

Abstract
The lack of effective therapies for treating tuberculosis (TB) is a global health problem. While Mycobacterium tuberculosis is notoriously resistant to most available antibiotics, we identified synthetic short cationic antimicrobial peptides that were active at low micromolar concentrations (less than 10 μM). These small peptides (averaging 10 amino acids) had remarkably broad spectra of antimicrobial activities against both bacterial and fungal pathogens and an indication of low cytotoxicity. In addition, their antimicrobial activities displayed various degrees of species specificity that were not related to taxonomy. For example, Candida albicans and Staphylococcus aureus were the best surrogates to predict peptide activity against M. tuberculosis, while Mycobacterium smegmatis was a poor surrogate. Principle component analysis of activity spectrum profiles identified unique features associated with activity against M. tuberculosis that reflect their distinctive amino acid composition; active peptides were more hydrophobic and cationic, reflecting increased tryptophan with compensating decreases in valine and other uncharged amino acids and increased lysine. These studies provide foundations for development of cationic antimicrobial peptides as potential new therapeutic agents for TB treatment.
INTRODUCTION
Mycobacterium tuberculosis is the causative agent of tuberculosis (TB), one of the world's major health problems. Current vaccines and chemotherapeutic measures are limited in their efficacy and are failing to prevent spread of the disease. The emergence of M. tuberculosis strains resistant to the few frontline drugs that are currently available makes it very difficult, too often impossible, to cure fatal infections. Cationic antimicrobial peptides (CAMPs) produced by the immune system can help resist infection. Since many CAMPs have rapid bactericidal activity against a broad range of microbes and there is a low probability of pathogens acquiring resistance (1), they represent promising new avenues for antibacterial drug development, especially for multidrug-resistant strains (2, 3).
Thousands of CAMPs have evolved within bacteria, plants, and animals (2, 4). Indeed, many antimicrobial peptides are under clinical development for treating a wide variety of diseases (2). While a single mechanism does not underlie all effects of CAMPs, their bactericidal activities often depend on their abilities to permeabilize membranes by creating pores or disrupting the organization of the lipid bilayer. Partial depolarization of the cytoplasmic membrane by CAMPs interferes with electron transport and oxidative metabolism, leading to cell death (5, 6). CAMP binding to ATP may mediate additional effects on metabolism (7–10). Directly or indirectly, exposure to CAMPs reduces the activities of essential processes, including synthesis of protein, DNA, RNA, and the bacterial cell envelope.
Bacteria have evolved resistance systems to avoid the antimicrobial effects of CAMPs. Anionic groups in the outer or inner membranes can be neutralized to prevent cationic antimicrobial peptides from binding (11–13). The activities of peptides and their species specificity are also determined by resistance systems that directly recognize structural motifs in specific CAMPs and then bind, degrade, or extrude them from the membrane (1). Membrane transporters also serve to extrude CAMPs from the cytoplasmic or inner membrane compartments (14–16). The proliferation of intracellular pathogens within host cells is often dependent on their abilities to sense and activate these resistance systems (17).
Our understanding of the amino acid sequence requirements that underlie the antimicrobial activities of CAMPs has been accelerated by the development of high-throughput assays and methods to analyze their physiochemical characteristics (18–20). Physiochemical characteristics of amino acids have been defined by dozens of quantitative structure-activity relationships (QSARs), including various descriptors of hydrophobicity, hydrophilicity, isoelectric point, charge, mass, volume, and other physiochemical characteristics (21). Although none of the individual descriptors were predictive of activity, QSAR analyses showed that antimicrobial activity was determined, to various extents, by combinations of these basic features (21–24). These analyses identified common features of CAMPs that make them active against a broad range of microbes and provided initial indications of specificity. In fact, several studies have shown that peptides can have different activities against representative pathogenic bacteria (20–22); however, this has never been statistically analyzed for possible correlations of their activities against different organisms or with their physiochemical properties. The possible activities of synthetic CAMPs against Mycobacterium species have not been reported.
The intrinsic drug resistance of M. tuberculosis, along with its low growth rate and requirements for biocontainment, has discouraged efforts to develop artificial small CAMPs effective against M. tuberculosis. M. tuberculosis is often described as being CAMP tolerant. In fact, many natural CAMPs are active against M. tuberculosis only at rather high concentrations that are not practical for TB treatment (25–30). Furthermore, CAMPs can be cytotoxic to mammalian cells, and long peptides are relatively expensive to produce. In this study, we took advantage of libraries enriched for CAMPs active against Pseudomonas aeruginosa (19–22) and devised screens to identify those having activity against M. tuberculosis. We report, for the first time, short, nontoxic, artificial CAMPs that inhibit M. tuberculosis growth in vitro at low micromolar concentrations.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Mycobacterium smegmatis mc26, M. tuberculosis H37Rv, and the M. tuberculosis lux strain (31) were routinely cultivated at 37°C in Difco Middlebrook 7H9 broth (ammonium sulfate, 0.5 g/liter; disodium phosphate, 2.5 g/liter; monopotassium phosphate, 1.0 g/liter; sodium citrate, 0.1 g/liter; magnesium sulfate, 0.05 g/liter; calcium chloride, 0.0005 g/liter; zinc sulfate, 0.001 g/liter; copper sulfate, 0.001 g/liter; l-glutamic acid, 0.5 g/liter; biotin, 0.0005 g/liter; ferric ammonium citrate, 0.04 g/liter; and pyridoxine, 0.001 g/liter) supplemented with 10% (vol/vol) BD BBL Middlebrook ADC enrichment (sodium chloride, 8.5 g/liter; bovine albumin [fraction V], 50.0 g/liter; dextrose, 20.0 g/liter; and catalase, 0.03 g/liter), 0.2% glycerol, and 0.05% (vol/vol) tyloxapol. Bacterial strains Escherichia coli UB1005 (F− nalA37 metB1), a wild-type Salmonella enterica serovar Typhimurium strain, wild-type Pseudomonas aeruginosa H103, Enterococcus faecalis ATCC 29212, Staphylococcus aureus ATCC 25923, a clinical isolate of Staphylococcus epidermidis, and Candida albicans were obtained from R. E. W. Hancock (Department of Microbiology and Immunology, University of British Columbia); Staphylococcus epidermidis was obtained from B. Dill (Department of Microbiology and Immunology, University of British Columbia); and Candida albicans was obtained from D. Speert (Department of Medicine, University of British Columbia). All nonmycobacterial strains, including C. albicans, were grown in Mueller-Hinton (MH; Difco) broth or MH agar at 37°C. Peptide activity against M. smegmatis was assayed in 7H9* medium (see below under “Medium composition”). M. tuberculosis screens and peptide antimicrobial activity assays were performed in 7H9* broth medium. Peptide activity against all other microbial pathogens was determined using MH broth (Difco) that was non-cation adjusted.
Medium composition.
Some cations, such as Ca2+, Mg2+, and Na+, can inhibit the antimicrobial activity of cationic peptides (18, 32). Among other salts, commercially available 7H9 medium contains 50 mg/liter of magnesium sulfate (physiological concentrations are 10 to 12.5 mg/liter) and 0.5 mg/liter calcium chloride (40- to 50-fold lower than physiological concentrations). In addition, ADC supplement contains 8.5 g/liter (145 mM) sodium chloride, while concentrations of 100 mM Na+ inhibited peptide activity (33). Based on this information, we developed a new medium to test peptide activity against mycobacteria. The components of either standard 7H9 or ADC supplement were individually prepared, autoclaved, and combined in the appropriate proportions. Both magnesium sulfate and sodium chloride were excluded from the 7H9 medium and ADC supplement, respectively. This new medium (7H9*) supported growth rates of M. tuberculosis and M. smegmatis similar to those of standard commercially available 7H9 (data not shown).
Peptide libraries.
A flowchart of the screening program used to select the most potent anti-M. tuberculosis peptides is outlined in Fig. 1. Peptide library PL-A included peptides enriched for higher activity against P. aeruginosa but also had peptides serving as internal negative controls. PL-A was synthesized on cellulose using the SPOT technology (34). A glycine linker was used to achieve maximum peptide density (35). After synthesis, the peptides were cleaved from the cellulose support by ammonia gas overnight. The peptide spots were punched out, transferred into polypropylene 96-well microtiter plates, and dissolved in 200 μl distilled autoclaved water (18). The plates were foil sealed and gently shaken overnight at room temperature. Peptides were then immediately used for the screen or stored at −20°C. To minimize costs for the screen, the peptides were not purified or further analyzed. Typically, the yield of a peptide per cm2 of cellulose using a glycine linker is 0.8 to 1.9 μmol (35). Peptide libraries PL-B and PL-C were combined to generate peptide library PL-D. PL-D was synthesized using standard Fmoc chemistry on resin and high-performance liquid chromatography (HPLC) purified to >85% purity. Peptides were weighed to obtain the final stock concentration, but concentration was corrected based on the measured absorption of the peptides at 280 nm, calculated using the amino acids W, F, C, and Y.
Fig 1.

Flowchart of the screening program used to select antimycobacterial peptides. Peptide library A (PL-A) was composed of 253 peptides from several published (19–22) and unpublished libraries, chosen for their antibacterial activities against Pseudomonas aeruginosa (note that 214 of these peptides qualified as positive hits against P. aeruginosa by the more stringent criteria used in previous studies [24]. The 39 nonactives were introduced as internal negative controls). Peptides in PL-A were screened against M. smegmatis and M. tuberculosis. Those active against M. tuberculosis, representing a range of amino acid compositions and sequences, were chosen for further analyses as PL-B (20 peptides). This pool was combined with 29 peptides selected for activities against P. aeruginosa and other representative pathogenic microbes (PL-C; CAMPs active against P. aeruginosa [unpublished data]) to generate PL-D. The antimicrobial activities of the 49 peptides in PL-D were quantified against a broad range of pathogenic microbes.
Antimicrobial peptide activity assays.
The mycobacterial screen was performed in a final volume of 100 μl 7H9* in sterile 96-well, round-bottom polypropylene microtiter plates (Costar). Cell viability was assessed using the resazurin assay as previously described (31, 36). Briefly, M. smegmatis and M. tuberculosis cells were incubated in the presence of peptides for 3 or 8 days and then in the presence of resazurin for 1 or 2 days, respectively. A change from blue to pink indicated bacterial growth. The activities of PL-D peptides against M. tuberculosis were determined using a luciferase-based assay (luminescence) (31). Growing cultures were transferred from polypropylene microtiter plates to detection polystyrene plates (Costar), and an equal volume of Bright Glo Luciferase assay reagent (Promega) was then added to the plates. After a 15-min incubation, luminescence was measured in a PerkinElmer Tropix luminometer using a 1-s exposure. MIC90 was calculated as the drug concentration that reduced luminescence by at least 90%, compared to untreated samples. Isoniazid (a benchmark anti-TB standard) was used for calibration assays in 7H9* medium. Various conditions for luminescence detection were optimized: the best signal with the highest dynamic range between growth and no-growth conditions was obtained after 72 h of incubation with 105 cells/ml inoculum and a 100-μl volume per well. Under these conditions, the MIC of isoniazid for the M. tuberculosis lux strain in 7H9* medium was 0.025 μg/ml, similar to levels previously established (37) and consistent with our resazurin assay. These conditions were then used to determine the antimicrobial activity of the PL-D peptides. Peptides were introduced to M. tuberculosis lux cultures in a 2-fold serial dilution format. As internal controls, isoniazid and drug-free wells were also included in every peptide assay plate. MIC values were the mean of two independent biological determinations. To measure antimicrobial activity of the PL-D peptides against other microbial pathogens, 2-fold serial dilutions of the peptides were introduced to cultures growing exponentially (2 × 105 to 7 × 105 cells/ml) in polypropylene 96-well plates. After 18 h of incubation at 37°C, the MIC was reported for the lowest concentration of peptide where no growth was observed (38). PL-D activity against M. smegmatis was determined by the resazurin assay.
Assay for peptide cytotoxicity against human THP-1 cells.
Frozen stocks of THP-1 cells (39) (ATCC TIB-202) were thawed and cultured in RPMI medium with 1% glutamine, 10% fetal bovine serum (FBS), 10,000 units/ml penicillin G (sodium salt), and 10 mg/ml streptomycin sulfate in 0.85% saline. Cells were passaged a maximum of 5 times and maintained without antibiotics. Four thousand cells per well were induced to differentiate in RPMI medium containing 1% glutamine, 80 ng/ml phorbol myristate acetate (PMA), and 10% FBS without antibiotics and left to adhere to the culture dish for 24 h. Black, clear-bottom 96-well microtiter plates were used. After cell differentiation, medium was removed from the wells and 2-fold serial dilutions of peptides were added in 100 μl of phenol red-free RPMI medium containing 10% FBS. Cells were incubated in the presence of the peptides for 72 h, after which 20 μl of resazurin (0.01%) was added to each well. After 4 h of incubation, fluorescence (excitation wavelength [lexc] = 535 nm; emission wavelength [lem] = 590 nm) was read to determine the viability of the cells; the 50% inhibitory concentration (IC50) was calculated relative to that of untreated cells.
Activity classes.
A binary classification term (“positive” and “negative”) was assigned to each peptide in each library and against each species. The resazurin assay was used to determine the activity of PL-A peptides against M. tuberculosis and M. smegmatis. This colorimetric redox indicator is based on a blue-to-pink color conversion. Peptides that fully inhibited cell growth (blue) or that partially inhibited cell growth (violet) for a given peptide concentration were classified as “positive,” and if a peptide did not inhibit cell growth, it was classified as “negative” (pink). A minimum dilution factor of >20 (corresponding to about 50 μM) was adopted to restrict the number of selected positive peptides. For activity analysis of the PL-D peptides, “positive” was defined as a MIC of <10 μM and “negative” as a MIC of >20 μM. This approach was used to identify and analyze descriptors for class differences (e.g., in amino acid distribution). Species with less than 10 peptides of class “negative” or “positive” were excluded from analyses of amino acid composition.
Data analysis and statistics.
The activities against P. aeruginosa and the descriptor features were analyzed by a methodology described in reference 24. In addition, the similarities of logarithmic MIC values against different microbes were analyzed using principal component analysis (PCA) to determine whether peptides act as broad-spectrum antibiotics against all species, groups of species, or individual species. PCA computes aggregated features of orthogonal linear combinations of the original features. The linear combinations were optimized and sorted so that each combination preserved maximal information content measured by the percentage of explained variance in descending order. In addition, the coefficients of the linear combinations (factor loadings) explain how the aggregated feature was computed. An additional correlation analysis was done using Pearson correlation coefficients for the logarithmic MIC (LMIC) values and the aggregated features from PCA. The correlations between aggregated features and LMIC were used to show the directions of the coefficients of the linear combination. This made interpretation of similar or different MICs for different species and aggregated features (positive or negative correlations) or missing relations (correlations near zero) possible. In addition, the aggregated features can be interpreted as tendencies in the complete library to have broad-spectrum activities (similar correlations of an aggregated feature to all LMICs), specific activities (correlation to only LMIC), or grouped activities (positive or negative correlation of specific LMIC groups). The complete analysis was performed by the peptide extension of the MATLAB toolbox Gait-CAD (40). The differences in amino acid distributions of peptides between classes “positive” and “negative” were tested using a (nonparametric) Wilcoxon rank sum test (equivalent to a Mann-Whitney U test) with a significance level of alpha = 0.05. It was Bonferroni corrected to an alpha value of 0.0025 with 20 amino acids.
RESULTS
Identification of CAMPs active against M. tuberculosis.
Based on studies showing that some natural CAMPs have moderate activities against M. tuberculosis (25–28) and with the availability of artificial short CAMP libraries (19–22), we explored the interesting possibility that some might target M. tuberculosis (our screening strategy is summarized in Fig. 1). Our principle peptide library (PL-A) was screened using a qualitative test for activities against M. smegmatis and M. tuberculosis. Since M. tuberculosis and P. aeruginosa each have their own distinctive envelope structures that provide selective permeability barriers and resistance to different sets of antibiotics, it was surprising to find that nearly one of every three peptides (82/253 [32.4%]) inhibited growth of M. tuberculosis, including several that were active at low micromolar concentrations (highest concentration tested was ca. 50 μM). Even more were active against M. smegmatis (a total of 131). Interestingly, only 50 out of 253 peptides were active against both M. tuberculosis and M. smegmatis (Fig. 1). Twenty peptides from this library (PL-A) with various degrees of activity against M. tuberculosis were then combined with an independent library of 29 peptides designed for activity against P. aeruginosa (PL-C). The MICs of these 49 peptides (PL-D) were quantified using an M. tuberculosis lux strain (Table 1). Twelve peptides had a MIC90 lower than 5 μM; peptide WKWLKKWIK was the most potent, with a MIC90 of 1 μM. To our knowledge, this is the first report of artificial CAMPs with potencies against M. tuberculosis lower than 5 μM.
Table 1.
Antimicrobial activity of selected peptides (library PL-D)
| Sequence | MIC (μM)a |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| M. tuberculosis | M. smegmatis | P. aeruginosa | E. coli | S. enterica serovar Typhimurium | C. albicans | S. epidermidis | S. aureus | E. faecalis | |
| WKWLKKWIK | 1.1 | 1.9 | 2.9 | 5.8 | 11.5 | 5.8 | 0.7–0.3 | 0.7 | 11.5 |
| ILRWKWRWWRWRR | 2.4 | 2.4 | 5.7 | 1.4 | 2.9 | 5.7 | 0.7 | 0.7 | 2.9 |
| ILPWKWRWWKWRR | 2.6 | 2.2 | 5.7 | 1.4 | 5.7 | 2.9 | 0.7 | 0.7 | 2.9 |
| RWRRKWWWW | 2.8 | 5.7 | 11.2 | 1.4 | 2.8 | 5.6 | 0.7 | 0.7 | 2.8 |
| WRKFWKYLK | 3.0 | 1.5 | 6.0 | 0.8 | 6.0 | 3.0 | 0.8 | 0.8 | 6.0 |
| KRWWKWWRR | 3.1 | 4.9 | 11.1 | 2.8 | 5.6 | 5.6 | 0.7 | 0.7 | 11.1 |
| RRWWKWWWR | 3.6 | 4.8 | 5.4 | 2.7 | 2.7 | 5.4 | 0.7 | 0.7 | 5.4 |
| KIWWWWRKR | 4.1 | 5.4 | 42.3 | 5.3 | 10.6 | 10.6 | 1.3 | 1.3 | 10.6 |
| RLWWWWRRK | 4.1 | 2.4 | 16.6 | 4.1 | 8.3 | 4.1 | 0.5 | 2.1 | 4.1 |
| KWKWWWRKI | 4.2 | 6.4 | 4.6 | 4.6 | 4.6 | 4.6 | 1.2 | 1.2 | 4.6 |
| RIRRWKFRW | 4.3 | 1.4 | 42.6 | 5.3 | 5.3 | 21.3 | 1.3 | 1.3 | 5.3 |
| RLKRWWKFL | 4.5 | 3.0 | 9.8 | 1.2 | 4.9 | 9.8 | 1.2 | 2.4 | 9.8 |
| RWWRWRKWW | 4.6 | 4.6 | 10.3 | 2.6 | 5.2 | 2.6 | 0.6 | 0.6 | 5.2 |
| KRWWWWRFR | 5.2 | 5.2 | 46.9 | 2.9 | 5.9 | 5.9 | 0.7 | 0.7 | 11.7 |
| KRWWRKWWR | 5.3 | 5.3 | 43.2 | 2.7 | 5.4 | 5.4 | 0.7 | 0.7 | 10.8 |
| RRWWRWVVW | 5.6 | 11.4 | 2.6 | 0.7 | 2.6 | 10.4 | 0.7 | 1.3 | 2.6 |
| WFKMRWWGR | 5.9 | 2.2 | 46.1 | 5.8 | 23.1 | 11.5 | 2.9 | 2.9 | 11.5 |
| KFKWWRMLI | 6.1 | 15.3 | 4.6 | 0.6 | 2.3 | 4.6 | 0.6 | 1.2 | 2.3 |
| FIKWKFRWWKWRK | 7.4 | 3.7 | 6.0 | 1.5 | 1.5 | 3.0 | 0.7 | 0.7 | 1.5 |
| RIKRWWWWR | 7.8 | 4.6 | 86.3 | 2.7 | 2.7 | 5.4 | 1.3 | 1.3 | 10.8 |
| RWRWWWRVY | 8.0 | 6.7 | 13.2 | 1.6 | 3.3 | 13.2 | 1.6 | 3.3 | 3.3 |
| HQFRFRFRVRRK | 8.2 | 4.1 | 32.0 | 8.0 | 4.0 | 8.0 | 1.0 | 2.0 | 16.0 |
| LKRRWKWWI | 8.8 | 5.1 | 19.1 | 2.4 | 9.6 | 19.1 | 1.2 | 2.4 | 9.6 |
| RRRIKIRWY | 8.9 | 1.5 | 38.2 | 0.6 | 9.5 | 19.1 | 1.2 | 2.4 | 9.5 |
| RLWWKIWLK | 9.0 | 3.8 | 22.5 | 2.8 | 11.3 | 5.6 | 1.4 | 1.4 | 5.6 |
| KRRWRIWLV | 9.1 | 2.7 | 3.0 | 3.0 | 3.0 | 12.0 | 1.5–0.7 | 1.5 | 3.0 |
| FFIYVWRRR | 11.9 | 1.3 | 15.3 | 3.8 | 7.6 | 7.6 | 1.0 | 1.9 | 3.8 |
| IRMRIRVLL | 13.7 | 0.3 | 16.0 | 2.0 | 4.0 | 16.0 | 2.0 | 4.0 | 16.0 |
| RWWRKIWKW | 16.6 | 8.3 | 2.2 | 8.8 | 8.8 | 4.4 | 1.1 | 2.2 | 4.4 |
| LRFILWWKR | 17.0 | 4.2 | 25.7 | 3.2 | 6.4 | 6.4 | 1.6 | 1.6 | 6.4 |
| RWWIRIRWH | 17.0 | 1.6 | 10.7 | 1.3 | 2.7 | 10.7 | 1.3 | 1.3 | 2.7 |
| RRRWWKLMM | 17.6 | 2.2 | 11.0 | 2.7 | 11.0 | 21.9 | 2.7–1.4 | 5.5 | 21.9 |
| LRRWIRIRW | 17.7 | 2.2 | 21.4 | 0.7 | 5.4 | 10.7 | 1.3–0.6 | 1.3 | 5.4 |
| RKFRWWVIR | 17.8 | 3.7 | 21.9 | 2.7.0 | 5.5 | 21.9 | 1.4–0.5 | 1.4 | 10.9 |
| WKIVFWWRR | 23.3 | 4.4 | 21.9 | 1.4 | 2.7 | 5.5 | 0.7 | 1.4 | 2.7 |
| RQRRVVIWW | 24.7 | 1.9 | 10.7 | 1.3 | 5.3 | 21.4 | 1.3 | 2.7 | 5.3 |
| RRWRVIVKW | 24.7 | 2.3 | 2.7 | 1.3 | 2.7 | 5.4 | 0.7 | 1.3 | 2.7 |
| RRWKIVVIRWRR | 24.8 | 6.2 | 5.6 | 1.4 | 2.8 | 11.1 | 0.3 | 0.7 | 1.4 |
| RLWRIVVIRVKR | 25.9 | 4.3 | 7.1 | 1.8 | 3.5 | 7.1 | 0.4 | 0.9 | 1.8 |
| RLRRIVVIRVFR | 29.8 | 5.0 | 8.0 | 4.0 | 4. | 16.0 | 0.5 | 1.0 | 2.0 |
| VRLRIRVRVIRK | 30.1 | 1.6 | 2.0 | 2.0 | 2.0 | 8.0 | 1.0 | 1.0 | 4.0 |
| RRYHWRIYI | 33.2 | 16.6 | 12.5 | 1.6 | 6.3 | 12.5 | 3.1 | 3.1 | 12.5 |
| RKWKIKWYW | 34.5 | 2.9 | 5.2 | 0.7 | 2.6 | 10.5 | 1.3 | 2.6 | 5.2 |
| YRLRVKWKW | 36.0 | 1.5 | 14.3 | 3.6 | 7.1 | 3.6 | 0.9 | 1.8 | 7.1 |
| WKWRVRVTI | 51.5 | 0.8 | 5.4 | 1.3 | 2.7 | 10.8 | 1.3 | 2.7 | 2.7 |
| RTKKWIVWI | 78.1 | 9.8 | 11.6 | 1.4 | 5.8 | 23.1 | 2.9 | 5.8 | 11.6 |
| NWRKLYRRK | 109.3 | 38.3 | >86.5 | 86.5 | >86.5 | 86.5 | 43.3 | 86.5 | >86.5 |
| YKFRWRIYI | 130.0 | 16.2 | >92.8 | 2.9 | >92.8 | >92.8 | 1.4 | 2.9 | 2.9 |
| KRKKRFKWW | 141.0 | 8.8 | 165–80.3 | 20.6 | 20.6 | 41.2 | 2.6 | 2.6 | 10.3 |
| Median | 9.0 | 4.1 | 11.2 | 2.6 | 5.3 | 8.0 | 1.2 | 1.4 | 5.3 |
In vitro assays for M. tuberculosis and M. smegmatis are described in Materials and Methods and presented as MIC90 values. For all others, the MIC was defined by no visual growth. Peptides originating from PL-C are shown in bold.
Comparison of CAMP activities against diverse microbes.
To evaluate antimicrobial specificity, PL-D peptides were then assayed for their activities against M. smegmatis and 7 representative Gram-negative, Gram-positive, and fungal pathogens, including P. aeruginosa, Escherichia coli, Salmonella enterica serovar Typhimurium, Candida albicans, Streptococcus epidermidis, Staphylococcus aureus, and Enterococcus faecalis (Table 1). Originally selected for their activities against P. aeruginosa, these peptides proved to be active against a broad range of microbes. It is also important to note that the average MICs of PL-D peptides varied about 10-fold between different microbes. The most sensitive bacteria were S. aureus (median MIC, 1.4 μM) and S. epidermis (median MIC, 1.2 μM); P. aeruginosa (median MIC, 11.2 μM) and M. tuberculosis (median MIC, 9.0 μM) were much more resistant.
Correlation analyses provided further evidence for broad-spectrum activities of the peptides. Pairwise comparisons of each peptide's logarithmic MIC value against each individual species revealed significant correlations of activities between species. Significant positive correlations were observed between 18 of the 36 pairs (Fig. 2A). The degree of correlation varied, providing initial evidence for species-specific peptides. While many peptides were active against M. smegmatis, this group of peptides had the lowest net correlation with other bacteria (including M. tuberculosis); M. smegmatis was the least attractive surrogate for M. tuberculosis screening for antimicrobial peptides. Surprisingly, S. aureus and C. albicans were the best candidates for an M. tuberculosis surrogate. These easy-to-handle and fast-growing strains could be exploited for further peptide-based TB drug development.
Fig 2.

Correlations of CAMP activities between diverse microbes. Peptides in PL-D were assayed for their MICs against 9 microbes, and their patterns of activities were compared using correlation analyses and principle component analyses. (A) Correlation analyses were used to compare peptide activities (logarithmic MIC) against pairs of microbes using a range of correlations (Pearson coefficients). Green indicates positive correlation; white, no correlation; red, negative correlation (not found). All statistically significant positive correlation coefficients between different species with P values of <0.05 (after Bonferroni correction of 36 pairs with P values of <0.0014) are indicated by +. (B) Principle component analysis (PCA) was used for multiple comparisons to identify shared (aggregated) features. The first three factor loadings correlated with the logarithmic MIC of peptides in PL-D. The first feature contains 49.7%, the first two features contain 66.5%, and the first three features contain 76.2% of the information. The first three aggregated features were used to do PCA on peptides in PL-D using Pearson correlation coefficients. Correlations are indicated by colors: green, positive; red, negative; white, no correlation.
A principal component analysis (PCA) documented increased activities of peptides against certain microbes or groups of microbes. PCA is a statistical method to simplify multiple comparisons in order to identify shared patterns as aggregated features (Fig. 2B). The first of three aggregated features, containing 49.7% of the information, probably reflects activity against all bacteria (all bacteria had similar values) determined by shared essential functions that are inactivated by multiple peptides. The second feature contributed 16.8% of the information and differentiated peptides more active against P. aeruginosa, S. enterica serovar Typhimurium, E. coli, and E. faecalis (positive values) from those more active against M. tuberculosis (negative values); the third aggregated feature reflected properties of peptides active against M. smegmatis. M. tuberculosis did not correlate with aggregated feature 3, again revealing significant differences between M. smegmatis and M. tuberculosis.
Molecular descriptors that define antimicrobial specificity.
PL-A peptides initially selected for their activities against P. aeruginosa are enriched for amino acids with cationic charges and hydrophobicity (23). Analyses were therefore done to determine whether these parameters were further increased in subgroups of peptides that were selected for their relatively high (positive groups in Fig. 3) or low (negative groups in Fig. 3) activities against P. aeruginosa, M. smegmatis, or M. tuberculosis. Analyses of these physiochemical properties rely on grouping amino acids based on similar charges (isoelectric points) or hydrophobicities (Hopp-Woods values) into 3 “terms” (low, medium, or high) and plotting peptide sequences (SEQ) as a function of their charges and hydrophobicities using specific subsets of amino acids that were either cationic (high pI, term 3) or hydrophobic (term 1, low Hopp-Woods values). These analyses confirmed that peptide activities against all bacteria were generally associated with cationic, hydrophobic amino acids and uncovered novel characteristics of peptides that were more active against M. tuberculosis. Distributions of positive and negative peptides were highly overlapping in the case of M. smegmatis, i.e., differences could not be readily visualized. In contrast, positives and negatives were separated in the case of M. tuberculosis. Peptides more active against M. tuberculosis were associated with sequences having 30 to 70% hydrophobic amino acids (TERM 1, SEQ Hopp-Woods values of 0.3 to 0.7) and 40% to 60% cationic amino acids (TERM 3, SEQ isoelectric point values of 0.4 to 0.6). Consistent with these observations, the subsets of peptides more active against any of the nine indicator microbes had similar amino acid compositions (see Fig. S1 in the supplemental material), with notable differences in peptides that were active against M. tuberculosis.
Fig 3.
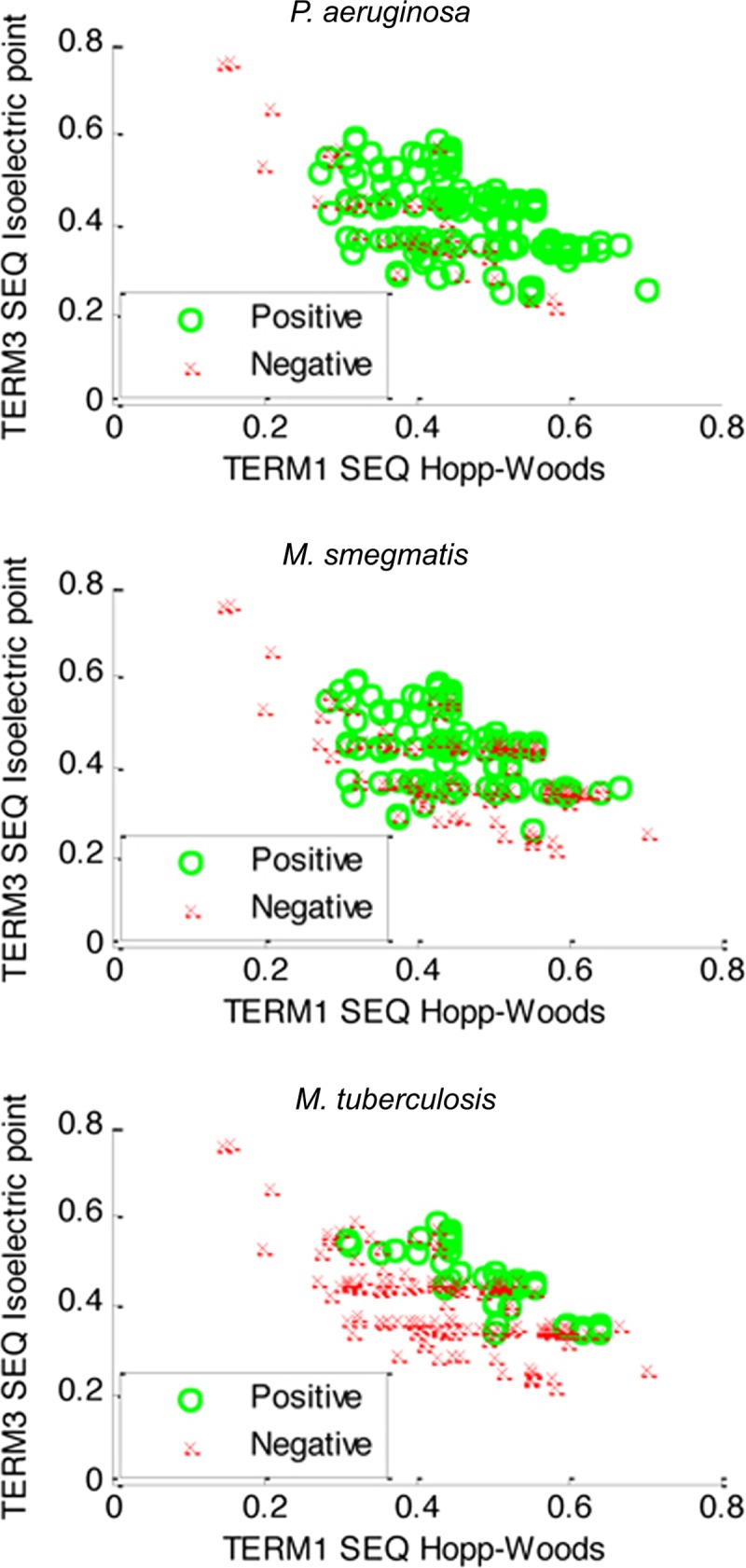
Scatterplot describing how the charges and hydrophobicity properties of peptides relate to their activities in three bacteria. Activities (positive or negative; described in Materials and Methods) of peptides in PL-A against P. aeruginosa (top), M. smegmatis (middle), and M. tuberculosis (bottom) are plotted as a function of the charge of the sequence (SEQ) (third term of the Hopp-Woods scale, isoelectric point) and its hydrophobicity (first term of the Hopp-Woods scale) (23).
Amino acid composition analyses of PL-A and PL-D peptides having high (“positive”) or low (“negative”) activities against M. tuberculosis further clarified why some peptides were more active against M. tuberculosis. The large number of peptides available for analysis in PL-A allowed the identification of statistically significant differences in amino acid composition (Table 2; see also Fig. S2 in the supplemental material). Positives had less alanine (0 versus 4%), isoleucine (6 versus 12%), leucine (2 versus 5%), and valine (2 versus 12%), and more tryptophan (39 versus 18%) and lysine (15 versus 11%). In contrast, the same analysis of peptide activities in M. smegmatis showed no significant differences in amino acid composition (Table 2; see also Fig. S2 in the supplemental material). Similar analyses of PL-D showed that stronger positives (redefined as MIC values of <10 μM), preselected from PL-A for their activities against M. tuberculosis, had significantly different amino acid compositions (analyses done using a Wilcoxon rank sum test [equivalent to a Mann-Whitney U test] with a significance level of alpha = 0.05 [Bonferroni corrected to 0.0025 with 20 amino acids]). In agreement with the analysis of PL-A, those having higher activity against M. tuberculosis had significantly elevated proportions of tryptophan (W; P = 0.0001) and decreased proportions of valine (V; P = 0.0004). In other microbes, differences in tryptophan and valine compositions were not obvious (see Fig. S1 in the supplemental material) and could not be statistically evaluated due to the low numbers of negative peptides (see Table S1 in the supplemental material). These remarkable differences in the activities of peptides against M. smegmatis compared to M. tuberculosis are likely due to differences in their intrinsic resistance systems and cell envelope properties.
Table 2.
Influence of amino acid composition on activities against M. tuberculosis and M. smegmatis in the PL-A peptide librarya
| Amino acid | Proportion (%) |
P value (Wilcoxon rank sum test) | |
|---|---|---|---|
| Positive | Negative | ||
| M. tuberculosis | |||
| A | 0 | 4 | 1.80E−07 |
| C | 0 | 0 | NS |
| D | 0 | 0 | NS |
| E | 0 | 0 | NS |
| F | 3 | 2 | NS |
| G | 0 | 1 | NS; Bonferroni corrected (0.00270357) |
| H | 1 | 0 | NS |
| I | 6 | 12 | 3.15E−07 |
| K | 15 | 11 | NS; Bonferroni corrected (0.00322959) |
| L | 2 | 5 | 0.000113558 |
| M | 0 | 1 | NS; Bonferroni corrected (0.00583051) |
| N | 0 | 0 | NS |
| P | 1 | 1 | NS |
| Q | 0 | 0 | NS |
| R | 30 | 28 | NS |
| S | 0 | 1 | NS; Bonferroni corrected (0.014283) |
| T | 0 | 1 | NS; Bonferroni corrected (0.00787191) |
| V | 2 | 12 | 1.32E−12 |
| W | 39 | 18 | 7.99E−19 |
| Y | 1 | 2 | NS |
| M. smegmatis | |||
| A | 3 | 2 | NS |
| C | 0 | 0 | NS |
| D | 0 | 0 | NS |
| E | 0 | 0 | NS |
| F | 2 | 3 | NS |
| G | 1 | 1 | NS |
| H | 0 | 0 | NS |
| I | 10 | 11 | NS |
| K | 13 | 12 | NS |
| L | 4 | 4 | NS |
| M | 1 | 1 | NS |
| N | 0 | 0 | NS |
| P | 0 | 1 | NS |
| Q | 0 | 0 | NS |
| R | 30 | 27 | NS; Bonferroni corrected (0.00545506) |
| S | 0 | 1 | NS |
| T | 0 | 1 | NS; Bonferroni corrected (0.00392398) |
| V | 9 | 8 | NS |
| W | 25 | 25 | NS |
| Y | 1 | 3 | NS; Bonferroni corrected (0.0449885) |
See Materials and Methods for definitions of positive and negative activity. M. tuberculosis, 82 positive, 171 negative; M. smegmatis, 131 positive, 122 negative. NS, not significant.
Determination of peptide cytotoxicity against human THP-1 cells.
CAMP cytotoxicity to human cells is also a significant concern for their development as clinical antibacterial agents (41). Initial cytotoxicity studies were carried out using THP-1 (39), a human cell line that can be induced to a differentiated, macrophage-like state. Differentiated THP-1 cells are widely used in tuberculosis research and for compound toxicity testing (42). The cytotoxicity of peptides was measured using differentiated THP-1 cells and resazurin as an indicator of their viability. Many peptides in PL-D did not generate significant toxicity using this assay. Results were expressed as selectivity indices, i.e., the ratio of in vitro activity against M. tuberculosis relative to THP-1 toxicity (either 50% or 90% inhibition, SI50 or SI90). SI50 ranged from 3 to 256 with an average of 79, and SI90 ranged from 1 to 51 with an average of 17 (see Table S2 in the supplemental material).
DISCUSSION
In M. tuberculosis, envelope structure and self-defense systems provide resistance to the majority of commonly used antibiotics as well as CAMPs (43). These intrinsic resistance systems were believed to restrict the use of CAMPs in treating TB (30). However, after screening libraries of peptides selected for activities against P. aeruginosa, we were surprised to find that at least 30% had significant activities against M. tuberculosis, including 26 with very low MIC values (1 to 10 μM). Furthermore, screens of P. aeruginosa-active peptides against diverse microbes showed that they had a broad spectrum of antimicrobial activity associated with their positive charge (term 1) and hydrophobicity (term 3; Fig. 3). However, our studies revealed that microbes each had their own characteristic levels of sensitivity to CAMPs in our libraries. The median MICs of peptides (PL-A) against individual pathogens varied about 10-fold (Table 1); some microbes, including M. tuberculosis and P. aeruginosa, had significantly higher average levels of CAMP resistance. Furthermore, individual peptides had their own spectra of maximal activity against certain microbes or groups of microbes. We were somewhat surprised to find that these individual activity spectra were not simple reflections of microbial taxonomy. For example, activities against C. albicans (a fungus) and various bacteria correlated better than most interbacterial pair comparisons. These observations led us to hypothesize that the activities of CAMPs were determined largely by commonly occurring genetic variations that provide resistance rather than stable genomic elements that define bacterial taxa. This model is further supported by analyses of CAMP activities in Mycobacterium species.
While mycobacterial envelopes have conserved structural elements that serve as effective permeability barriers to most antibacterial agents, envelope composition and corresponding drug resistance spectra are species dependent (43). Their envelopes are composed of multiple layers of hydrophilic and hydrophobic polymers that serve as permeability barriers (2, 8, 44). Their membranes contain anionic phosphatidylglycerol (PG) that can be converted to a cationic form by lysinylation (PG-L), thus limiting CAMP binding or penetration and providing resistance to antimicrobial CAMPs that it encounters within the macrophage (30, 45). As in other bacteria, the presence of such modifications or transporters, as well as specificity of corresponding regulatory systems, may determine CAMP resistance in different mycobacteria that have become adapted to different environments. This concept may explain heterogeneity in the resistance of M. tuberculosis and M. smegmatis to different subsets of CAMPs.
Despite the fact that M. tuberculosis is viewed as being tolerant to natural CAMPs, our studies identified some synthetic CAMPs that were highly active. Furthermore, comparisons of the activities of CAMPs against M. tuberculosis and M. smegmatis showed that levels of sensitivity to CAMPs were not a specific characteristic of an individual mycobacterial species. In fact, activities of various CAMPs against M. tuberculosis correlated better with unrelated microbes than with its close relative, M. smegmatis. M. smegmatis has been successfully employed as a surrogate model for M. tuberculosis in drug screening programs. However, a recent report suggests that it may not be as predictive as previously thought (46). Our data also document limitations of using M. smegmatis for CAMP screening.
Both the microbe-specific CAMP activities and their broad-spectrum antimicrobial activities are apparently dependent on the enriched content of hydrophobic and cationic amino acids. Scatterplots showed that more active peptides had similar average masses and descriptors of either charge (term 1) or hydrophobicity (term 3) in all microbes (Fig. 3). Further dissection of these foundational physiochemical characteristics revealed that amino acid composition was a determinant of increased activity against M. tuberculosis. The original P. aeruginosa-active libraries (PL-A and PL-C) contained high proportions of K, R, and W (see Table S3 in the supplemental material) and had an average length of 10 amino acids. Peptides that were most active against M. tuberculosis had further increased proportions of tryptophan, which was offset by decreased proportions of some uncharged amino acids (valine, alanine, isoleucine, and leucine). The fact that these differences in amino acid compositions were not observed in the same analyses of peptides more active against other organisms supports PCA analyses indicating that unique features determine a degree of specificity against M. tuberculosis. Furthermore, many of these peptides had low levels of toxicity against macrophage-like human THP-1 cells. Additional cytotoxicity studies using multiple cell lines will be needed to select the best candidates for in vivo testing.
Our studies showed that the activities of these peptides had significant specificity for M. tuberculosis and that this was associated with specific amino acid compositions. Together, these insights will be used to improve peptide sequence design to increase their activities and specificity against M. tuberculosis, including drug-resistant strains, while minimizing cytotoxicity. In the future, better understandings of the modes of action, resistance mechanisms, and pharmacological properties of CAMPs are needed to accelerate their development as anti-infective therapies.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jonas van den Berg and Gaye Sweet for technical assistance and Bob Hancock for providing access to peptides and peptide sequence libraries during the early stages of this project. We also thank the University of British Columbia (UBC) Facility for Infectious Disease and Epidemic Research (FINDER) for technical support.
This work was supported by grants from The Canadian Institute of Health Research (MOP-82745 and MOP-82855 to C.J.T.), the British Columbia Lung Association (C.J.T.), and the BioInterfaces program of the Helmholtz Association (R.M., M.R., and K.H.).
Footnotes
Published ahead of print 11 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00175-13.
REFERENCES
- 1. Peschel A, Sahl HG. 2006. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 4:529–536 [DOI] [PubMed] [Google Scholar]
- 2. Fjell CD, Hiss JA, Hancock RE, Schneider G. 2012. Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discov. 11:37–51 [DOI] [PubMed] [Google Scholar]
- 3. Marr AK, Gooderham WJ, Hancock RE. 2006. Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr. Opin. Pharmacol. 6:468–472 [DOI] [PubMed] [Google Scholar]
- 4. Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389–395 [DOI] [PubMed] [Google Scholar]
- 5. Spindler EC, Hale JD, Giddings TH, Jr, Hancock RE, Gill RT. 2011. Deciphering the mode of action of the synthetic antimicrobial peptide Bac8c. Antimicrob. Agents Chemother. 55:1706–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fantner GE, Barbero RJ, Gray DS, Belcher AM. 2010. Kinetics of antimicrobial peptide activity measured on individual bacterial cells using high-speed atomic force microscopy. Nat. Nanotechnol. 5:280–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schneider T, Kruse T, Wimmer R, Wiedemann I, Sass V, Pag U, Jansen A, Nielsen AK, Mygind PH, Raventos DS, Neve S, Ravn B, Bonvin AM, De Maria L, Andersen AS, Gammelgaard LK, Sahl HG, Kristensen HH. 2010. Plectasin, a fungal defensin, targets the bacterial cell wall precursor lipid II. Science 328:1168–1172 [DOI] [PubMed] [Google Scholar]
- 8. Brogden KA. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3:238–250 [DOI] [PubMed] [Google Scholar]
- 9. Kragol G, Lovas S, Varadi G, Condie BA, Hoffmann R, Otvos L., Jr 2001. The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry 40:3016–3026 [DOI] [PubMed] [Google Scholar]
- 10. Hilpert K, McLeod B, Yu J, Elliott MR, Rautenbach M, Ruden S, Burck J, Muhle-Goll C, Ulrich AS, Keller S, Hancock RE. 2010. Short cationic antimicrobial peptides interact with ATP. Antimicrob. Agents Chemother. 54:4480–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gunn JS. 2008. The Salmonella PmrAB regulon: lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol. 16:284–290 [DOI] [PubMed] [Google Scholar]
- 12. Ernst CM, Peschel A. 2011. Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol. Microbiol. 80:290–299 [DOI] [PubMed] [Google Scholar]
- 13. Li M, Cha DJ, Lai Y, Villaruz AE, Sturdevant DE, Otto M. 2007. The antimicrobial peptide-sensing system aps of Staphylococcus aureus. Mol. Microbiol. 66:1136–1147 [DOI] [PubMed] [Google Scholar]
- 14. Shafer WM, Qu X, Waring AJ, Lehrer RI. 1998. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc. Natl. Acad. Sci. U. S. A. 95:1829–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pranting M, Negrea A, Rhen M, Andersson DI. 2008. Mechanism and fitness costs of PR-39 resistance in Salmonella enterica serovar Typhimurium LT2. Antimicrob. Agents Chemother. 52:2734–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tzeng YL, Ambrose KD, Zughaier S, Zhou X, Miller YK, Shafer WM, Stephens DS. 2005. Cationic antimicrobial peptide resistance in Neisseria meningitidis. J. Bacteriol. 187:5387–5396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kraus D, Peschel A. 2008. Staphylococcus aureus evasion of innate antimicrobial defense. Future Microbiol. 3:437–451 [DOI] [PubMed] [Google Scholar]
- 18. Hilpert K, Hancock RE. 2007. Use of luminescent bacteria for rapid screening and characterization of short cationic antimicrobial peptides synthesized on cellulose using peptide array technology. Nat. Protoc. 2:1652–1660 [DOI] [PubMed] [Google Scholar]
- 19. Hilpert K, Elliott MR, Volkmer-Engert R, Henklein P, Donini O, Zhou Q, Winkler DF, Hancock RE. 2006. Sequence requirements and an optimization strategy for short antimicrobial peptides. Chem. Biol. 13:1101–1107 [DOI] [PubMed] [Google Scholar]
- 20. Hilpert K, Volkmer-Engert R, Walter T, Hancock RE. 2005. High-throughput generation of small antibacterial peptides with improved activity. Nat. Biotechnol. 23:1008–1012 [DOI] [PubMed] [Google Scholar]
- 21. Fjell CD, Jenssen H, Hilpert K, Cheung WA, Pante N, Hancock RE, Cherkasov A. 2009. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 52:2006–2015 [DOI] [PubMed] [Google Scholar]
- 22. Cherkasov A, Hilpert K, Jenssen H, Fjell CD, Waldbrook M, Mullaly SC, Volkmer R, Hancock RE. 2009. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem. Biol. 4:65–74 [DOI] [PubMed] [Google Scholar]
- 23. Mikut R, Hilpert K. 2009. Interpretable features for the activity prediction of short antimicrobial peptides using fuzzy logic. Int. J. Pept. Res. Ther. 15:129–137 [Google Scholar]
- 24. Mikut R. 2010. Computer-based analysis, visualization, and interpretation of antimicrobial peptide activities. Methods Mol. Biol. 618:287–299 [DOI] [PubMed] [Google Scholar]
- 25. Miyakawa Y, Ratnakar P, Rao AG, Costello ML, Mathieu-Costello O, Lehrer RI, Catanzaro A. 1996. In vitro activity of the antimicrobial peptides human and rabbit defensins and porcine leukocyte protegrin against Mycobacterium tuberculosis. Infect. Immun. 64:926–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sonawane A, Santos JC, Mishra BB, Jena P, Progida C, Sorensen OE, Gallo R, Appelberg R, Griffiths G. 2011. Cathelicidin is involved in the intracellular killing of mycobacteria in macrophages. Cell. Microbiol. 13:1601–1617 [DOI] [PubMed] [Google Scholar]
- 27. Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zugel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL. 2006. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311:1770–1773 [DOI] [PubMed] [Google Scholar]
- 28. Linde CM, Hoffner SE, Refai E, Andersson M. 2001. In vitro activity of PR-39, a proline-arginine-rich peptide, against susceptible and multi-drug-resistant Mycobacterium tuberculosis. J. Antimicrob. Chemother. 47:575–580 [DOI] [PubMed] [Google Scholar]
- 29. Jiang Z, Higgins MP, Whitehurst J, Kisich KO, Voskuil MI, Hodges RS. 2011. Anti-tuberculosis activity of alpha-helical antimicrobial peptides: de novo designed L- and D-enantiomers versus L- and D-LL-37. Protein Pept. Lett. 18:241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maloney E, Stankowska D, Zhang J, Fol M, Cheng QJ, Lun S, Bishai WR, Rajagopalan M, Chatterjee D, Madiraju MV. 2009. The two-domain LysX protein of Mycobacterium tuberculosis is required for production of lysinylated phosphatidylglycerol and resistance to cationic antimicrobial peptides. PLoS Pathog. 5:e1000534 doi:10.1371/journal.ppat.1000534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ramon-Garcia S, Ng C, Anderson H, Chao JD, Zheng X, Pfeifer T, Av-Gay Y, Roberge M, Thompson CJ. 2011. Synergistic drug combinations for tuberculosis therapy identified by a novel high-throughput screen. Antimicrob. Agents Chemother. 55:3861–3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bowdish DM, Davidson DJ, Hancock RE. 2005. A reevaluation of the role of host defence peptides in mammalian immunity. Curr. Prot. Pept. Sci. 6:35–51 [DOI] [PubMed] [Google Scholar]
- 33. Hancock RE, Sahl HG. 2006. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24:1551–1557 [DOI] [PubMed] [Google Scholar]
- 34. Hilpert K, Winkler DF, Hancock RE. 2007. Peptide arrays on cellulose support: SPOT synthesis, a time and cost efficient method for synthesis of large numbers of peptides in a parallel and addressable fashion. Nat. Protoc. 2:1333–1349 [DOI] [PubMed] [Google Scholar]
- 35. Kamradt T, Volkmer-Engert R. 2004. Cross-reactivity of T lymphocytes in infection and autoimmunity. Mol. Divers. 8:271–280 [DOI] [PubMed] [Google Scholar]
- 36. Montoro E, Lemus D, Echemendia M, Martin A, Portaels F, Palomino JC. 2005. Comparative evaluation of the nitrate reduction assay, the MTT test, and the resazurin microtitre assay for drug susceptibility testing of clinical isolates of Mycobacterium tuberculosis. J. Antimicrob. Chemother. 55:500–505 [DOI] [PubMed] [Google Scholar]
- 37. Brennan PJ, Young DB. 2008. Handbook of anti-tuberculosis agents. Tuberculosis 88:85–170 [DOI] [PubMed] [Google Scholar]
- 38. Wiegand I, Hilpert K, Hancock RE. 2008. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3:163–175 [DOI] [PubMed] [Google Scholar]
- 39. Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. 1980. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 26:171–176 [DOI] [PubMed] [Google Scholar]
- 40. Mikut R, Burmeister O, Braun S, Reischl M. 2008. The open source Matlab toolbox Gait-CAD and its application to bioelectric signal processing, p 109–111 Proc. DGBMT-Workshop Biosignalverarbeitung, Potsdam [Google Scholar]
- 41. Huang Y, Huang J, Chen Y. 2010. Alpha-helical cationic antimicrobial peptides: relationships of structure and function. Prot. Cell 1:143–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pick N, Cameron S, Arad D, Av-Gay Y. 2004. Screening of compounds toxicity against human monocytic cell line-THP-1 by flow cytometry. Biol. Proced. Online 6:220–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nguyen L, Thompson CJ. 2006. Foundations of antibiotic resistance in bacterial physiology—the mycobacterial paradigm. Trends Microbiol. 14:304–312 [DOI] [PubMed] [Google Scholar]
- 44. Wilmes M, Cammue BP, Sahl HG, Thevissen K. 2011. Antibiotic activities of host defense peptides: more to it than lipid bilayer perturbation. Nat. Prod. Rep. 28:1350–1358 [DOI] [PubMed] [Google Scholar]
- 45. Ortalo-Magne A, Lemassu A, Laneelle MA, Bardou F, Silve G, Gounon P, Marchal G, Daffe M. 1996. Identification of the surface-exposed lipids on the cell envelopes of Mycobacterium tuberculosis and other mycobacterial species. J. Bacteriol. 178:456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stanley SA, Grant SS, Kawate T, Iwase N, Shimizu M, Wivagg C, Silvis M, Kazyanskaya E, Aquadro J, Golas A, Fitzgerald M, Dai H, Zhang L, Hung DT. 2012. Identification of novel inhibitors of M. tuberculosis growth using whole-cell based high-throughput screening. ACS Chem. Biol. 7:1377–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.