

Abstract
Previous studies indicate that while transgenic mice with ATXN1[30Q]-D776-induced disease share pathological features caused by ATXN1[82Q] having an expanded polyglutamine tract, they fail to manifest the age-related progressive neurodegeneration seen in spinocerebellar ataxia type 1. The shared features include morphological alterations in climbing fiber (CF) innervation of Purkinje cells (PCs). To further investigate the ability of ataxin-1 (ATXN1) to impact CF/PC innervation, this study used morphological and functional approaches to examine CF/PC innervation during postnatal development in ATXN1[30Q]-D776 and ATXN1[82Q] cerebella. Notably, ATXN1[30Q]-D776 induced morphological alterations consistent with the development of the innervation of PCs by CFs being compromised, including a reduction of CF translocation along the PC dendritic tree, and decreased pruning of CF terminals from the PC soma. As previously shown for ATXN1[82Q], ATXN1[30Q]-D776 must enter the nucleus of PCs to induce these alterations. Experiments using conditional ATXN1[30Q]-D776 mice demonstrate that both the levels and specific timing of mutant ATXN1 expression are critical for alteration of the CF–PC synapse. Together these observations suggest that ATXN1, expressed exclusively in PCs, alters expression of a gene(s) in the postsynaptic PC that are critical for its innervation by CFs. To investigate whether ATXN1[30Q]-D776 curbs the progressive disease in ATXN1[82Q]-S776 mice, we crossed ATXN1[30Q]-D776 and ATXN1[82Q]-S776 mice and found that double transgenic mice developed progressive PC atrophy. Thus, the results also show that to develop progressive cerebellar degeneration requires expressing ATXN1 with an expanded polyglutamine tract.
Introduction
Refinement of initial polyneuronal synaptic input to achieve the adult innervation pattern in which each Purkinje cell (PC) is contacted by only one climbing fiber (CF) requires a number of important developmental events, including refinement of select synapses and elimination of nonfunctional or redundant connections. These changes result in a precise neural circuitry that is required for proper function and established the cerebellum as a model system for investigating these processes (Ramon ý Cajal, 1911; O'Leary et al., 1971; Mason et al., 1990). In mouse cerebellum, CF development proceeds through three distinct stages in early postnatal life. Shortly after birth, multiple CFs form somatic “pericellular nests” over a PC soma (Altman, 1972). Subsequently, from postnatal day 3–7 (p3–p7), a single CF is strengthened (Crepel, 1982; Kano et al., 1995; Hashimoto and Kano, 2003). The end of this phase is marked by the “winner” CF's axon translocating onto the PC's apical dendrite. In the final phase, p12–p21, somatic synapses from both the “winner” and “loser” CFs are eliminated (Hashimoto et al., 2009). By p21, the majority of PCs are innervated by a single CF whose synapses are located on the PC's primary, secondary, and tertiary dendrites.
Expansion of the polyglutamine tract in the protein ataxin-1 (ATXN1) results in the fatal progressive neurodegenerative disorder, spinocerebellar ataxia type 1 (SCA1) (Orr et al., 1993). Mice expressing expanded ATXN1[82Q] in PCs exhibit progressive PC pathology and ataxic gait (Burright et al., 1995; Clark et al., 1997), similar to that seen in SCA1 patients (Landis et al., 1974; Matilla-Dueñas et al., 2008). In addition to expansion of the polyglutamine tract, other features of ATXN1 are critical for pathogenesis and severity of disease. Among these are the translocation of mutant ATXN1 to the nucleus and phosphorylation of ATXN1 at S776 (Klement et al., 1998; Emamian et al., 2003). In particular, a potentially phospho-mimicking aspartic acid amino acid at position 776 enhances pathogenesis of ATXN1[82Q] and transforms wild-type (Wt) ATXN1[30Q] into a pathogenic protein (ATXN1[30Q]-D776) (Duvick et al., 2010). Compromising cerebellar development is another component that contributes to severity of SCA1 in adult mice. Mice in which expression of ATXN1[82Q] was delayed during postnatal development display a much less severe form of disease in adults than mice in which mutant ATXN1 was expressed before the completion of cerebellar postnatal development (Serra et al., 2006).
Interestingly, adult mice expressing disease-causing forms of ATXN1 in their PCs display alterations in CF innervation that include a lack of CF terminals on PC secondary and tertiary dendrites and a reduction in the CF–PC synaptic strength (Duvick et al., 2010; Barnes et al., 2011). In particular, the increased number of CF terminals present on the PC soma in ATXN1[30Q]-D776 mice is reminiscent of the CF “pericellular nests” that form transiently during early postnatal cerebellar development. Thus, these mice offer an opportunity to examine the regulation of CF innervation of PCs and assess the extent to which alteration in this circuitry might contribute to disease in transgenic models of SCA1.
Materials and Methods
Transgenic mice.
Mice were housed under specific pathogen-free condition in an Association for Assessment and Accreditation of Laboratory Animal Care approved facility and managed by Research Animal Resources. ATXN1[30Q]-S776 (line A02), ATXN1[82Q]-S776 (line B05), and ATXN1[30Q]-D776 (line L2) mice were used (Burright et al., 1995; Duvick et al., 2010). In order for transgenic lines to have comparable levels of transgene expression, all lines were used in the hemizygous configuration except ATXN1[30Q]-S776 mice that were homozygous for the transgene (denoted as ATXN1[30]-S776/S776). For PC volume modeling, mice were crossed with Pcp2-GFP BAC mice (Gong et al., 2003). ATXN1[30Q]-K772T mice were generated by adding a point mutation to the original construct used to make the ATXN1[30Q]-D776 mice (Duvick et al., 2010) using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene #200521). The transgene was linearized with SalI, gel isolated, purified by ethanol precipitation, and suspended in injection buffer (10 mm Tris-Cl, pH 8.0; 0.1 mm EDTA) at a concentration of 4 ng/ml. Conditional ATXN1[30Q]-D776 (cATXN1[30Q]-D776) mice were similarly generated by adding a point mutation to TRE-SCA1[30Q] construct, which were cloned in the same manner as TRE-SCA1[82Q] mice (Zu et al., 2004). The transgene was linearized with HindIII and XhoI and prepared as above. Embryo injections were performed by the Mouse Genetics Laboratory, University of Minnesota. PCR and Southern blot analyses were used to identify transgene-positive animals. Mice were given 1.5 mg/ml doxycycline (dox) in a 4% sucrose solution in their drinking water. In all experiments involving the use of mice, animals of either sex were used.
All animal experimentation was approved by the Institutional Animal Care and Use Committee of the University of Minnesota and was conducted in accordance with the National Institutes of Health Principles of Laboratory Animal Care (86–23, revised 1985) and the American Physiological Society Guiding Principles in the Use of Animals.
Stereotaxic injections in the inferior olive to anterogradely label CF.
CF anterograde labeling was performed as previously described (Duvick et al., 2010). Briefly, mice were anesthetized by intramuscular injection of a ketamine and xylazine mixture (100 mg of ketamine and 10 mg of xylazine per kilogram). The dorsal skin of the head was shaved and animals were placed into a stereotaxic frame (Model 963LS; David Kopf Instruments). The obex of the brainstem was exposed and a glass pipette glued to a Hamilton syringe was inserted into the inferior olive. Then 150 nl of 4% Alexa Fluor 488 or Alexa Fluor 568-conjugated dextran (10,0000 MW; Invitrogen) was bilaterally injected using a microinjector (Model 5000; David Kopf Instruments). Three to five days later, mice were perfused and brains were processed as described below.
Histology and immunostaining.
Animals were deeply anesthetized and transcardially exsanguinated with PBS, pH 7.4, and perfused using 10% formalin (30 ml). Brains were postfixed overnight in 10% formalin and subsequently placed in 30% sucrose in PBS at 4°C for 48 h before sectioning. The cerebellum was sagittally sectioned into 50-μm-thick sections using a freezing sliding microtome. Epitopes were exposed using antigen retrieval by boiling sections four times for 10 s each in 0.01 m urea. The sections were blocked overnight in 2% normal donkey serum and 0.3% Triton X-100 in PBS. All subsequent staining was performed in 2% normal donkey serum and 0.3% Triton X-100 in PBS. The following primary antibodies were used: goat anti-calbindin (SC-7691; Santa Cruz Biotechnology) at 1:250 and mouse anti-vesicular glutamate transporter 2 (VGLUT2) (MAB5504; Millipore) at 1:1000.
Sections were incubated for 24 h in primary antibodies at 4°C. Following incubation, the sections were washed three times in PBS and exposed to secondary antibodies (DyLight 488, Dylight 549, and Dylight 649; Jackson ImmunoResearch) for 24 h at 4°C. Last, sections were washed three times in PBS and mounted onto charged slides (ColorFrost Plus; Fisher Scientific). Fluorescently labeled tissue was imaged using a confocal Olympus 1000 IX inverted microscope.
Disease severity scale.
As an assessment of disease status the following disease severity scale was used (Oz et al., 2010): 0.5, near-normal but molecular layer (ML) somewhat thinner; 1, mild changes including heterotopic PCs, vacuoles in PCs, thinning of ML, largely confined to the posterior lobules; 2, similar to 1 but more widespread, heterotopic PCs more numerous and often higher in the ML; 3, widespread ML thinning, numerous heterotopic PCs involving anterior lobules nearly as frequently as posterior, mild PC loss, primarily in the posterior lobules; 4, severe disorganization of cerebellar cortex with generalized severe atrophy of ML and frequent heterotopic PCs and PC loss.
Quantification of PC length and CF extension distance.
PC length and CF extension into the PC dendrites were measured using 20× confocal images from 50 μm cerebellar sections stained for calbindin and VGLUT2 from p14, p17, p21, and 5-, 12-, and 20-week-old animals. Confocal image stacks of 10 μm depth and step size of 2 μm were captured from lobule VI and analyzed using the imaging program, Fluoview Viewer 1.7. The PC length was measured as the distance from the base of the PC soma to tip of the PC dendrite. CF extension into the molecular layer of lobule VI was measured from the base of the PC soma to the tip of the CF arbor, as previously described (Duvick et al., 2010). Six separate measurements of PC length and CF extension were taken from each image of lobule VI and averaged. On average, 18 measurements were obtained from three midline sections from lobule VI of each animal.
CF terminals are represented by VGLUT2-labeled puncta of coarse size and proximal to the dendritic shaft (Miyazaki et al., 2004). For the p14 and p17 time points, we only included puncta that meet these criteria in the quantification for CF extension. At least three animals were used for each genotype at each time point.
Imaris quantification of CF—PC soma contacts.
Mid-sagittal sections (50 μm) of cerebellum from mice 5 weeks old were immunostained for calbindin and VGLUT2 and mounted onto charged slides. Confocal images (60×) of the PC layer in the primary fissure with 10 μm depth and step size of 2 μm were captured and imported into the software program Imaris (Bitplane). This software allowed the objective counting of the somatic contacts based upon size and fluorescent intensity. Using the Spot application of Imaris, PC bodies were individually isolated by the “region of interest” function (ROI); average ROI dimensions: X = 50 μm, Y = 70 μm, Z = 10 μm. PC VGLUT2 somatic contacts were identified and counted using an “estimated spot diameter” of 2.3 μm. Therefore, puncta <2.3 μm was not counted. Intensity “thresholds” were automatically calculated. The output was recorded as the number of the VGLUT2-positive boutons per PC soma. Greater than 10 images per genotype were analyzed with Imaris and each image had an average of 5 PCs per image.
VGLUT1 and PC somatic spines were counted following the same staining procedure above except VGLUT1 was substituted for VGLUT2 immunostaining. Single frame 100× images were captured of individual PCs in lobule VI. For VGLUT1 quantitation, VGLUT1 puncta only on the bottom two-thirds of the PC soma was counted to avoid the VGLUT1 puncta on the dendrites of the PC. To quantitate PC somatic spines, individual PCs of lobule VI from calbindin immunostained sections were imaged at 100×. The PC soma was scored based upon visual identification of spine protrusions; a minimum of three spines per PC were required to consider the PC soma spine positive.
Imaris PC volume modeling.
Using the transgenic lines that express green fluorescent protein (GFP) in PCs by virtue of being crossed to PCP2-GFP BAC mice (Gong et al., 2003), 100× confocal images of PC somas with anterogradely labeled CFs and immunostained for VGLUT2 were collected and imported into Imaris to generate an isosurface representing the total intensity of the fluorescence per pixel. The isosurface is a digital representation of the fluorescence expression within a volume of space. The isosurfaces were generated for each fluorescent channel captured (PC, CF, CF terminals).
For the PCs, the source channel was defined as “Smooth” and had a “Surfaces Area Detail Level” of 0.25 μm. Thresholding was set to “Absolute Intensity,” and manually adjusted to render a digital volume representative of the tissue-specific immunofluorescence signal. The final image was filtered using the “Number of Voxels” parameter to eliminate extraneous volume modeling of adjacent PC somata or background staining. CFs and VGLUT2-positive terminals were processed similarly except the Surface Area Detail Level was lowered to <0.2 μm, depending on signal intensity.
Animal preparation and surgical procedure for optical imaging and electrophysiology.
Experimental details of the anesthetized mouse preparation and in vivo optical imaging technique were performed in adult mice as previously described (Barnes et al., 2011). Mice were anesthetized by intramuscular injection of acepromazine (0.1 mg/g) and by intraperitoneal injection of urethane (2 mg/g). Animals were placed into a stereotaxic frame and were mechanically ventilated. A craniotomy exposed a portion of the left cerebellar hemisphere. A durotomy was performed to expose the caudal medulla and dorsal surface of the brainstem to permit placement of a stimulating electrode into the inferior olivary complex. Finally, a watertight chamber was made to encompass all areas of the exposed brain and was filled with Ringer's solution gassed with 95% O2 and 5% CO2.
CF stimulation was accomplished by stereotaxically inserting a tungsten microelectrode (1–3 MΩ) into the right inferior olive. The contralateral inferior olive (CIO) electrode was initially lowered 1.4 mm into the brainstem from the dorsal surface near the obex. Each subsequent test site was 200 μm ventral, penetrating to a final depth of 2.0 mm. Stimulations occurred at 10 Hz for 10 s (100 μs pulse width, 200 μA) and were done in quadruplicate at each depth.
Optical imaging.
Exposed cerebellar cortices were initially imaged with a Quantix cooled charge-coupled device camera with 12-bit digitization (Roper Scientific) attached to a modified Nikon epifluorescence microscope fitted with a 4× objective. A 100W xenon–mercury lamp (Hamamatsu Photonics) with a direct current (DC) controlled power supply (Opti Quip) was used as the light source. A bandpass excitation filter (455 ± 35 nm), an extended reflectance dichroic mirror (500 nm), and a >515 nm long-pass emission filter were used for imaging flavoprotein autofluorescence (Reinert et al., 2004).
To test functional integrity of the cerebellar cortex, the autofluorescence response was first assessed by stimulating parallel fibers. This was accomplished by lowering the stimulating electrode (1–3 MΩ) until it touched the surface of the cerebellum and applying a stimulus (100 pulses delivered at 10 Hz or 100 Hz, 200 μA, 100 μs). As long as the characteristic beam-like response with an intensity of ∼1–2% ΔF/F was elicited, the CIO was subsequently stimulated.
Optical imaging data analysis.
Each series of images consisted of 110 frames of 200 ms duration. Stimulation began after a 15 frame control period, allowing the 10 frames that preceded stimulation onset to be averaged and serve as the control average. The temporal change in fluorescence (ΔF/F) was accomplished in several steps. First, the control average for a stack was subtracted from each individual frame. Each difference image was then divided by the control average.
Optical responses were quantified using ∼5–8 circles with areas of 137 pixels (∼13,700 μm2) as ROIs that uniformly sampled from within the medial-most parasagittal optical band. Nine frames centered about the peak ΔF/F for each ROI within the medial band were averaged and used to quantify the CIO-evoked response. Only sites within the medial band were used for quantification due to its large area, robust response, and reliability of being evoked. It was defined as the region in Crus II that extended ∼1100 μm from the vermis-hemisphere boundary.
For each mouse, the depth in the inferior olive at which the maximal optical response was evoked was used to generate an optical stack average based on four separate stimulations. Individual composite responses within a genotype were then averaged and a composite response was created for each genotype by averaging nine frames at the peak ΔF/F.
Electrophysiology.
Conventional extracellular electrophysiological techniques using glass recording microelectrodes (2 m NaCl, 2–5 MΩ) were used to record CIO-evoked field potentials (FPs; Eccles et al., 1966; Reinert et al., 2004; Gao et al., 2006; Barnes et al., 2011). The ROI in which FPs were recorded was defined as the area on Crus II that extended laterally ∼1100 μm from the vermis-hemisphere boundary (for description, see Barnes et al., 2011). A total of 16 pulses were delivered to the CIO at 1 Hz for each individual recording site and were subsequently averaged. Excitation of the CF–PC circuitry by CIO stimulation produced a biphasic, negative–positive potential recorded on the cortical surface that typically reversed at greater depth. Evoked potentials were recorded in increments of ∼100 μm until reversal. Upon completion of recording, marking lesions were created at the tip of the electrode in the CIO by passing 100 μA of DC three times for 1 s per pulse. Last, animals were perfused and whole brains were harvested. The tissue was incubated in 30% sucrose for ∼48 h before sectioning and identification of CIO lesions.
Triple labeling for multiple innervation analysis.
Cerebellar sections with anterogradely labeled CF were antibody immunostained for calbindin and VGLUT2 as described above. Confocal images of individual PC somata with a synapsing anterogradely labeled CF were captured at 60× with a zoom of 3. Calbindin, anterogradely labeled CF, and VGLUT2 were sequentially captured on separate channels. To identify all synaptic contacts between the anterogradely labeled CFs and unlabeled CFs, the entire PC soma was captured in a z-stack projection made up of 1 μm slices. The Z-projections of captured images were examined in Fluoview Viewer 1.7 (Olympus). The merged projections were analyzed and multiple innervation was quantified by identifying any lone VGLUT2 synapses that did not colocalize with the anterogradely labeled CF. A threshold of three VGLUT2 synapses on PC soma was required to score a PC as being multiply innervated. Statistical analysis was performed using a χ2 test with 3 df.
Reverse transcription quantitative PCR Analysis of ATXN1 Expression.
Total RNA was isolated from dissected cerebella using TRIzol Reagent (Life Technologies) following the manufacturer's protocols. Random-primed cDNA was generated from 1 μg of RNA using the SuperScript VILO cDNA Synthesis Kit (Life Technologies) according to manufacturer's protocols. Reverse transcription quantitative PCRs were completed using LightCycler 480 Probes Master Mix and hydrolysis probe #67 (Roche) following manufacturer's protocols. The following primers were generated to human ATXN1: 5′-AGAGATAAGCAACGACCTGAAGA-3′ (forward), 5′-CCAAAACTTCAACGCTGACC-3′ (reverse). Universal ProbeLibrary Mouse GAPD Gene Assay (Roche) was used as a control.
Statistical analyses.
For electrophysiological data, Spike2 (CED) software was used to record FPs. The potential with the largest initial negativity (CF-N1) was quantified as the peak value of the trough relative to baseline and was used for comparison between animals. Responses for every animal were normalized to the average CF-N1 value in 12-week-old FVB mice. Statistical analysis of CIO-evoked FPs was performed using a within-subjects ANOVA followed by a Bonferroni post hoc test to determine whether there were significant differences between genotypes. Corrected p values were based on the number of genotypes compared for each statistical analysis and were used for each dataset. In all other cases, statistical analysis was performed with a one-way ANOVA, followed by a Bonferroni post hoc test to determine whether there were statistical differences between genotypes (p < 0.05). All data are reported as mean ± SEM.
Results
CF extension along PC dendrites during development in ATXN1 mice
During normal cerebellar postnatal development, CFs progressively extend along the dendrites of PCs toward the pial surface (Kano and Hashimoto, 2009). We followed the developmental profile of CF extension along PC dendrites using cerebellar sections from p14, p17, p21, and 5- and 12-week-old ATXN1 transgenic mice immunostained for VGLUT2 that labels postsynaptic CF terminals (Freemeau et al., 2001) and for calbindin that labels PCs (Clark et al., 1997). CF extension was calculated by dividing the CF length, measured from the base of the PC soma to the furthest point of VGLUT2 staining in the PC dendrites, by the length of the PC, which was measured from the base of the PC soma to the pial surface.
At p14 extension of CFs along the PC dendritic trees in all ATXN1 mice was similar, reaching ∼70% of the distance to the pial surface (Fig. 1A,B). As postnatal development proceeded in homozygous ATXN1[30Q]-S776/ATXN1[30Q]-S776 (referred to as ATXN1[30Q]-S776/S776) and Wt FVB mice there was a progressive increase in CF extension along PC dendrites, reaching a maximum of 80 and 85%, respectively, at 5 weeks of age that remained out to 12 weeks of age.
Figure 1.

Altered CF development in SCA1-affected mice. A, Immunofluorescent confocal images taken at 60× from p14 (top) and 5 weeks of age (bottom) cerebellar sections immunostained with calbindin to identify PCs (red) and VGLUT2 to identify CF terminals (green). B, Time course of distal extension profile of CF terminals onto the PC dendrites at different stages in development. The Wt FVB and ATXN1[30Q]-S776/S776 mice have a steady progression until 5 weeks of age. SCA1 mice show no progression (ATXN1[30Q]-D776/+) or retarded progression (ATXN1[82Q]-S776/+). Number of animals (n) assessed for each genotype are Wt FVB: p14, 4; p17, 4; p21, 4; 5 weeks, 3; 12 weeks, 5; ATXN1[30Q]-S776/S776: p14, 4; p17, 4; p21, 3; 5 weeks, 3; 12 weeks, 7; ATXN1[82Q]-S776/+: p14, 3; p17, 3; p21, 3; 5 weeks, 3; 12 weeks, 6; and ATXN1[30Q]-D776/+: p14, 3; p17, 4; p21, 4; 5 weeks, 3; 12 weeks, 4. At 7 weeks cATXN1[30Q]-D776 gene-on, n = 3; and cATXN1[30Q]-D776 gene-off/gene-on, n = 4. Data are represented as mean ± SEM. *, denotes a significant difference compared with the Wt FVB controls (p < 0.01, Bonferroni post hoc comparison). The 12 weeks of age time point data are provided as a point of reference from Duvick et al., 2010.
Extension of CFs along the PC dendritic tree in mice expressing ATXN1[82Q]-S776 in their PCs was not significantly delayed compared with Wt FVB mice (Fig. 1A,B). By 5 weeks of age, CFs in the ATXN1[82Q]-S776 mice extended along the PC dendritic tree although not quite to the degree seen in nontransgenic Wt FVB cerebella. Between 5 and 12 weeks of age CFs in ATXN1[82Q]-S776 animals regressed from 81.7 to 77.4% (p < 0.01, ANOVA followed by Bonferroni post hoc test) reflecting induction of the degenerative stage of disease (Duvick et al., 2010).
While at p14 ATXN1[30Q]-D776 mice showed a pattern of CF innervation similar to Wt FVB and ATXN1[30Q]-S776/S776 mice, with subsequent postnatal development the CFs in these mice showed no further extension along the PC dendritic tree. Moreover, from 5 to 12 weeks of age CF extension retracted from 73.6 to 68.9% in ATXN1[30Q]-D776 mice (p < 0.01) (Fig. 1A,B). These results indicate that expression of ATXN1[30Q]-D776 in PCs compromised developmental ascension of CF terminals along the PC dendritic tree and as disease progressed CF terminals on the proximal PC dendritic tree degenerated.
Disrupted pruning of CF synapses from the PC soma
During development, CF synapses are pruned from the PC soma as the dominant CF grows upward along the proximal PC dendritic shaft (Hashimoto et al., 2009). Previous work revealed that a disruption in CF extension along PC dendrites is often accompanied by a decreased arborization of CF axons as well as decreased elimination of CF synapses from the PC soma (Takagishi et al., 2007). To examine these processes further we first visualized individual CFs using an anterograde tracer, fluorescently labeled dextran, injected into the CIO. In Wt FVB and ATXN1[30Q]-S776/S776 mice, a single unbranched CF axon passed over the PC soma and apical dendrite (Fig. 2A, top row). In contrast, in ATXN1[30Q]-D776 mice the CF axon contained a large number of varicosities unevenly distributed over the PC body and apical dendrite. Similarly, but to a lesser degree, ATXN1[82Q]-S776 mice displayed increased CF axonal area at the PC soma and apical dendrites.
Figure 2.
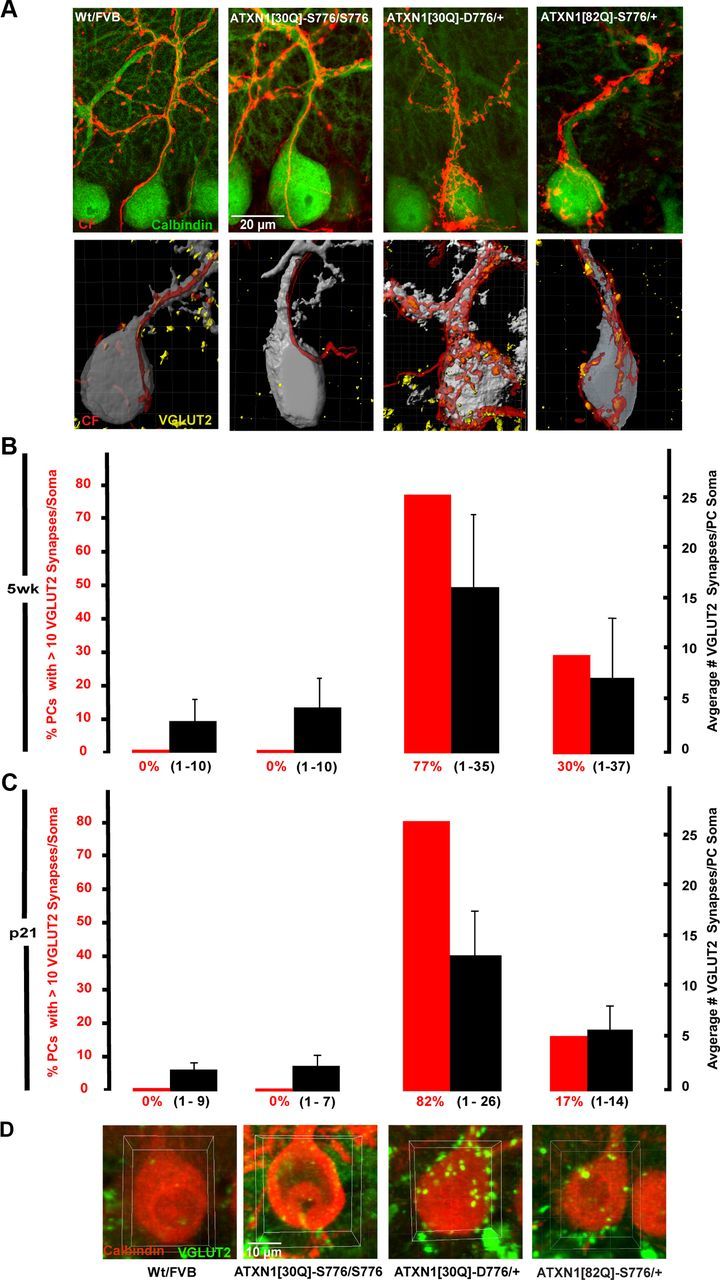
Irregular morphology and impaired CF synapse elimination in adult ATXN1[30Q]-D776 mice. A, Representative immunofluorescent confocal images taken at 100× of cerebellar sections with anterograde tracer-labeled CFs (red) and calbindin-labeled PCs (green) (top). Digital representation of the PC–CF interaction from cerebellar sections with GFP-expressing PCs (gray), anterogradely labeled CF (red), and immunolabeled CF terminals (yellow) (bottom). Note the colocalization between anterogradely labeled CF and VGLUT2 puncta in both control and mutant mice. B, Graph shows number of PCs with >10 VGLUT2 synapses (left axis, red bars) and average number of VGLUT2 synapses per PC soma (right axis, black bars) in mice 5 weeks of age. The Wt FVB (n = 65 PCs) and ATXN1[30Q]-S776/S776 (n = 56) mice show no PCs with >10 VGLUT2 somatic synapses. ATXN1[30Q]-D776/+ (n = 56) mice fail to eliminate the most somatic synapses. The data are represented as mean ± SEM. C, Graph shows number of VGLUT2 synapses on the PC cell bodies in mice 21 d of age: Wt FVB (n = 54 PCs), ATXN1[30Q]-S776/S776 (n = 52), ATXN1[30Q]-D776/+ (n = 54), and ATXN1[82Q]-S776/+ (n = 51). D, Representative 100× images of isolated PCs (red) and VGLUT2 (green) with area of interest defined (yellow box).
To evaluate whether the CF enlargements in ATXN1[30Q]-D776 and ATXN1[82Q]-S776 mice reflected synaptic contacts with PCs, images of VGLUT2-stained sections were imported into the Imaris software package to generate 3D rendered digital images of confocal z-stacks. The 3D images reveal colocalization of the anterogradely labeled CFs (red) and VGLUT2-immunoreactive puncta (yellow) on GFP-expressing PCs (gray) (Fig. 2A, bottom row). The CF terminals in Wt FVB and ATXN1[30Q]-S776/S776 mice localized to the apical dendrites and increased in number as the CF extended along the PC dendrite into the ML. VGLUT2-positive puncta were restricted to the more distal dendrites. In contrast, ATXN1[30Q]-D776 mice had a large number of CF synapses on the PC soma and apical dendrite. ATXN1[82Q]-S776 mice also displayed PCs with an increased number of CF synapses contacting the soma.
CF terminals on the PC soma were quantified using confocal images immunostained for VGLUT2 and calbindin from 5 week old mice (Fig. 2B). Two measurements were taken: the average number of VGLUT2 puncta per PC and the percentage of PCs with >10 VGLUT2 puncta per soma. As shown in Figure 2B, Wt FVB and ATXN1[30Q]-S776/S776 mice had a comparable presence of VGLUT2-positive puncta per PC soma. At 5 weeks of age, Wt FVB mice and ATXN1[30Q]-S776/S776 mice had an average of 3 and 4.4 VGLUT2 puncta per cell body (black bars), respectively (Fig. 2B). Neither line showed PCs with >10 VGLUT2 positive on the soma (red bars). ATXN1[82Q]-S776 mice had an average of 7.6 VGLUT2-positive puncta per PC soma (p < 0.0001, ATXN1[82Q]-S776 vs Wt FVB, ANOVA followed by Bonferroni post hoc test) and 30% of PCs somata were occupied by >10 VGLUT2-positive puncta (Fig. 2B) (p < 0.0001 for FVB vs ATXN1[82Q]-S776). The most remarkable result was found in ATXN1[30Q]-D776 mice that have an increase in VGLUT2-positive CF terminals by two measurements. First, the percentage of PCs with >10 VGLUT2-positive puncta was 77 in ATXN1[30Q]-D776 mice versus 0 in Wt FVB and ATXN1[30Q]-S776/S776 mice. Second, the average number of VGLUT2-positive puncta per PC soma was 16.5 in ATXN1[30Q]-D776 mice versus 3 in the Wt FVB mice (p < 0.0001, Bonferroni post hoc test) and 4.4 in the ATXN1[30Q]-S776/S776 mice (Fig. 2B) (p < 0.0001, Bonferroni post hoc test). These data indicate that pruning of CF terminals from PC somata was compromised by PC expression of ATXN1[30Q]-D776 and ATXN1[82Q]-S776.
To determine whether the retention of CF synapses on PC somata reflected an alteration in the developmental pruning of CF-PC synapses or a degeneration of the maintenance of CF–PC synapses already established during development, we examined CF synapses at p21. Similar to 5 weeks of age, Wt FVB and ATXN1[30Q]-S776/S776 mice at p21 had an average of 2.3 and 2.7 VGLUT2 puncta per cell body (black bars), respectively, and no PCs with >10 VGLUT2 puncta (red bars) (Fig. 2C). ATXN1[30Q]-D776 and ATXN1[82Q]-S776 mice had significant retention of VGLUT puncta, with an average of 13.8 in ATXN1[30Q]-D776 mice (p < 0.0001 vs Wt FVB) and 5.8 ATXN1[82Q]-S776 mice (p < 0.0001, Wt FVB vs ATXN1[30Q]-D776 and ATXN1[30Q]-D776, Bonferroni post hoc test). ATXN1[30Q]-D776 and ATXN1[82Q]-S776 mice also had a significant number of PCs with >10 VGLUT2 puncta per soma, 83 and 11%, respectively. These data suggest that the CF–PC somatic puncta observed in ATXN1[30Q]-D776 and ATXN1[82Q]-S776 mice are the result of an alteration in the developmental pruning of CF–PC synapses. Further, we examined Sca1−/− mice (Matilla et al., 1998) and found no CF synapses on the PC somata (data not shown), suggesting the CF alterations in ATXN1[30Q]-D776 and ATXN1[82Q]-S776 mice reflect a gain-of-function of mutant ATXN1.
Using flavoprotein autofluorescence optical imaging (Reinert et al., 2004, 2007), we previously showed that PC activation by CFs following stimulation of the CIO was markedly reduced in adult ATXN1[82Q]-S776 mice (Barnes et al., 2011), a finding replicated here in Figure 3. Interestingly, ATXN1[30Q]-D776 CFs retained a greater ability to activate PCs following stimulation of the CIO (Fig. 3A,C). Although to a lesser level than found in Wt nontransgenic animals, CIO stimulation evoked a stronger postsynaptic response in ATXN1[30Q]-D776 than in ATXN1[82Q]-S776 mice. Notably, the response detected in ATXN1[30Q]-D776 PCs was similar to that observed in homozygous ATXN1[30Q]-S776/S776 PCs that have an enhanced level of ATXN1 expression comparable to the level of transgenic expression seen in ATXN1[30Q]-D776 mice. The reduction in all genotypes was significantly different from Wt FVB mice. Also, the reduction in ATXN1[82Q]-S776 mice was significantly different from both ATXN1[30Q]-D776 and ATXN1[30Q]-S776/S776 mice (p < 0.01, ANOVA followed by Bonferroni post hoc test).
Figure 3.
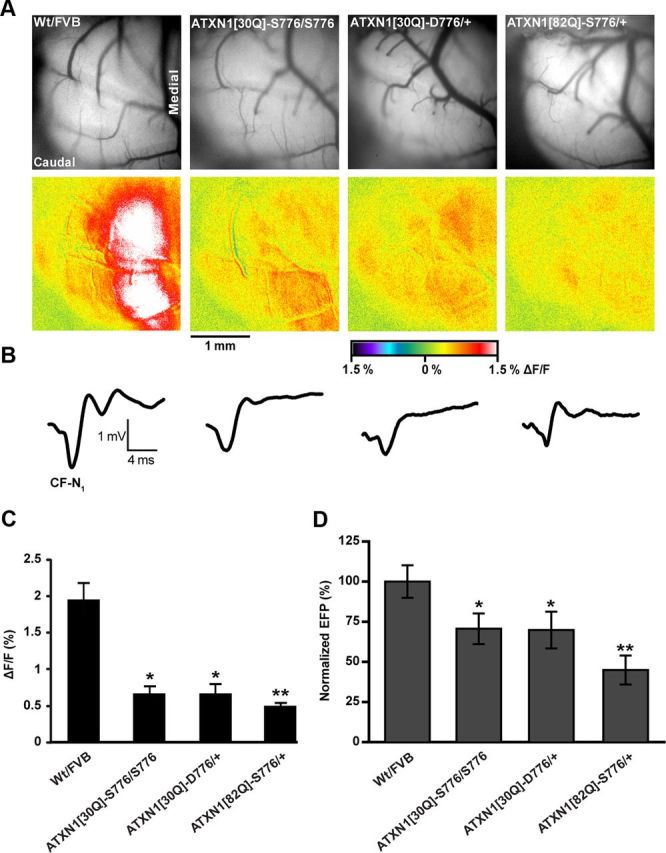
PC response evoked from CIO stimulation in mice 12 weeks of age. A, Background immunofluorescent images from exposed in vivo cerebellum (upper row) and compilation images of autofluorescent flavoprotein responses evoked by CIO stimulation (lower row). B, Representative FPs evoked by CIO stimulation. C, Quantification of flavoprotein autofluorescence responses evoked from CIO stimulation. Wt FVB: N = 11 animals, n = 60 observations for autofluorescent images; ATXN1[30Q]-S776/S776: N = 6, n = 30 observations; ATXN1[30Q]-D776/+: N = 6, n = 30 observations; ATXN1[82Q]-S776/+: N = 4, n = 32 observations. D, Quantification of CF-mediated negativity (CF-N1) of the FP showing greatest reduced response to CF stimulation in ATX1[82Q]-S776/+ mice. Wt FVB: N = 5, n = 14 observations; ATXN1[30Q]-S776/S776: N = 6, n = 30 observations; ATXN1[30Q]-D776/+: N = 6, n = 30 observations; ATX1[82Q]-S776/+: N = 4, n = 30 observations. Wt FVB and ATXN1[82Q]-S776/+ data points and images are provided as reference points from Barnes et al., 2011. *, denotes genotypes are statistically different from Wt/FVB and ATX1[82Q]-S776/+, p < 0.01. **, denotes genotype is statistically different from Wt FVB, ATXN1[30Q]-S776/S776, and ATXN1[30Q]-D776/+; p < 0.01.
As a second measure of the CF–PC circuitry, in vivo extracellular FPs were recorded from PCs during CIO stimulation in Wt FVB, ATXN1[30Q]-S776/S776, ATXN1[30Q]-D776, and ATXN1[82Q]-S776 mice. CIO stimulation evokes excitatory postsynaptic potentials in an all-or-none manner in PCs (Granit and Philips, 1956; Eccles et al., 1966). The FPs evoked a negative–positive wave form and the initial negativity reflects the PC activation due to CF stimulation (Eccles et al., 1966). Using this method, the maximal negativity was quantified from a minimum of 4 mice from each line at 12 weeks of age. The initial negativity in Wt FVB mice was the largest of the four lines (Fig. 3B,D). In ATXN1[30Q]-S776/S776 and ATXN1[30Q]-D776 mice, the initial negativity was reduced 25% from that recorded in Wt FVB mice (Fig. 3B,D). As observed using flavoprotein optical imaging, ATXN1[82Q]-S776 mice had the largest reduction (51% less than Wt FVB) in the CF-evoked PC response (p < 0.001, ANOVA followed by Bonferroni post hoc test) (Fig. 3B,D).
Multiple CF innervation of PCs in ATXN1 mice
Normal pruning of CF terminals from the PC soma results in only one CF innervating each PC (Kano et al., 1995; Hashimoto and Kano, 2003; Kakizawa et al., 2003). Given the evidence that CF-terminal pruning was disrupted in ATXN1[30Q]-D776 mice, we sought to examine whether PCs in ATXN1[30Q]-D776 mice were innervated by multiple CFs (Fig. 4). This was accomplished using a triple labeling morphological approach (Hashimoto et al., 2009). Cerebellar sections were obtained in which a single CF was labeled with an anterograde tracer injected into the CIO (green), VGLUT2 immunostaining was used to visualize all CF postsynaptic terminals (red), and calbindin immunostaining to visualize all PCs (gray). Using this approach, if a PC was innervated by a single CF, then all VGLUT2 puncta (red) on its soma would colocalize with the anterogradely labeled CF (green) and would appear yellow. In contrast, PCs innervated by more than one CF would have CF terminals (red) that did not colocalize with the anterogradely labeled CF (green).
Figure 4.

Twelve-week-old ATXN1[30Q]-D776 PCs are multiply innervated. Immunofluorescent images of PC somata (gray) were examined to detect PCs with lone VGLUT2-positive CF synapses (red, white arrows) that do not colocalize with the anterograde tracer labeled CFs (green). A, A Wt FVB PC with no VGLUT2-positive puncta. B, A CF multiply innervated mouse model, L7-PKCI/+ PC with VGLUT2-positive somatic synapses (white arrows). C, An ATXN1[30Q]-D776/+ PC with VGLUT2-positive somatic synapses (white arrows). D, An ATXN1[82Q]-D776/+ PC with no VGLUT2-positive puncta. E, Graph showing the percentage of PCs per genotype that have lone red VGLUT2-positive synapses (p = 0.002, χ2 test). Greater than three mice were used for each genotype; Wt FVB: n = 20 PCs examined; L7-PKCI/+: n = 40; ATXN1[30Q]-D776/+: n = 39; ATXN1[82Q]-S776/+: n = 43.
In Wt FVB cerebellum all CF terminals colocalized with the anterograde labeled CF (i.e., there were no red terminals), demonstrating the normal adult innervation pattern (Fig. 4A,E). In contrast, as a positive control for PCs that are innervated by multiple CFs, mice overexpressing an inhibitor of protein kinase C (L7-PKCI) (De Zeeuw et al., 1998), 30% of the PCs examined had multiple CF terminals (red) on their soma that did not colocalize with the anterogradely labeled CF (Fig. 4B,E). Although to a lesser degree than the L7-PKCI mice, 15% of PCs in ATXN1[30Q]-D776 mice had a number of VGLUT2-positive puncta that did not colocalize with the anterogradely labeled CF (p = 0.002, χ2 test) (Fig. 4C,E). The ATXN1[82Q]-S776 mice had no significant evidence of PCs with supernumerary CF innervation (Fig. 4D,E). These morphological findings provide evidence for multiple innervation of PCs by CFs in ATXN1[30Q]-D776 mice.
Expansion of parallel fiber spine territory in ATXN1[30Q]-D776 mice
Several studies demonstrated that new spines appear on proximal dendrites of PCs following loss of CF innervation (Sotelo et al., 1975; Rossi et al., 1991; Bravin et al., 1999., Kakizawa et al., 2005). This led to the hypothesis that formation of spines by PCs is an “intrinsic mechanism” and that CF innervation inhibits spine formation on proximal regions of PCs and, thus, inhibits invasion by parallel fibers (Sotelo, 1978; Bravin et al., 1999; Strata 2002; Bosman and Konnerth, 2009).
To evaluate whether the genesis of PC spines and the territory of parallel fiber innervation were altered in ATXN1[30Q]-D776 mice, calbindin immunolabeled sections from 12 week old mice were imaged at high magnification (260–700×). As expected, Wt FVB PC somata exhibited a smooth membrane with spines restricted to the tertiary dendrites (Fig. 5A, left). In contrast, ATXN1[30Q]-D776 PCs showed an abundant number of spines on their apical dendrite and soma (Fig. 5A, middle). To determine whether the PC somatic spines were innervated by parallel fibers, sections were immunostained for VGLUT1, a marker of parallel fiber presynaptic terminals (Freemeau et al., 2001). As shown in Figure 5A, right, the somatic spines on PCs in ATXN1[30Q]-D776 mice colocalized with VGLUT1 indicating innervation by parallel fibers. Quantification of the VGLUT1 showed a 600% increase in puncta on the PC somata in ATXN1[30Q]-D776 mice compared with Wt FVB controls (p < 0.001, Student's t test). These data suggest that the abnormal somatic PC spines have parallel fiber synapses in the territory normally restricted to CF innervation (Fig. 5B). Spines on ATXN1[30Q]-D776 PC somata and proximal dendritic shafts were detected on 60% before onset of CF regression as early as 5 weeks of age, remained out to at least one year of age (Fig. 5C,E), and were not found on PCs in ATXN1[82Q]-S776 mice to a high degree at any age (Fig. 5D,E). Thus, while PC expression of ATXN1[82Q]-S776 impacted CF somatic pruning similar to ATXN1[30Q]-D776, it did not lead to the supernumerary CF innervation or atypical PC spines observed in ATXN1[30Q]-D776 mice.
Figure 5.
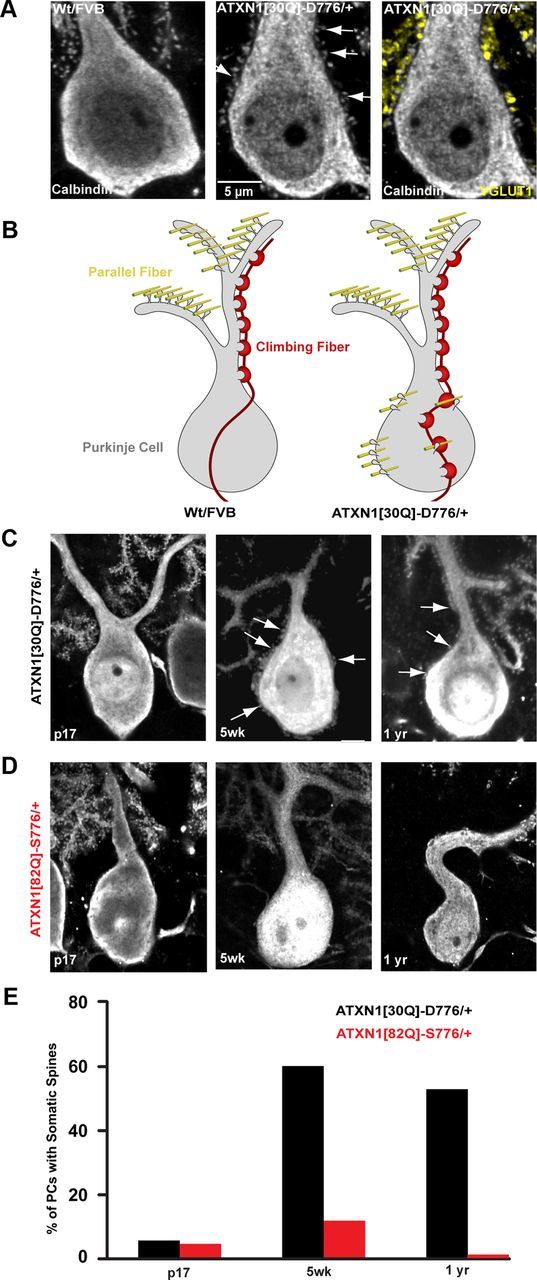
Abnormal parallel fiber distribution in ATXN1[30Q]-D776 mice. A, 350× immunofluorescent images of calbindin-labeled PCs somata from Wt FVB, left and ATXN1[30Q]-D776/+ mice, middle and right. Atypical spines can be seen protruding from the PC soma and apical dendrite in ATXN1[30Q]-D776/+ (white arrows, middle). Colocalization between the presynaptic parallel fiber–terminal marker terminal marker, VGLUT1 (yellow, right), and somatic PC spines. B, Diagram depiction of the disruption of CF (red) and parallel fiber synapses (yellow) and their relationship to PC spines in Wt FVB and ATXN1[30Q]-D776/+ mice. C, D, 150× immunofluorescent images of spine development in SCA1 mice. ATXN1[30Q]-D776/+ mice (C) display somatic spines at 5 weeks of age and remain out to 1 year of age. D, No atypical spines were detected at 5 weeks of age in ATXN1[82Q]-S776/+ mice. E, Time course of the percentage of PCs displaying somatic spines. ATXN1[30Q]-D776/+: n = 50 PCs examined; ATXN1[82Q]-S776/+: n = 63.
Nuclear localization is critical for ATXN1[30Q]-D776-induced disruption of CF synapse elimination
ATXN1 normally is able to shuttle between the cytoplasm and nucleus (Irwin et al., 2005), with nuclear localization of mutant ATXN1 being critical for disease pathogenesis (Klement et al., 1998; Barnes et al., 2011). To determine whether the disruption of CF synaptic elimination from the PC soma also requires nuclear localization of ATXN1[30Q]-D776, we generated transgenic mice expressing ATXN1[30Q]-D776 with a dysfunctional nuclear localization signal by replacing the Lys at position 772 with a Thr (Klement et al., 1998). Since pathogenesis in transgenic mice is directly related to the amount of ATXN1 expressed, we first compared the level of ATXN1 in cerebellar lysates prepared from ATXN1[30Q]-D776 and ATXN1[30Q]-D776-K772T mice. Figure 6A shows a Western blot demonstrating that the amount of ATXN1 did not differ significantly (replicated with three animals, p > 0.05, Student's t test).
Figure 6.
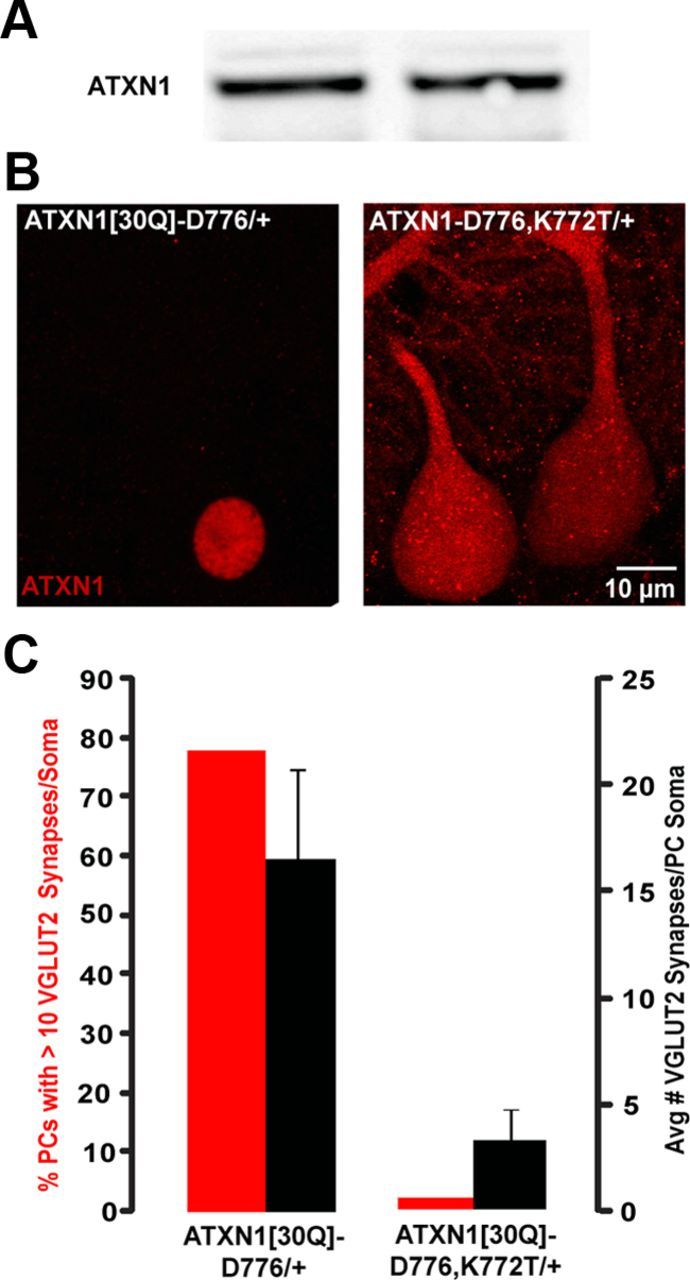
Nuclear localization of ATXN1[30Q]-D776 is critical for disruption of CF synapse elimination. A, Western blot of ATXN1 protein levels in cerebellar lysates. ATXN1[30Q]-D776/+ and ATXN1[30Q]-D776-K772T/+. B, 100× immunofluorescent images of ATXN1 localization at PC layer. C, Graph of average number of VGLUT2 synapses per PC soma (right axis, black bars) and number of PCs with >10 VGLUT2 synapses (left axis, red bars) in ATXN1[30Q]-D776-K772T and ATXN1[30Q]-D776/+ mice.
Next, the ability of the K772T substitution to alter the subcellular distribution of ATXN1[30Q]-D776 in PCs was examined by immunostaining. Immunofluorescent staining specific for human ATXN1 showed that ATXN1[30Q]-D776 localized exclusively to the nucleus of PCs (Fig. 6B). In contrast, ATXN1 was distributed widely throughout the PC cytoplasm in ATXN1[30Q]-D776, K772T mice (Fig. 6B).
To assess whether blocking entry of ATXN1[30Q]-D776 into the nucleus of PCs restored normal CF/PC development, the presence of CF terminals on PC somata was determined at 5 weeks of age in ATXN1[30Q]-D776, K772T mice. Figure 6C shows that by both percentage of PCs with >10 VGLUT2 puncta and average number of VGLUT2-positive puncta per PC soma, CF terminals in ATXN1[30Q]-D776, K772T mice were pruned to a degree similar to that found in Wt FVB and ATXN1[30Q]-S776/S776 mice (Fig. 2). Therefore, we conclude that nuclear localization of ATXN1[30Q]-D776 is critical for its ability to disrupt the pruning of CF synapses from PC soma.
Expression of mutant ATXN1 during cerebellar development is critical for ATXN1[30Q]-D776-induced alterations to CF extension along PC dendrites
To determine whether changes observed at CF–PC synapses in ATXN1[30Q]-D776 mice reflect an effect of ATXN1 during a critical period of development, we generated a conditional ATXN1[30Q]-D776 mouse model (cATXN1[30Q]-D776). Using the tetracycline-regulated system (Gossen and Bujard, 1992; Zu et al., 2004), ATXN1[30Q]-D776 expression was controlled by administration of the tetracycline derivative, dox. In the absence of dox, ATXN1[30Q]-D776 expression is induced. In the presence of dox, ATXN1[30Q]-D776 expression is suppressed. When cerebellar mRNA was isolated from mice treated with dox for 1 d, ATXN1[30Q]-D776 was undetectable. Likewise, ATXN1[30Q]-D776 expression was restored to previous levels 1 week after halting dox treatment (data not shown).
For these studies, we examined CF extension using cerebellar sections from three groups of mice. The cATXN1[30Q]-D776 gene-on control group was not administered dox from birth until they were killed at 7 weeks of age. The cATXN1[30Q]-D776 gene-off/gene-on group was treated with dox from birth until p21, at which time the gene was turned on and expressed for an additional 7 weeks of age. Using this strategy, both groups of mice express ATXN1[30Q]-D776 for ∼7 weeks. In the cATXN1[30Q]-D776 gene-on the expression period included the first three postnatal weeks as a critical period of cerebellar development while in cATXN1[30Q]-D776 gene-off/gene-on mice expression was delayed until after cerebellar development is complete (Goldowitz and Hamre, 1998).
Cerebellar sections labeled with calbindin and VGLUT2 from mice in the cATXN1[30Q]-D776 gene-on group showed a high degree of both PC and CF terminal atrophy (Fig. 7A, left). Conversely, cerebellar sections from mice in the cATXN1[30Q]-D776 gene-off/gene-on group showed healthy PC dendrites with a high density of CF terminals (Fig. 7A, middle). CF extension reached an average of 52% in the cATXN1[30Q]-D776 gene-on, significantly lower from both the cATXN1[30Q]-D776 gene-off/gene-on mice (Fig. 7A, middle) and the cATXN1[30Q]-D776 gene-off mice (Fig. 7A, right) (p < 0.01 and p < 0.001, ANOVA followed by Bonferroni post hoc test). In the gene cATXN1[30Q]-D776 gene-off/gene-on mice, the CF terminals extended to 70% and it was not significantly different from cATXN1[30Q]-D776 gene–off mice (p > 0.05, ANOVA followed by Bonferroni post hoc test) (Fig. 7B), suggesting that ATXN1[30Q]-D776 affects CF extensions during development when the distal CF terminals are being established.
Figure 7.

CF–PC extension is restored when ATXN1[30Q]-D776 is not expressed during cerebellar development. A, 60× immunofluorescent images from primary fissure showing restored PC morphology (green) and CF extension (red) in mice not expressing ATXN1[30Q]-D776 during the first 3 weeks of age. B, Graph of distal extension of CF terminals onto the PC dendrites in cATXN1[30Q]-D776 mice. cATXN1[30Q]-D776 off: N = 4; cATXN1[30Q]-D776 gene-off/gene-on: N = 3 animals; cATXN1[30Q]-D776 on: N = 4. C, Graph of ATXN1 expression during development in ATXN1[30Q]-D776/+ and cATXN1[30Q]-D776, ATXN1[82Q]-S776, and ATXN1[30Q]-D776-K772T/+mice. Data are normalized to ATXN1 expression in ATXN1[30Q]-D776/+ mice at p7. *p < 0.05, **p < 0.01, ***p < 0.001.
Of note is the difference observed between the ATXN1[30Q]-D776 and cATXN1[30Q]-D776 gene-on mice. ATXN1[30Q]-D776 mice retained CF synapses on the PC soma to a remarkable extent (Fig. 2), while in cATXN1[30Q]-D776 gene-on mice CF–PC somatic synapses were not evident (data not shown). Further, CF extension in cATXN1[30Q]-D776 gene-on mice at 7 weeks of age was lower than that in ATXN1[30Q]-D776 mice at any age. Considering that mutant ATXN1 expression during development was critical for affecting the CF–PC synapses (Fig. 7), we examined the developmental expression of the respective transgenes at p7, p9, p11, p14, and p21. Expression of cATXN1[30Q]-D776 gene-on mRNA was relatively constant through development (Fig. 7C). Alternatively, ATXN1[30Q]-D776 mice expression rose dramatically between p9 and p11 (Fig. 7C). These data further support the hypothesis that mutant ATXN1 during development is critical for the alteration of CF–PC synapse and that the specific timing of expression during development alters the phenotypic profile.
Disease severity in mice expressing ATXN1[30Q]-D776 and ATXN1[82Q]-S776
Although both ATXN1[30Q]-D776 and ATXN1[82Q]-S776 mice develop severe ataxia, ATXN1[30Q]-D776 animals fail to develop the progressive PC disease seen in ATXN1[82Q]-S776 mice (Duvick et al., 2010). To determine whether expression of ATXN1[30Q]-D776 dampens the progressive disease in ATXN1[82Q]-S776 mice, we crossed ATXN1[30Q]-D776 and ATXN1[82Q]-S776 mice and assessed PC pathology. The double transgenic mice retained CF somatic synapses to an extent similar to ATXN1[30Q]-D776, with an average of 10.8 VGLUT2 per PC soma versus 13, respectively (p > 0.05, ANOVA followed by Bonferroni post hoc test) (Fig. 8A). Additionally, transgenic ATXN1[30Q]-D776/ATXN1[82Q]-S776 mice had a comparable proportion of PCs with VGLUT2 puncta >10 relative to ATXN1[30Q]-D776 mice, 61.5 versus 48.9%, respectively (Fig. 8A). Next, as a measure of PC health, we assessed PC dendrite atrophy by measuring molecular layer thickness (Zu et al., 2004). ATXN1[30Q]-D776, ATXN1[82Q]-S776, and ATXN1[30Q]-D776/ATXN1[82Q]-S776 mice all had a significantly reduced molecular layer thickness compared with Wt FVB mice (p < 0.001) (Fig. 8B,C). Last, the global severity was assessed in each disease model. Using this approach, ATXN1[30Q]-D776/ATXN1[82Q]-S776 exhibited the greatest severity score that was significantly higher than ATXN1[30Q]-D776 mice (p < 0.001, ANOVA followed by Bonferroni post hoc test) (Fig. 8D). These data suggest that the increased number of VGLUT2 puncta present on the PC soma in mice expressing ATXN1[30Q]-D776 along with ATXN1[82Q]-S776 is insufficient to “protect” PCs from the progressive degeneration induced by ATXN1[82Q]-S776.
Figure 8.

Mice expressing both ATXN1[30Q]-D776 and ATXN1[82Q]-S776 display increased severity of disease at 9 months of age. A, Graph shows number of PCs with >10 VGLUT2 synapses (left axis, red bars) and average number of VGLUT2 synapses per PC soma (right axis, black bars) in mice 5 weeks of age. ATXN1[30Q]-D776/ATXN1[82Q]-S776 PC somata retain VGLUT2 synapses to a similar extent see in ATXN1[30Q]-D776/+ mice. Greater than three mice were used for each genotype; Wt FVB: n = 74 PCs examined; ATXN1[30Q]-D776/+: n = 52; ATXN1[82Q]-S776/+: n = 73; ATXN1[30Q]-D776/ATXN1[82Q]-S776: n = 50. B, 60× immunofluorescent images of calbindin-labeled PCs (red) and VGLUT2-labeled CF synapses (green). C, Graph shows the average molecular thickness. Wt FVB: N = 5 mice; ATXN1[30Q]-D776/+: N = 7; ATXN1[82Q]-S776/+: N = 8; ATXN1[30Q]-D776/ATXN1[82Q]-S776: N = 6. D, Graph of pathological severity score. Wt FVB: N = 5 mice; ATXN1[30Q]-D776/+: N = 7; ATXN1[82Q]-S776/+: N = 8; ATXN1[30Q]-D776/ATXN1[82Q]-S776: N = 6. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
In this study we show that PC-specific expression of two pathogenic forms of ATXN1, ATXN1[82Q]-S776 and ATXN1[30Q]-D776, result in developmental abnormalities in the innervation of PCs by CFs (Fig. 9). In ATXN1[30Q]-D776 mice there is a more substantive disruption in CF development that includes a failure of CFs to extend along PC dendrites during postnatal development, a dramatic retention of CF synapses on PC somata, and innervation of PCs by CFs from multiple inferior olivary neurons in adults.
Figure 9.

CF development and regression in affected SCA1 mice. Normal development of CF/PC synapses is depicted in the top row. At early stages (p7) each PC is innervated by multiple CFs from the inferior olive (IO). As development proceeds (p14) activity of one CF strengthens, its terminals translocate up the proximal dendritic shaft of the PC. At 5 weeks of age, and into the adult, one CF maintains its synapses with the PC dendritic tree segregated from the synaptic territory of parallel fibers. In ATXN1[82Q]-S776 mice development is mostly normal except that translocation of the dominant CF terminals up the PC dendritic tree is delayed and some of its terminals remain on the PC soma (5 weeks). As disease progresses in ATXN1 mice CF terminals regress from the PC dendritic tree (12 week old adult). In ATXN1[30Q]-D776 mice development is more substantially altered. Although one CF seems to be become dominant and translocates its terminals up the PC dendritic tree (p14) subsequent terminal translocation is more severely compromised and pruning of terminals from the PC soma fails to occur (5 weeks). As in ATXN1[82Q]-S776 mice, CF terminals that translocated up the PC dendritic tree regress with age (12 week old adult). DCN, deep cerebellar nuclei.
Normally, early in the postnatal life of a mouse each PC soma is innervated by multiple CFs from different inferior olivary neurons. During the first postnatal week the somatic synapses of one CF functionally become stronger (Hashimoto and Kano, 2003). Subsequently, the terminals of this strong CF translocate up from the cell body and along the proximal PC dendritic shaft. During the late phase in development, the dominant CF begins its climb up the PC dendritic tree. During the second postnatal week, the synapses on the PC soma from both the dominant and other weaker CFs are eliminated. By the end of the third postnatal week one CF forms hundreds of synapses along a PC primary dendrite, with essentially no synapses remaining on the PC soma (Cesa and Strata, 2009; Kano and Hashimoto, 2009). The capacity of ATXN1 to disrupt CF innervation is shown here to depend upon the timing of ATXN1 expression during development. In ATXN1[30Q]-D776 cerebella, CF translocation is normal at p14 and subsequently CF terminals fail to translocate beyond that point and the number of CF synapses on PC somata remain high. The failure of CFs to develop properly after p14 is synchronized with the expression of ATXN1[30Q]-D776 increasing dramatically after p9. During this period, the late phase CF development has begun and ATXN1[30Q]-D776 expression altered the normally proceeding CF translocation and CF synapse elimination. Further, using a cATXN1[30Q]-D776 model we show that by delaying expression until p21, CF extension is restored compared with mice that express ATXN1 during development, supporting the hypothesis that ATXN1 expression during development alters CF–PC innervation. Unlike the ATXN1[30Q]-D776 model, cATXN1[30Q]-D776 mice express ATXN1 as early as p7 and these mice display a dramatically decreased CF extension compared with the other SCA1 models. The expression of ATXN1 at p7 in conditional ATXN1[30Q]-D776 mice resulted in improved CF–PC somatic synapse elimination. These data reinforce the hypothesis that the altered CF development is dependent upon the temporal pattern of ATXN1 expression during development.
Other additional phenotypic differences observed between the models studied provide insight into ATXN1's effect on normal CF development. Previous studies indicate that activity and the selective functional strengthening of one CF is critical for proper CF translocation along the PC dendritic tree and elimination of CF terminals from the PC soma (Hashimoto and Kano, 2003). The innervation of PCs by CFs in the ATXN1[82Q]-S776 mice appeared to proceed normally to 5 weeks of age, aside from a small increase in the number of CF terminals on their PC somata. Unlike the ATXN1[30Q]-D776 mice, the normal CF extension beyond p14 in ATXN1[82Q]-S776 mice is correlated with the proper selection of a single dominant CF. This indicates that the normal CF translocation to p14 seen in ATXN1[30Q]-D776 mice is not sufficient to ensure elimination of CF synapses from the PC soma or the proper strengthening of a single CF. These data support the hypothesis that the late phase of CF synapse elimination from the PC soma (Kano and Hashimoto, 2009) is linked to CFs completing their climb up the PC dendrite.
The ATXN1[30Q]-D776 mice manifested another characteristic associated with altered CF innervation of PCs that was not detected in ATXN1[82Q]-S776 cerebella. From 5 weeks to 1 year of age, the ATXN1[30Q]-D776 PC soma and the proximal dendritic shaft displayed postsynaptic spines that were, at least in part, innervated by parallel fibers. Hyperspiny PCs innervated by parallel fibers are also found in mice with inferior olive lesions (Sotelo et al., 1975) as well as in mice with loss of the P/Q-type Ca2+ a1A channel protein (Miyazaki et al., 2004). These results led to the concept that PCs form spines by an intrinsic mechanism and that CF innervation acts to inhibit spine formation (Sotelo, 1978). Yet, in ATXN1[30Q]-D776 mice the PC somata remain morphologically and to some degree functionally innervated by CFs, suggesting that CF synapses alone are not sufficient to inhibit spine formation. Normally, CF and parallel fiber synapses are confined to distinct areas of the PC dendritic tree. These two afferents appear to be in competition for each other's synaptic territory such that if the innervation territory of one is compromised, the innervation territory of the other is enhanced (Bosman and Konnerth, 2009). The observation that the ATXN1[30Q]-D776-induced alteration in CF development was associated with an increase in the territory innervated by parallel fibers is consistent with the idea that these two afferents compete for innervation of the PC. We suggest that the failure of the CF terminals to complete their translocation along the PC dendrites in ATXN1[30Q]-D776 mice inhibits the competitive interaction between parallel fibers and CFs for innervation territory on PCs. Thus, in ATXN1[30Q]-D776 mice parallel fibers are able to invade the proximal dendritic shaft and soma in conjunction with the activation of spine formation. These results are another demonstration that CF translocation along the dendritic tree and elimination of synapses from the soma are linked.
As shown in Figure 9, we propose that PC-specific expression of mutant ATXN1 induces changes to developmental pathways that are dependent upon translocation of the CF onto PC dendrites. The results presented here and previously for ATXN1[82Q]-K772T (Klement et al., 1998) show that mutant ATXN1 must enter the nucleus of PCs to induce these alterations. This suggests that ATXN1[82Q]-S776 and ATXN1[30Q]-D776 alter the expression of a gene(s) in the postsynaptic PC that are critical for its innervation by CFs. While the mechanisms are unclear, we speculate that mutant ATXN1 alters CF development and CF synaptic elimination via PC-specific signaling molecules. For example, PC trophic factors are known to affect CF synapse elimination. Insulin growth factor-1 expressed in PCs influences CF synapse elimination during critical periods of development (Kakizawa et al., 2003). Future profiling experiments could elucidate whether ATXN1[30Q]-D776 PCs and inferior olivary neurons differentially express proteins involved in CF development relative to Wt FVB animals. While both forms of ATXN1 alter CF development, we find it intriguing that the ATXN1[30Q]-D776 phenotype is more severe relative to ATXN1[82Q]-S776. Perhaps phosphorylation at S776 is critical for this phenotype. If so, substitution of an Asp at position 776 would result in a constitutively phosphorylated state no longer able to be dephosphorylated. Thus, the more severe phenotype induced by ATXN1[30Q]-D776 might reflect the disruption of the normal dynamic phosphorylation and dephosphorylation reactions. Regardless, we suggest that the ATXN1[30Q]-D776 mice offer a tool to further unravel postsynaptic pathways in PCs that have a critical role in the plasticity of developing CF–PC synapses and how these pathways impact PC viability.
Both ATXN1[82Q]-S776 and ATXN1[30Q]-D776 cerebella showed a regression of CF terminals from PC dendritic trees from 5 to 12 weeks of age. Thus, in these two affected ATXN1 transgenic lines CF terminal regression from PC dendrites was a prominent pathological feature of disease. ATXN1[30Q]-D776 mice are as neurologically compromised and develop a similar degree of PC dendritic atrophy out to 12 weeks of age as found in ATXN1[82Q]-S776 animals. We suggest that the changes in CF extension contribute to the decreased response to inferior olive stimulation in ATXN1[82Q]-S776 and ATXN1[30Q]-D776 cerebella as shown by flavoprotein imaging and extracellular FP recordings. However, the milder phenotype in ATXN1[30Q]-D776 mice is due to the increased number of somatic CF contacts that will provide greater activation of PCs than in the ATXN1[82Q]-S776. Yet, we also observed a reduction in CF response in the ATXN1[30Q]-S776 homozygous mice where CF extension was not altered. This likely reflects the mild pathological changes in PCs with Wt ATXN1 overexpression (Fernandez-Funez et al., 2000). Thus, the PC response to CF stimulation whether assessed by flavoprotein imaging or extracellular FPs is a very sensitive indicator of PC health.
One additional feature of PCs expressing ATXN1[30Q]-D776 that distinguishes them from those expressing ATXN1[82Q]-S776 is the absence of progressive atrophy with increasing age (Duvick et al., 2010). Thus, ATXN1[30Q]-D776 mice were crossed with ATXN1[82Q]-S776 animals to test whether aspects of the ATXN1[30Q]-D776 phenotype, e.g., retention of CF terminals on the PC soma, provide a protective function(s) sufficient to impact the progressive PC disease seen in ATXN1[30Q]-D776 mice. While the double transgenic mice kept features of ATXN1[30Q]-D776 mice, such as retention of a large number of CF synapses on PC soma, expression of ATXN1[30Q]-D776 failed to dampen the progressive PC atrophy out to 9 months of age. If anything, PC degeneration was enhanced in ATXN1[30Q]-D776/ATXN1[82Q]-S776-expressing mice compared with age-matched ATXN1[82Q]-S776 animals (Fig. 8D). This finding indicates that the absence of progressive neurodegeneration in ataxic ATXN1[30Q]-D776 mice reflects the absence of an expanded polyglutamine tract. These results provide further evidence that induction of the progressive neurodegeneration of SCA1 is dependent on expressing ATXN1 with an expanded polyglutamine tract.
Footnotes
We thank Orion Rainwater for his expertise in maintaining the mouse colony. Supported in part by National Institutes of Health Grants NS022920 and NS045667 (H.T.O.), NS18332 (T.J.E.), NS048944 (T.J.E.), NS062561 (J.A.B.), and NS062158 (T.J.E., H.T.O.).
The authors declare no competing financial interests.
References
- Altman J. Postnatal development of the cerebellar cortex in the rat. II. Phases in the maturation of Purkinje cells and of the molecular layer. J Comp Neurol. 1972;145:399–463. doi: 10.1002/cne.901450402. [DOI] [PubMed] [Google Scholar]
- Barnes JA, Ebner BA, Duvick LA, Gao W, Chen G, Orr HT, Ebner TJ. Abnormalities in the climbing fiber-purkinje cell circuitry contribute to neuronal dysfunction in ATXN1[82Q] Mice. J Neurosci. 2011;31:12778–12789. doi: 10.1523/JNEUROSCI.2579-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosman LW, Konnerth A. Activity-dependent plasticity of developing climbing fiber-Purkinje cell synapses. Neuroscience. 2009;162:612–623. doi: 10.1016/j.neuroscience.2009.01.032. [DOI] [PubMed] [Google Scholar]
- Bravin M, Morando L, Vercelli A, Rossi F, Strata P. Control of spine formation by electrical activity in the adult cerebellum. Proc Natl Acad Sci U S A. 1999;96:1704–1709. doi: 10.1073/pnas.96.4.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burright EN, Clark HB, Servadio A, Matilla T, Feddersen RM, Yunis WS, Duvick LA, Zoghbi HY, Orr HT. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995;82:937–948. doi: 10.1016/0092-8674(95)90273-2. [DOI] [PubMed] [Google Scholar]
- Cesa R, Strata P. Axonal competition in the synaptic wiring of the cerebellar cortex during development and in the mature cerebellum. Neuroscience. 2009;162:624–632. doi: 10.1016/j.neuroscience.2009.02.061. [DOI] [PubMed] [Google Scholar]
- Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, Matilla A, Zoghbi HY, Orr HT. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17:7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepel F. Regression of functional synapses in the immature mammalian cerebellum. Trends Neurosci. 1982;5:266–269. [Google Scholar]
- de Chiara C, Menon RP, Strom M, Gibson TJ, Pastore A. Phosphorylation of S776 and 14–3-3 binding modulate ataxin-1 interaction with splicing factors. PLoS One. 2009;4:e8372. doi: 10.1371/journal.pone.0008372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zeeuw CI, Hansel C, Bian F, Koekkoek SK, van Alphen AM, Linden DJ, Oberdick J. Expression of a protein kinase C inhibitor in Purkinje cells blocks cerebellar LTD and adaptation of the vestibulo-ocular reflex. Neuron. 1998;20:495–508. doi: 10.1016/s0896-6273(00)80990-3. [DOI] [PubMed] [Google Scholar]
- Duvick L, Barnes J, Ebner B, Agrawal S, Andresen M, Lim J, Giesler GJ, Zoghbi HY, Orr HT. SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron. 2010;67:929–935. doi: 10.1016/j.neuron.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Llinís R., Sasaki K. The excitatory synaptic action of climbing fibres on the purkinje cells of cells of the cerebellum. J Physiol. 1966;182:268–296. doi: 10.1113/jphysiol.1966.sp007824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, Clark HB, Orr HT. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38:375–387. doi: 10.1016/s0896-6273(03)00258-7. [DOI] [PubMed] [Google Scholar]
- Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, McCall A, Canal I, Orr HT, Zoghbi HY, Botas J. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- Freemeau RT, Jr, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- Gao W, Chen G, Reinert KC, Ebner TJ. Cerebellar cortical molecular layer inhibition is organized in parasagittal zones. J Neurosci. 2006;26:8377–8387. doi: 10.1523/JNEUROSCI.2434-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldowitz D, Hamre K. The cells and molecules that make a cerebellum. Trend Neurosci. 1998;21:375–382. doi: 10.1016/s0166-2236(98)01313-7. [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granit R, Philips CG. Excitatory and inhibitory processes acting upon individual Purkinje cells of cerebellum in cats. J Physiol. 1956;133:520–547. doi: 10.1113/jphysiol.1956.sp005606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Kano M. Functional differentiation of multiple climbing fiber inputs during synapse elimination in the developing cerebellum. Neuron. 2003;38:785–796. doi: 10.1016/s0896-6273(03)00298-8. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Ichikawa R, Kitamura K, Watanabe M, Kano M. Translocation of a “winner” climbing fiber to the Purkinje cell dendrite and subsequent elimination of “losers” from the soma in developing cerebellum. Neuron. 2009;63:106–118. doi: 10.1016/j.neuron.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Irwin S, Vandelft M, Howell JL, Graczyk J, Orr HT, Truant R. RNA association and nucleocytoplasmic shuttling by ataxin-1. J Cell Sci. 2005;118:233–242. doi: 10.1242/jcs.01611. [DOI] [PubMed] [Google Scholar]
- Kakizawa S, Yamada K, Iino M, Kano M. Effects of insulin-like growth factor 1 on climbing fibre synapse elimination during cerebellar development. Eur J Neurosci. 2003;17:545–554. doi: 10.1046/j.1460-9568.2003.02486.x. [DOI] [PubMed] [Google Scholar]
- Kakizawa S, Miyazaki T, Yanagihara D, Iino M, Watanabe M, Kano M. Maintenance of presynaptic function by AMPA receptor-mediated excitatory postsynaptic activity in adult brain. Proc Natl Acad Sci U S A. 2005;102:19180–19185. doi: 10.1073/pnas.0504359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Hashimoto K. Synapse elimination in the central nervous system. Curr Opin Neurobiol. 2009;19:154–161. doi: 10.1016/j.conb.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Chen C, Abeliovich A, Aiba A, Kurihara H, Watanabe M, Inoue Y, Tonegawa S. Impaired synapse elimination during cerebellar development in PKCg mutant mice. Cell. 1995;83:1223–1231. doi: 10.1016/0092-8674(95)90147-7. [DOI] [PubMed] [Google Scholar]
- Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- Landis DMD, Rosenberg RN, Landis DM, Schut L, Nyhan WL. Olivopontocerbellar degeneration clinical and ultrastructural abnormalities. Arch Neurol. 1974;31:295–307. doi: 10.1001/archneur.1974.00490410043003. [DOI] [PubMed] [Google Scholar]
- Mason CA, Christakos S, Catalano SM. Early climbing fiber interactions with Purkinje cell in the postnatal mouse cerebellum. J Comp Neurol. 1990;297:77–90. doi: 10.1002/cne.902970106. [DOI] [PubMed] [Google Scholar]
- Matilla A, Roberson ED, Banfi S, Morales J, Armstrong DL, Burright EN, Orr HT, Sweatt JD, Zoghbi HY, Matzuk MM. Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation. J Neurosci. 1998;18:5508–5516. doi: 10.1523/JNEUROSCI.18-14-05508.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilla-Dueñas A, Goold R, Giunti P. Clinical, genetic, molecular, and pathophysiological insights into spinocerebellar ataxia type 1. Cerebellum. 2008;7:106–114. doi: 10.1007/s12311-008-0009-0. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Hashimoto K, Shin HS, Kano M, Watanabe M. P/Q-type Ca2+ channel a1A regulates synaptic competition on developing cerebellar Purkinje cells. J Neurosci. 2004;24:1734–1743. doi: 10.1523/JNEUROSCI.4208-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary JL, Inukai J, Smith JM. Histogenesis of the cerebellar climbing fiber in the rat. J Comp Neurol. 1971;142:377–391. doi: 10.1002/cne.901420307. [DOI] [PubMed] [Google Scholar]
- Orr HT, Chung MY, Banfi S, Kwiatkowski TJ, Jr, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LP, Zoghbi HY. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- Oz G, Nelson CD, Koski DM, Henry PG, Marjanska M, Deelchand DK, Shanley R, Eberly LE, Orr HT, Clark HB. Noninvasive detection of presymptomatic and progressive neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci. 2010;30:3831–3838. doi: 10.1523/JNEUROSCI.5612-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon ý Cajal S. Histolgie du systéme nerveux de l'homme et des vertébrés. Paris: A. Malone; 1911. [Google Scholar]
- Reinert KC, Dunbar RL, Gao W, Chen G, Ebner TJ. Flavoprotein autofluorescence imaging of neuronal activation in the cerebellar cortex in vivo. J Neurophysiol. 2004;92:199–211. doi: 10.1152/jn.01275.2003. [DOI] [PubMed] [Google Scholar]
- Reinert KC, Gao W, Chen G, Ebner TJ. Flavoprotein autofluorescence imaging in the cerebellar cortex in vivo. J Neurosci Res. 2007;85:3221–3232. doi: 10.1002/jnr.21348. [DOI] [PubMed] [Google Scholar]
- Rossi F, van der Want JJ, Wiklund L, Strata P. Reinnervation of cerebellar Purkinje cells by climbing fibres surviving to a subtotal lesion of the inferior olive in the adult rat. Synaptic organization on reinnervated Purkinje cells. J Comp Neurol. 1991;308:536–554. doi: 10.1002/cne.903080404. [DOI] [PubMed] [Google Scholar]
- Serra HG, Duvick L, Zu T, Carlson K, Stevens S, Jorgensen N, Lysholm A, Burright E, Zoghbi HY, Clark HB, Andresen JM, Orr HT. RORα-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell. 2006;127:697–708. doi: 10.1016/j.cell.2006.09.036. [DOI] [PubMed] [Google Scholar]
- Sotelo C. Purkinje cell ontogeny: formation and maintenance of spines. Progr Brain Res. 1978;48:149–170. doi: 10.1016/S0079-6123(08)61021-3. [DOI] [PubMed] [Google Scholar]
- Sotelo C, Hillman DE, Zamora AJ, Llinás R. Climbing fiber deafferentation: its action on Purkinje cell dendritic spines. Brain Res. 1975;98:574–581. doi: 10.1016/0006-8993(75)90374-1. [DOI] [PubMed] [Google Scholar]
- Strata P. Dendritic spines in Purkinje cells. Cerebellum. 2002;1:230–232. doi: 10.1080/147342202320883533. [DOI] [PubMed] [Google Scholar]
- Takagishi Y, Hashimoto K, Kayahara T, Watanabe M, Otsuka H, Mizoguchi A, Kano M, Murata Y. Diminished climbing fiber innervation of Purkinje cells in the cerebellum of myosin Va mutant mice and rats. Dev Neurobiol. 2007;67:909–923. doi: 10.1002/dneu.20375. [DOI] [PubMed] [Google Scholar]
- Zu T, Duvick LA, Kaytor MD, Berlinger MS, Zoghbi HY, Clark HB, Orr HT. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J Neurosci. 2004;24:8853–8861. doi: 10.1523/JNEUROSCI.2978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]