HIV-1NL4-3 was initially exposed to 0.0007 μM GRL-0519 and underwent 37 passages, after which the concentration of GRL-0519 was found to have increased 19-fold (0.0131 μM) compared to that at the initiation of selection. Judging from the amounts of p24 Gag protein produced in the culture medium (up to ∼282 ng/ml), the replicative capacity of HIV-1NL4-3 at passage 37 (HIV519RP37) was thought to have been reasonably well maintained. Compared to the kinetics of the emergence of variants resistant to APV, the emergence of GRL-0519- and DRV-resistant variants was substantially delayed (Fig. 2). Of note, HIV-1 variants resistant to APV and capable of replicating at >5 μM emerged by passage 20, and variants resistant to GRL-0529, replicating at >1 μM, emerged by passage 6, while it became fairly difficult to increase the concentrations of DRV and GRL-0519 around and beyond passage 20, since the virus populations ceased to replicate with further increased concentrations.
Fig 2.

In vitro selection of PI-resistant HIV-1 variants. HIV-1NL4-3 was propagated in MT-4 cells in the presence of increasing concentrations of amprenavir (●), darunavir (△), GRL-0519 (▲), or GRL-0529 (○). Each passage of virus was conducted in a cell-free manner.
The protease-encoding region of the proviral DNA isolated from infected MT-4 cells was cloned and sequenced at passages 6, 13, 22, 29, and 37 under GRL-0519 selection. The sequences of the region cloned and the percent frequency of identical sequences at each passage are depicted in Fig. 3. By passage 13, the wild-type protease gene sequence was seen in 7 of 19 clones, although an N37S substitution was noted in 10 of the 19 clones. However, by passage 22 and beyond, N37S disappeared, and the virus had mostly acquired K43I and A71T substitutions. As the passages proceeded, greater numbers of amino acid substitutions emerged. At passage 29, V82I substitution was seen in 16 of 22 clones, and V82I became dominant by passage 37 (25 of 26 clones). An L33V substitution was also observed in 4 of the 26 clones by passage 37. Reportedly, APV-resistant HIV-1 variants contain V32I, I50V, I54L/M, L76V, I84V, and L90M substitutions (30, 31), and such substitutions were also identified in the present study (data not shown). However, no such APV resistance-associated amino acid substitutions emerged during the GRL-0519 selection (Fig. 3). Additionally, HIV-1 selected with GRL-0519 (HIV519RP37) acquired the following Gag amino acid substitutions: E17K and V84A in the matrix (MA) region and G61E and D152N in the CA region.
Fig 3.

Amino acid sequences of the protease-encoding regions of HIV-1NL4-3 variants selected in the presence of GRL-0519. The amino acid sequences of protease, deduced from the nucleotide sequence of the protease-encoding region of each proviral DNA isolated at each indicated time, are shown. The amino acid sequence of the wild-type HIV-1NL4-3 protease is illustrated at the top as a reference.
HIV519RP37 fails to replicate at a low concentration of GRL-0519.
Since the replicative capacity of HIV519RP37 was thought to be reasonably well maintained despite the presence of GRL-0519, as mentioned above, we determined the replication kinetics of HIV519RP37 and HIV-1NL4-3. As shown in Fig. 4A, HIV-1NL4-3 failed to replicate in the presence of as little as 0.005 μM GRL-0519 during the entire culture period of 7 days. However, HIV519RP37 was capable of replicating in the presence of 0.005 and 0.01 μM GRL-0519, and the amount of p24 produced in the culture medium reached the amount without GRL-0519 by day 7. However, HIV519RP37 failed to replicate in the presence of 0.015 μM GRL-0519, and no p24 was detected throughout the culture period (Fig. 4B).
Fig 4.
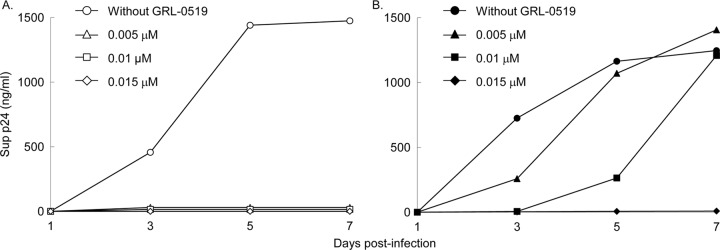
Replication kinetics of HIV-1NL4-3 and HIV519RP37. MT-4 cells (3.2 × 105) were exposed to a HIV-1NL4-3 or HIV-1519RP37 preparation containing 10 ng/ml p24 in 6-well culture plates for 3 h, and the MT-4 cells were washed with fresh medium and divided into 4 fractions, each cultured with or without GRL-0519 (final concentration of MT-4 cells, 104/ml; drug concentrations, 0, 0.005, 0.01, and 0.015 μM). The amount of p24 in each culture flask was measured every 2 days for up to 7 days, once at each time point.
Roles of 3 amino acid substitutions that emerged during selection with GRL-0519 in GRL-0519's anti-HIV-1 activity.
We also examined the roles of 3 substitutions that emerged during the GRL-0519 selection experiment, K43I, A71T, and V82I, using single-amino-acid substitutions carrying HIV-1NL4-3 variants. First, we determined the exact EC50 of GRL-0519 against HIV519RP37 using a p24 assay. GRL-0519 suppressed the replication of HIV519RP37 with an EC50 of 6.0 nM, 12-fold different from the EC50 against HIV-1NL4-3 (Table 5). Next, we determined EC50s of GRL-0519 against 3 single-amino-acid substitution-containing HIV-1NL4-3 variants (HIV-1K43I, HIV-1A71T, and HIV-1V82I). GRL-0519 inhibited the replication of HIV-1K43I, HIV-1A71T, and HIV-1V82I with EC50s of 1.2, 1.4, and 3.2 nM, respectively (differences were 2-, 3-, and 6-fold compared to those against HIV-1NL4-3, respectively) (Table 5).
Table 5.
Roles of 3 amino acid substitutions that emerged during selection with GRL-0519 on GRL-0519's anti-HIV-1 activity
| Virusa | GRL-0519 EC50 (nM)b |
|---|---|
| HIV-1NL4-3 | 0.5 ± 0.1 |
| HIV-1519RP37 | 6.0 ± 1.4 (12) |
| HIV-1K43I | 1.2 ± 0.2 (2) |
| HIV-1A71T | 1.4 ± 0.6 (3) |
| HIV-1V82I | 3.2 ± 0.1 (6) |
HIV-1K43I, HIV-1A71T, and HIV-1V82I were created using a HIV-1NL4-3 plasmid clone.
The EC50 values were determined with MT-4 cells, employing a p24 assay. The data shown represent mean values (±1 SD) derived from the results of two or four independent experiments. The numbers in parentheses represent the fold changes of EC50s compared to the value against HIV-1NL4-3.
In vitro selection of a mixture of 8 HIV-1MDR variants resistant to GRL-0519.
As described above and as shown in Fig. 2, the emergence of HIV-1 variants resistant to GRL-0519 was much delayed, and it took as many as 37 passages (37 weeks) for the virus to acquire its replicative activity in the presence of low concentrations (∼0.01 μM). Thus, we attempted to select out GRL-0519-resistant variants using a mixture of 8 multidrug-resistant clinical isolates (HIV-1MDR) as a starting HIV-1 population, as previously described (28, 32). We performed an additional selection experiment with MT4 cells and a mixture of 8 HIV-1MDR variants as a starting virus population, with APV, DRV, and GRL-0519 and -0529. Similar to the results of selection experiments using MT-4 cells and HIV-1NL4-3, the emergence of HIV-1 strains capable of replicating in the presence of GRL-0519 was significantly delayed compared to the cases with APV and DRV (Fig. 5).
Fig 5.

In vitro selection using a mixture of 8 multi-PI-resistant HIV-1MDR isolates. A mixture of 8 different multi-PI-resistant clinical HIV-1 isolates was propagated in MT-4 cells in the presence of increasing concentrations of amprenavir (●), darunavir (△), GRL-0519 (▲), or GRL-0529 (○). Each passage of virus was conducted in a cell-free manner. The amino acid substitutions identified in the protease-encoding regions of 2 different multi-PI-resistant clinical isolates compared to the consensus type B sequence cited from the Los Alamos database include L10I, I15V, E35D, N37E, K45R, I54V, L63P, A71V, V82T, L90M, I93L, and C95F in HIV-1MDR/A and L10R, N37D, M46I, I62V, L63P, A71V, G73S, V77IV82T, L90M, and I93L in HIV-1MDR/SS. The amino acid substitutions of the other 6 HIV-1MDR variants are given in Table 3, note a.
Structural analyses of GRL-0519 interactions with protease.
We have recently reported the crystal structure depicting the binding mode and interactions of GRL-0519 with protease (17). GRL-0519 has a tris-THF moiety as its P2 ligand, and DRV has a bis-THF group as its P2 ligand (Fig. 1). Another difference is that GRL-0519 has a methoxybenzene as a P2′ ligand, whereas DRV has an aniline. We analyzed the crystal structure of GRL-0519 with protease and compared it with the structure of protease complexed with DRV. We found that the oxygens of the first and second THF rings of GRL-0519 form hydrogen bond interactions with the backbone amide nitrogens of Asp-29 and Asp-30 in the S2 binding pocket of the protease (Fig. 6A). The urethane NH of GRL-0519 forms a hydrogen bond with Gly-27. The hydroxyl group of GRL-0519 forms hydrogen bonds with the catalytic Asp-25 and Asp-25′. The carbonyl oxygen and the sulfonamide oxygen of GRL-0519 form polar interactions with Ile-50 and Ile-50′ in the protease flap through a bridging water molecule. These interactions are also seen for DRV (Fig. 6B). In fact, the polar interactions with Gly-27, Ile-50, and Ile-50′, as well as with Asp-25 and Asp-25′, are also seen for other protease inhibitors (33, 34). The oxygen of the methoxybenzene (P2′ ligand) of GRL-0519 forms a hydrogen bond with the backbone amide nitrogen of Asp-30′ in the S2′ site of protease. This interaction at the S2′ site is different in the case of DRV. DRV has an aniline as the P2′ ligand, and it forms polar interactions with the backbone carbonyl oxygen of Asp-30′. The third THF ring of the tris-THF has water-mediated interactions with Thr-26 and Arg-8 involving 6 bridging water molecules. We also analyzed the van der Waals interactions between GRL-0519 and protease (Fig. 7). The methoxy moiety of GRL-0519 makes good van der Waals contacts with the Asp-29′ and Asp-30′ residues at the S2′ site of protease (Fig. 7A). The contacts seem stronger than the interactions of the corresponding amine group of DRV (Fig. 7B). Analysis of the crystal structure of protease complexed with DRV had suggested that there is room for an extra THF moiety to make additional contacts with protease. The crystal structure examined in the present study confirms that the tris-THF fully occupies the binding cavity and forms better van der Waals contacts with protease than the bis-THF of DRV (Fig. 7C and D). Contact (C) is defined by the following formula: C = D12/(R1 + R2), where D12 is the distance between atoms 1 and 2 and R1 and R2 are the van der Waals radii of atoms 1 and 2. A good contact is defined as follows: 1.30 > C > 0.89.
Fig 6.

Hydrogen bond interactions of GRL-0519 and darunavir with protease. The interactions between protease–GRL-0519 (PDB ID, 3OK9) and protease-DRV (PDB ID, 2IEN) complexes as determined from their respective crystal structures were analyzed. (A and B) Similar interactions between protease–GRL-0519 (A) and protease-DRV (B) complexes. GRL-0519 has polar interactions with Asp-29, Asp-30, Gly-27, Asp-25, and Asp-25′. It has polar interactions with Ile-50 and Ile-50′ through a bridging water molecule. The protease-DRV complex also has these polar interactions. Both inhibitors have polar interactions with different backbone atoms of Asp-30′ in the S2′ site of the protease. GRL-0519 interacts with the backbone amide nitrogen, whereas DRV interacts with the carbonyl oxygen. (C and D) The interactions of tris-THF (C) and bis-THF (D) moieties with water molecules highlight differences in the polar interactions of the two inhibitors. The additional THF ring of GRL-0519 interacts with an extra water molecule and has four more polar interactions than DRV. The figures shown here were made with Maestro version 9.3 (Schrödinger, LLC, New York, NY, 2012).
Fig 7.

van der Waals interactions of GRL-0519 and darunavir with protease. GRL-0519 is shown as sticks (green carbons), and the van der Waals surfaces of selected moieties of GRL-0519 and its complexed protease are shown in gray and blue, respectively. DRV is shown as sticks (gray carbons), and the van der Waals surfaces of selected moieties of DRV and its complexed protease are shown in yellow and plum, respectively. (A and B) van der Waals interactions between the benzomethoxy of GRL-0519 and Asp-29′ and Asp-30′ of protease (A) and interactions between the aniline of DRV and Asp-29′ and Asp-30′ of protease (B). The surface interactions suggest that the methoxy group of GRL-0519 makes stronger van der Waals interactions with Asp-29′ and Asp-30′ of protease than does DRV. (C and D) The molecules are rotated to show the van der Waals surface interactions of the tris-THF (C) and bis-THF (D) moieties with protease. The tris-THF group of GRL-0519 forms better van der Waals contacts at the S2 site of protease than the bis-THF of DRV. The figures were made with Maestro version 9.3 (Schrödinger, LLC, New York, NY, 2012).
The tris-THF moiety increases water-mediated polar interactions with protease.
We further analyzed the polar interactions around the tris-THF ring of GRL-0519. The crystal structure shows that there are several water molecules that form polar interactions with GRL-0519 and bridge polar interactions with protease (Fig. 6C). Three water molecules, Wat-1, Wat-2, and Wat-3, form a tight network of hydrogen bonds among themselves and mediate polar interactions between GRL-0519 and the active-site residues. Wat-1 directly bridges the polar interactions between the oxygen of the second THF of GRL-0519 and Asp-29. The oxymethyl oxygen of the third THF ring of GRL-0519 enhances the strength of these networks of hydrogen bonds by its polar interactions with Wat-3 and Wat-4 (Fig. 6C). Wat-2 and Wat-3 also bridge the polar interaction between the oxymethyl of the third THF and Arg-87. There is one polar interaction between the third oxymethyl and Arg-8′ through Wat-3 and another network of polar interactions through Wat-4, Wat-5, and Wat-6. The crystal structure of DRV indicates the presence of five water molecules in this region compared to six present for GRL-0519 (Fig. 6D). The tris-THF of GRL-0519 and the additional water molecule are responsible for forming a total of 18 hydrogen bonds in this region compared to 14 for DRV, which has a bis-THF group as its P2 ligand.
DISCUSSION
GRL-0519, which contains a unique cyclic ether-derived nonpeptide P2 ligand, tris-THF, and a sulfonamide isostere, suppressed the replication of a wide spectrum of wild-type HIV-1 and HIV-2 strains with extremely low EC50s (Table 1). GRL-0519 was highly potent against a variety of multidrug-resistant clinical HIV-1 isolates with EC50s ranging from 0.0008 to 0.0034 μM, while the existing FDA-approved PIs examined either failed to suppress the replication of those isolates or required much higher concentrations for viral inhibition (Table 3). GRL-0519 also efficiently blocked, with an EC50 of 0.03 μM, the replications of a highly DRV-resistant variant (HIVDRVRP20), to which the three PIs (APV, ATV, and LPV) had EC50s of >1 μM and DRV had an EC50 of 0.255 μM (Table 3). Moreover, GRL-0519 exerted potent activity against laboratory PI-selected HIV-1 variants (except HIV-1APVR5μM) with significantly low EC50s (Table 2). GRL-0519 was less potent against HIV-1APVR5 μM, with an EC50 of 38.0 nM (a 76-fold difference), presumably due to the structural resemblance between GRL-0519 and APV, both of which contain a sulfonamide isostere (Fig. 1).
In an attempt to explain why GRL-0519 showed such potent activity against both wild-type and drug-resistant variants, we analyzed the crystal structure of the protease–GRL-0519 complex with protease (PDB ID, 3OK9). GRL-0519 has strong polar interactions with multiple regions of protease (Fig. 6A and C). The oxygens from two of the THF rings of GRL-0519 have strong polar interactions with the backbone amide nitrogens of Asp-29 and Asp-30. GRL-0519 also forms hydrogen bonds with Gly-27 and with the side chains of the catalytic aspartates, Asp-25 and Asp-25′. The sulfonamide oxygen and the carbonyl oxygen form polar contacts with Ile-50 and Ile-50′ in the flap through the bridging water molecule. Comparison of the crystal structures of protease complexes with GRL-0519 and protease complexes with DRV highlights the similarities and differences in interactions between these two inhibitors. The two oxygens from the bis-THF of DRV form hydrogen bond interactions with the backbone amide nitrogens of Asp-29 and Asp-30 (Fig. 6B). DRV also forms polar interactions with Gly-27, Asp-25, and Asp-25′ and polar contacts with the protease flap through the bridging water molecule. However, there are important differences in the interactions of GRL-0519 and DRV with protease. An additional water molecule (Wat-3 in Fig. 6C) around the tris-THF ring of GRL-0519 is observed in the crystal structure. This water molecule forms hydrogen bond interactions with the third THF ring of GRL-0519 and enhances the polar contact through a network of hydrogen bonds with Asp-29, Thr-26, Arg-87, and Arg-8′. There are four additional polar contacts arising out of the presence of the third THF ring of GRL-0519 and Wat-3 compared to the water-mediated polar interactions in this region of the protease complexes with DRV (Fig. 6D). Even though both GRL-0519 and DRV form polar interactions with the backbone atoms of Asp-30′ in the S2′ site, they interact with different sets of atoms (Fig. 6A and B). GRL-0519 forms the hydrogen bond with the amide nitrogen, whereas DRV forms polar contact with the carbonyl oxygen of Asp30′. The van der Waals surface interactions of GRL-0519 and DRV are also different (Fig. 7A to D). The tris-THF and methoxy group of GRL-0519 form stronger van der Waals contacts with the S2 and S2′ sites of protease, respectively. These interactions are stronger than the corresponding interactions of the bis-THF and aniline groups of DRV.
It should be noted that GRL-0519, as previously reported (35), blocks the dimerization of HIV-1 protease monomer subunits more potently, by at least 10-fold, than DRV, as examined by a fluorescence resonance energy transfer (FRET)-based HIV-1 expression assay that uses cyan (CFP) and yellow (YFP) fluorescent protein-tagged protease monomers (17). Considering that the dimerization of HIV-1 protease subunits is an essential process for its acquisition of proteolytic activity, which plays a critical role in the maturation and replication of the virus (36), the potent activity of GRL-0519 to block protease dimerization should also contribute to the greater antiviral potency of GRL-0519 than of DRV. Taken together, the stronger polar and nonpolar contacts of GRL-0519 with protease, as observed in its crystal structure, in addition to its potent activity to block protease dimerization, are likely to be responsible for its much more potent antiviral activity than that of DRV.
In our study, all the PIs examined showed no significant reduction of antiviral activity with the addition of HSA (Table 4). In contrast, the addition of AAG substantially reduced the antiretroviral activities of APV, ATV, and DRV by more than 9-fold, and their EC50s increased to 286.3, 32.7, and 33.8 nM, respectively. However, the reduction with GRL-0519 was only 4-fold, and its absolute EC50 was as low as 2.4 nM (Table 4). AAG is an acute-phase protein, and its concentration can increase upon injury, surgery, inflammation, malignancy, and infection, including HIV-1 infection (29, 37). Therefore, this feature of GRL-0519 may represent an advantage for its potential clinical application.
In our HIV-1NL4-3 selection experiment using GRL-0519, the emergence of GRL-0519-resistant variants was substantially delayed compared to other PIs and DRV. The use of a mixture of multiple-drug-resistant HIV-1 isolates can expedite the emergence of variants resistant to the drug used for the selection in vitro through homologous recombination and should reflect what occurs within individuals harboring a number of drug-resistant HIV-1 species (quasispecies) (28, 32). Thus, in this study, we also employed the mixture of 8 different multi-PI-resistant clinical isolates as a starting viral population (Fig. 5). In the present study, by passage 37, 3 major amino acid substitutions (K43I, A71T, and V82I) were identified in PR of HIV-1NL4-3. The residue V82 is located in the vicinity of the binding pocket of the protease and forms van der Waals contact with GRL0519. The V82A substitution is reportedly associated with resistance against various protease inhibitors (33, 34). However, K43 and A71 are distal from the inhibitor binding pocket and have no direct association with GRL0519. Thus, it is likely K43I and A71T are secondary substitutions. It is particularly noteworthy that we failed to select the A28S amino acid substitution in the present selection experiment with GRL-0519. In our previous studies of potent PIs, such as TMC-126 and GRL-1398, containing a paramethoxy group in the P2′ site, the A28S amino acid substitution was identified as a resistant variant (19, 25). In this regard, the combination of tris-THF as the P2 ligand and the paramethoxy moiety at P2′ seems to have prevented the selection of the A28S substitution as a resistant variant.
The tris-THF moiety has more interactions with the S2 site of the protease than the bis-THF present in either TMC-126 or DRV. Also, the p-OCH3 moiety seems to have more favorable van der Waals interactions with the S2 site than DRV (Fig. 7). It would be reasonable to expect that the combination of tris-THF and p-OCH3 may increase the activity, but the antiviral data suggest that GRL-0519 and TMC-126 have essentially the same antiviral activity (see Table S1 in the supplemental material). For any potential antiretroviral agents (including protease inhibitors) to exert their antiretroviral activity even in vitro, multiple factors are involved. They include (i) structural stability in culture medium, as well as in the cytoplasm; (ii) permeability into cells; and (iii) compartmentalization. Moreover, such potential antiretroviral agents have to tightly bind to the active site of the target viral protein (i.e., the protease active site) but should not bind to cellular proteins critical to the survival and functionality of the cells. It is possible that while GRL-0519 has greater interactions derived from the presence of the tris-THF group, those interactions are not directly reflected in the ultimate antiretroviral activity.
Since the GRL-0519-selected variant, HIV519RP37, was thought to be substantially replication competent, the replication kinetics of HIV-1NL4-3 and HIV519RP37 were compared in the presence and absence of GRL-0519. The data showed that HIV-1NL4-3 failed to replicate in the presence of 0.005 μM GRL-0519 and that HIV519RP37 did the same in the presence of 0.015 μM GRL-0519 throughout the 7-day culture period (Fig. 4). The concentration range of 0.005 to 0.015 μM is relatively easily achieved in the clinical use of various PIs. For example, the peak and nadir plasma levels of DRV were ∼7.1 and 3.4 μM when 400 and 100 mg of DRV/RTV were administered twice daily for 7 days (38). Considering that the EC50s of GRL-0519 are extremely low, ranging from 0.0005 to 0.030 μM (Tables 1 to 4), and that GRL-0519's selectivity index of 63,714 is highly favorable compared to other conventional PIs examined in this study (Table 1), both the anti-HIV potency and safety of GRL-0519 could be favorable, although the efficacy and emergence of adverse effects should be ultimately determined by controlled clinical trials.
In conclusion, GRL-0519 possesses a number of fairly favorable features for the development of the compound as a potential therapeutic for HIV-1 infection and AIDS. However, its oral bioavailability, pharmacokinetics/pharmacodynamics, and biodistribution remain to be determined, and further investigation is warranted.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by a Grant for Global Education and Research Center Aiming at the Control of AIDS (Global Center of Excellence, supported by Monbu-Kagakusho); Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan; a Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Reemerging Infectious Diseases (Renkei Jigyo, no. 78; Kumamoto University) of Monbu-Kagakusho (H.M.); the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (H.M.); and by a grant from the National Institutes of Health (GM53386; A.K.G.).
Footnotes
Published ahead of print 12 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02189-12.
REFERENCES
- 1. Edmonds A, Yotebieng M, Lusiama J, Matumona Y, Kitetele F, Napravnik S, Cole SR, Van Rie A, Behets F. 2011. The effect of highly active antiretroviral therapy on the survival of HIV-infected children in a resource-deprived setting: a cohort study. PLoS Med. 8:e1001044 doi:10.1371/journal.pmed.1001044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lohse N, Hansen AB, Gerstoft J, Obel N. 2007. Improved survival in HIV-infected persons: consequences and perspectives. J. Antimicrob. Chemother. 60:461–463 [DOI] [PubMed] [Google Scholar]
- 3. Mitsuya H, Maeda K, Das D, Ghosh AK. 2008. Development of protease inhibitors and the fight with drug-resistant HIV-1 variants. Adv. Pharmacol. 56:169–197 [DOI] [PubMed] [Google Scholar]
- 4. Walensky RP, Paltiel AD, Losina E, Mercincavage LM, Schackman BR, Sax PE, Weinstein MC, Freedberg KA. 2006. The survival benefits of AIDS treatment in the United States. J. Infect. Dis. 194:11–19 [DOI] [PubMed] [Google Scholar]
- 5. De Clercq E. 2002. Strategies in the design of antiviral drugs. Nat. Rev. Drug Discov. 1:13–25 [DOI] [PubMed] [Google Scholar]
- 6. Siliciano JD, Siliciano RF. 2004. A long-term latent reservoir for HIV-1. J. Antimicrob. Chemother. 54:6–9 [DOI] [PubMed] [Google Scholar]
- 7. Simon V, Ho DD. 2003. HIV-1 dynamics in vivo: implications for therapy. Nat. Rev. Microbiol. 1:181–190 [DOI] [PubMed] [Google Scholar]
- 8. Carr A. 2003. Toxicity of antiretroviral therapy and implications for drug development. Nat. Rev. Drug Discov. 2:624–634 [DOI] [PubMed] [Google Scholar]
- 9. Fumero E, Podzamczer D. 2003. New patterns of HIV-1 resistance during HAART. Clin. Microbiol. Infect. 9:1077–1084 [DOI] [PubMed] [Google Scholar]
- 10. Grabar S, Weiss L, Costagliola D. 2006. HIV infection in older patients in the HAART era. J. Antimicrob. Chemother. 57:4–7 [DOI] [PubMed] [Google Scholar]
- 11. Hirsch HH, Kaufmann G, Sendi P, Battegay M. 2004. Immune reconstitution in HIV-infected patients. Clin. Infect. Dis. 38:1159–1166 [DOI] [PubMed] [Google Scholar]
- 12. Little SJ, Holte S, Routy JP, Daar ES, Markowitz M, Collier AC, Koup RA, Mellors JW, Connick E, Conway B, Kilby M, Wang L, Whitcomb JM, Hellmann NS, Richman DD. 2002. Antiretroviral-drug resistance among patients recently infected with HIV. N. Engl. J. Med. 347:385–394 [DOI] [PubMed] [Google Scholar]
- 13. Naggie S, Hicks C. 2010. Protease inhibitor-based antiretroviral therapy in treatment-naive HIV-1-infected patients: the evidence behind the options. J. Antimicrob. Chemother. 65:1094–1099 [DOI] [PubMed] [Google Scholar]
- 14. Ghosh AK, Kincaid JF, Cho W, Walters DE, Krishnan K, Hussain KA, Koo Y, Cho H, Rudall C, Holland L, Buthod J. 1998. Potent HIV protease inhibitors incorporating high-affinity P2-ligands and (R)-(hydroxyethylamino)sulfonamide isostere. Bioorg. Med. Chem. Lett. 8:687–690 [DOI] [PubMed] [Google Scholar]
- 15. Ghosh AK, Krishnan K, Walters DE, Cho W, Cho H, Koo Y, Trevino J, Holland L, Buthod J. 1998. Structure based design: novel spirocyclic ethers as nonpeptidal P2-ligands for HIV protease inhibitors. Bioorg. Med. Chem. Lett. 8:979–982 [DOI] [PubMed] [Google Scholar]
- 16. Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. 2003. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 47:3123–3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghosh AK, Xu CX, Rao KV, Baldridge A, Agniswamy J, Wang YF, Weber IT, Aoki M, Miguel SG, Amano M, Mitsuya H. 2010. Probing multidrug-resistance and protein-ligand interactions with oxatricyclic designed ligands in HIV-1 protease inhibitors. ChemMedChem 5:1850–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shirasaka T, Kavlick MF, Ueno T, Gao WY, Kojima E, Alcaide ML, Chokekijchai S, Roy BM, Arnold E, Yarchoan R, Mitsuya H. 1995. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc. Natl. Acad. Sci. U. S. A. 92:2398–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoshimura K, Kato R, Kavlick MF, Nguyen A, Maroun V, Maeda K, Hussain KA, Ghosh AK, Gulnik SV, Erickson JW, Mitsuya H. 2002. A potent human immunodeficiency virus type 1 protease inhibitor, UIC-94003 (TMC-126), and selection of a novel (A28S) mutation in the protease active site. J. Virol. 76:1349–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoshimura K, Kato R, Yusa K, Kavlick MF, Maroun V, Nguyen A, Mimoto T, Ueno T, Shintani M, Falloon J, Masur H, Hayashi H, Erickson J, Mitsuya H. 1999. JE-2147: a dipeptide protease inhibitor (PI) that potently inhibits multi-PI-resistant HIV-1. Proc. Natl. Acad. Sci. U. S. A. 96:8675–8680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ghosh AK, Leshchenko S, Noetzel M. 2004. Stereoselective photochemical 1,3-dioxolane addition to 5-alkoxymethyl-2(5H)-furanone: synthesis of bis-tetrahydrofuranyl ligand for HIV protease inhibitor UIC-94017 (TMC-114). J. Org. Chem. 69:7822–7829 [DOI] [PubMed] [Google Scholar]
- 22. Maeda K, Yoshimura K, Shibayama S, Habashita H, Tada H, Sagawa K, Miyakawa T, Aoki M, Fukushima D, Mitsuya H. 2001. Novel low molecular weight spirodiketopiperazine derivatives potently inhibit R5 HIV-1 infection through their antagonistic effects on CCR5. J. Biol. Chem. 276:35194–35200 [DOI] [PubMed] [Google Scholar]
- 23. Nakata H, Amano M, Koh Y, Kodama E, Yang G, Bailey CM, Kohgo S, Hayakawa H, Matsuoka M, Anderson KS, Cheng YC, Mitsuya H. 2007. Activity against human immunodeficiency virus type 1, intracellular metabolism, and effects on human DNA polymerases of 4′-ethynyl-2-fluoro-2′-deoxyadenosine. Antimicrob. Agents Chemother. 51:2701–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Amano M, Koh Y, Das D, Li J, Leschenko S, Wang YF, Boross PI, Weber IT, Ghosh AK, Mitsuya H. 2007. A novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI), GRL-98065, is potent against multiple-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 51:2143–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ide K, Aoki M, Amano M, Koh Y, Yedidi RS, Das D, Leschenko S, Chapsal B, Ghosh AK, Mitsuya H. 2011. Novel HIV-1 protease inhibitors (PIs) containing a bicyclic P2 functional moiety, tetrahydropyrano-tetrahydrofuran, that are potent against multi-PI-resistant HIV-1 variants. Antimicrob. Agents Chemother. 55:1717–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tojo Y, Koh Y, Amano M, Aoki M, Das D, Kulkarni S, Anderson DD, Ghosh AK, Mitsuya H. 2010. Novel protease inhibitors (PIs) containing macrocyclic components and 3(R),3a(S),6a(R)-bis-tetrahydrofuranylurethane that are potent against multi-PI-resistant HIV-1 variants in vitro. Antimicrob. Agents Chemother. 54:3460–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaminski GA, Friesner EA. 2001. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 105:6474–6487 [Google Scholar]
- 28. Koh Y, Amano M, Towata T, Danish M, Leshchenko-Yashchuk S, Das D, Nakayama M, Tojo Y, Ghosh AK, Mitsuya H. 2010. In vitro selection of highly darunavir-resistant and replication-competent HIV-1 variants by using a mixture of clinical HIV-1 isolates resistant to multiple conventional protease inhibitors. J. Virol. 84:11961–11969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schon A, Ingaramo M, Freire E. 2003. The binding of HIV-1 protease inhibitors to human serum proteins. Biophys. Chem. 105:221–230 [DOI] [PubMed] [Google Scholar]
- 30. Marcelin AG, Affolabi D, Lamotte C, Mohand HA, Delaugerre C, Wirden M, Voujon D, Bossi P, Ktorza N, Bricaire F, Costagliola D, Katlama C, Peytavin G, Calvez V. 2004. Resistance profiles observed in virological failures after 24 weeks of amprenavir/ritonavir containing regimen in protease inhibitor experienced patients. J. Med. Virol. 74:16–20 [DOI] [PubMed] [Google Scholar]
- 31. Young TP, Parkin NT, Stawiski E, Pilot-Matias T, Trinh R, Kempf DJ, Norton M. 2010. Prevalence, mutation patterns, and effects on protease inhibitor susceptibility of the L76V mutation in HIV-1 protease. Antimicrob. Agents Chemother. 54:4903–4906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aoki M, Danish ML, Aoki-Ogata H, Amano M, Ide K, Koh Y, Mitsuya H. 2012. The loss of protease dimerization inhibition activity of tipranavir (TPV) and its association with HIV-1 acquisition of resistance to TPV. J. Virol. 86:13384–13396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wlodawer A, Vondrasek J. 1998. Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 27:249–284 [DOI] [PubMed] [Google Scholar]
- 34. Wlodawer A, Erickson JW. 1993. Structure-based inhibitors of HIV-1 protease. Annu. Rev. Biochem. 62:543–585 [DOI] [PubMed] [Google Scholar]
- 35. Koh Y, Matsumi S, Das D, Amano M, Davis DA, Li J, Leschenko S, Baldridge A, Shioda T, Yarchoan R, Ghosh AK, Mitsuya H. 2007. Potent inhibition of HIV-1 replication by novel non-peptidyl small molecule inhibitors of protease dimerization. J. Biol. Chem. 282:28709–28720 [DOI] [PubMed] [Google Scholar]
- 36. Dunn BM, Goodenow MM, Gustchina A, Wlodawer A. 2002. Retroviral proteases. Genome Biol. 3:reviews3006.1-reviews3006.7. doi:10.1186/gb-2002-3-4-reviews3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fournier T, Medjoubi NN, Porquet D. 2000. Alpha-1-acid glycoprotein. Biochim. Biophys. Acta 1482:157–171 [DOI] [PubMed] [Google Scholar]
- 38. Boffito M, Miralles D, Hill A. 2008. Pharmacokinetics, efficacy, and safety of darunavir/ritonavir 800/100 mg once-daily in treatment-naïve and -experienced patients. HIV Clin. Trials 9:418–427 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.